

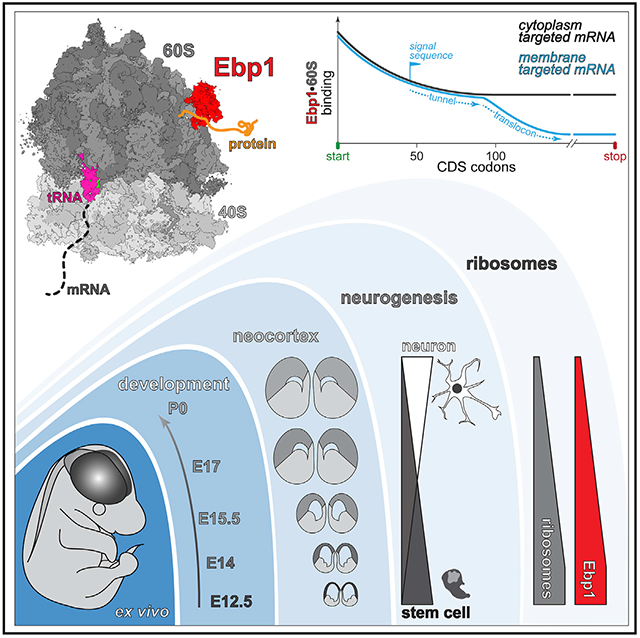
SUMMARY
Protein synthesis must be finely tuned in the developing nervous system as the final essential step of gene expression. This study investigates the architecture of ribosomes from the neocortex during neurogenesis, revealing Ebp1 as a high-occupancy 60S peptide tunnel exit (TE) factor during protein synthesis at near-atomic resolution by cryoelectron microscopy (cryo-EM). Ribosome profiling demonstrated Ebp1-60S binding is highest during start codon initiation and N-terminal peptide elongation, regulating ribosome occupancy of these codons. Membrane-targeting domains emerging from the 60S tunnel, which recruit SRP/Sec61 to the shared binding site, displace Ebp1. Ebp1 is particularly abundant in the early-born neural stem cell (NSC) lineage and regulates neuronal morphology. Ebp1 especially impacts the synthesis of membrane-targeted cell adhesion molecules (CAMs), measured by pulsed stable isotope labeling by amino acids in cell culture (pSILAC)/bioorthogonal noncanonical amino acid tagging (BONCAT) mass spectrometry (MS). Therefore, Ebp1 is a central component of protein synthesis, and the ribosome TE is a focal point of gene expression control in the molecular specification of neuronal morphology during development.
Graphical Abstract
In Brief
Kraushar et al. visualize protein synthesis in the developing mouse brain at near-atomic resolution. Ebp1 binds the 60S tunnel exit to regulate translation initiation and N-terminal peptide elongation proteome-wide. Ebp1 is particularly abundant in early-born neocortex neural stem cells and regulates neuronal morphology, impacting cell adhesion molecule synthesis.
INTRODUCTION
Proteostasis, the fine-tuned balance of protein homeostasis, is fundamental in establishing the molecular landscape of the nervous system. The demand for spatially targeted and precisely timed protein synthesis is exceptionally high in mammalian nervous system development, where amorphous neural stem cells (NSCs) generate intricate neuronal morphology through targeted gene expression (Holt et al., 2019; Jayaraj et al., 2020; Jung et al., 2014) . This is particularly true in the evolutionarily advanced mammalian neocortex, the central neuronal circuit of complex cognition in the brain (Silbereis et al., 2016). Concordantly, the nervous system is uniquely susceptible to abnormal proteostasis, a major driver of neurodevelopmental and neurodegenerative disease (Bosco et al., 2011; Kapur et al., 2017; Sossin and Costa-Mattioli, 2019). How proteostasis is achieved, therefore, stands as a crucial question toward understanding neurogenesis in the neocortex.
The neurogenic phase of stem cell maturation in neocortical development follows a trajectory largely conserved across mammalian species (DeBoer et al., 2013; Molyneaux et al., 2007) (Figure 1A). NSCs lining the lateral cortical ventricular zone (VZ) initially divide symmetrically to expand the cellular pool. NSC divisions then transition to yield newly born neurons, which progressively and sequentially undergo superficial migration, ultimately forming a layered cortical plate (CP) composed of structurally and functionally distinct neurons. In mice, lower layer neocortical neurons appear at approximately embryonic day 12.5 (E12.5), with the switch to upper layer formation at E15.5. By postnatal day 0 (P0), neurogenesis is largely complete, with ongoing ventricular stem cell divisions yielding glial cells. The elaboration of intricate neuronal morphology during this developmental window requires tight regulation of the neurite outgrowth and synaptic proteome (Holt et al., 2019; Jung et al., 2014), a fine-tuned balance of membrane proteins like cell adhesion molecules (CAMs) that establish neuronal connectivity (de Wit and Ghosh, 2016).
Figure 1. Ebp1 Is a Highly Associated Cofactor of the Neocortex Ribosome across Development.

(A) Schematic of the experimental system to measure the architecture of active protein synthesis (polysomal ribosomes) from the neocortex across embryonic and early postnatal neurogenesis.
(B) Analytic density gradient fractionation of A260-normalized neocortex lysates, measuring the relative abundance of ribosomal subunits, 80S ribosomes, and polysomes. A260 curves plotted as mean ± SD across replicate fractionations (n = 2–3) for each stage, baseline (1.0) centered at onset of 40S peak.
(C) Statistical comparison of ∑A260 within gray marked regions in (B), shown as mean ± SD with significance testing by one-way ANOVA and Dunnett’s post hoc test versus E12.5. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(D) MS analysis (n = 3) of neocortex polysomal complexes across development, scatterplots comparing E12.5 with each subsequent stage for enrichment of Ebp1, ribosomal proteins (RPs) of the large (Rpl) and small (Rps) subunits, and translation-associated proteins (GO: 0006417). See also Figures S1 and S2A.
(E) Neocortex expression of Ebp1, Rpl, Rps, and translation-associated genes measured in total steady-state levels by RNA-seq (left, n = 2) and MS (right, n = 3) across developmental stages. Median expression is plotted ± SD; one-way ANOVA and Bonferroni corrected post hoc test versus E12.5, p < 0.05. Significantly changing levels versus E12.5 (true) are shown as filled circles and non-significant (false) values as empty circles.
(F) Western blot probing for Ebp1 (Ebp1CT, Ebp1NT) in total neocortex lysates compared to full-length recombinant Ebp1-His, along with the RP uL30 and Gapdh; full blots are shown in Figures S3A and S3B.
(G) Jitter plots comparing the median stoichiometry of Rpl and Rps (centered at 0) with Ebp1 and translation-associated proteins in total, 80S, and polysomes at E12.5. Other stages are shown in Figure S2B.
(H) Western blot analysis (top) of Ebp1 enrichment in free, 80S, and polysome fractions across development compared to Gapdh and uL30. Quantification (bottom, n = 2 blots) of Ebp1 and uL30 levels versus E12.5 is shown, and values represent mean ± SD (t test for significance: *p < 0.05; ***p < 0.001).
See also Figures S3C-S3E.
Analysis of the molecular landscape in the developing neocortex has largely focused on transcriptional regulation (Lein et al., 2017; Silbereis et al., 2016), with the neocortical transcriptome coming into focus recently at the single-cell level (single-cell RNA sequencing [scRNA-seq]) (Nowakowski et al., 2017; Telley et al., 2019; Yuzwa et al., 2017). However, the ultimate output of gene expression is protein, and bridging the neocortical transcriptome to proteome is the current challenge. The ribosome is the gatekeeper of the proteome, poised at the final essential step of gene expression as the macromolecular hub of protein synthesis, at the crossroads of gene expression in cellular proliferation, differentiation, and disease (Kraushar et al., 2016; Mills and Green, 2017; Shi and Barna, 2015; Teixeira and Lehmann, 2019). However, the architecture of ribosomal complexes and proteostasis control in neocortical development remain unknown.
In this study, we analyze the molecular architecture of native ribosome complexes from the mammalian neocortex during developmental neurogenesis at near-atomic resolution. We find that the ErbB3-binding protein 1 (Ebp1) participates in high-occupancy binding to the 60S subunit of both actively translating and non-translating ribosomes through high-affinity interactions with the peptide tunnel exit (TE) surface in the embryonic and perinatal neocortex. Ebp1’s function in protein synthesis during nervous system development is unknown. Ebp1 abundance across developmental stages scales directly with dynamic ribosome levels and is cell-type specific; Ebp1 is dominantly expressed in early-born NSCs compared to later-born NSCs and post-mitotic neurons in contrast to other exit tunnel cofactors. Ebp1-ribosome interaction occurs in the cytoplasm of NSCs in the neocortical VZ at early embryonic stages when ribosomal complex levels are highest and persists in post-mitotic neurons of the expanding CP as steady-state ribosome levels decline. With Ebp1 selective ribosome profiling (SeRP), we show that Ebp1’s highest occupancy on actively translating ribosomes is during start codon initiation, with knockdown resulting in ribosome accumulation at the AUG. Ebp1 binding is maintained during elongation, especially during synthesis of N-terminal peptides throughout the proteome, until translocon signal sequences for membrane targeting emerge from the 60S tunnel and putatively engage competition for a common binding surface with SRP/Sec61. Ebp1 maintains neuronal proteostasis, especially impacting the synthesis of membrane-targeted CAMs as measured by pulsed stable isotope labeling by amino acids in cell culture (pSILAC)/bioorthogonal noncanonical amino acid tagging (BONCAT) mass spectrometry (MS). Concordantly, in vivo embryonic Ebp1 knockdown selectively in early-born neocortical NSCs results in dysregulated membrane morphology during neuronal maturation. This study is the first near-atomic resolution analysis of protein synthesis in the nervous system, positioning Ebp1 and the 60S peptide TE as a focal point of gene expression control during neurogenesis.
RESULTS
Ebp1 Is a High-Occupancy Translation Cofactor Proportional to Dynamic Ribosome Levels during Neocortex Development
To analyze the architecture of neocortical ribosome complexes across development, we first optimized a protocol to purify actively translating ribosomes ex vivo rapidly and stably without the use of chemical inhibitors that bias its conformational state, capturing the full repertoire of integral translation cofactors. Initial analytical sucrose density gradients revealed that global ribosome levels are dynamic across neocortex development (Figure 1B). High levels of 80S ribosomes (monosomes) and chains of multiple 80S actively translating mRNA (polysomes) predominate at E12.5–E14, transitioning to a lower steady state from E15.5 to P0 (Figure 1C). This decrease is not wholly accounted for by the availability of individual subunits in the cytoplasm, as 40S-60S levels decrease marginally. Thus, ribosomal complexes exist at elevated levels during early neocortical neurogenesis and transition to a lower steady state at later stages.
We next performed MS analysis of 80S and polysomes, in addition to corresponding total lysates, across neocortex developmental stages. Sample reproducibility was observed in hierarchical clustering of the MS data (Figure S1). Results from the neocortical polysome MS are shown in Figure 1D, comparing protein levels at E12.5 with each subsequent developmental stage. As expected, core ribosomal proteins (RPs) are the most abundant proteins in polysomes, including RPs of the large 60S (Rpl) and small 40S (Rps) subunits. Translation-associated proteins (Gene Ontology [GO]: 0006417) associate to varying degrees with polysomes throughout development. Unexpectedly, we observed Ebp1 co-purifying at levels approaching the RPs themselves in polysomes, higher than any other translation-associated protein. Ebp1 is metazoan-specific, broadly expressed across cell types, and was largely studied in the context of cancer (Nguyen et al., 2018). Ebp1 was observed to play only a niche role in protein synthesis, promoting internal ribosome entry site (IRES)-dependent translation of a specific viral mRNA (Pilipenko et al., 2000) and suppressing eIF2a phosphorylation in conditions of cellular stress (Squatrito et al., 2006), by unknown mechanisms. Thus, we were intrigued by Ebp1’s exceptionally high polysome levels and observed a similarly robust association with 80S complexes (Figure S2A). Furthermore, Ebp1 is among the most abundant proteins measured in total neocortical lysates across development (Figure S2A).
