

The ensemble Stokes' shift gives the average single-molecule linewidth for quantum-confined nanomaterials.
Abstract
For quantum-confined nanomaterials, size dispersion causes a static broadening of spectra that has been difficult to measure and invalidates all-optical methods for determining the maximum photovoltage that an excited state can generate. Using femtosecond two-dimensional (2D) spectroscopy to separate size dispersion broadening of absorption and emission spectra allows a test of single-molecule generalized Einstein relations between such spectra for colloidal PbS quantum dots. We show that 2D spectra and these relations determine the thermodynamic standard chemical potential difference between the lowest excited and ground electronic states, which gives the maximum photovoltage. Further, we find that the static line broadening from many slightly different quantum dot structures allows single-molecule generalized Einstein relations to determine the average single-molecule linewidth from Stokes’ frequency shift between ensemble absorption and emission spectra.
INTRODUCTION
Electronically excited molecules and materials can store energy (1, 2), generate power (3, 4), or emit light (1, 4). For homogeneous materials such as bulk crystalline semiconductors, the thermodynamic maximum photovoltage can be determined from the rate constants for photon absorption and emission as a function of frequency (4). Determination uses a generalization of Einstein’s rate theory for absorption, stimulated emission, and spontaneous emission from gas-phase line spectra (5) to thermalized, homogeneously broadened condensed-phase spectra (6–11). In condensed phases, thermalization shifts the emission spectrum to lower frequency than the absorption spectrum, this is known as Stokes’ shift. The homogeneous generalized Einstein relations connect the equilibrium Stokes’ shift to the linewidth. These also connect the rate constants for absorption and emission to the standard chemical potential that determines the maximum photovoltage. These relations are widely applicable to impurity ions in crystals (11) and bulk crystalline semiconductors (3, 4). In contrast, the relations have appeared to be inapplicable to molecular spectra (10); we suggest that distinct isomers (7, 12) or protonation states (7) in the ionic dye solutions used to test the relations may contribute to both this apparent inapplicability (10) and the apparent inhomogeneity of some dyes in photon echo experiments (12). The rate constant measurements needed to determine the standard chemical potential are not easy (4), and the approach is not valid if spectra are broadened by static inhomogeneities (10), such as the conformational heterogeneity of photosynthetic proteins or the size distribution of quantum dots.
A quantum dot is a semiconductor nanocrystal large enough to have bulk lattice properties but small enough that three-dimensional (3D) quantum confinement increases its bandgap above the bulk bandgap (13). Nanocrystal applications benefit from narrow size and structure distributions (14). With particle-in-a-box quantum confinement, the size distribution from a synthesis generates a static bandgap distribution that is diagnostic for size dispersion. For polydisperse samples, the resulting broadening of the absorption spectrum has successfully guided synthetic efforts to reduce the size dispersion. But even with narrow size distributions a few atomic layers wide (15–18), colloidal nanocrystals have an astonishing variety of atomic structures (14), emission spectra of single quantum dots differ from each other (18), and there is no homogeneous spectrum that is the same for every quantum dot. Instead, a quantum dot sample has a static inhomogeneous distribution of bandgaps and an average single-dot lineshape for each bandgap called the dynamical lineshape (19). The dynamical lineshape arises from the motions of electrons, holes, spins, ligands, and vibrations of the atomic lattice. Every homogeneous lineshape can be correctly called a dynamical lineshape, but dynamical lineshapes are not always homogeneous. For historical reasons, dynamical lineshapes have often been called homogeneous even when single-molecule spectroscopy has shown that the sample does not have a homogeneous lineshape. Unexpectedly, nonlinear spectroscopy has shown that the dynamical emission linewidth can be modified by a capping shell (20). As a result, in contrast to previous assumption, the total static and dynamic linewidth is not a reliable gauge for comparing size dispersion between samples with narrow static size distributions.
This realization, together with the thousands of electron microscopy image analyses required to characterize a size distribution, has motivated use of nonlinear and single-molecule spectroscopies to characterize dynamical linewidths in quantum-confined nanomaterials (17–20). By comparison to size/shape distributions from electron microscopy, femtosecond 2D Fourier transform (2DFT) spectroscopy (Fig. 1) has been proven to accurately measure bandgap inhomogeneity in quantum dot samples (19). With size/shape deviations on the order of an atomic layer, the bandgap distribution is quite accurately Gaussian (19). Because absorption and emission both contribute to 2D spectra (21–23), 2D separation of static inhomogeneity provides a way to test relations between dynamical absorption and emission spectra and use dynamical spectra to measure the standard chemical potential of excited states in heterogeneous samples.
Fig. 1. The quantum dot size distribution diagonally elongates 2D spectra.
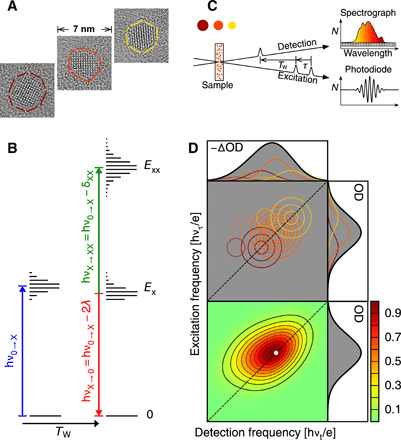
(A) Transmission electron microscopy (TEM) images show the crystal lattices of three PbS dots from the ligand-covered ensemble. Maroon, orange, and yellow outline larger, average, and smaller dots, respectively. (B) Quantum dot energy bands (ground 0, exciton X, and bi-exciton XX) and optical transitions at thermal quasi-equilibrium. Exciton absorption has frequency ν0 → X; after a sufficient waiting time Tw, exciton stimulated emission has frequency νX → 0 = ν0 → X − 2λ/h and exciton to bi-exciton absorption has frequency νX → XX = ν0 → X − δXX/h. (C) The 2D pulse sequence uses two collinear pump pulses separated by an excitation delay τ, followed, after Tw, by a noncollinear probe pulse. The change in probe spectrum is detected by a spectrograph. (D) At fixed Tw, 2D spectra are obtained by Fourier transforming detected probe spectra with respect to excitation delay τ and displayed as contour maps of optical density change as a function of excitation and detection frequency. 2D signals from three dot sizes are colored to match TEM image outlines (top). Smaller dots are excited and detected at higher frequencies, diagonally elongating the ensemble 2D spectrum (bottom).
Here, we formulate single-molecule generalized Einstein relations and present an equation for the 2D spectrum in terms of single-molecule Einstein B coefficient spectra. We record high-accuracy macroscopic 2D spectra and transform them into these Einstein B coefficient 2D spectra by removing pulse and propagation effects. We test the generalized Einstein relations on PbS quantum dot ensembles from two different synthetic procedures by developing least squares fit algorithms for 2D spectra. Dynamical absorption and emission spectra from such 2D fits provide the result we sought, a determination of the standard chemical potential change.
We were surprised to find a second remarkable result from these 2D fits. Quantum dot heterogeneity gives an ensemble-average Stokes’ shift equal to the Stokes’ shift for quantum dots with the average bandgap. This Stokes’ shift and the single-molecule generalized Einstein relations can determine the dynamical linewidth. Together, these two results prove that the dynamical linewidth and static heterogeneity can be determined from ensemble absorption and emission spectra. We validate this determination method using literature data from fluorescence correlation spectroscopy (17).
RESULTS
Single-molecule generalized Einstein relations
The Einstein coefficients are single-molecule rate coefficients that quantify strengths for transitions between two quantum mechanical levels (5, 24). Einstein implicitly assumed that degenerate quantum states within each level equilibrate rapidly. For broadened spectra, the frequency spectra of the Einstein coefficients are related after the excited band reaches internal thermal quasi-equilibrium, where equilibrium holds within each band but not necessarily between bands. For example, within a few picoseconds after absorption of light, quasi-Fermi levels are established for each electronic band in bulk semiconductors (4), and electronically excited molecules vibrationally thermalize in condensed phases (25).
We begin by defining rate coefficients for three elementary rate processes with broadband spectra. The probability per unit time that a molecule j in the ground band 0 will absorb a photon and make a transition to the excited band X is
| (1A) |
where bj,0 → X(ν, p, T) is the Einstein absorption B coefficient spectrum (absorption-b), u(ν) is the electromagnetic energy density per unit frequency ν, p is the pressure, and T is the absolute temperature. The probability per unit time that radiation will stimulate a molecule j in the excited band X to emit a photon into the same radiation mode and make a transition to the ground band 0 is
| (1B) |
where bj, X → 0(ν, p, T) is the Einstein stimulated emission B coefficient spectrum (emission-b). Last, in the absence of radiation, the probability per unit time that a molecule j in the excited band X will spontaneously emit a photon and make a transition to the ground band 0 is
| (1C) |
where ai, X → 0(ν, p, T) is the Einstein spontaneous emission A coefficient spectrum (emission-a). The total probability per unit time that a molecule j in the excited band X will emit a photon and make a transition to the ground band 0 is .
With the assumptions of Boltzmann statistics, thermal quasi-equilibrium within each band, and a constant refractive index n, the generalized Einstein relations connect absorption and emission transitions between the same two bands
| (2A) |
| (2B) |
where h is Planck’s constant, c is the speed of light in vacuum, k is Boltzmann’s constant, and is the difference in thermodynamic standard chemical potential between the excited band X and the ground band 0 for molecule j (9). is an intrinsic property of a single molecule and its environment, specified by solvent, pressure, and temperature; (26) it is independent of the standard states. The first relation is derivable from quantum electrodynamics (27) by assuming kinetic rates for spontaneous and stimulated emission in a transparent and nondispersive medium. The second relation is derivable from the first by applying detailed balance [equal forward (absorption) and reverse (total emission) rates ] to single-molecule kinetics at thermal equilibrium. Equilibrium requires both a Planck blackbody radiation spectrum inside the refractive medium [equation 301 of (28)] and a Boltzmann population ratio (26) between bands, . The vacuum Einstein relations are recovered with delta function lines bX→0(ν) = BνX→0δ(ν − νX→0) and , where gX is the degeneracy of level X. Each Einstein coefficient is equal to the frequency integral of its spectrum.
Although the relationship between absorption and emission rate coefficients is obtained from a consideration of thermal equilibrium, it is valid whenever thermal quasi-equilibrium holds within each band. Lifetime broadening precludes thermal quasi-equilibrium, so Eq. 2B does not apply to lifetime broadened Lorentzian lineshapes in atoms. It can be violated if emission originates from two or more partially equilibrated bands (for example, excitons and defects) but the violation is diagnostically useful. The frequency-dependent exponential in Eq. 2B predicts that the quasi-equilibrium stimulated emission peak will always be shifted to a lower frequency than the absorption peak and that this shift becomes evident for linewidths comparable to the thermal energy kT (the Supplementary Materials illustrate this for Gaussian lineshapes); this frequency difference between peak maxima for b-spectra is Stokes’ shift.
The difference in standard chemical potential is the difference in standard Gibbs free energy per molecule; where NA is Avogadro’s number. In reactions that produce fuels, such as photosynthesis (1, 2), it gives the driving force for the chemical equilibrium constant. At constant pressure and temperature,
| (3) |
where Δu is the change in molecular internal energy, is the change in molecular volume, and Δs is the change in molecular entropy (26). For photo-excitation, the pressure-volume term is usually negligible [based on the pressure dependence of PbS quantum dot absorption (29), changes in lattice spacing and compressibility give a shift of ≈10−6 eV at atmospheric pressure]. Standard molecular entropy increases arise from an increased number of electron/hole/spin configurations and their intraband splittings, from changes in lattice and ligand vibrations, from changes in nanocrystal volume, and from the solvent. The difference in chemical potential depends on the difference in standard chemical potential and on the population ratio (26)
| (4) |
In solid state physics, Δμ0 → X is the light-dependent difference between quasi-Fermi levels in the conduction and valence bands (4, 30). In photovoltaics, sustained nonequilibrium population of the excited state can generate a voltage V = Δμ0 → X/e, where e is the elementary charge. At thermal equilibrium, Δμ0 → X = 0 and the voltage is zero; the voltage that can be generated under saturating light (NX ∼ N0) is given by the difference in standard chemical potential.
Effects of a bandgap distribution on 1D and 2D spectra
The absorption and stimulated emission cross sections are proportional to the ensemble average of the corresponding B coefficient spectrum
| (5) |
The ensemble average denoted by angle brackets is an average over quantum dots j with different sizes and shapes, which gives rise to static inhomogeneous broadening of the absorption and emission spectra. Equation 2B is valid for single quantum dots but does not hold for ensemble-averaged quantum dot spectra because quantum dots of different size have different chemical potentials. The previously known relation (8–11) between macroscopic stimulated emission and absorption cross sections with the same form as Eq. 2B holds only for homogeneous samples. The Beer’s law absorbance or optical density spectrum for an isolated electronic transition is proportional to the absorption cross section
| (6) |
where OD is the optical density, N0 is the molecular number density in band 0, and ℓ is the sample path length. The transformation of absorbance and photoluminescence (PL) spectra into absorption and emission 〈b〉 is shown below in Materials and Methods.
The absorptive Einstein B coefficient 2D spectrum of the ensemble is an average of single-molecule 2D spectra (Fig. 1D), each of which is a 2D product of the excitation probability spectrum with the sum of transition probability spectra for the three detected transitions in Fig. 1B
| (7) |
where the pressure and temperature dependence has been suppressed and NX is the number density of the excited band X, which depends on the waiting time Tw. ντ is the excitation frequency and νt is the detection frequency. In Eq. 7, bj,0→X(ντ) is the absorption probability for the excitation step, which depletes the ground band 0 and populates the single-exciton band X. Excitation changes the optical density of the sample in three ways: depleting 0 causes a reduction in 0 → X absorption [more detected photons, positive signal bj,0→X(νt)]; populating X causes both X → 0 single exciton stimulated emission [more detected photons, positive signal bj, X→0(νt)] and X → XX exciton to bi-exciton absorption [fewer detected photons, negative signal bj, X→XX(νt)] (23). 2D spectra from each single-molecule add together to give the ensemble 2D spectrum. Fig. 1 shows how 2D spectra can separate the dynamical broadening perpendicular to the diagonal from the total broadening parallel to the diagonal, allowing a test of the generalized Einstein relation between B coefficient spectra. Furthermore, a single 2D spectrum also puts the dynamical absorption and emission b on the same intensity scale, allowing determination of the change in standard chemical potential upon excitation.
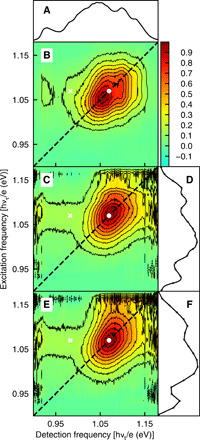
Fig. 2. Comparison between experimental and fit Einstein B coefficient 2D spectra for the partially ligand-covered PbS quantum dot sample.
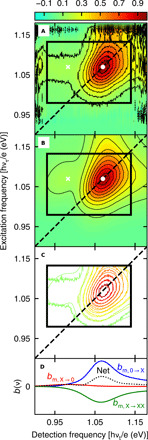
The color bar at the top is used to show signal as a fraction of maximum signal. (A) Experimental 2D spectra with black contours at 10 to 90% of maximum. The rectangle encloses the fitted region. The dashed line shows the diagonal, the white dot marks the absorption-〈b〉 maximum, and the white cross marks the emission-〈b〉 maximum. (B) Least squares fit to the 2D spectrum using eq. S9. (C) Overlaid 10 to 90% contours of the experimental (2.4% noise) and fit (smooth) 2D spectra. (D) The net signal (black dotted curve) and three dynamical contributions for the most probable quantum dot size and bandgap (m): positive reduced exciton absorption (blue curve), positive exciton stimulated emission (red curve), and negative exciton to bi-exciton absorption (green curve).
High-accuracy 2D spectra show stimulated emission and enable quantitative fitting
For 2D experiments, colloidal PbS quantum dots with a bandgap of 1.07 eV were prepared by two different air and moisture free syntheses: one sample was synthesized from lead chloride and sulfur in oleylamine (15) and capped with 3.1 oleate ligands/nm2 (covered sample); the second sample was synthesized from lead oleate and substituted thiourea (16) and capped with 1.1 oleate ligands/nm2 (part-covered sample). Figure S1 shows electron microscopy images and histograms that reveal the size and shape distribution (table S1). The transformation of experimental absorbance and PL spectra into Einstein 〈b〉 transition probability lineshapes for absorption and stimulated emission is illustrated in Materials and Methods. Thermally equilibrated 2D spectra are measured for air and moisture-free PbS quantum dots in a sealed, interferometrically stable spinning sample cell (31) that rotates to a fresh sample for every laser shot, thus eliminating contamination of the 2D spectra by photocharging (32). Without the usual measures to reduce absorption or red-shift emission (33), this enables the observation of single-exciton stimulated emission needed to test the generalized Einstein relations. For high accuracy, the 2D spectra are recorded using a set of excitation pulse pair delays obtained by step-scanning a Mach-Zehnder interferometer (fig. S2) that is actively stabilized to ±0.6 nm pathlength difference at each step. As recorded, each 2D spectrum is attenuated by the pulse spectra and sample absorption. Materials and Methods shows the transformation to an Einstein B coefficient 2D spectrum through division by pulse spectra and propagation functions.
Figure 2A shows an experimental Einstein B coefficient 2D spectrum of PbS quantum dots with partial ligand coverage at Tw= 1 ps waiting time, after intraband relaxation (34, 35). The most notable aspects of the experimental 2D spectrum in Fig. 2A are the strong stimulated emission around the white cross and the absence of the usual region with net negative signal arising from absorption by the excited state (19). These features were revealed by air-free synthesis and elimination of repetitive excitation with the spinning sample cell (31). Since stimulated emission by itself can only shift the 2D maximum to lower detection frequency, as in Fig. 1D, the slight shift of the 2D maximum to higher detection frequency (see the contours around the white dot) indicates that exciton to bi-exciton absorption lies only slightly to lower detection frequency than the depleted ground state absorption. Partial cancelation between these two overlapping and oppositely signed signals makes stimulated emission appear stronger.
An algorithm was developed to least-squares fit the 2D spectra to a model containing the three signal contributions in Fig. 1B and Eq. 7. With size/shape deviations on the order of an atomic layer on each face, the bandgap distribution is quite accurately Gaussian, as shown by transmission electron microscopy and 2D spectroscopy (19). Starting from the ensemble absorption and emission, the algorithm (flowchart in fig. S3) first finds dynamical absorption and emission by deconvolution with the static bandgap distribution [a Gaussian with standard deviation (SD) Δstatic]. The next step shifts (δXX) and broadens (by convolution with a Gaussian of SD ΔXX) the dynamical absorption to get the dynamical exciton to bi-exciton absorption. We use eq. S5 to sum these three terms with variable strengths for emission (wX → 0) and exciton to bi-exciton absorption (wX → XX) and use eq. S9 as an algorithmically convenient form for the 2D spectrum in Eq. 7. The product 2D spectrum in Eq. 7 is then ensemble-averaged over the bandgap distribution, and all five parameters are optimized. This fit always matches the ensemble absorption and stimulated emission spectra exactly—only the 2D spectrum is treated as data to be fit. The fit (Fig. 2, B and C) has a reduced chi-squared of 1.04. All five parameters are necessary (fig. S4), and the 2D width (fig. S5) provides evidence for bi-exciton broadening (Fig. 2D). Table 1 has parameters and error bars for 2D spectra, including a later waiting time and the covered sample (fig. S6).
Table 1. Best fit parameters for 2D spectra of the two PbS quantum dot samples.