To examine the global trajectory of neocortical Ebp1 and core RP gene expression across development, we next analyzed total lysates by RNA-seq (Figure 1E). Ebp1 mRNA steadily decreases after E12.5, while Rpl and Rps mRNA decreases lag behind at E17. However, corresponding MS measurements revealed total Ebp1 protein levels decline abruptly at E15.5 along with total Rpl and Rps levels in the neocortex, suggesting their protein levels are regulated in concert, with protein changes anticipating mRNA changes for the RPs. MS findings were confirmed by western blot analysis of total neocortex lysates (Figures 1F, S3A, and S3B), showing that levels of Ebp1 are highest in the early prenatal neurogenic period and decrease at E15.5, with the lowest Ebp1 levels occurring in the postnatal period. The timing of Ebp1, Rpl, and Rps total protein decreases coincides with the timed decrease of global ribosome levels measured by density gradient fractionation (Figures 1B and 1C).
Ebp1 has been previously reported as a full-length 48-kDa protein (“p48”) and a 42-kDa isoform (“p42”) generated by Ebp1 mRNA splicing (Liu et al., 2006). Western blot findings with a C-terminal targeting antibody (Ebp1CT) recognizing both long and short isoforms (Figures 1F and S3A) and a N-terminal-specific antibody (Ebp1NT) recognizing only full-length Ebp1 (Figures 1F and S3B), compared to polyhistidine-tagged full-length recombinant Ebp1 (Ebp1-His), showed that the dominant isoform of Ebp1 in neocortical development is full length.
The core of the eukaryotic 80S ribosome is a macromolecular machine consisting of ~79 RPs on a scaffold of four rRNAs, with translation-associated proteins transiently binding to catalyze and modulate ribosomal functions. We next calculated the stoichiometry between translation-associated cofactors and core RPs in neocortical ribosomes across development, in addition to their balance in total steady state (Figures 1G and S2B). In contrast to the majority of core RPs, translation-associated proteins are maintained at a wide range of total steady-state levels, and their association with purified ribosomal complexes tends to be substantially sub-stoichiometric. At all stages, Ebp1’s total steady-state level is similar to RPs (0.7–1.5 total stoichiometry) and is the 80S and polysome cofactor with the highest association, in the range of 0.4–0.6 80S stoichiometry and 0.2–0.3 polysome stoichiometry.
Given that Ebp1 is sub-stoichiometric in 80S and polysome complexes but near stoichiometric in total, a substantial proportion of its total levels are likely extra-ribosomal. To test this, we next measured the balance of ribosome-associated Ebp1 compared to “free” extra-ribosomal Ebp1 in neocortical development (Figures 1H, S3C, and S3D). Results showed that, indeed, at each developmental stage, the majority of Ebp1 is extra-ribosomal in contrast to the RP uL30 (Figure S3E), consistent with Ebp1 being a ribosome cofactor rather than a core protein. Furthermore, comparing E12.5 with subsequent stages showed a decrease of Ebp1 in ribosomal fractions beginning at E15.5 to P0 that mirrors changes in the RP uL30 (Figure 1H). In contrast, free extra-ribosomal Ebp1 is maintained over time.
Taken together, these data suggest that Ebp1 associates with both neocortical 80S and polysomes, maintaining a high and consistent stoichiometry, in concert with decreasing ribosome levels across development. The unusual abundance of Ebp1-ribosome association suggests that Ebp1 may play a more central role in neocortical translation rather than niche for a small subset of transcripts or during transient conditions as previously reported (Pilipenko et al., 2000; Squatrito et al., 2006).
Ebp1 Is Enriched in Early-Born NSCs and Localizes throughout the Cytoplasm
As the above observations were in bulk neocortex tissue, we next analyzed the cellular expression of Ebp1 in neocortex development with scRNA-seq data (Telley et al., 2019) measuring the transcriptome of early- and late-born NSCs maturing into lower and upper layer neurons, respectively (Figure 2A). Strikingly, Ebp1 mRNA is particularly enriched in early-born NSCs, with levels decreasing abruptly during neuronal differentiation and in the later-born NSC pool. Likewise, Rpl and Rps mRNA levels decline with differentiation; however, the Rpl and Rps expression patterns are more generic in NSCs, regardless of birthdate.
Figure 2. Ebp1 Is Enriched in Early-Born NSCs and Localizes Throughout the Neuronal Cytoplasm.

(A) Expression heatmaps of Ebp1 compared to averaged Rpl and Rps family mRNA enrichment in scRNA-seq analysis of the developing mouse neocortex, derived from (Telley et al., 2019). Relative expression in apical progenitor (AP) NSCs during differentiation into mature neurons (N4d) is shown on the y axis, corresponding to NSC birthdates E12, E13, E14, and E15 on the x axis.
(B) Immunohistochemistry analysis of Ebp1 the developing neocortex ventricular zone (VZ) and cortical plate (CP). Early-born NSCs in the VZ generate lower layer (LL) neurons, while later-born NSCs in the VZ generate upper layer (UL) neurons. Axonal white matter (WM); DAPI staining (gray). Zoomed images (inset, left) correspond to the VZ and leading-edge of the CP at each stage, quantified signal/area (n = 5-7) in each region of interest (inset, right heatmap). AU, arbitrary units. See also Figures S4A and S4B.
(C) Immuno-electron microscopy with anti-Ebp1NT immunogold labeling (black dots) in the neocortex at E12.5, E15.5, and P0. Neural stem cells (NSCs; blue nuclei) and neurons (N, red nuclei). Nucleoli (n), mitochondria (m, green), endoplasmic reticulum (er), dendrite (D), plasma membrane (arrows).
(D) Quantification of (C), comparing the cytoplasmic versus nuclear distribution of Ebp1 (n = 5–64 cells per condition) in VZ stem cells and CP neurons, with 1° antibody leave-out control. See also Figures S4C and S4D. Significance testing by Welch ANOVA, *p < 0.001.
(E) Primary neuronal cultures from the E12.5 neocortex, immunocytochemistry at div 0, 2, 4, and 5 for Nestin, Nex:Cre;tdTomato, and Ebp1. Growing neurites and distal growth cones are indicated (arrows).
Consistent with the scRNA-seq data, immunohistochemistry analysis across developmental stages (Figures 2B, S4A, and S4B) demonstrated particularly high cytoplasmic Ebp1 protein enrichment in the VZ and nascent CP at E12.5–E14. Ebp1 is persistent in maturing neurons laminating the CP at later stages, albeit at lower levels. Interestingly, Ebp1 enrichment in the P0 VZ that contains early gliogenic progenitor cells (DeBoer et al., 2013; Molyneaux et al., 2007) is substantially lower than in the neurogenic E12.5 VZ. This enrichment in neural progenitors may relate to the observation that Ebp1 is particularly enriched in neurons compared to astroglia in the postnatal period (Ko et al., 2017). Thus, Ebp1 enrichment is specific to cell type, differentiation status, and NSC birthdate in the neocortex.
To assess subcellular Ebp1 localization at higher resolution, we next analyzed the neocortex at E12.5, E15.5, and P0 by immunoelectron microscopy (immuno-EM), probing for Ebp1 with both Ebp1NT (Figure 2C) and Ebp1CT (Figure S4C) antibodies. Quantification of Ebp1 immunogold labeling demonstrated almost exclusively cytoplasmic signal (Figures 2D and S4D), occurring in clusters throughout the cytoplasm most abundantly in early-born NSCs in the VZ and their daughter neurons in the nascent CP at E12.5. Lower cytoplasmic Ebp1 levels were measured in CP neurons at later stages, and VZ glial progenitors at P0, concordant with immunohistochemistry data (Figures S2B and S4A). Ebp1 was largely absent from nuclei, including the nucleolus and nuclear membrane, and was not observed in mitochondria, in strict proximity to the endoplasmic reticulum (ER), or at the plasma membrane. Thus, Ebp1 may bind the ribosome in the cytoplasm, rather than as a subunit assembly or export factor in the nucleus, consistent with prior observations regarding Ebp1 retention in the cytoplasm (Bradatsch et al., 2007).
Ebp1 was also observed in dendrites of maturing neurons at P0 (Figure 2C), suggesting Ebp1 localizes throughout cytoplasmic compartments as neocortical NSCs mature into neurons. We next sought to visualize Ebp1 localization during the progressive differentiation of early-born neocortical NSCs into post-mitotic neurons undergoing neurite outgrowth. Primary cultures were prepared from the E12.5 neocortex of Nex:Cre;Ai9 mice (Turko et al., 2019), which label post-mitotic pyramidal neurons, followed by immunohistochemical analysis of Ebp1 expression (Figure 2E). Ebp1 is enriched in cytoplasmic foci co-localizing with nestin labeling in NSCs at days in vitro 0 (DIV0) and persists in differentiating Nex-positive neurons at DIV2–DIV4. Ebp1 puncta are visualized in robust neuronal protrusions by DIV5 and particularly apparent with further magnification of neurites and growth cones, including the most distal aspects of extending processes, consistent with prior observations in hippocampal neurons (Ko et al., 2017; Kwon and Ahn, 2011).
Ebp1 Binds the 60S Peptide TE in Actively Translating and Inactive Ribosomes
To analyze the architecture of neocortical ribosome complexes and visualize the physiologic binding mode of Ebp1 at near-atomic resolution, 80S and polysomes were purified by sucrose density gradient fractionation from P0 neocortex lysates, pooled together, and frozen on grids for cryoelectron microscopy (cryo-EM). Micrographs confirmed the presence of both 80S and polysome complexes in the sample (Figure 3A). High-resolution cryo-EM data collection (Figure S5) and initial single-particle reconstruction yielded a map of the complete 80S, along with extra-ribosomal density (red) adjacent to the 60S peptide TE (Figure 3B). Fitting the crystal structure of Ebp1 (Kowalinski et al., 2007; Monie et al., 2007) to the extra-ribosomal density unequivocally identified Ebp1 in complex with the neocortical 60S. Robust density was present for nearly the entire N terminus, identifying the full-length isoform of Ebp1 is bound. Ebp1 forms a concavity above the TE vestibule with a porous interface, including gaps (~28 Å at the widest point) that may permit peptide chain exit. The mouse neocortex 80S core structure was found to be otherwise highly conserved with previously solved human (Behrmann et al., 2015) and rabbit (Flis et al., 2018) structures, with Ebp1 density the greatest distinction.
Figure 3. Ebp1 Binds the 60S Tunnel Exit (TE) in Actively Translating and Inactive 80S Complexes.

(A) Cryo-electron micrograph of pooled monosome and polysome complexes from P0 mouse neocortical lysates ex vivo.
(B) Cryo-EM maps of (A) with extra-ribosomal density conforming to mouse Ebp1 (PDB: 2V6C) over the 60S TE (side view, top image; aerial view, bottom image). N-terminal Ebp1 residues (NT, black ribbon) corresponding to full-length “p48” Ebp1. See also Figures S5-S7.
(C) Actively translating (left: classical state with A/A and P/P tRNAs) and non-translating (right: rotated state with eEF2) 80S-Ebp1 complexes.
(D) Model of the Ebp1 binding surface at the 60S peptide TE, including 60S rRNA helices H24, H53, H59, and 60S RPs eL19, uL23, uL24, and uL29.
(E) Aerial view of the Ebp1 footprint (red outline) over the 60S peptide TE, with rRNA helices and RP model surfaces colored as in (D); residues/nucleosides making electrostatic interactions with Ebp1 are highlighted (yellow).
(F) 2D structure diagram of Ebp1 domains adapted from Kowalinski et al. (2007), orienting Ebp1 on the ribosome surface, with binding domains highlighted (yellow).