Asymmetrical error bars are given in parentheses for each fit parameter and result. The change in standard chemical potential is given for quantum dots with the most probable bandgap, . For reference, bi-exciton broadening increases the exciton to bi-exciton lineshape FWHM by 12.44 meV in the best fit for the covered sample at Tw = 1 ps. The half-width at half maximum (HWHM) is used to quantify the dynamical linewidth for the 1S-1S transition because its high frequency side is overlapped by transitions to higher energy states.
| Fit parameters | Part-covered, Tw = 1 ps | Covered, Tw = 1 ps | Part-covered, Tw = 5 ps |
| Δstatic/e (meV) | 23.9 (−0.5,+0.5) | 27.4 (−0.7, +0.7) | 23.8 (−0.5, +0.5) |
| δXX/e (meV) | 7.4 (−2.8, +3.7) | 4.8 (−3.5, +9.3) | 5.8 (−2.6, +3.5) |
| ΔXX/e (meV) | 19.7 (−3.4, +3.2) | 16.3 (−7.3, +7.6) | 15.6 (−2.7, +2.9) |
| wX → 0 | 6.9 (−1.9, +2.2) | 7.2 (−4.4, +6.5) | 5.2 (−2.0, +2.1) |
| wX → XX | 49.9 (−2.8, +8.8) | 49.4 (−9.6, +20.8) | 52.5 (−5.0, +9.0) |
| Fit results | |||
| χ2 | 2901 | 6256 | 7864 |
| 1.04 | 1.99 | 2.83 | |
| HWHMdyn/e(meV) | 36.0 (−0.4, +0.4) | 42.6 (−0.6, +0.5) | 36.4 (−0.6, +0.7) |
| (eV) | 0.97 (−0.01, +0.01) | 0.97 (−0.03, +0.02) | 0.97 (−0.02, +0.01) |
| (eV) | 1.070 | 1.069 | 1.070 |
| (eV) | 0.982 | 0.985 | 0.982 |
The ensemble linewidths are dominated by dynamical broadening. It has been reported that passivating ZnS shells can reduce the dynamical linewidths for CdSe quantum dots by up to one-third (20). We find that differences in shape and ligand coverage between samples change the dynamical broadening by about one-sixth of the dynamical linewidth, increasing the dynamical half width at half maximum from 36 meV (part-covered) to 43 meV (fully covered). The two samples naturally differ in static size dispersion broadening, with Δstatic/e = 24 versus 27 meV. All remaining parameters agree within error for both samples, which is consistent with intrinsic properties of PbS quantum dots.
Where comparison is possible, agreement with prior work is good. The shift δXX/e = 7 meV of the exciton to bi-exciton transition agrees with a prior measurement using two-photon excitation (36). In bulk PbS, the valence and conduction bands both have an eightfold degeneracy when electronic momentum, angular momentum, and spin are included. The ground state, with a full valence band and empty conduction band, has only one electron-spin configuration. The exciton, with one hole in the valence band and one electron in the conduction band, has 64 = 8 × 8 electron-hole-spin configurations (37, 38). According to a widely used degeneracy model (39) with electron-hole-spin configuration splittings that are small relative to the thermal energy kBT, the relative strengths of depleted ground state absorption, exciton to bi-exciton absorption, and stimulated emission should be w0 → X : wX → XX : wX → 0= 64:49:1 in PbS quantum dots. The ground to exciton and exciton to bi-exciton absorption strengths match the predicted 64:49 ratio.
Table 1 also presents internally consistent new results. Since the Stokes’ shift generally arises from a combination of electron-hole-spin configuration splittings and exciton-phonon interactions (40), the degeneracy model must implicitly attribute the Stokes’ shift of ~4kBT to exciton-phonon coupling. However, stimulated emission is about seven times stronger than predicted by the degeneracy model. This stronger emission indicates that relaxation down to energy levels that are lowered by electron-hole-spin configuration splitting and bright in emission plays an important role in red-shifting the emission by more than the thermal energy. The splittings are a property of the exciton and may depend on the atomic structure of an individual quantum dot (41). On the basis of the above explanation of the excess emission strength from electron-hole-spin configuration splittings, we expect broadening in PbS quantum dots because of the increase from 64 electron-hole-spin configurations in the exciton band to 784 in the bi-exciton band. Such broadening of the exciton to bi-exciton transition is known in quantum wells (42). The best fit bi-exciton broadening is (ΔXX/e ~ 18 meV). The bi-exciton broadening is expected to be approximately the same as the broadening of the exciton transition from electron-hole-spin configuration splittings, so the overall dynamical linewidth is consistent with previously reported exciton-phonon couplings for vibrations of the nanocrystal lattice (35). Since the high-accuracy 2D spectra do not show a bare negative signal from the exciton to bi-exciton transition, these remarkably consistent bi-exciton parameters are a key result of the least squares fit algorithm developed here. The dynamical absorption and emission spectra are found to agree with the generalized Einstein relations; each predicts the maximum for the other within 5 meV. At the most probable bandgap, the change in standard chemical potential is eV for both samples.
Tests of the generalized Einstein relations
A second fit of the 2D spectra (flowchart in fig. S7) enforced the generalized Einstein relations as a constraint. In this second fit, the dynamical absorption and stimulated emission spectra are related by Eq. 2B, and the ensemble absorption lineshape, ensemble stimulated emission lineshape, and 2D spectrum are all data to be fit. The results are shown in Fig. 3A for the center of the static distribution. The dynamical emission lineshape is quite asymmetric, with a strong high-energy shoulder. A similar asymmetry has been reported in single-molecule PbS/CdS core/shell quantum dot emission spectra and attributed to intraband splittings (38). Calculations (37) have predicted a corresponding asymmetry in absorption. The 2D spectra reveal a weak low-energy shoulder in dynamical absorption that corresponds to the dynamical emission maximum and shows that the high-energy dynamical emission shoulder corresponds to the dynamical absorption maximum.
Fig. 3. Einstein B coefficient spectra of part-covered PbS quantum dots obtained by simultaneously fitting absorption, emission, and 2D spectra with a Gaussian static bandgap distribution while enforcing the single-molecule generalized Einstein relations.
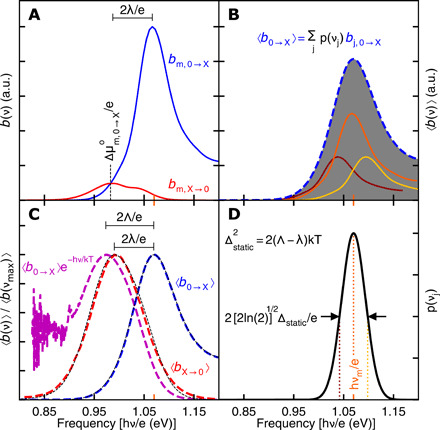
(A) The dynamical absorption-b (blue curve) and stimulated emission-b (red curve) are plotted on the same scale for the most probable bandgap hνm (orange tickmarks). Dynamical absorption and emission become equal at the most probable change in standard chemical potential () between ground and exciton bands. (B) The ensemble-averaged absorption spectrum (blue dashed curve ) and three probability-weighted dynamical absorption spectra (maroon, orange, and yellow curves) in the average. (C) The ensemble-averaged absorption (blue dashed curve) and generalized Einstein relations can test for static broadening by calculating a hypothetical homogeneous stimulated emission spectrum (purple dashed curve) that overestimates the Stokes’ shift (2Λ). Comparison to the ensemble-averaged stimulated emission spectrum (red dashed curve) and Stokes’ shift (2λ) indicates static broadening. The black dotted curves show the ensemble-averaged absorption and stimulated emission spectra calculated from the dynamical spectra, which have Stokes’ shift (2λ) marked above. (D) The Gaussian probability distribution of static bandgaps is centered on the most probable bandgap hνm with variance . Dotted vertical lines mark static bandgaps for the three dynamical absorption spectra in (B). a.u., arbitrary units.
The change in standard chemical potential, , can be found from Eq. 2B as the photon energy at which dynamical absorption and emission have equal b. This determination in Fig. 3A is possible because 2D spectroscopy puts the dynamical absorption and emission spectra on a common intensity scale (eq. S10 gives the connection to the weights in Table 1). = 0.98 eV from Fig. 3A is coincidentally equal to the dynamical emission maximum and agrees with the unconstrained fit result within error. As a point of reference, using a Gaussian linewidth with SD Δvib = 25 meV for the lattice vibrations or phonons and the three bright levels from the calculation in (38) to calculate the absorption and emission spectra of the bright exciton would give a lower emission weight of wX→0 ≈ 5 and put the standard chemical potential change about 20 meV below the emission maximum. Given the perfectly faceted quantum dots in (38), this is remarkable agreement. Compared to the photon energy at the absorption maximum, the standard chemical potential change for this three bright level model is lowered about 12 meV by lattice vibrations and ligand motions (an internal energy effect) and about 80 meV by intraband relaxation (divided almost evenly between energetic and entropic effects) among the energetically split levels of the bright exciton.
The change in standard chemical potential also affects the spontaneous emission rate, as can be seen from Eqs. 1C and 2. Using the emission weight from the 2D spectra, the calculated radiative lifetime (see Materials and Methods) for the bright exciton is about 200 ns, consistent with an initial fast PL decay in figure 3D of (17). Without any changes in the emission spectrum, the spontaneous emission rate becomes slower as the bright exciton equilibrates with the nonradiating dark exciton. After 1 μs, the PL decay has a single-exponential lifetime of about 2 μs (17). This 2-μs lifetime matches the radiative lifetime calculated for thermal quasi-equilibrium among all 64 exciton states (the levels from (38) give an emission weight wX→0 ≈ 0.7). This agreement indicates a parallel with the long radiative lifetime in PbSe quantum dots, which arises from quasi-equilibrium with dark states (37). Slowing of the radiative rate by a factor of 10 corresponds to lowering the standard chemical potential by about 50 meV as the bright exciton equilibrates with the dark exciton. In other words, the standard chemical potential of the quasi-equilibrated exciton is 50 meV lower than the standard chemical potential of the bright exciton measured here.
Using ensemble absorption and emission to determine inhomogeneity
The single-molecule generalized Einstein relations can be directly applied to ensemble spectra as a test for static inhomogeneity. Figure 3B illustrates ensemble-averaging with the bandgap distribution in Fig. 3D. Figure 3C compares the ensemble absorption and emission 〈b〉 from this constrained fit to experiment. Figure 3C also shows the ensemble emission lineshape predicted under the hypothesis of thermal quasi-equilibrium and homogeneity using Eq. 2B: 〈bX→0(ν)〉 ∝ 〈b0→X(ν)〉 exp(−hν/kT). For this hypothesis test, zero baseline levels are needed to avoid exponential amplification of background or noise over the signal. If the absorption and emission lineshapes are mutually incompatible with Eq. 2B, the incompatibility is diagnostic of partially equilibrated emission (for example, exciton emission faster than quasi-equilibrium with a defect or trap). On the other hand, if the lineshapes are compatible except for their Stokes’ shift, comparison between the hypothetical Stokes’ shift calculated from the ensemble spectra (2Λ) and the experimental Stokes’ shift (2λ) can quantify the bandgap distribution of the ensemble. Figure 3C compares this hypothetical homogeneous emission lineshape to experiment. Since the ensemble absorption is broader than dynamical absorption, this test overestimates the ensemble Stokes’ shift as 2Λ. As can be seen by comparing Fig. 3 (A and C), the observed ensemble Stokes’ shift matches the dynamical Stokes’ shift for the center of the bandgap distribution and is not appreciably altered by the static inhomogeneity. This overestimate of the Stokes’ shift provides an easily measurable, model-free signature of static inhomogeneity in an ensemble.
Simple quantitative relationships are specific to the common case of Gaussian lineshapes. For a Gaussian dynamical absorption lineshape with variance (so that Δdyn is its SD), the dynamical stimulated emission will have a Gaussian lineshape with the same width and a Stokes’ shift of 2λ that obey
| (8) |
Equation 8 is a consequence of the generalized Einstein relations for any Gaussian dynamical lineshape and says that larger dynamical linewidths necessarily arise from larger Stokes’ shifts. Neglecting variation in the Stokes’ shift with bandgap, convolution with a Gaussian static lineshape with variance gives Gaussian ensemble absorption and emission lineshapes with the same increased variance
| (9) |
Because the high-frequency side of the lowest exciton absorption typically has an asymmetrical shape caused by transitions to higher exciton states, the ensemble full width at half maximum (FWHM) for emission gives the more reliable . The experimental Stokes’ shift of 2λ can be used to calculate the dynamical lineshape variance from Eq. 8. Together, the ensemble and dynamical variances determine the static line broadening using Eq. 9.
Alternatively, the static line broadening can be determined from
| (10) |
as illustrated in Fig. 3D, which relies on Gaussian convolution in a different way. Eqs. 8 to 10 independently determine all three linewidths and can be combined to act as a consistency check for Gaussian line broadening. The variance, FWHM and Stokes’ shift also increase or decrease together for other lineshapes. Practically, Eq. 9 is useful for ensemble absorption and emission lineshapes that approximate Gaussians with the same linewidth in between their maxima, while Eq. 10 requires that 2Λ be calculated by applying Eq. 2B to a high signal to noise lineshape. For the PbS quantum dots studied here, the latter route determines (2Λ)/e = 112 meV, so that Eq. 10 gives Δstatic/e = 24 meV, which agrees with the 2D estimate within error.
We have tested these relationships using Gaussian dynamical emission spectra from the literature. The test for photon-correlation Fourier spectroscopy of PbS quantum dots (17) is shown in fig. S8, and tables S2 and S3 list the data points plotted in fig. S8. For the four smaller PbS quantum dot sizes with dynamically broadened emission spectra (17), the ensemble Stokes’ shift and ensemble emission linewidth exemplify the generalized Einstein relation result for homogeneous Gaussian lineshapes in Eq. 8. The generalized Einstein relations also reveal that four ensembles of larger size PbS quantum dots, for which photon correlation Fourier spectra were not reported in (17), have previously unrecognized static size distribution broadening.
DISCUSSION
Outlook for using the Stokes’ shift to quantify static and dynamic linewidths
For the two different PbS quantum dot samples with the same bandgap studied here, the dynamical linewidths account for over 90% (Δdyn/Δens) of the ensemble emission linewidths. However, the dynamical linewidths differ by more than 15% between the ligand-covered and part-covered samples. This comparison shows that ensemble linewidths cannot be used to assess size distributions with a width of a few atomic layers, so new methods are needed.
The single-molecule generalized Einstein relations between absorption and thermalized emission spectra of the same transition relate the Stokes’ shift between Einstein b lineshapes to the dynamical linewidth. Use of the ensemble-averaged Stokes’ shift to determine the dynamical linewidth in heterogeneous semiconductor nanomaterials is likely to be widely practical for five reasons: First, bulk semiconductors typically attain the required thermal quasi-equilibrium within a few picoseconds; second, nanomaterial emission lifetimes are usually long enough that the measured time-integrated PL is dominated by emission at thermal quasi-equilibrium; third, quantum-confined nanomaterial Stokes’ shifts are usually small enough that the overlap between absorption and emission spectra is dominated by the same transition so that the generalized Einstein relations apply; fourth, with these smaller Stokes’ shifts, static ensemble broadening increases the absorption-emission overlap to permit numerically reliable cross-estimates of ensemble absorption and emission maxima from the generalized Einstein relation as a test of the homogeneity hypothesis; fifth, the semi-infinite variety of nanomaterial structures generates inhomogeneous distributions of the static bandgap that are nearly Gaussian for the same reasons the central limit theorem generates Gaussian distributions, simplifying the ensemble-dynamical-static lineshape relationship.
This conclusion does not contradict the well-known principle that a nonlinear spectroscopic measurement is required to differentiate homogeneous and inhomogeneous broadening. The required nonlinear spectroscopic measurement in the generalized Einstein relation/Stokes’ shift method presented here is measurement of the emission spectrum. PL is a two-photon process with the great advantage that it can be easily measured on commercial spectrofluorometers.
Outlook for excited state chemical potentials
The change in standard chemical potential for creation of a bright exciton is an intrinsic property of the quantum dot, independent of surface coverage to within 10-meV experimental uncertainty. Such independence is expected for the bulk (13) but seems remarkable for a 4-nm diameter quantum dot in which about one-third of the atoms lie on the surface. With picosecond thermalization, this chemical potential controls exciton dynamics. Figure 1B illustrates a nonequilibrium decrease in the internal energy of the bright exciton as it relaxes to thermal quasi-equilibrium during the waiting time.
Within a picosecond, the 2D spectra become stationary, indicating that energetic relaxation among bright levels is complete. On the same time scale, the decay of the cross-polarized transient grating signal (34, 35) indicates complete exciton-spin relaxation for the bright levels. Together, these two observations show that the bright exciton is at internal thermal quasi-equilibrium within a picosecond, but they are silent about the dark exciton. The standard chemical potential change (or the closely related stimulated emission strength) shows that the bright exciton has not yet appreciably equilibrated with the dark exciton levels. Bright-dark exciton equilibration requires separate electron and hole angular momentum/spin relaxation. In CdSe quantum dots, electron-spin flips are reported to be over an order of magnitude slower than exciton-spin flips (43). It is not yet clear whether separate electron or hole total angular momentum relaxation in PbS quantum dots is slow enough to explain the few hundred nanosecond relaxation from the bright exciton to a more slowly emitting mixture of bright and dark excitons in thermal quasi-equilibrium with each other [in principle, defect (17) or trap states could also play a role]. However, relaxation between bright and dark excitons must be much slower than picoseconds to explain the stronger than expected single exciton emission in the 2D spectra, which arises from the bright exciton. During bright-dark equilibration, the 2D spectra would measure a nonequilibrium chemical potential; at longer waiting times, the 2D spectra would measure the standard chemical potential change for creating an exciton.
The standard chemical potential change for creation of an exciton generalizes electronic degeneracy factors to incorporate intraband splittings from quantum-confined electron-hole-spin interactions (even if greater than the thermal energy), changes in phonons, ligand, and solvent rearrangement. Although changes in quantum dot volume can affect all three terms in Eq. 3, these volume-change contributions cancel in the chemical potential (the change in chemical potential equals the maximum nonexpansion work). As can be seen from Eqs. 1C and 2, the change in standard chemical potential alters the spontaneous emission rate, which will alter thermalized (44) excitation energy transport.
2D spectroscopy allows measurement of the standard chemical potential for transient excited species present at a particular waiting time of interest. Thermalized excitation energy transport in quantum dot arrays can take place over tens of picoseconds (44), which is after the bright exciton has reached thermal quasi-equilibrium but before bright exciton relaxation toward equilibrium with the dark exciton. In these circumstances, the appropriate description of excitation energy transport uses the standard chemical potential for creation of a bright exciton.
Since the work of Förster on excited state proton transfer (45) and of Marcus on excited state charge transfer (46), photochemical equilibria have been treated using approximate estimates of standard internal energy changes from spectra. For understanding such photochemical equilibria, the necessary quantity has always been the standard free energy change, which is given by the standard chemical potential change. The use of 2D spectra to measure excited state thermodynamics through the standard chemical potential solves the previously known inhomogeneity roadblock to use of the generalized Einstein relations. It has also revealed a previously unknown problem for their use that arises from time-dependent nonequilibrium mixtures of metastable species. As seen here for PbS quantum dots, such nonequilibrium mixtures can have time-varying radiative rates that would frustrate attempts to measure their thermodynamics through spontaneous emission. 2D spectroscopy can solve this problem by measuring stimulated emission at specified waiting times. After quantum decoherence, nonequilibrium statistical mechanics and irreversible thermodynamics (47) govern reactions. 2D spectroscopic sensitivity to entropy opens up study of photochemical thermodynamics and relaxation in heterogeneous systems such as nanomaterials, polymers, glasses, and proteins. Just as femtosecond 2D spectroscopy enabled study of quantum coherent dynamics in heterogeneous systems (48–50), the Einstein B coefficient 2D spectra and single-molecule generalized Einstein relations presented here enable study of the quasi-equilibrium thermodynamics that govern the efficiency of photovoltaics and light-emitting diodes in heterogeneous materials.