To disentangle the ribosome conformational states bound by Ebp1, we proceeded with hierarchical multiparticle sorting and 3D classification of both large and small scale heterogeneity intrinsic to the data (Behrmann et al., 2015; Loerke et al., 2010) (Figure S6). Ribosome complexes in both the rotated and classical conformations were first sorted, including populations with (1) eEF2 and (2) eEF2+P/E tRNA in the rotated state and populations with (3) A/A+P/P tRNAs, (4) E/E tRNA, and (5) without tRNAs in the classical state. Within each of these five states, a strategy of modified focused classification was utilized to separate sub-states with and without Ebp1, yielding 10 total classes. Across all states, Ebp1 was bound to 48% of ribosomes, with ~50% binding to each of the five sub-states.
We proceeded with high-resolution refinement of Ebp1-bound and unbound populations in the inactive rotated state with eEF2 (3.1 Å global resolutions) and the active classical state with A/A+P/P tRNAs (3.3 Å global resolutions) (Figure 3C). When Ebp1 was bound, the structural conformations of both Ebp1 and the 60S binding surface were identical between active and inactive ribosomes. These data indicate Ebp1 binds to both actively translating and non-translating neocortical ribosome states with approximately equal probability, high occupancy, and identical conformations.
The near-atomic resolution of our data (Figures S5 and S7) permitted modeling of the entire neocortical Ebp1-60S complex. Figure 3D visualizes the peptide TE surface in proximity to Ebp1, including four RPs (eL19, uL23, uL24, and uL29) and three rRNA helices (H24, H53, and H59). An aerial view of the Ebp1 footprint over the TE surface highlights the 60S RP residues and rRNA nucleosides making electrostatic interactions with Ebp1 (Figure 3E), demonstrating that Ebp1 contacts the immediate TE surface. The neocortical Ebp1-60S complex establishes previously unassigned functions to Ebp1 structural domains (Figure 3F; adapted from Kowalinski et al., 2007), where binding by Ebp1’s insert domain and α5 helix positions β sheets 1, 3, 4, 5, 7, and 13 directly over the TE.
Ebp1 Binding Requires a Conserved 60S Helix H59-H53 Swinging Latch Mechanism
Multiparticle sorting of our data into Ebp1-bound and unbound states enabled identification of 60S structural changes facilitating Ebp1 interactions with an internal negative control (Figure S6). Dynamic interactions occur with helix H59 of 28S rRNA (Figures 4A and S8A). In the Ebp1-bound state, the tip of H59 undergoes a backbone rearrangement enabled by a 235° flip of H59 G-2690, releasing contact with H53 G-2501, G-2502, and C-2513 as seen in the canonical unbound state, resulting in H59 G-2690 transitioning to intra-helical base stacking interactions. This “swinging latch” mechanism further includes a 73° flip of H59 U-2687, with the base reaching into a pocket of Ebp1’s insert domain (Figure S8B), locking Ebp1 into position. This particular movement of H59 U-2687 was previously observed for binding of the yeast nuclear export (Bradatsch et al., 2007) and peptide tunnel quality control (Greber et al., 2016) factor Arx1 to the 60S, thus representing a conserved binding mechanism. However, unlike Arx1, Ebp1 binding does not appear to require stabilization by rRNA expansion segment ES27 on the solvent side (Greber et al., 2016), which we confirmed by 3D-variability calculations (Penczek et al., 2006) and independent map reconstructions with alternative methods (Punjani et al., 2017; Scheres, 2012; data not shown), thus representing a distinction in its binding mode.
Figure 4. Ebp1-60S Binding Utilizes a Conserved H59 Latch Mechanism and Is Incompatible with Simultaneous Binding of Other TE Cofactors.

(A) 60S rRNA H59 and H53 models in conformations with and without Ebp1, adjacent to the Ebp1 insert domain. See also Figures S8A and S8B.
(B–D). Ebp1-60S binding interface in detail, with interacting residues highlighted for Ebp1 (gray) and the 60S (yellow). See also Figures S8C-S8E.
(E–G) Global alignment of Ebp1, Metap2, and Arx1 (top, ribbon), likewise when viewed from within the 60S tunnel (bottom, electrostatic potential map) from the perspective of emerging nascent chain. See also Figure S8F.
(H) Aerial view with overlapping footprints of eukaryotic TE binding factors superimposed on the neocortex 60S. Accession numbers are as follows: Metap2, PDB: 1KQ9; Arx1, PDB: 5APN; Sec61, PDB: 3J7R; SRP, PDB: 6FRK; Ltn1, PDB: 3J92; NatA, PDB: 6HD7; Ttc5, PDB: 6T59; RAC, EMDB: 6105; NAC, EMDB: 4938.
(I) Jitter plots comparing the median stoichiometry of Rpl and Rps (centered at 0) with Ebp1 and other TE cofactors in total, 80S, and polysomes at E12.5 and P0. See also Figure S9.
(J) Ebp1-60S binding affinity assay (Figure S10C), with independent replicate experiments (white and gray circles) and curve best fit to the data. 60S concentration (blue line) maintained at a constant 100 nM.
(K) Ebp1-60S binding dynamics assessed by pelleting assay and western blot. Binding pellet signal for (1) super-saturating Ebp1-His, (2) native Ebp1 in RRL, and (3) competition between added Ebp1-His and native Ebp1 in RRL. Arrow, native Ebp1; star, Ebp1-His signal.
Protrusion of H59 U-2687 into the insert domain of Ebp1 is stabilized by hydrogen bonds with the backbones of Y-255 and G-256, in addition to S-267, and π-stacking interactions with F-266 (Figures 4B and S8C). Reorientation of H59 brings the adjacent U-2688 in proximity to R-271, yielding further hydrogen bond stabilization. The RP eL19 confers stability to both the flexible loop and α6 helix in Ebp1’s insert domain (Figures 4B, 4C, S8C and S8D). Hydrogen bonding occurs between Ebp1 Q-254 and Y-255 with eL19 N-34 and N-36, respectively, with a particularly prominent role for Ebp1 R-263 coordination by eL19 Q-39 and Q-40.
The Ebp1 α6 and α8 binding interfaces at the TE rim further include 28S rRNA H53 and uL23 (Figures 4C and S8D). The intra-molecular interaction between Ebp1 K-258 and Y-255 is reinforced by H53 C-2505 and uL23 E-84. Ebp1 α6/8 interactions with uL23 further include hydrogen bonds between Ebp1 methionines 259 and 291 with uL23 K-88 and E-91, respectively, while Ebp1 K-287 is coordinated by uL23 L-147, D-148, and N-151, with N-151 also contacting Ebp1 R-290. Contact between Ebp1 and uL29, in contrast, is less robust and mediated by van der Waals interactions.
Finally, 5.8S rRNA H24 and uL24 tether the Ebp1 α5 domain on the opposite side (Figures 4D and S8E). Ebp1 K-211 is in proximity to H24 A-383 and C-384, while Ebp1 D-207 is immediately opposite uL24 N-91 and T-93.
Ebp1-60S Binding Is Incompatible with Simultaneous Binding of Other Eukaryotic Peptide TE Cofactors
Ebp1, Metap2, and Arx1 share a common β-α—α-β insert domain that facilitates 60S binding, a “pita-bread” β6 fold motif positioned over the peptide TE, and a solvent-side α4 motif (Figures 4E-4G). In the event of Ebp1 or Metap2 binding, emerging peptide chain would encounter a deep, electronegative pocket; however, Metap2 β6 fold residues catalyzing aminopeptidase activity (Nonato et al., 2006) are absent in Ebp1 (Kowalinski et al., 2007; Monie et al., 2007), rendering Ebp1 catalytically inactive. Furthermore, the Ebp1 α5 domain facilitating electrostatic contacts with H24 and uL24 is absent in Metap2 (Figure S8F); however, a Metap2-60S structure has not yet been solved. In contrast, the yeast Arx1 pita-bread fold binds FG repeat domains of nuclear membrane nucleoporins (Bradatsch et al., 2007) and threads Rei1 into the peptide tunnel to probe the 60S as a quality-control step (Greber et al., 2016), with contrasts to the metazoan Ebp1 described previously (Bradatsch et al., 2007). Thus, the binding of these distinct TE factors creates unique structural and electrochemical environments for emerging peptide chains.
The binding of Ebp1 would be sterically incompatible with the simultaneous docking of other 60S TE cofactors, competing for limited real estate surrounding an emerging nascent peptide chain (Figure 4H). The footprint of Ebp1 is shown superimposed on the footprints of Metap2 (Nonato et al., 2006) and Arx1 (Greber et al., 2016), in addition to the ER targeting machinery SRP (Kobayashi et al., 2018) and Sec61 (Voorhees et al., 2014); the Ltn1-NEMF ubiquitin ligase complex (Shao et al., 2015); the N-terminal acetyltransferase NatA (Knorr et al., 2019); the ribosome-associated complex (RAC) coupling nascent-chain elongation and folding (Zhang et al., 2014); the nascent polypeptide-associated complex (NAC) preventing ER mistargeting and protein aggregation (Gamerdinger et al., 2015; Shen et al., 2019); and Ttc5, a negative-feedback sensor of tubulin synthesis (Lin et al., 2020). Ebp1 is among the most abundant of these TE factors in the neocortex (Figure 4I), comparable to NAC, and the Hspa8 subunit of RAC. Early in development, however, Ebp1 is the most highly associated with 80S and polysomes; later in development, as Ebp1 and ribosome levels decline, only Hspa8 supersedes Ebp1 in 80S and polysomes. Furthermore, the neocortical cell type and temporal specificity of Ebp1 enrichment is in contrast to some TE cofactors, such as Ttc5, while similar to others, such as RAC (Figure S9). Dynamic enrichment of Ebp1 versus other TE cofactors may represent the differential regulation of protein synthesis in response to the unique demands of particular stages in neurogenesis.
Ebp1 Binds the 60S with High Affinity and Active Turnover
Given the competition for a common TE surface, we next sought to measure the affinity and dynamics of Ebp1-60S binding. We first confirmed the specificity of Ebp1 for the 60S subunit of both neocortical and rabbit reticulocyte (RRL) ribosomes, in the absence of mRNA (Figures S10A and S10B). We then determined the relative affinity range of Ebp1-60S binding (Figures 4J and S10C). The curve best fit to data (r2 = 0.99) indicates Ebp1 reaches a Kd(app) at ~124 nM, with saturated Ebp1-60S binding at ~200 nM, relative to 100 nM 60S. These data indicate Ebp1 binds the 60S with high affinity, reaching saturation at ~2-fold excess Ebp1 over the 60S. The 0.7–1.5 stoichiometry of total steady-state Ebp1 compared to the core ribosome measured by MS (Figures 1G and S2B) along with a high relative affinity may account for the high degree of Ebp1-ribosome association measured in the neocortex across development.
We next tested whether Ebp1 binding undergoes dynamic turnover by reconstitution of the following binding conditions in parallel: (1) saturating levels of recombinant Ebp1-His in the presence of rabbit 60S, (2) RRL containing native Ebp1, and (3) saturating Ebp1-His added to RRL (Figure 4K). Native Ebp1 in RRL co-pelleted with the ribosome as did Ebp1-His to the 60S, undergoing dynamic binding demonstrated by the nearly complete turnover of native Ebp1 with saturating Ebp1-His. The dynamic turnover of the Ebp1-60S binding mode may permit emerging peptide chain motifs to recruit other TE cofactors and displace Ebp1.