Limitations
The equations presented here do not include a wavelength-dependent refractive index or allow for appreciable absorption over distances on the order of one wavelength (as occurs in metals). A quantitative determination of static inhomogeneous broadening from 1D spectra using Eqs. 8 and 9 requires a single electronic transition with Gaussian lineshapes. This excludes poly-disperse samples in which the lowest exciton peak is only a shoulder. A smaller size dispersion that is still large enough to cause a systematic increase in the Stokes’ shift with bandgap broadens ensemble absorption more than ensemble emission (the Supplementary Materials provide an estimate of this effect); in such cases, replacing convolution with a linear integral equation might improve accuracy. Because errors are amplified by the exponential, directly testing a homogeneity hypothesis with Eqs. 2B and 10 requires accurate background subtraction that yields a zero baseline for frequencies below the absorption band. This method can usefully predict an emission peak for samples in which the lowest exciton transition is only a shoulder in absorption. Eqs. 2B and 10 rely on absorbance and PL spectra that overlap each other with all emitting bands in mutual quasi-equilibrium. For example, partial equilibration between the exciton and a defect or trap state with overlapping emission can cause a diagnostically useful failure to capture one lineshape from the other, as can an inhomogeneous departure from proportionality between the ensemble-averaged spontaneous emission rate spectrum and the time-integrated PL spectrum (see the Supplementary Materials). Briefly, the 1D generalized Einstein relation/ensemble Stokes’ shift method can determine dynamical and static line broadening for high-quality samples with small size dispersion, where simply comparing the width of the first exciton peak fails. 2D spectra can resolve inhomogeneities to recover asymmetrical dynamical lineshapes and static bandgap distributions so long as the waiting time exceeds the time required to attain thermal quasi-equilibrium. Determination of the standard chemical potential of the excited band requires a 2D spectrum without cumulative excitation effects to characterize single-exciton stimulated emission.
Conclusions
Using 2D spectra to test single-molecule generalized Einstein relations led to two methods for characterizing heterogeneous materials. Single-molecule generalized Einstein relations between absorption and emission show that the Stokes’ shift between ensemble absorption and emission spectra can quantify dynamical linewidths and static disorder in heterogeneous materials. Here, differences in shape and ligand coverage can alter the intrinsic dynamical linewidth by more than the static size dispersion alters the ensemble linewidths. As a result, the Stokes’ shift estimates should replace use of the ensemble-averaged absorption linewidth for estimating size dispersion broadening. Further, the single-molecule generalized Einstein relations can be used to measure the chemical potential of excited bands by 2D spectroscopy. This measurement was made possible by using air-free quantum dot synthesis and shot-to-shot sample exchange, which led to strong stimulated emission in the 2D spectra. For determining the chemical potential, 2D experiments replace separate absolute measurements of absorption and emission, which are restricted to homogeneous materials. 2D spectra probe both absorption and emission in a single measurement that resolves inhomogeneity and is thus applicable to disordered materials. The relative absorption and emission strengths in 2D spectra provide a contact-free determination of the standard chemical potential, which gives the maximum photovoltage a disordered material can generate.
MATERIALS AND METHODS
Materials
Low bandgap PbS nanocrystals were chosen so that a reported defect state (17) lies above the lowest exciton and is not involved in the experiment. The ligand-covered sample of lead sulfide (PbS) nanocrystals with lowest electronic absorption peak at 1.074 eV was synthesized by hot injection of elemental sulfur in oleylamine into lead (II) chloride in oleylamine (15) with a modified washing procedure (51). For literature comparison, the usual assumption of spherical nanocrystals gives a nominal oleate ligand surface coverage of ~3.1/nm2. Chloride also passivates these nanocrystal surfaces. The partially ligand-covered sample of lead sulfide (PbS) nanocrystals with the lowest electronic absorbance peak at 1.072 eV was synthesized from lead oleate and a thiourea (16). Some oleate capping ligands were removed with 1,2-bis(dimethylphosphino)ethane in toluene solution for a nominal oleate surface coverage of ~1.09/nm2. Annular dark field–scanning transmission electron microscopy images of both “covered” and “part-covered” samples show faceted nanocrystals (fig. S1). Table S1 lists similar average area equivalent circular diameters of 4.29(11) and 4.27(9) nm, respectively. The covered sample has a smaller major/minor axis projection ratio (~1.14) than the part-covered sample (~1.26). Optical experiments were performed on clear solutions of colloidal nanocrystals in tetrachloroethylene.
Methods
Absorbance spectra and conversion to Einstein absorption B spectra
Linear absorbance spectra (52) of covered and part-covered PbS nanocrystals were recorded in both the spinning sample cell used for 2D spectroscopy (0.27-mm pathlength) and the cuvette (10-mm pathlength) used for PL spectroscopy. The 400 to 1500 nm wavelength range was scanned with a 1-nm wavelength sampling interval. Accurate calculation of hypothetically homogeneous emission from absorbance requires that the ensemble absorption lineshape decay to zero baseline at frequencies below the peak. Figure 4 shows the transformation from absorption and PL spectra as a function of wavelength to absorption and stimulated emission Einstein B coefficient lineshapes for the part-covered sample.
Fig. 4. Transformation of ensemble absorbance and PL spectra as a function of wavelength into absorption and stimulated emission Einstein B coefficient lineshapes as a function of frequency.
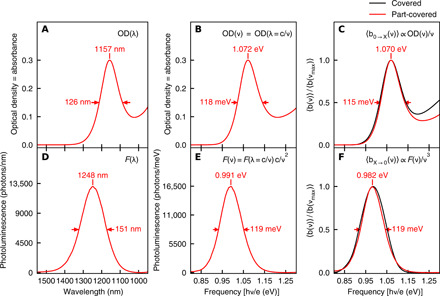
The 1S-1S maximum and FWHM are marked in each panel. (A) Optical density (= decadic absorbance) spectrum as a function of wavelength. (B) Optical density spectrum as a function of frequency. (C) Einstein B absorption coefficient lineshape. (D) PL (= spontaneous emission) spectrum as a function of wavelength. (E) Spontaneous emission spectrum as a function of frequency 〈aX → 0(ν)〉 ∝ F(ν). (F) Einstein B stimulated emission coefficient lineshape. The right column compares the ensemble-averaged Einstein B coefficient absorption lineshapes (C) and stimulated emission lineshapes (F) for the ligand-covered (black curve) and part-covered (red curve) PbS nanocrystals. Each lineshape is peak normalized.
The net absorption of light by a macroscopic sample is usually quantified with the decadic absorbance spectrum or optical density spectrum
| (11) |
where I0(λ) is the incident spectral irradiance, Iℓ(λ) is the transmitted spectral irradiance, and λ is the vacuum wavelength [not to be confused with the Stokes’ shift (2λ) in Eqs. 8 and 10. Irradiance is sometimes called intensity. The optical density wavelength spectrum is calculated as OD(λ) = − log10[Iℓ(λ)/I0(λ)]. The spectral irradiance conversion from vacuum wavelength to frequency is
| (12) |
The negative sign in the first equality arises from the reversal of axis direction between wavelength and frequency. For calculating the optical density frequency spectrum, the change of variables factor dλ/dν common to the numerator and denominator in the irradiance ratio cancels, so that the optical density frequency spectrum is
| (13) |
Slight changes in shape between Fig. 4 (A and B) arise solely from nonlinear distortion of the horizontal axis in the conversion from wavelength to frequency, ν = c/λ. The quantum dot solutions are optically clear and assumed not to scatter light, so that attenuation arises only from absorption. In the frequency range with only ground to exciton absorption, Eqs. 5 and 6 give
| (14) |
where N0 is the number density of molecules in the ground band 0 (units: molecules/volume), ℓ is the sample cell pathlength, c is the speed of light in vacuum, n is the refractive index of the sample medium, and h is Planck’s constant. Changes in shape between Fig. 4 (B and C) arise from the division by frequency when converting between optical density and absorption B coefficient spectrum in Eq. 14. For determination of the Stokes’ shift, it is not necessary to know the quantum dot concentration to obtain the Einstein 〈b〉 lineshape. The lineshape can be normalized to its maximum peak height as in Fig. 4C. Alternatively, a lineshape normalized to unit area is needed to determine relative transition strengths from the 2D spectrum (see the Supplementary Materials).
If other absorption transitions contribute to the optical density spectrum, the absorption for the photoluminescent transition can be obtained from the optical density spectrum using an experimental procedure from (53). At a minimum, experimental measurement of the ensemble Stokes’ shift (2λ) requires a maximum in this lineshape, although determination of 2(Λ − λ) in Eq. 10 will be distorted if the minimum on the high-frequency side is not separated from the maximum by the width of the static inhomogeneous lineshape. A zero baseline below the bandgap is required for use of Eq. 10.
PL spectra and conversion to Einstein stimulated emission B spectra
Steady-state PL spectra of covered and part-covered PbS nanocrystals were recorded on diluted nanocrystal solutions in tetrachloroethylene using an 850-nm excitation light source with 30-nm FWHM bandwidth (54). The part-covered quantum dot samples were diluted in tetrachloroethylene to ODmax(1S ‐ 1S) = 0.27 at the bandgap in the 10-mm cuvette (the covered sample had ODmax(1S ‐ 1S) = 0.20). PL quantum yields were 35% (ligand-covered) and 15% (part-covered). PL spectra were detected in a perpendicular or side-view geometry and recorded at a wavelength sampling interval of 5 nm; the spectrum from a nominally identical cuvette containing tetrachloroethylene was subtracted as a background.
After accounting for wavelength-dependent spectrograph sensitivity, the time-integrated emission spectrum, Fdet(λ), has units of photons per wavelength. The PL spectrum is then corrected for the inner-filter, self-absorption, or reabsorption effect (52). Assuming a narrow excitation beam in the center of the square cell, the perpendicularly detected emission spectrum is related to the true emission spectrum by equation 37 of (55)
| (15) |
Self-absorption red-shifts the detected PL maximum from its true position by ~4 to 5 meV. The corrected PL wavelength spectrum on the left-hand side of Eq. 15 is shown in Fig. 4D.
The time-integrated PL frequency spectrum is an irradiance spectrum and must be converted from wavelength to frequency using Eq. 12, so that
| (16) |
The PL spectrum on the left-hand side of Eq. 16 has units of photons per frequency. Changes in shape between Fig. 4 (D and E) arise from both the nonlinear distortion of the horizontal axis (which also occurs between Fig. 4, A and B) and the frequency-dependent change of variables factor between wavelength and frequency in Eq. 16.
For homogeneous materials, rapid thermal quasi-equilibrium implies both that the emission spectrum is independent of the excitation wavelength (56) and that the Einstein A spontaneous emission spectrum is proportional to the time-integrated PL spectrum
| (17) |
The requirement of thermal quasi-equilibrium excludes partially equilibrated emission from excitons and defect states not at thermal quasi-equilibrium with each other, but quasi-equilibrium emission from either state alone will obey a generalized Einstein relation with its absorption. Heterogeneous materials require different considerations, addressed in the Supplementary Material, to guarantee that the ensemble average is undistorted in the time-integrated spectrum so that 〈aj, X → 0(ν, p, T)〉j ∝ F(ν), which combined with Eq. 2A gives
| (18) |
Equation 2A shows that the shape of a spontaneous emission spectrum is distorted by a ν3 factor that does not appear in stimulated emission spectra. Changes in shape between Fig. 4 (E and F) arise from the resulting frequency-dependent conversion from spontaneous to stimulated emission lineshapes in Eq. 18. A zero baseline is needed on the high-frequency side of this lineshape if it is used in the generalized Einstein relation to predict the absorption lineshape.
The radiative lifetime
The spontaneous emission rate is given by Eq. 1C. Substituting the stimulated emission b-spectrum for the spontaneous emission a-spectrum using Eq. 2A gives
| (19) |
(The pressure and temperature arguments are suppressed in this subsection). Using the integrated Einstein B coefficient and area normalized lineshape , Eq. 19 can be rewritten as
| (20) |
Using Eq. 5, the integrated Einstein B coefficient can be rewritten as
| (21) |
where Δν0 → X is the absorption linewidth and is the absorption cross section maximum at ν0 → X. The first approximation becomes an equality for homogeneously broadened spectra, in which case inserting Eq. 21 into Eq. 20 generalizes the Strickler-Berg relation (7) by eliminating the Condon approximation. Using both approximations gives a simple estimate of the radiative rate
| (22) |
where the ratio of B coefficients in Eq. 21 has been replaced by the corresponding ratio of weights from Table 1. Using the absorption linewidth from Fig. 4C, = 4.18 × 10−20 m2 calculated from the molar extinction coefficient in (57), a solvent refractive index of n = 1.5, and the frequencies from Table 1, the simple estimate gives a radiative lifetime of 2 × 10−7 s for the bright exciton at internal thermal quasi-equilibrium. The ratio in brackets, which is not included in the Strickler-Berg relation (7), extends the radiative lifetime by about an order of magnitude. The actual lifetime can be shortened through quenching or lengthened through equilibrium with unquenched dark states.
2D spectra and conversion to Einstein B 2D spectra
A femtosecond Ti:sapphire regenerative amplifier operating at 1-kHz repetition rate pumps a noncollinear optical parametric amplifier (58) to generate the femtosecond short-wave infrared pulses used for 2D spectroscopy. The pulses are compressed down to ~16 fs by a deformable mirror grating compressor using an adaptive algorithm (58, 59). The pulses used for this experiment have energies of 3.6 μJ with a stability better than 0.4%. 2DFT spectra were recorded using the pump-probe geometry with the 2D spectrometer shown in fig. S2. The 2D spectrometer incorporates an actively stabilized Mach-Zehnder interferometer to generate the excitation pump-pulse pair (pulses a and b) with a delay, τ. The interferometer achieves a pathlength difference stability of less than ±0.6 nm. A noncollinear detection probe pulse c is used to measure the change in absorption after a waiting time Tw (60–62). The signal is modulated only in amplitude as a function of the time delay between the pump-pulse pair τ, which results in purely absorptive 2D spectra.
Nonlinear optical experiments on the sample of PbS nanocrystals were performed using a custom-built spinning sample cell (31). The sample temperature was 291 K under the ~84 kPa ambient atmospheric pressure in Boulder, CO. The optical density of the sample solutions was adjusted to around 0.3 in ~270-μm pathlength. The spinning sample cell keeps the sample air-free with ultra high vacuum compatible seals and completely exchanges the sample between laser shots. Using the beam diameter of 100 μm, pulse energy of 10.2 nJ for each pump pulse in the pump pulse pair, pulse energy of 10.2 nJ for the probe pulse at the sample position, pulse spectrum, and the estimated frequency-dependent extinction coefficient (57), the average excitation probability was calculated to be ~9%, so over 90% of the signal arises from singly excited quantum dots.
The pump pulse-pair interferogram is recorded during the 2D scan and Fourier transformed to retrieve the pump pulse-pair spectrum Iab(ν) (the absolute value of the complex-valued Fourier transform) and the spectral phase offset between pump pulses (its phase) (63). This also allows phase correction of the 2D spectra for both the Mach-Zehnder interferometer phase difference and the delay sampling offset without any adjustable parameters (see below). For each 2D spectrum, the waiting time Tw between the probe pulse c and the last pulse of the pump-pulse pair (a or b) is fixed. The probe pulse, c, and the signal are coupled into a single-mode fiber entering a spectrograph with an InGaAs detector array. The interference between the signal and probe pulse c is directly recorded as a function of the detection wavelength λt. The probe + signal interference spectrum is corrected for the wavelength sensitivity of the spectrograph to generate spectra with units of photons per pixel. The change of variables transformation in Eq. 12 generates probe + signal interference frequency spectra with units of photons per frequency. The excitation frequency (ντ) axis is indirectly obtained by Fourier transforming the array of directly detected probe spectrum over a two-sided scan from positive to negative τ (60, 64). By using the 1D procedure for two-sided interferograms (63) at each detection frequency, the 2D spectrum is phase-corrected with the excitation frequency-dependent spectral phase offset between pump pulses obtained from the pump pulse-pair interferogram.
2D spectra of PbS quantum dots at waiting times from Tw = 0 to 500 fs evolve rapidly as a function of the waiting time. In particular, the stimulated emission Stokes’ shift develops during this range of waiting times. Waiting times of 1 and 5 ps were chosen for this study because the shape of the 2D spectrum appeared stationary and because a 1-ps waiting time exceeds the reported exciton-spin relaxation times for similarly sized PbS quantum dots (34, 35), so the bright exciton was expected to be in thermal quasi-equilibrium.
In the partially collinear geometry shown in Fig. 1C, the experimentally measured 2DFT spectra with units of photons per (frequency)2 are attenuated 2D spectra, denoted by in (65). Attenuated 2D spectra are distorted by the photon number spectra of the excitation and detection pulses as they propagate through and are absorbed by the sample. After all coherence has decayed, the experimental 2D spectra can be exactly transformed to a dimensionless Einstein B representation that eliminates the propagation and absorptive distortions and the distortions from the pulse spectra
| (23) |
where OD(ν) is the linear optical density (decadic absorbance) spectrum of the sample from Eq. 13, and Iab and Ic are pulse photon number spectra for the pump pulse-pair and the probe pulse, respectively (dimensions = photons per frequency) at the sample entrance. The term on the first line of the right-hand side of Eq. 23 transforms the 2D spectrum along the excitation frequency axis to remove the filtering effect of the pump pulse spectrum, including the attenuation of the pump pulse as it propagates through the sample. This attenuation factor is precisely division by the average of the pump pulse spectrum over the sample length (64–66). The term on the second line of the right-hand side of Eq. 23 transforms the 2D spectrum along the detection frequency axis to remove the combined filtering effect of the probe pulse spectrum, probe attenuation, and signal attenuation along the detection axis. Since attenuation of the probe has the same effect as attenuation of the signal, this attenuation factor is precisely division by the transmitted probe spectrum (64–66). [An equivalent last step for processing 2D spectra recorded with the fully noncollinear geometry would require division by the electric field spectra of separate pulses c (probe) and d (detection)—most 2D spectra are shown with only the latter division and thus depend on both the pulse spectra and the sample properties.] The resulting 2D spectrum on the left-hand side of Eq. 23 is called the Einstein B coefficient representation and goes beyond the ideal representation of 2D spectra (66) in dividing the ideal 2D spectrum by the pulse spectra at the sample entrance. Like the 1D absorption cross section or molar extinction coefficient, the Einstein B 2D spectrum is a microscopic property of the sample that is independent of the macroscopic pulse spectra and sample absorbance used to record it.
After this transformation, the 2D spectra can be modeled using only the Einstein b spectra in Eq. 7. An example 2D spectrum before and after conversion to the Einstein B representation is shown in Fig. 5. Division by the low-amplitude wings of the pulse spectra eventually diverges, so the data can only be fit within the region where the noise does not blow up appreciably. For the excitation frequency, the boundaries of this region are chosen as the half maxima of the pump pulse spectrum. For the detection frequency, the boundaries are chosen as the high-energy half maximum and the low-energy quarter maximum of the probe pulse spectrum. This unequal boundary (marked in Fig. 2) is due to the differences in resolution between the two axes and the contribution of emission signals along only the detection axis. The Einstein B 2D spectra are filtered outside this boundary region to minimize visual distraction in Figs. 2 and 5.
Fig. 5. Transformation of experimental attenuated 2D spectra into Einstein B 2D spectra.
(A) Probe or detection pulse spectrum measured with the InGaAs spectrograph. (B) Real-valued 2D correlation spectrum of the part-covered sample at 1-ps waiting time. This attenuated 2D spectrum is filtered by the pulse spectra and distorted by sample absorption. (C) Einstein B 2D spectrum from Eq. 23 using division by the probe pulse spectrum along both frequency axes. (D) Transmitted probe pulse spectrum with frequency sampling points corresponding to sampling points of the 2D spectrum marked as circles. (E) Einstein B 2D spectrum from Eq. 23 using division by the spectrograph probe pulse spectrum along the detection axis and the interferogram-detected pump pulse-pair spectrum along the excitation axis. (F) Excitation pulse-pair spectrum from the Fourier transform of the excitation pulse-pair interferogram collected during the 2D experiment – frequency sampling points (circles) match the sampling points of the 2D spectrum. White dots on 2D spectra mark the 1S-1S absorption lineshape maximum; white diagonal crosses mark the 1S-1S stimulated emission lineshape maximum. Black contours are at 10% intervals and color changes are at 1% intervals (color bar at top right).