Start Codon Initiation and N-Terminal Peptide Synthesis Are Regulated Proteome-wide by Ebp1
To interrogate the specific translation activity of Ebp1-bound ribosomes at high resolution proteome-wide, we performed Ebp1 SeRP (Schibich et al., 2016), deep sequencing of ribosome-protected mRNA fragments, in mouse neuronal cultures (Neuro2a). We first confirmed that, like the neocortex, Neuro2a cells dominantly express full-length Ebp1 (Figure S11A), which associates specifically with 80S and polysomes (Figures S11B and S11C). Comparison of overall mRNA enrichment in the Ebp1-ribosome immunoprecipitation (IP) interactome with the total translatome (Figure S12) demonstrated a high degree of correlation (Figure 5A), consistent with high-occupancy ribosome binding by Ebp1 during the synthesis of a large, highly generalized cohort of transcripts. Highlighting the subset of mRNAs with ≥1.5-fold enrichment in Ebp1-ribosome IP versus the total translatome demonstrated a cellular compartment-specific translation preference (Figure 5B). Ebp1-ribosome complexes are more likely to be engaged in the translation of proteins localizing to the nucleus and other intracellular compartments while disfavoring membrane-targeted proteins of the ER.
Figure 5. Ebp1-Ribosome Complexes Engage in Translation Initiation and Elongation, with High Occupancy prior to N-Terminal Membrane Targeting.

(A) Correlation between the neuronal Ebp1-ribosome interactome and total translatome measured by SeRP (n = 2); mRNAs with RPKM enrichment ≥1.5-fold are highlighted. See also Figures S11 and S12.
(B) Gene Ontology (GO) analysis of mRNAs enriched in the Ebp1-ribosome interactome versus total translatome from (A).
(C) Proteome-wide metagene read density of the Ebp1-ribosome interactome versus total translatome over the coding sequence, aligned to the start (left) or stop (right) codon, plotted as mean with 95% confidence intervals (CIs). See also Figure S12C.
(D) Metagene plots as in (C), separated by subcellular protein localization.
(E) Cytoplasmic and signal peptide-containing protein metagene plots aligned to the start or stop codon (left figure), highlighting the relative enrichment at 70 codons (gray dashed line). Metagene plot for signal peptide-containing proteins aligned to the C-terminal codon of the signal sequence (right figure), with 60S tunnel transit region 40 codons downstream (gray box).
(F) Metagene plot for transmembrane domain (TMD)-containing proteins with (left figure) and without (right figure) an upstream translocon signal peptide, aligned to the C-terminal codon of the first TMD.
(G) Metagene read density distribution comparing Ebp1 knockdown versus control neuronal ribosome profiling (n = 3), separated by subcellular protein localization, and aligned to the start or stop codon. See also Figure S13.
(H) Ebp1 knockdown and control ribosome P-site count metagene plots (95% CI), aligned to the start or stop codon, for cytoplasmic and signal peptide-containing mRNAs. Inset right: scaled to highlight relative differences at the start codon P-site.
(I) Ebp1 knockdown/control fold change ribosome P-site counts at the start, stop, and adjacent codons (±3 nt) in the CDS of cytoplasmic and signal peptide-containing mRNAs. All ribosome positions shown for both mRNA groups are significantly different than control conditions (hypothesis siEbp1/control ≠ 1; 95% CI; p < 0.001), in addition to the significant difference annotated in the figure with *p = 0.03. Significance testing by t test.
Proteome-wide metagene analysis (Figure 5C) shows that, on average, Ebp1-ribosome interaction is highest during initiation at the start codon. Proteins ultimately targeted to distinct cellular sub-compartments during translation, including the cytoplasm and ER, share this characteristic enrichment (Figure 5D), consistent with highly comprehensive Ebp1-ribosome transcript interaction at the AUG. During elongation in the coding sequence across protein subsets, Ebp1 occupancy progressively declines until ~120 codons, when steady-state binding is established and maintained until the stop codon.
However, for ER-targeted proteins, a distinct, lower steady state of Ebp1-ribosome interaction is established from ~70–120 codons into the coding sequence (Figures 5D and 5E). Translation is targeted to the ER by 60S TE binding proteins SRP and Sec61, which facilitate recruitment and membrane insertion, respectively, of ribosome-nascent-chain complexes by interaction with emerging translocon signal sequences and transmembrane domains (TMDs) (Jan et al., 2014; Schibich et al., 2016). Such membrane-targeting domains commonly emerge within this N-terminal window, after traversing the mammalian 60S tunnel that is ~40 amino acids in length (Jan et al., 2014). Alignment to the C-terminal codon of signal peptides demonstrated that decreased Ebp1 interaction occurs ~40 amino acids downstream (Figure 5E). Such signal sequences upstream of a TMD lead to Ebp1 displacement before the first TMD is translated, whereas a first TMD without an upstream signal sequence likewise results in Ebp1 displacement after ~40 codons (Figure 5F). The competition between Ebp1 and SRP/Sec61 for a common binding surface at the 60S TE (Figure 4H) suggests that Ebp1 is displaced by SRP/Sec61 when recruited by membrane-targeting nascent-chain motifs.
To further interrogate the translation-specific function of Ebp1, we next performed ribosome profiling of Neuro2a cells in Ebp1 knockdown and control conditions (Figure S13). Metagene plots demonstrated an accumulation of ribosomes at N-terminal codons with Ebp1 knockdown, in particular for ER-targeted, transmembrane, and signal peptide-containing proteins, where ribosome accumulation extends from the start codon until ~10–15 codons downstream (Figure 5G). P-site alignment of ribosome-protected fragments and normalization to mapped reads per gene more precisely demonstrated the accumulation of ribosome occupancy at the start codon in Ebp1 knockdown conditions for both cytoplasmic and signal peptide-containing proteins (Figures 5H and 5I). After the start codon, Ebp1 knockdown further leads to ribosome accumulation during the elongation of early N-terminal amino acids, particularly for signal peptide-containing proteins, as seen by ribosomal P-site occupancy (Figure 5I).
Given high-occupancy Ebp1-ribosome binding at the start codon and early N-terminal peptides, with differential occupancy during N-terminal peptide discrimination, the impact of Ebp1 depletion further demonstrates a role for Ebp1 during active protein synthesis at the initiation and early-elongation phases proteome-wide.
Ebp1 Regulates the Morphology and Proteome of the Neuronal Membrane
Since we observed particularly high Ebp1 enrichment in early-born NSCs of the developing neocortex (Figures 2A-2D and S4A), we next sought to study the cellular effect of Ebp1 knockdown in early-born NSCs during their maturation into neocortical neurons in vivo. In utero electroporation (IUE) of a shEbp1 knockdown or control plasmid along with a CAG-GFP transfection reporter at E12 in the VZ was followed by analysis at E16 during neuronal maturation in the CP (Figure 6A). Analysis at E16 demonstrated increased branching of neuronal processes in shEbp1 conditions compared to control, as normal pyramidal neuron projections include a single unbranched axon extending toward basal white matter tracts, along with an apical dendrite oriented toward the pial surface. Tracing the morphology of transfected neurons (Figure 6B) highlighted the impact of Ebp1 depletion on neurite outgrowth at various neurite lengths, with Sholl analysis (Figure 6C) demonstrating a significantly increased branch number in shEbp1 conditions, an ~2-fold increase for proximal segments. Importantly, this increased branching phenotype was rescued by co-electroporation with an Ebp1 overexpression plasmid (oeEbp1) in addition to shEbp1, with neuronal morphology tracing and branching analysis quantified as indistinguishable from control conditions.
Figure 6. Ebp1 Regulates Neocortical Neuronal Morphology during Development and the Synthesis of Membrane-Targeted Cell Adhesion Molecules (CAMs).

(A) E12 in utero electroporation (IUE) of NSCs followed by neuronal analysis at E16, comparing shEbp1 and scrambled shRNA control, and rescue by co-electroporation with Ebp1 overexpression (oeEbp1). Co-electroporation with CAG-GFP visualizes transfected cells, shown magnified (bottom), including basally projecting axons (arrows) forming white matter tracts below upper (UL) and lower (LL) layers.
(B) Morphology tracing GFP labeled neurons in control, shEbp1, and rescue sh+oeEbp1 conditions from (A).
(C) Sholl analysis of (B), comparing branching per unit distance from the soma (top figure) and sum total (bottom figure) (n = 15 cells per condition). Values represent mean ± SD, with significance testing by one-way ANOVA with Bonferroni corrected post hoc test versus control (*p < 0.01).
(D) Schematic of the strategy to measure both chronic proteostasis and acute protein synthesis responses to Ebp1 knockdown in Neuro2a cells with pSILAC and BONCAT MS, respectively. AHA pulsed for 4 h.
(E) pSILAC- and pSILAC-AHA-labeled protein levels in siEbp1 relative to non-targeting siRNA control in biological replicates (n = 2) with label swab. Ebp1 levels were below the MS quantification threshold in siEbp1 conditions and thus not plotted. The number of significantly changing proteins at ≥2-, 1.5-, and 1.25-fold change thresholds are shown (dotted lines), in addition to the total number of proteins measured (top left). See also Figure S14.
(E′) Significantly changing proteins measured in common between pSILAC and pSILAC-AHA datasets at 1.25-fold-change thresholds.
(F) GO pathway analysis of proteins in (E) with ≥ 2-fold change in Ebp1 knockdown conditions.
(G) Metagene enrichment plots of the Ebp1-interactome (Ebp1-IP) and Ebp1 knockdown ribosome distribution translating L1cam mRNA, aligned to the start and stop codons, plotted as mean with 95% CIs.
To interrogate the potential function of Ebp1 in maintaining proteostasis during neuronal differentiation and neurite outgrowth, we again performed Ebp1 small interfering RNA (siRNA) knockdown in Neuro2a cells, and grew cultures in low serum conditions to induce maturation of neuronal morphology (Evangelopoulos et al., 2005). The impact of Ebp1 depletion on acute protein synthesis and chronic proteostasis in Neuro2a was measured by MS (Eichelbaum et al., 2012; Howden et al., 2013) with a combination of pSILAC (Schwanhäusser et al., 2009) and BONCAT (Dieterich et al., 2006). SILAC isotopes labeled all newly made proteins throughout Ebp1 knockdown for longitudinal proteome changes, while pulse labeling with a methionine analog (L-azidohomoalaine [AHA]) captured a snapshot of newly synthesized proteins at the nadir of Ebp1 levels (Figure 6D). Importantly, Ebp1 levels were below the quantification threshold in siEbp1 conditions, confirming robust knockdown.
Results showed that Ebp1 knockdown impacts 19% of the pSILAC measured proteome and 27% of the pSILAC-AHA measured proteome, with ≥1.25-fold change (Figures 6E and 6E’). Correlation with RNA-seq data in Neuro2a Ebp1 knockdown conditions from the above ribosome profiling analysis (Figures 5G-5I) showed that changes in steady-state mRNA were not sufficient to account for the majority of changes measured at the protein level (Figure S14A). CAMs are highly represented among the proteins most strongly impacted by Ebp1 (≥2-fold change) (Figure 6F), such as L1cam, Mcam, Cadm1, and Cdh15, which play a critical role in neurite outgrowth and synaptogenesis (de Wit and Ghosh, 2016). Such membrane-targeted proteins may be particularly susceptible to Ebp1 depletion, given the role of Ebp1 during initiation and elongation at the N terminus.
Proteins like L1cam were found to change in common between the pSILAC and pSILAC-AHA datasets (Figure 6E’), concordant with direct and protein-synthesis-specific regulation by Ebp1. Analysis of L1cam translation by Ebp1-ribosome complexes underscores the dynamic interactions of Ebp1 at the 60S TE during initiation and elongation of L1cam peptides (Figure 6G), with high occupancy at the start and N terminus that include fluctuations in binding, transitioning to generally lower occupancy downstream. Ebp1 knockdown redistributes ribosome occupancy in the L1cam coding sequence with a trend approximately opposite to Ebp1 binding in some regions of the coding sequence, such as at ~120 codons, with the notable exception at the start, consistent with elevated start codon occupancy with Ebp1 depletion. Given the sensitivity of N-terminal dynamics for proper membrane-targeted translation, the efficient translation of proteins like L1cam may be compromised in Ebp1 deficient conditions.
DISCUSSION
Taken together, this study analyzes the architecture of protein synthesis in the developing neocortex at high-resolution, positioning Ebp1 among 60S TE cofactors to regulate neuronal proteostasis in the molecular specification of morphology during neurogenesis. With a multidisciplinary approach, we demonstrate that Ebp1 is a chief component, rather than a niche regulator, of the protein synthesis machinery (Figure 7). Ebp1 participates in the initiation and elongation phases of translation in the neuronal cytoplasm, with high-occupancy 60S TE binding during start codon initiation and N-terminal peptide synthesis proteome-wide, reaching a dynamic binding equilibrium during elongation unless displaced by translocon targeting motifs. Ebp1 abundance is cell-type and temporally specific, enriched in the early-born NSC pool, in direct proportion to the transient abundance of ribosomal complexes at this early developmental stage. During NSC differentiation, Ebp1 particularly impacts the synthesis of CAMs that are essential components of the neuronal membrane and morphology. The metazoan specificity and broad cellular expression of Ebp1 points toward its central role as a translation regulator in evolutionarily advanced species.
Figure 7. Model of Ebp1 Function in Protein Synthesis and Neurodevelopment.

For details, see text. ER, endoplasmic reticulum.
Ebp1 and the 60S TE
60S TE cofactors compete for a common binding surface to engage proteins synthesized by a modular macromolecular machine (Deuerling et al., 2019; Kramer et al., 2019). While the regime of Rp1 and Rps mRNA expression appears to follow generally elevated levels in all neocortical NSCs compared to their daughter neurons, there is a great diversity of TE cofactor expression patterns in the developing neocortex (Figure S9). Ebp1 is particularly enriched in early-born NSCs, similar to RAC subdomains, but in contrast to Metap2, Ltn1, or NAC. Modulating the balance of TE cofactors (Figures 4H and 4I) may be a key determinant of cell-type-specific proteostasis, gatekeepers at the very moment a nascent protein emerges from the tunnel.
In our simple model (Figure 7), Ebp1 binding to the 60S in the cytoplasm may be a “default state” during translation initiation and early elongation proteome-wide before nascent chain exits the TE. Ebp1 binding with active turnover establishes a dynamic equilibrium throughout elongation, depending on competition from other TE cofactors if recruited by their associated nascent-chain moieties. Future work will determine in more detail how Ebp1 acts in coordination with SRP (Chartron et al., 2016; Jan et al., 2014; Schibich et al., 2016) and NAC (Gamerdinger et al., 2015) to organize subcellular targeting and N-terminal processing. Ebp1 may have an initial competitive advantage, given its abundance in the neocortical cytoplasm relative to other TE factors, high-affinity interaction with the 60S, and permissive binding requirements, including both translating and non-translating ribosomes.
Limitations
Whether Ebp1’s role in active and inactive complexes is linked or distinct remains unclear; for example, Ebp1 may play a role in 60S recycling for reinitiation or protect a reserve of inactive, dormant ribosomes available to participate in translation. Exploring such possibilities may help clarify the phenotype of Ebp1 knockdown, which yields increased ribosome occupancy during both start codon initiation generally and in the transition to elongation for signal peptide-containing proteins in particular. For example, increased ribosome profiling signal at the start codon may reflect 48S pre-initiation complexes waiting for 60S joining (Archer et al., 2016) or 80S complexes stalled before the transition to elongation.
While this paper was in revision, a structure of Ebp1 bound to inactive 80S from HeLa cells was published (Wild et al., 2020), in agreement with our data for interactions surrounding the TE rim. However, the study shows rRNA expansion segment ES27L interacting with Ebp1 on the solvent side, which we do not visualize as a binding requirement, which is a discrepancy of unknown significance but a possible distinction in mouse brain ribosome expansion segment structure. Furthermore, Ebp1’s potential direct interaction with nascent peptide chain and/or recruitment of other ribosome cofactors remains to be established. Our cryo-EM analysis of native Ebp1-ribosome complexes includes ribosomes engaging with the entire translated proteome, with our Ebp1 SeRP data demonstrating highly generalized transcript sequence binding by these complexes. Thus, nascent-chain density is lacking entirely at the TE vestibule outside the tunnel, secondary to heterogeneity intrinsic to the data, where peptides can assume numerous conformations. Future studies in a more homogeneous system will be required to interpret potential Ebp1/nascent-chain interactions at high resolution.
Neuronal Translation and the Neuronal Membrane
Many of the proteins impacted by Ebp1 are membrane associated, particularly cell-cell adhesion pathways regulating neuronal protrusions, with neocortical Ebp1 knockdown resulting in increased neurite branching. How Ebp1 depletion ultimately impacts protein output and membrane architecture is unclear but may be multifaceted and interconnected if the 60S TE environment is compromised. Depletion of a global ribosome cofactor like Ebp1 could impact many translation steps, either directly or indirectly, at the mRNA and protein levels, including mRNA degradation coupled to translation (Pelechano et al., 2015), the efficiency of translation initiation and elongation, and/or protein degradation that may occur as a result of misfolding, mistargeting, and ER stress (Martínez et al., 2018).
Ribosomes locally translate mRNAs in neuronal protrusions (Hafner et al., 2019; Zappulo et al., 2017) where Ebp1 is also present (Figures 2C and 2E), and several Ebp1-regulated proteins are predominantly translated in neuronal protrusions (Figure S14B). In conjunction with dynamic Ebp1 abundance, regulation may further derive from shifts in global ribosome levels that transition during neocortical neurogenesis (Figure 1), in line with previous observations of RP downregulation in the mouse forebrain during neural tube closure between E8.5 and E10.5 (Chau et al., 2018) and further between E13 and P0 (Kraushar et al., 2015). Global shifts in steady-state ribosome levels may reflect the dynamics of cellular homeostasis (Delarue et al., 2018; Mills and Green, 2017; Sinturel et al., 2017) in neocortex development. The subcellular and coordinated actions of Ebp1-ribosome complexes are interesting directions for future study.
Transcriptional control has been the principal focus in gene expression analysis during neocortex development (Silbereis et al., 2016), and recent excellent work has advanced this analysis to the single-cell level (Nowakowski et al., 2017; Telley et al., 2019; Yuzwa et al., 2017). However, while these studies assign transcriptional signatures to cell subtypes, they also strongly suggest that generic gene expression programs are refined by successive layers of regulation (Cadwell et al., 2019), such as post-transcriptional mechanisms and extrinsic signals (Kraushar et al., 2016). Our present work indicates binding events at the 60S TE during translation constitute a locus of control during neurogenic gene expression.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, christian.spahn@charite.de (C.M.T.S.).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Requests for code generated during this study, i.e., not otherwise referenced in the Method Details and Key Resources Table, may be directed to and will be fulfilled by the Lead Contact, christian.spahn@charite.de (C.M.T.S.). Data have been deposited in publicly available repositories as indicated in the Method Details and Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Ebp1CT | rabbit, Abcam | ab35424; RRID:AB_732061 |
| anti-Ebp1NT | rabbit, Millipore | ABE43; RRID:AB_10616223 |
| anti-eEF2 | rabbit, Cell Signaling | 2332S; RRID:AB_10693546 |
| anti-Gapdh | mouse, Millipore | MAB374; RRID:AB_2107445 |
| anti-GFP | chicken, Abcam | ab13970; RRID:AB_300798 |
| anti-Map2 | chicken, Millipore | AB5543; RRID:AB_571049 |
| anti-Nestin | mouse, Millipore | MAB353; RRID:AB_94911 |
| anti-Rpl7 (uL30) | rabbit, Abcam | ab72550; RRID:AB_1270391 |
| anti-Rps5 (uS7) | mouse, Santa Cruz | sc-390935; RRID:AB_2713966 |
| Gold-conjugated-anti-rabbit IgG | goat, Nanoprobes | 2003; RRID:AB_2687591 |
| HRP-anti-rabbit-Light Chain | mouse, Dianova | 211-032-171; RRID:AB_2339149 |
| HRP-anti-mouse-Heavy Chain | goat, Millipore | 71045; RRID:AB_11211441 |
| 488-anti-chicken | donkey, Jackson ImmunoResearch | 703-545-155; RRID:AB_2340375 |
| 488-anti-rabbit | donkey, Jackson ImmunoResearch | 711-545-152; RRID:AB_2313584 |
| 594-anti-mouse | donkey, Jackson ImmunoResearch | 715-585-150; RRID:AB_2340854 |
| 647-anti-chicken | donkey, Jackson ImmunoResearch | 703-605-155; RRID:AB_2340379 |
| Recombinant DNA | ||
| Control siRNA (non-targeting) | Dharmacon | D-001810-10-05 |
| Homo sapiens siPa2g4 siRNA | Dharmacon | SMARTpool ON-TARGETplus #5036, #L008860-00-0005 |
| Luciferase reporter pSPUTK-luc+ | Rakwalska and Rospert, 2004 | |
| Mus musculus Pa2g4 cDNA | Source BioScience | IRAVp968A0190D |
| Mus musculus shPa2g4 shRNA | Sigma Mission | TRCN0000236756, RefSeq NM_011119 |
| Mus musculus siPa2g4 siRNA | Dharmacon | SMARTpool ON-TARGETplus #18813, #L-042883-01-0005 |
| pCAGIG (pCAG-IRES-GFP) | Ambrozkiewicz et al., 2018 | |
| pET-28a(+) | Novagen | 69864-3 |
| pSuper-Neo-GFP | OligoEngine | VEC-pBS-0006 |
| pSuper-Neo-GFP-sh-Scramble | Ambrozkiewicz et al., 2018 | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile | CHEMSOLUTE | 2697 |
| Acetonitrile (Alkyne-agarose enrichment) | Sigma-Aldrich | 271004 |
| Acetylated Bovine Serum Albumin (BSA-c) | Aurion | 900.022 |
| Agarose | Sigma-Aldrich | A9539 |
| Alkyne-agarose beads | Click-Chemistry Tools | 1033 |
| Ammonium bicarbonate (ABC) | Sigma-Aldrich | 9830 |
| B27 | Thermo Fisher | 17504044 |
| Bovine serum albumin | Sigma-Aldrich | A3294 |
| Copper(II) sulfate pentahydrate | Sigma-Aldrich | 209198 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| DAPI (Nuc Blue, Molecular Probes) | Invitrogen | R37606 |