The pulse spectrum associated with each axis must be used when converting the attenuated representation of the 2D spectrum into the Einstein B 2D photon number spectrum. The excitation frequency axis is indirectly measured through the Fourier transform, whereas the detection frequency axis is directly resolved using the spectrograph. Unlike the excitation axis intensities, which only depend on accurate time delay control of the pump pulse pair, the detection axis intensities depend on coupling into the single-mode fiber in a consistent manner. Fine differences between pump and probe paths in the 2D interferometer make their pulse spectra slightly different. The spectra also differ slightly between the two pump arms of the Mach-Zehnder interferometer, as discussed in (67). Using probe pulse spectra to approximate pump spectra in Eq. 23 can generate artifacts in the Einstein B representation of the experimental 2D spectrum. As shown in Fig. 5C, this approximation causes an apparent kink across the 2D spectrum near (hντ/e)= 1.116 eV excitation. Figure 5E shows the Einstein B 2D photon number spectrum calculated using the pump pulse-pair spectrum Iab(ντ) from the Fourier transform of the pump pulse-pair interferogram measured during the 2D scan. The 2D spectrum is obviously smoother when the pump pulse-pair spectrum is used for division along the excitation frequency axis. This smoother 2D spectrum also leads to a quantitatively better model fit; the optimal reduced chi squared is =1.56 when approximating the pump pulse-pair spectrum by the probe pulse spectrum versus 1.04 when using the pump pulse-pair spectrum.
Acknowledgments
We thank J. T. Hynes, A. Nozik, R. Skodje, and D. H. Son for commenting on draft manuscripts and J. C. Johnson (NREL) for measuring the PL spectra. Funding: This material is based upon work of J.R. and D.M.J. supported by the Air Force Office of Scientific Research under AFOSR award no. FA9550-18-1-0211 and by the NSF under award number CHE-1800523, work of D.B. and S.D.P. supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under award number DE-FG02-07ER15912, and work of I.R. and J.S.O. supported by the NSF under award number CHE-1710352. Author contributions: D.M.J. and J.S.O. conceptualized the comparison study; D.B. and I.R. synthesized and characterized quantum dots; S.D.P., J.R., and D.B. performed 2D experiments; J.R. analyzed 2D spectra; D.B. and D.M.J. checked the ensemble generalized Einstein relation for lineshapes; D.M.J. and J.R. determined inhomogeneity from absorption and emission; J.R. and D.M.J. drafted the manuscript with contributions from all co-authors; all authors reviewed and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabf4741/DC1
REFERENCES AND NOTES
- 1.Ross R. T., Calvin M., Thermodynamics of light emission and free-energy storage in photosynthesis. Biophys. J. 7, 595–614 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.R. E. Blankenship, Molecular Mechanisms of Photosynthesis (Blackwell Science Ltd., 2002). [Google Scholar]
- 3.Shockley W., Queisser H. J., Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 32, 510–519 (1961). [Google Scholar]
- 4.P. Würfel, U. Würfel, Physics of Solar Cells (Wiley-VCH, ed. 3, 2016). [Google Scholar]
- 5.A. Einstein, in Sources of Quantum Mechanics, B. L. van der Waerden, Ed. (Dover, 1967), pp. 63–77. [Google Scholar]
- 6.Stepanov B. I., A universal relation between the absorption and luminescence spectra of complex molecules. Sov. Phys.-Dokl. 2, 81–84 (1957). [Google Scholar]
- 7.Strickler S. J., Berg R. A., Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 37, 814–822 (1962). [Google Scholar]
- 8.McCumber D. E., Einstein relations connecting broadband emission and absorption spectra. Phys. Rev. 136, A954–A957 (1964). [Google Scholar]
- 9.Ross R. T., Radiative lifetime and thermodynamic potential of excited states. Photochem. Photobiol. 21, 401–406 (1975). [Google Scholar]
- 10.Van Metter R. L., Knox R. S., On the relation between absorption and emission spectra of molecules in solution. Chem. Phys. 12, 333–340 (1976). [Google Scholar]
- 11.Band Y. B., Heller D. F., Relationships between the absorption and emission of light in multilevel systems. Phys. Rev. A 38, 1885–1895 (1988). [DOI] [PubMed] [Google Scholar]
- 12.Park S., Joo T., Diffractive optics based three-pulse photon echo peak shift studies of spectral diffusion in polar liquids: Evidence for long lived frequency correlations. J. Chem. Phys. 131, 164508 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Hagfeldt A., Graetzel M., Light-induced redox reactions in nanocrystalline systems. Chem. Rev. 95, 49–68 (1995). [Google Scholar]
- 14.Kim B. H., Heo J. H., Kim S., Reboul C. F., Chun H., Kang D., Bae H., Hyun H., Lim J., Lee H., Han B., Hyeon T., Alivisatos A. P. A., Ercius P., Elmlund H., Park J. P., Critical differences in 3D atomic structure of individual ligand-protected nanocrystals in solution. Science 368, 60–67 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Weidman M. C., Beck M. E., Hoffman R. S., Prins F., Tisdale W. A., Monodisperse, air-stable PbS nanocrystals via precursor stoichiometry control. ACS Nano 8, 6363–6371 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Hendricks M. P., Campos M. P., Cleveland G., Jen-La Plante I., Owen J. S., A tunable library of substituted thiourea precursors to metal sulfide nanocrystals. Science 348, 1226–1230 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Caram J. R., Bertram S. N., Utzat H., Hess W. R., Carr J. A., Bischof T. S., Beyler A. P., Wilson M. W. B., Bawendi M. G., PbS nanocrystal emission is governed by multiple emissive states. Nano Lett. 16, 6070–6077 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Dong Y., Qiao T., Kim D., Parobek D., Rossi D., Son D. H., Precise control of quantum confinement in cesium lead halide perovskite quantum dots via thermodynamic equilibrium. Nano Lett. 18, 3716–3722 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Park S. D., Baranov D., Ryu J., Cho B., Halder A., Seifert S., Vajda S., Jonas D. M., Bandgap inhomogeneity of a PbSe quantum dot ensemble from two-dimensional spectroscopy and comparison to size inhomogeneity from electron microscopy. Nano Lett. 17, 762–771 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Cui J., Beyler A. P., Coropceanu I., Cleary L., Avila T. R., Chen Y., Cordero J. M., Heathcote S. L., Harris D. K., Chen O., Cao J., Bawendi M. G., Evolution of the single-nanocrystal photoluminescence linewidth with size and shell: Implications for exciton−phonon coupling and the optimization of spectral linewidths. Nano Lett. 16, 289–296 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Hybl J. D., Albrecht A. W., Gallagher Faeder S. M., Jonas D. M., Two-dimensional electronic spectroscopy. Chem. Phys. Lett. 297, 307–313 (1998). [Google Scholar]
- 22.Mukamel S., Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 51, 691–729 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Jonas D. M., Two-dimensional femtosecond spectroscopy. Annu. Rev. Phys. Chem. 54, 425–463 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Hilborn R. C., Einstein coefficients, cross sections, f values, dipole moments, and all that. Am. J. Phys. 50, 982–986 (1982). [Google Scholar]
- 25.Balevičius V. Jr., Wei T., Di Tommaso D., Abramavicius D., Hauer J., Polívka T., Duffy C. D. P., The full dynamics of energy relaxation in large organic molecules: From photo-excitation to solvent heating. Chem. Sci. 10, 4792–4804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.J. G. Kirkwood, I. Oppenheim, Chemical Thermodynamics (McGraw-Hill, 1961). [Google Scholar]
- 27.C. Cohen-Tannoudji, J. Dupont-Roc, G. Grynberg, Atom-Photon Interactions: Basic Processes and Applications (Wiley-Interscience, 1992). [Google Scholar]
- 28.M. Planck, The Theory of Heat Radiation (Dover Press, ed. Translation of the Second (1912) German Edition, 1959). [Google Scholar]
- 29.Bian K., Richards B. T., Yang H., Bassett W., Wise F. W., Wang Z., Hanrath T., Optical properties of PbS nanocrystal quantum dots at ambient and elevated pressure. Phys. Chem. Chem. Phys. 16, 8515–8520 (2014). [DOI] [PubMed] [Google Scholar]
- 30.C. Kittel, H. Kroemer, Thermal Physics (W. H. Freeman, ed. 2, 1980). [Google Scholar]
- 31.Baranov D., Hill R. J., Ryu J., Park S. D., Huerta-Viga A., Carollo A. R., Jonas D. M., Interferometrically stable, enclosed, spinning sample cell for spectroscopic experiments on air-sensitive samples. Rev. Sci. Instrum. 88, 014101 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Qin W., Guyot-Sionnest P., Evidence for the role of holes in blinking: Negative and oxidized CdSe/CdS dots. ACS Nano 6, 9125–9132 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Fan F., Voznyy O., Sabatini R. P., Bicanic K. T., Adachi M. M., McBride J. R., Reid K. R., Park Y.-S., Li X., Jain A., Quintero-Bermudez R., Saravanapavanantham M., Liu M., Korkusinski M., Hawrylak P., Klimov V. I., Rosenthal S. J., Hoogland S., Sargent E. H., Continuous-wave lasing in colloidal quantum dot solids enabled by facet-selective epitaxy. Nature 544, 75–79 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Johnson J. C., Gerth K. A., Song Q., Murphy J. E., Nozik A. J., Scholes G. D., Ultrafast exciton fine structure relaxation dynamics in lead chalcogenide nanocrystals. Nano Lett. 8, 1374–1381 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Masia F., Langbein W., Moreels I., Hens Z., Borri P., Exciton dephasing in lead sulfide quantum dots by X-point phonons. Phys. Rev. B 83, 201309 (2011). [Google Scholar]