| Dithiothreitol (DTT) | Sigma-Aldrich/Roche | DTT-RO |
| Dithiothreitol (DTT) (Alkyne agarose enrichment) | BioMol | 40010.25 |
| DMEM | GIBCO | 31966047 |
| DMEM - methionine free | Sigma-Aldrich | D0422 |
| DNase-I | Roche | 4716728001 |
| Ebp1 recombinant protein | mouse, this paper | |
| EcoRI restriction enzyme | New England Biolabs | R0101 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | E-5143 |
| Ethylene glycol bis(β-aminoethylether) tetraacetic acid (EGTA) | Roth | 3054 |
| Epoxy embedding medium Epon 812 | Sigma-Aldrich | 45345 |
| Ethanol | J.T. Baker | 8025 |
| Fetal Bovine Serum | GIBCO | 10270106 |
| Fetal Bovine Serum - dialyzed | PAN-Biotech | P30-2102 |
| Fluoromount-G | Southern Biotech | 0100-01 |
| Formic acid | Sigma-Aldrich | 33015 |
| Glutamax | Thermo Fisher | 35050-038 |
| Glutaraldehyde | Sigma-Aldrich | G5882 |
| HEPES | Sigma-Aldrich | 391338 |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 |
| Iodoacetamide (IAA) | Sigma-Aldrich | I6125 |
| KCl | Roth | 6781.1 |
| L-Arginine:HCl (13C6, 99%; 15N4, 99%) (Arg-10) | Cambridge Isotope Labs | CNLM-539 |
| L-Arginine:HCl (13C6, 99%) (Arg-6) | Cambridge Isotope Labs | CLM-2265 |
| L-azidohomoalaine (AHA) | Anaspec | AS-63669 |
| L-Lysine:2HCl (13C6, 99%; 15N2, 99%) (Lys-8) | Cambridge Isotope Labs | CNLM-291 |
| L-Lysine:2HCl (4,4,5,5-D4, 96-98%) (Lys-4) | Cambridge Isotope Labs | DLM-2640 |
| Lead citrate | Fluka | GA10655 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher | 13778075 |
| Liquid ethane, grade 3.5 | Linde GmbH | |
| Lysyl endopeptidase (LysC) | Wako | 12505061 |
| Methanol | Merck Millipore | 1.06009.2511 |
| MgCl2 | Ambion | AM9530G |
| Nanogold silver enhancement | Nanoprobes | |
| Neurobasal medium | Thermo Fisher | 21103049 |
| Neurobasal custom medium (-met / -arg/ -lys) | GIBCO | 041-96642M |
| Normal goat serum | PAN-Biotech | P30-1002 |
| Osmium tetroxide (OsO4) | Polysciences | 0972A |
| Papain | Sigma-Aldrich | P4762 |
| Paraformaldehye (PFA) | Sigma-Aldrich | P6148 |
| Penicillin-Streptomycin | Thermo Fisher | 15140-122 |
| Phusion High-Fidelity DNA polymerase | Thermo Fischer | F-530XL |
| Phenylmethyl sulphonyl fluoride (PMSF) | Roth | 6367 |
| Poly-L-Lysine | Sigma-Aldrich | P1399 |
| Protease Inhibitor Cocktail Set III, EDTA-Free | Calbiochem/Sigma-Aldrich | 539134 |
| Protease Inhibitor cOmplete EDTA-free | Roche | 5056489001 |
| Rabbit reticulocyte lysate nuclease-treated | Promega | L4960 |
| ReproSil-Pur C18-AQ 3-μm resin | Dr. Maisch GmbH | r13.aq |
| RNasin Plus RNase inhibitor | Promega | N2615 |
| RNase-I | Thermo Fisher | EN0601 |
| SeeBlue Plus2 Prestained Protein Ladder | Thermo Fisher | LC5925 |
| SILAC-DMEM | PAN-Biotech | P04-02505 |
| Sodium borohydride (NaBH4) | Sigma-Aldrich | 452882 |
| Sodium deoxycholate | Sigma-Aldrich | D6750 |
| Sodium dodecyl sulfate (SDS) | Roth | 2326.1 |
| Sodium L-ascorbate | Sigma-Aldrich | A7631 |
| Spermidine●3HCl | Sigma-Aldrich | S2501 |
| Spermine●4HCl | Sigma-Aldrich | S2876 |
| Sucrose | Sigma-Aldrich | S0389 |
| SUPERase-In RNase inhibitor | ThermoFisher | AM2694 |
| T4 PNK | New England Biolabs | M0201S |
| Tris-HCl | Roth | 9090.3 |
| Tris(3-hydroxypropyltriazolylmethyl) amine (THPTA) | Sigma-Aldrich | 762342 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| TRIzol-LS | Invitrogen | 10296010 |
| Trypsin | Promega | V511A |
| TurboDNase | Thermo Fisher | AM2238 |
| Tween | Sigma-Aldrich | P9416 |
| Uranyl acetate | Merck | 1.08473.0100 |
| Urea | Sigma-Aldrich | 51459 |
| Vectashield Antifade Mounting Medium | Vector Laboratories | H-1000 |
| Critical Commercial Assays | ||
| Amersham ECL Prime | GE Healthcare | RPN2232 |
| Dynabeads | Life Technologies | 10008D |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | New England BioLabs | E7420L |
| NEXTflex Small RNA-seq Kit v3 | Bio Scientific | NOVA-5132-06 |
| RNA Clean & Concentrator-25 Kit | Zymo Research | R1017 |
| RiboZero Kit | Illumina | 20037135 |
| TruSeq Stranded mRNA Kit | Illumina | 20020594 |
| Zymoclean Gel DNA Recovery Kit | Zymo Research | D4007/D4008 |
| Deposited Data | ||
| Neocortex total lysate, 80S, polysome mass spectrometry | this paper | ProteomeXchange PXD014841 |
| Neuro2a pSILAC/AHA mass spectrometry | this paper | ProteomeXchange PXD014740 |
| Neocortex total lysate RNA sequencing | this paper | NIH GEO: GSE157425 |
| Cryo-EM maps of the P0 neocortical ribosome | this paper | Worldwide Protein Data Bank EMD-10321 |
| Atomic model of the P0 neocortical 60S●Ebp1 complex | this paper | Worldwide Protein Data Bank PDB: 6SWA |
| Ebp1-selective Ribosome Profiling | this paper | NIH GEO: GSE157425 |
| Ebp1-knockdown Ribosome Profiling | this paper | NIH GEO: GSE157425 |
| Ebp1-knockdown RNaseq | this paper | NIH GEO: GSE157425 |
| Experimental Models: Cell Lines | ||
| Neuro2a | Thermo Fisher | RRID: CVCL_0470 |
| Experimental Models: Organisms/Strains | ||
| CD1 WT mice | Charles River | N/A |
| Nex:Cre;Ai9 mice | Turko et al., 2019 | N/A |
| NMRI WT mice | Charles River and Janvier Labs | N/A |
| Software and Algorithms | ||
| Andromeda | Cox et al., 2011 | N/A |
| APBS | Jurrus et al., 2018 | N/A |
| CCP4Interface CONTACT | Potterton et al., 2003 | N/A |
| CLUSTAL Omega MSA (1.2.4) | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| COOT | Emsley and Cowtan, 2004 | N/A |
| CTFfind4 | Mindell and Grigorieff, 2003 | N/A |
| DAVID | Huang et al., 2009 | N/A |
| EMAN2 | Tang et al., 2007 | N/A |
| EPU | FEI Company | N/A |
| ERRASER | Chou et al., 2013 | N/A |
| FIJI | Schindelin et al., 2012 | https://fiji.sc/ |
| GraphPad Prism 7 | GraphPad Software Inc | https://www.graphpad.com/ |
| IBAQ | Schwanhäusser et al., 2011 | N/A |
| Illustrator | Adobe Creative Cloud | N/A |
| Image stitching plugin (FIJI) | Preibisch et al., 2009 | N/A |
| Leginon | Carragher et al., 2000; Suloway et al., 2005 | N/A |
| LFQ | Cox et al., 2014 | N/A |
| MaxQuant | Cox and Mann, 2008 | N/A |
| MolProbity | Chen et al., 2010 | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus | N/A |
| MotionCor2 | Zheng et al., 2017 | N/A |
| Neurite Tracer plugin (FIJI) | Longair et al., 2011 | N/A |
| Perseus | Tyanova et al., 2016 | N/A |
| PHENIX | Adams et al., 2010 | N/A |
| Photoshop | Adobe Creative Cloud | N/A |
| Plastid CS | Dunn and Weissman, 2016 | N/A |
| RiboseQC v1.1 | https://github.com/ohlerlab/RiboseQC | N/A |
| Sholl analysis plugin (FIJI) | Ferreira et al., 2014 | N/A |
| SPHIRE/SPARX | Moriya et al., 2017 | N/A |
| SPIDER | Frank et al., 1996 | N/A |
| STAR | Dobin et al., 2013 | N/A |
| TopHat2 | Kim et al., 2013 | N/A |
| UCSF Chimera | Pettersen et al., 2004 | N/A |
| UCSF ChimeraX | Goddard et al., 2018 | N/A |
| Primers | ||
| Ebp1-His forward (recombinant protein) | Eurofins | 5′AATTCCATGGGCCACCATCACCATCA CCATTCGGGCGAGGACGAGCAAC3′ |
| Ebp1-His reverse (recombinant protein) | Eurofins | 5′TTAAGGATCCTTAGTCCCCAGCTTCA TTTTCTTC3′ |
| Ebp1-HA forward (overexpression plasmid) | Eurofins | 5′gtctcatcattttggcaaagATGTACCCATA CGATGTTCCAGATTACGCTTCGGG CGAAGACGAG3′ |
| Ebp1-HA reverse (overexpression plasmid) | Eurofins | 5′cggccgcgatatcctcgaggTCAGTCCCC AGCTCCATTC3′ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were utilized in the embryonic (E12.5, E14, E15.5, E17) and early post-natal (P0) period, inclusive of both male and female sexes in each litter without discrimination. All experiments and associated procedures involving animals in this study were conducted in compliance with the welfare guidelines of the Landesamt für Gesundheit und Soziales (LAGeSo) Berlin and Charité Universitätsmedizin Berlin under certified protocols (Spahn Lab: T0267/15; Vida Lab: T0215/11; Tarabykin Lab: G00206/16, G0054/19), and the Rutgers-Robert Wood Johnson Medical School Institutional Animal Care and Use Committee (IACUC) (Rasin Lab: I12-065-10). Timed pregnant wild-type (WT) CD-1 mice were obtained from the Charles River Company and utilized for all experiments, with two exceptions: (1) for primary neocortical cell cultures and immunocytochemistry (Figure 2E), homozygous Nex:Cre females (C57BL/6) were crossed with hemizygous Ai9 males (C57BL/6J) to produce Nex:Cre;Ai9 mice as described previously (Turko et al., 2019), labeling post-mitotic glutamatergic neocortical neurons with tdTomato (protocol T0215/11); (2) for in utero electroporation (Figures 6A-6C), NMRI WT (Charles River and Janvier Labs RRID:IMSR_TAC:nmri) mice were utilized (protocols G00206/16, G0054/19).
Cell lines
Mouse neuroblastoma Neuro2a cells were obtained from Thermo Fisher (RRID: CVCL_0470) for Ebp1-selective ribosome profiling, Ebp1-knockdown ribosome profiling and RNaseq, pulsed stable isotope labeling by amino acids in cell culture (pSILAC), and bioorthogonal noncanonical amino acid tagging (BONCAT) mass spectrometry experiments.
METHOD DETAILS
Neocortex dissection and lysis
For all experiments, embryonic (E12.5, E14, E15.5, E17) and postnatal (P0) mouse neocortices were dissected in a 4 °C room in ice-cold phosphate buffered saline (PBS; ThermoFisher #14040133), frozen as tissue pellets in 1.5 mL tubes on dry ice, and stored at −80 °C. Once sufficient stocks of tissue were generated, each experiment was performed in biological replicates, such that each replicate incorporated an equivalent number of neocortices pooled from distinct litters of mice to meet the input requirements. Frozen tissue pellets were gently lysed by cryogenic grinding on ice using a P1000 tip in 1.5 mL tubes, similar to prior studies (Kraushar et al., 2014, 2015), but with the following lysis buffer: 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, pH 7.4, supplemented with 20 mM Dithiothreitol (DTT), 0.04 mM Spermine, 0.5 mM Spermidine, 1x Protease Inhibitor cOmplete EDTA-free (Roche, 05056489001), 200 U/mL SUPERase-In RNase inhibitor (ThermoFisher, AM2694), 0.3% v/v IGEPAL CA-630 detergent (Sigma, I8896). Tissue lysates were clarified of membranes to post-nuclear, post-mitochondrial supernatants by centrifugation at 16100 xg for 10 minutes at 4 °C with a benchtop centrifuge, and directly applied to downstream analysis. Ribosomal content was estimated by A260 optical density units (ODU) with a NanoDrop 1000 Spectrophotometer. Two neocortical hemispheres (one brain) yields ~2 ODU at P0, 1 ODU at E15.5, and 0.5 ODU at E12.5.
Sucrose density gradient ultracentrifugation fractionation
Sucrose density gradients were prepared in Beckman Coulter Ultra-Clear Tubes; 344057 for preparative 5 mL 10%–50% gradients (for mass spectrometry, western blot), 344060 for quantitative/analytic 14 mL 5%–45% gradients. Base buffer consisted of 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, 20 mM Dithiothreitol (DTT), 0.04 mM Spermine, 0.5 mM Spermidine, 1x Protease Inhibitor cOmplete EDTA-free (Roche, 05056489001), 20 U/mL SUPERase-In RNase inhibitor (ThermoFisher, AM2694), pH 7.4, prepared with either 5 & 45% or 10 & 50% sucrose w/v. Overlaid 5 & 45% or 10 & 50% sucrose-buffer solutions were mixed to linearized gradients with a BioComp Gradient Master 107ip. Neocortical lysates were balanced to equivalent ODU and volume across samples for comparison in analytic gradients, 3 ODU for each biological replicate. Lysates were overlaid on gradients pre-cooled to 4 °C. 5%–45% gradients were centrifuged in a SW40 rotor (Beckman Coulter) for 5 hr, 4 °C, 25000 rpm; 10%–50% gradients were centrifuged in a SW55 rotor (Beckman Coulter) for 1 hr, 4 °C, 37000 rpm. Gradients were fractionated using a BioComp Piston Gradient Fractionator and Pharmacia LKB SuperFrac, with real-time A260 measurement by an LKB 22238 Uvicord SII UV detector recorded using an ADC-16 PicoLogger and associated PicoLogger software. Collected samples were stored at −80 °C for downstream analysis. Notably, with the lysis technique described in the above Method Details (0.3% v/v IGEPAL CA-630 detergent), only cytoplasmic and ER-associated mature ribosomal subunits and complexes were measured and fractionated. Analytic gradient analysis for 40S-60S, 80S, and polysome peaks was calculated as the sum of A260 values for each peak(s), with mean and standard deviation plotted across replicates (2-3 biological replicates per stage), and significance testing by ANOVA with Dunnett’s post hoc test performed in GraphPad Prism (https://www.graphpad.com/scientific-software/prism/).
Mass spectrometry analysis of neocortex total lysate, 80S, and polysomes
Sample preparation
Samples were prepared in biological triplicate (n = 3). 80S and polysomes samples were prepared by preparative 10%–50% sucrose density gradient ultracentrifugation as described in the Methods above. Notably, ribosome fractionations only included post-nuclear, post-mitochondrial, cytoplasmic and ER-associated mature ribosomal subunits and complexes. Total input lysates were lysed in RIPA buffer to ensure isolation of the entire cellular protein content. Each biological replicate incorporated 12 neocortices (6 animals) at P0, 18 neocortices (9 animals) at E17, 24 neocortices (12 animals) at E15.5, 30 neocortices (15 animals) at E14, and 36 neocortices (18 animals) at E12.5. Tissues were pooled such that each biological replicate included an equal number of neocortices derived from multiple distinct litters of embyros/pups.
Samples were processed essentially as described previously (Imami et al., 2018). Briefly, proteins were precipitated from input lysates, or directly from sucrose gradient fractions, with ethanol, then resuspended in 50 μL of 8 M urea and 0.1 M Tris-HCl, pH 8. Proteins were then reduced with 10 mM dithiothreitol (DTT) at room temperature for 30 min, and alkylated with 50 mM iodoacetamide (IAA) at room temperature for 30 min in the dark room. Protein digestion was first performed with lysyl endopeptidase (LysC) (Wako) at a protein-to-LysC ratio of 100:1 (w/w) at room temperature for 3 hr. Then, the sample solution was diluted to final concentration of 2 M urea with 50 mM ammonium bicarbonate (ABC). Trypsin (Promega) digestion was performed at a protein-to-trypsin ratio of 100:1 (w/w) under constant agitation at room temperature for 16 hr. Peptides were desalted with C18 Stage tips (Rappsilber et al., 2007) prior to LC-MS/MS analysis.
NanoLC-MS/MS analysis
Measurements were performed essentially as described previously with minor adjustments. Reversed-phase liquid chromatography was performed by employing an EASY nLC 1000 or 1200 (Thermo Fisher) using self-made fritless C18 microcolumns (Ishihama et al., 2002) (75 μm ID packed with ReproSil-Pur C18-AQ 3 mm resin, Dr. Maisch GmbH) connected on-line to the electrospray ion source (Proxeon) of a Q Exactive plus (Thermo Fisher). The mobile phases consisted of (A) 0.1% formic acid and 5% acetonitrile and (B) 0.1% formic acid and 80% acetonitrile. Peptides were eluted from the analytical column at a flow rate of 200 nL/min by altering the gradient: 5%–6% B in 2 min, 6%–8% B in 18 min, 8%–20% B in 80 min, 20%–33% in 80 min, 33%–45% B in 20 min, 45%–60% B in 2 min, 60%–95% B in 1 min. The Q Exactive plus instrument was operated in the data dependent mode with a full scan in the Orbitrap followed by top 10 MS/MS scans using higher-energy collision dissociation (HCD). The full scans were performed with a resolution of 70,000, a target value of 3x106 ions and a maximum injection time of 20ms. The MS/MS scans were performed with a 17,500 resolution, a 1x106 target value, and a 60 ms maximum injection time. The isolation window was set to 2 and normalized collision energy was 26. Ions with an unassigned charge state and singly charged ions were rejected. Former target ions selected for MS/MS were dynamically excluded for 30 s.
Processing of mass spectrometry data
All raw data were analyzed and processed by MaxQuant (v1.5.1.2) (Cox and Mann, 2008). Default settings were kept except that ‘match between runs’ was turned on. Search parameters included two missed cleavage sites, cysteine carbamidomethyl fixed modification and variable modifications including methionine oxidation, protein N-terminal acetylation and deamidation of glutamine and asparagine. The peptide mass tolerance was 4.5 ppm and the MS/MS tolerance was 20 ppm. Minimal peptide length of 7 amino acids was required. Database search was performed with Andromeda (Cox and Mann, 2008; Cox et al., 2011) against the UniProt/SwissProt mouse database (downloaded 11/2014) with common serum contaminants and enzyme sequences. The false discovery rate (FDR) was set to 1% at peptide spectrum match (PSM) level and at protein level. Protein quantification across samples was performed using the label-free quantification (LFQ) algorithm (Cox et al., 2014). A minimum peptide count required for LFQ protein quantification was set to two. Only proteins quantified in at least two out of the three biological replicates were considered for further analyses. LFQ intensities were log2-transformed and imputation for missing values was performed in Perseus (Tyanova et al., 2016) software based on a simulated normal distribution to represent low abundance values below the noise level (generated at 1.8 standard deviations of the total intensity distribution, subtracted from the mean, and a width of 0.3 standard deviations). Hierarchical clustering of the input, 80S, and polysome data for ANOVA significant proteins (FDR = 0.05) was done in Morpheus (https://software.broadinstitute.org/morpheus), with clustering based on one minus Pearson correlation using an average linkage method. Proteins whose abundance differed significantly among developmental stages were identified by multiple sample ANOVA test at a permutation-based FDR cutoff of 0.05. Log2 LFQ intensities were further z-transformed for only significantly changed proteins.
To estimate protein abundance within input and ribosome fractions, the intensity-based absolute quantification (iBAQ) algorithm was used (Schwanhäusser et al., 2011). Stoichoimetry jitter plots compared the median Rpl and Rps iBAQ value across replicates with each gene, plotting the log2 transformed ratio. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Vizcaíno et al., 2014) (http://proteomecentral.proteomexchange.org) via the PRIDE partner: PXD014841
RNA sequencing of total neocortex lysates
Total RNA was isolated from post-nuclear, post-mitochondrial, total neocortical lysates prepared as described above in biological duplicate (n = 2), with each replicate including the following number of neocortical hemispheres (animals) at each developmental stage: E12.5, 80 (40); E14, 60 (30); E15.5, 42 (21); E17, 40 (20); P0, 34 (17). Tissues were pooled such that each biological replicate included an equal number of neocortices derived from multiple distinct litters of embyros/pups. RNA was isolated with TRIzol-LS (Invitrogen, 10296010), and 1 μg of RNA per sample was used to prepare libraries with the TruSeq Stranded mRNA kit (Illumina, 20020594) according to manufacturer’s instructions. Sequencing was performed on a HiSeq4000. Reads were aligned to the mouse M12 genome using the splice aware aligner STAR (Dobin et al., 2013), and GENCODE (Frankish et al., 2019) gene annotation GRCm38.p5. We used the STAR parameters ‘–alignSJoverhangMin 8–alignSJDBoverhangMin 1–outFilterMismatchNmax 999–out-FilterMismatchNoverLmax 0.04–alignIntronMin 20’ and default otherwise. Gene-level counts were produced using the subread package, with duplicates and multimappers discarded. TPMs were calculated using the total exon length for each gene. Significantly changing levels over time of Ebp1, or the median value of Rpl, Rps, and translation-associated gene groups, was assessed by one-way ANOVA followed by Bonferroni corrected post hoc testing versus E12.5. RNaseq data have been deposited in the NIH Gene Expression Omnibus (GEO) (Edgar et al., 2002): GSE157425
Western blot
Analysis was performed with the NuPAGE (Invitrogen) western blot system according to the manufacturer’s protocol, including 4%–12% Bis-Tris NuPAGE gels (Invitrogen NP0321BOX, NP0322BOX, NP0323BOX), MES running buffer, and transfer onto nitrocellulose membranes (Amersham Protran 0.45 NC, GE Life Sciences, 10600002) with NuPAGE transfer buffer (NP0006) prepared with 10% methanol. All membranes were blocked in phosphate buffered saline with Tween (PBST; 0.5% Tween) prepared with 5% milk (w/v) for 20 minutes at room temperature, followed by overnight incubation with primary antibody at 4 °C in PBST-5% milk. Primary antibodies: anti-Ebp1CT (rabbit, Abcam, ab35424), anti-Ebp1NT (rabbit, Millipore, ABE43), anti-Gapdh (mouse, Millipore, MAB374), anti-Rpl7/uL30 (rabbit, Abcam, ab72550), anti-Rps5/uS7 (mouse, Santa Cruz, sc-390935). Membranes were then washed in PBST at room temperature, and HRP secondary antibodies applied in PBST with 5% milk for 1 hour at room temperature, and again washed in PBST before developing (Amersham ECL Prime Western Blotting Detection Reagent, GE Healthcare, RPN2232) and imaging (GE Amersham Imager 600). Secondary antibodies: HRP-anti-rabbit-Light Chain (mouse, Dianova, 211-032-171), HRP-anti-mouse-Heavy Chain (goat, Millipore, 71045).
Importantly, note that HRP-anti-Light Chain secondary antibody was used because probing with HRP-anti-Heavy Chain secondary antibody introduced a non-specific band (~50 kDa) just above Ebp1 signal (48 kDa), obscuring the interpretation of actual Ebp1 signal. Band molecular weights were compared to the SeeBlue Plus2 Prestained Standard Protein Ladder (Thermo Fisher, LC5925) as shown in each figure. Band signal intensity was measured using GE Amersham Imager 600 software, with significance testing by ANOVA with Dunnett’s post hoc test (≥3 comparisons), or two-tailed unpaired t test (≤2 comparisons), versus E12.5 with GraphPad Prism. Western blot signal for endogenous Ebp1 in lysates was compared to full-length recombinant Ebp1 with a N-terminal polyhistidine tag (Ebp1-His) as a marker, which was cloned in a pET-28a(+) backbone (Novagen #69864-3) and purified as described (Kowalinski et al., 2007). Sucrose density gradient fractions analyzed by western blot were controlled for equal loading by equivalent lysate ODU (3 ODU per sample) as the input across all purifications.
Single-cell RNA sequencing analysis
The transcriptional birthdate and differentiation maps for individual genes were acquired from the open source website associated with (Telley et al., 2019) (http://genebrowser.unige.ch/telagirdon/#query_the_atlas). Averaging the data across all Rpl and Rps mRNAs into combined single maps for these gene families was performed with the kind support of Ludovic Telley and the Denis Jabaudon lab with a customized in-house computational pipeline.