- 36.Gesuele F., Sfeir M. Y., Koh W. K., Murray C. B., Heinz T. F., Wong C. W., Ultrafast supercontinuum spectroscopy of carrier multiplication and biexcitonic effects in excited states of PbS quantum dots. Nano Lett. 12, 2658–2664 (2012). [DOI] [PubMed] [Google Scholar]
- 37.An J. M., Franceschetti A., Zunger A., The excitonic exchange splitting and radiative lifetime in PbSe quantum dots. Nano Lett. 7, 2129–2135 (2007). [Google Scholar]
- 38.Hu Z., Kim Y., Krishnamurthy S., Avdee I. D., Nestoklon M. O., Singh A. S., Malko A. V., Goupalov S. V., Hollingsworth J. A., Htoon H., Intrinsic exciton photophysics of PbS quantum dots revealed by low-temperature single nanocrystal spectroscopy. Nano Lett. 19, 8519–8525 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Trinh M. T., Sfeir M. Y., Choi J. J., Owen J. S., Zhu X., A hot electron−hole pair breaks the symmetry of a semiconductor quantum dot. Nano Lett. 13, 6091–6097 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Liptay T. J., Marshall L. F., Rao P. S., Ram R. J., Bawendi M. G., Anomalous Stokes shift in CdSe nanocrystals. Phys. Rev. B 76, 155314 (2007). [Google Scholar]
- 41.Poddubny A. N., Nestoklon M. O., Goupalov S. V., Anomalous suppression of valley splittings in lead salt nanocrystals without inversion center. Phys. Rev. B 86, 035324 (2012). [Google Scholar]
- 42.Langbein W., Hvam J. M., Biexcitonic bound and continuum states of homogeneously and inhomogeneously broadened exciton resonances. Phys. Status Solidi A 190, 167–174 (2002). [Google Scholar]
- 43.Ghosh T., Dehnel J., Fabian M., Lifshitz E., Baer R., Ruhman S., Spin blockades to relaxation of hot multiexcitons in nanocrystals. J. Phys. Chem. Lett. 10, 2341–2348 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Azzaro M. S., Le A. K., Wang H., Roberts S. T., Ligand-enhanced energy transport in nanocrystal solids viewed with two-dimensional electronic spectroscopy. J. Phys. Chem. Lett. 10, 5602–5608 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Förster T., Fluoreszenzspektrum und Wasserstoffionen-konzentration. Naturwissenschaften 36, 186–187 (1949). [Google Scholar]
- 46.Marcus R. A., Relation between charge transfer absorption and fluorescence spectra and the inverted region. J. Phys. Chem. 93, 3078–3086 (1989). [Google Scholar]
- 47.S. R. de Groot, P. Mazur, Non-Equilibrium Thermodynamics (Dover, 1984). [Google Scholar]
- 48.Scholes G. D., Fleming G. R., Chen L. X., Aspuru-Guzik A., Buchleitner A., Coker D. F., Engel G. S., van Grondelle R., Ishizaki A., Jonas D. M., Lundeen J. S., McCusker J. K., Mukamel S., Ogilvie J. P., Olaya-Castro A., Ratner M. A., Spano F. C., Whaley K. B., Zhu X., Utilizing coherence to enhance function in chemical and biophysical systems. Nature 543, 647–656 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Jonas D. M., Vibrational and nonadiabatic coherence in 2D electronic spectroscopy, the Jahn-Teller effect, and energy transfer. Annu. Rev. Phys. Chem. 69, 327–352 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Cao J., Cogdell R. J., Coker D. F., Duan H.-G., Hauer J., Kleinekathöfer U., Jansen T. L. C., Mančal T., Miller R. J. D., Ogilvie J. P., Prokhorenko V. I., Renger T., Tan H.-S., Tempelaar R., Thorwart M., Thyrhaug E., Westenhoff S., Zigmantas D., Quantum biology revisited. Sci. Adv. 6, eaaz4888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baranov D., Lynch M. J., Curtis A. C., Carollo A. R., Douglass C. R., Mateo-Tejada A. M., Jonas D. M., Purification of oleylamine for materials synthesis and spectroscopic diagnostics for trans isomers. Chem. Mater. 31, 1223–1230 (2019). [Google Scholar]
- 52.J. D. Ingle, S. R. Crouch, Spectrochemical Analysis (Prentice-Hall, 1988). [Google Scholar]
- 53.Ketskeméty I., Dombi J., Horvai R., Fluoreszenzemission, Absorption und Temperaturstrahlung von Lösungen. Ann. Phys. 463, 342–352 (1961). [Google Scholar]
- 54.Semonin O. E., Johnson J. C., Luther J. M., Midgett A. G., Nozik A. J., Beard M. C., Absolute photoluminescence quantum yields of IR-26 Dye, PbS, and PbSe quantum dots. J. Phys. Chem. Lett. 1, 2445–2450 (2010). [Google Scholar]
- 55.Crosby G. A., Demas J. N., Measurement of photoluminescence quantum yields. Review. J. Phys. Chem. 75, 991–1024 (1971). [Google Scholar]
- 56.Kennard E. H., On the interaction of radiation with matter and on fluorescent exciting power. Phys. Rev. 28, 672–683 (1926). [Google Scholar]
- 57.Moreels I., Lambert K., Smeets D., De Muynck D., Nollet T., Martins J. C., Vanhaecke F., Vantomme A., Delerue C., Allan G., Hens Z., Size-dependent optical properties of colloidal PbS quantum dots. ACS Nano 3, 3023–3030 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Brida D., Bonora S., Manzoni C., Marangoni M., Villoresi P., De Silvestri S., Cerullo G., Generation of 8.5-fs pulses at 1.3 μm for ultrabroadband pump-probe spectroscopy. Opt. Express 17, 12510–12515 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Zeek E., Maginnis K., Backus S., Russek U., Murnane M., Mourou G., Kapteyn H., Vdovin G., Pulse compression by use of deformable mirrors. Opt. Lett. 24, 493–495 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Gallagher Faeder S. M., Jonas D. M., Two-dimensional electronic correlation and relaxation spectra: Theory and model calculations. J. Phys. Chem. A 103, 10489–10505 (1999). [Google Scholar]
- 61.Grumstrup E. M., Shim S.-H., Montgomery M. A., Damrauer N. H., Zanni M. T., Facile collection of two-dimensional electronic spectra using femtosecond pulse-shaping technology. Opt. Express 15, 16681–16689 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Fuller F. D., Ogilvie J. P., Experimental implementations of two-dimensional Fourier transform electronic spectroscopy. Annu. Rev. Phys. Chem. 66, 667–690 (2015). [DOI] [PubMed] [Google Scholar]
- 63.R. J. Bell, Introductory Fourier Transform Spectroscopy (Academic Press, 1972). [Google Scholar]
- 64.Hybl J. D., Albrecht Ferro A., Jonas D. M., Two-dimensional Fourier transform electronic spectroscopy. J. Chem. Phys. 115, 6606–6622 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Yetzbacher M. K., Belabas N., Kitney K. A., Jonas D. M., Propagation, beam geometry, and detection distortions of peak shapes in two-dimensional Fourier transform spectra. J. Chem. Phys. 126, 044511 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Spencer A. P., Li H., Cundiff S. T., Jonas D. M., Pulse propagation effects in optical 2D Fourier-transform spectroscopy: Theory. J. Phys. Chem. A 119, 3936–3960 (2015). [DOI] [PubMed] [Google Scholar]
- 67.J. Ryu, thesis, University of Colorado, Boulder (2018). [Google Scholar]
- 68.Anderson N. C., Hendricks M. P., Choi J. J., Owen J. S., Ligand exchange and the stoichiometry of metal chalcogenide nanocrystals: Spectroscopic observation of facile metal-carboxylate displacement and binding. J. Am. Chem. Soc. 135, 18536–18548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segets D., Lucas J. M., Klupp Taylor R. N., Scheele M., Zheng H., Alivisatos A. P., Peukert W., Determination of the quantum dot band gap dependence on particle size from optical absorbance and transmission electron microscopy measurements. ACS Nano 6, 9021–9032 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Ihly R., Tolentino J., Liu Y., Gibbs M., Law M., The photothermal stability of PbS quantum dot solids. ACS Nano 5, 8175–8186 (2011). [DOI] [PubMed] [Google Scholar]
- 71.M. Chemnitz, thesis, Friedrich-Schiller Universität Jena (2019).
- 72.Micic O. I., Cheong H. M., Fu H., Zunger A., Sprague J. R., Mascarenhas A., Nozik A. J., Size-dependent spectroscopy of InP quantum dots. J. Phys. Chem. B 101, 4904–4912 (1997). [Google Scholar]
- 73.T. Förster, in Biological Physics, E. V. Mielczarek, E. Greenbaum, R. S. Knox, Eds. (American Institute of Physics, 1993), pp. 148–160. [Google Scholar]
- 74.T. Förster, in Modern Quantum Chemistry, O. Sinanoğlu, Ed. (Academic Press Inc., 1965), vol. III, pp. 93–137. [Google Scholar]
- 75.Braslavsky S. E., Fron E., Rodríguez H. B., San Román E., Scholes G. D., Schweitzer G., Valeur B., Wirz J., Pitfalls and limitations in the practical use of Förster’s theory of resonance energy transfer. Photochem. Photobiol. Sci. 7, 1444–1448 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Hill R. J., Courtney T. L., Park S. D., Jonas D. M., Lightweight hollow rooftop mirrors for stabilized interferometry. Opt. Eng. 52, 105103–105103 (2013). [Google Scholar]
- 77.Jones D. J., Potma E. O., Cheng J.-x., Burfeindt B., Pang Y., Ye J., Xie X. S., Synchronization of two passively mode-locked, picosecond lasers within 20 fs for coherent anti-Stokes Raman scattering microscopy. Rev. Sci. Instrum. 73, 2843–2848 (2002). [Google Scholar]
- 78.R. N. Bracewell, The Fourier Transform and its Applications (McGraw-Hill series in Electrical and Computer Engineering Circuits and Systems, McGraw Hill, ed. 3, 2000). [Google Scholar]
- 79.Roberts S. T., Loparo J. J., Tokmakoff A., Characterization of spectral diffusion from two-dimensional line shapes. J. Chem. Phys. 125, 084502 (2006). [DOI] [PubMed] [Google Scholar]
- 80.P. A. Jansson, Deconvolution: With Applications in Spectroscopy (Academic Press, 1984). [Google Scholar]
- 81.Choi Y., Sim S., Lim S. C., Lee Y. H., Choi H., Ultrafast biexciton spectroscopy in semiconductor quantum dots: Evidence for early emergence of multiple-exciton generation. Sci. Rep. 3, 3206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.P. R. Bevington, Data Reduction and Error Analysis for the Physical Sciences (McGraw-Hill, ed. 3, 2003). [Google Scholar]
- 83.A. Rohatgi, “WebPlotDigitizer” version 4.4 (2020); https://automeris.io/WebPlotDigitizer.
- 84.Mukamel S., Femtosecond optical spectroscopy: A direct look at elementary chemical events. Annu. Rev. Phys. Chem. 41, 647–681 (1990). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabf4741/DC1