Neocortex immunohistochemistry
Tissue processing and immunohistochemistry was performed similar to the previously described method (Kraushar et al., 2014). In brief, embryonic (E12.5, E14, E15.5, E17) and postnatal (P0) mouse brains were dissected at 4°C in ice cold PBS (ThermoFisher, 14040133), and initially immersion-fixed with 4% (w/v) paraformaldehyde (PFA) in PBS (PBS-PFA; pH 7.4) at room temperature for 30 min, followed by overnight PBS-PFA fixation at 4°C. Fixed brains were then embedded in 3.2% agarose-PBS, and coronally sectioned at 70 μm on a Leica vibratome (VT1000S). Sections of the anterior sensorimotor neocortex were collected, incubated in blocking solution (PBS, 10% normal donkey serum, 2% w/v BSA, 0.2% w/v glycine, 0.2% w/v lysine), then incubated overnight in probing solution with 0.4% Triton-X and primary antibody at 4°C. Primary antibodies: anti-Map2 (chicken, Millipore, AB5543), anti-Ebp1CT (rabbit, Abcam, ab35424), anti-Ebp1NT (rabbit, Millipore, ABE43). Samples were washed in PBS, then all secondary antibodies, Alexa 488 anti-rabbit (goat, Jackson ImmunoResearch) and Alexa 647 anti-chicken (goat, Jackson ImmunoResearch), were applied at 1:250 dilution in probing solution for 2 hr at room temperature, washed, incubated with DAPI (NucBlue, Molecular Probes, Invitrogen, R37606) for 10 min, and mounted with Vectashield. Confocal imaging was performed with an upright confocal microscope (FV-1000, Olympus), 20x air objective, maintaining constant parameters and setting across all images. Images were analyzed using the FIJI distribution of ImageJ software (Schindelin et al., 2012) (https://fiji.sc/), including the pairwise stitching plugin (Preibisch et al., 2009), maintaining constant LUT parameters across images. Ebp1 fluorescence intensity per unit area quantification was performed in FIJI (Analysis > Measure function), for 5-7 fields per region of interest (ventricular zone, cortical plate, lower layers, upper layers) per developmental stage, and heatmap representation of the data average in GraphPad Prism.
Immuno-electron microscopy
Neocortex was dissected at E12.5, E15.5, and P0 at 4°C as described above, and immersion fixed at 4°C in phosphate buffered saline (PBS) containing 4% PFA and 0.1% Glutaraldehyde overnight, followed by 24 hours incubation in 4% PFA-PBS, and finally stored in 1% PFA-PBS. In order to identify the subcellular localization of EBP1 protein in neocortical precursor/stem and neuronal cells at different developmental stages, we performed pre-embedding nanogold-silver enhanced immunolabeling for Ebp1.
Fixed brains were rinsed several times in PBS and sectioned on a Vibratome (Leica VT1000S) at 50-100 μm. Floating sections were washed again in PBS, followed by incubation in 0.1% sodium borohydride (NaBH4; Sigma-Aldrich, 452882) in PBS for 15 min to inactivate residual aldehyde groups. Sections were then washed with PBS several times until the solution was clear of bubbles. To improve reagent penetration, the sections were then treated with PBS containing 0.05% Triton X-100 for 30 min and then washed 3x with PBS. To avoid nonspecific binding, sections were incubated for 1 hr in blocking solution containing 5% normal goat serum (NGS; PAN Biotech, P30-1002), and 5% bovine serum albumin (BSA; Sigma-Aldrich, A3294) in PBS. All following immuno-incubations were done with gentle agitation, overnight at 4°C. After blocking, sections were incubated with primary antibodies: rabbit anti-Ebp1NT (rabbit, Millipore, ABE43) or rabbit anti-EBP1CT (rabbit, Abcam, ab35424) diluted in PBS containing 0.5% acetylated BSA (BSA-c, Aurion, 900.022). To ultimately validate the signal and rule out non-specific secondary labeling, we also prepared primary antibody leave-out control samples. After washes with PBS/BSA-c, sections were incubated in the secondary nanogold conjugated antibody (Nanoprobes, 2003) goat anti-rabbit IgG diluted 1:100 in PBS/BSA-c. To remove unbound secondary antibodies, sections were washed thoroughly with PBS/BSA-c and then with PBS. Subsequently, sections were post-fixed with 2% GA in PBS for 2 hr to crosslink nanogold in the tissue in order to prevent the loss of labeling during subsequent processing. Next, sections were washed several times in PBS and in double distilled water (ddH2O) and prepared for silver enhancement according to the manufacturer’s instruction (Nanoprobes). For structural stabilization, section were incubated with buffered 1% osmium tetroxide (OsO4; Polysciences, 0972A) for 1 hr and then washed in PBS followed by ddH2O. Sections were dehydrated in increasing concentrations of ethanol and flat-embedded in Epoxy embedding medium (Epon 812; Sigma-Aldrich, 45345) between two sheets of Aclar film (Plano, 10501-10). After resin polymerization at 60°C, small pieces of cortex were dissected, mounted on plastic stubs, and sectioned en face into 60-65 nm sections on an Ultramicrotome (Reichert Ultracut S, Leica) and mounted on 200-mesh Formvar-coated nickel grids (Plano, G2710N). Ultrathin sections were finally stained with 2% aqueous uranyl acetate (Merck, 1.08473.0100) for 2 min and with lead citrate (Fluka #GA10655) (Reynolds, 1963) for 30 s. Sections were imaged using a Zeiss TEM-912 equipped with a digital camera (Proscan 2K Slow-Scan CCD-Camera, Zeiss).
Quantification of immuno-labeling distribution between the nucleus and the cytoplasm in stem cells and recently born daughter neurons in the ventricular zone, and neurons in the cortical plate, was performed in Image-J and calculated as gold particles per μm2, with significance testing by Welch ANOVA in GraphPad Prism, and further comparison with primary antibody leave-out controls. Subcellular regions of interest were highlighted in the images with pseudo-color in Adobe Photoshop, as detailed in the guide by Eric Jay Miller (http://www.nuance.northwestern.edu/docs/epic-pdf/Basic_Photoshop_for_Electron_Microscopy_06-2015.pdf).
Primary neocortical culture and immunocytochemistry
Primary E12.5 neocortical cultures were prepared from Nex:Cre;Ai9 animals, as previously described (Turko et al., 2019). Briefly, dissected neocortex tissue was dissociated with Papain for 25 min (1.5 mg/ml) before trituration in bovine serum albumin (10 mg/ ml). Cells were then resuspended in Neurobasal (medium, supplemented with 1x B27, 1x Glutamax, and 100 U/ml Penicillin-Streptomycin). Dissociated cells were grown on 12 mm round, glass coverslips coated with Poly-L-Lysine (20 μg/ml) in 24-well plates. Cells were plated in 40 μl droplets at a concentration of 500 cells per μl (total: 20,000 cells per coverslip). Cultures were grown in humidified conditions at 37°C, 5% CO2. Cells were cultured for 5 days to allow for neural stem cell (NSC) differentiation into post-mitotic Nex-positive neurons.
At days in vitro 0, 2, 4 and 5 coverslips were fixed and analyzed by immunocytochemistry for Ebp1 expression in Nestin-positive NSCs and Nex-positive neurons as described previously (Turko et al., 2019). In brief, cells were fixed for 15 min in 4% paraformaldehyde (PFA), 4°C solution before subsequent washes in: 0.1 M phosphate buffered solution (PB) and phosphate buffered saline (PBS). All antibodies were diluted (1:1000) in PBS with 0.1% Triton X-100, and incubated overnight at 4°C on an orbital shaker. Primary antibodies: anti-Nestin (mouse, Millipore, MAB353), anti-Ebp1NT (rabbit, Millipore, ABE43). Secondary antibodies: Alexa Fluor 647-conjugated anti-mouse (goat, Jackson ImmunoResearch) and Alexa Fluor 488-conjugated anti-rabbit (goat, Jackson ImmunoResearch). DAPI was applied to visualize nuclei (NucBlue, Molecular Probes, Invitrogen, R37606). Coverslips were mounted on glass slides using Fluoromount-G (Southern Biotech, #0100-01). Images were captured on an upright confocal microscope (FV-1000, Olympus) using 30x silicon oil-immersion objective (1.05 NA, 0.8 mm WD). Images were analyzed using the FIJI distribution of ImageJ software, maintaining constant LUT parameters across images.
Cryo-electron microscopy
Sample and grid preparation
Pooled 80S and polysomal ribosomes were purified ex vivo by preparative 10%–50% sucrose density gradient ultracentrifugation from dissected frozen P0 mouse neocortex tissue as described above, but with the following adaptations optimizing for cryo-electron microscopy (cryo-EM). Frozen P0 mouse neocortex (32 animals, 64 neocortex hemispheres) were lysed by cryogenic pulverization with 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, pH 7.4, supplemented with 20 mM Dithiothreitol (DTT), 0.04 mM Spermine, 0.5 mM Spermidine, 1x Protease Inhibitor cOmplete EDTA-free (Roche #05056489001), 480 U/mL RNasin Plus RNase inhibitor (Promega, N2615), 0.3% v/v IGEPAL CA-630 detergent (Sigma, I8896), and 0.1 mg/mL cycloheximide (Sigma-Aldrich, C7698). Lysates were subjected to further passive lysis by incubation for 1 hr on ice to enhance lipid membrane dissociation, followed by lysate clarification as above. 10%–50% sucrose gradients in Beckman Coulter Ultra-Clear Tubes (344057) were prepared with a base buffer of 10 mM HEPES, 50 mM KCl, 5 mM MgCl2, to pH 7.4, supplemented with 20 mM Dithiothreitol (DTT), 0.04 mM Spermine, 0.5 mM Spermidine, 1x Protease Inhibitor cOmplete EDTA-free, 40 U/mL RNasin Plus RNase inhibitor, and 0.1 mg/mL cycloheximide. Samples were centrifuged in a SW55 rotor for 50 min at 37000 rpm, 4°C. Fractions corresponding to the 80S and polysomal peaks were collected, pooled, and diluted 1:1 v/v with 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, pH 7.4, supplemented with 20 mM Dithiothreitol (DTT), 0.04 mM Spermine, 0.5 mM Spermidine, 1x Protease Inhibitor cOmplete EDTA-free, and 0.1 mg/mL cycloheximide to dilute the sucrose concentration to ≤ 20%. Samples were then pelleted by ultracentrifugation in Beckman Coulter Ultra-Clear Tubes (344057) with a SW55 rotor for 50 min at 37000 rpm, 4°C. Pellets were resuspended in the same dilution buffer, testing for concentration and quality control by negative stain EM with 2% uranyl acetate. Samples were diluted 1:6 with resuspension buffer, and 3.6 μL of sample were applied to glow-discharged holey carbon grids (Quantifoil R3/3 100 Holey Carbon Films; 2 nm carbon; Micro Tools GmbH), blotted with a Vitrobot device (FEI) for 2-4 s at 4°C, and plunged in liquid ethane. Samples were stored in liquid nitrogen until imaging.
Cryo-EM data collection
Initial datasets were collected for sample quality control and low-resolution ribosome reconstruction on a 120 keV Tecnai Spirit cryo-EM (FEI; MPI Molecular Genetics, Berlin) equipped with a CMOS camera (TVIPS), with automated Leginon software (Carragher et al., 2000; Suloway et al., 2005). Projection images were then analyzed by 3-D reconstruction and unsupervised classification for intrinsic ribosomal structure heterogeneity in silico with SPIDER (Frank et al., 1996) as described previously (Behrmann et al., 2015; Loerke et al., 2010). These data revealed the presence of extra-ribosomal density at the 60S exit tunnel. To validate these findings, an independent biological replicate sample was re-prepared, with new grids frozen, and likewise imaged using the same protocol, yielding identical density at the 60S exit tunnel.
High-resolution data were collected on a 300 keV Titan Krios (FEI; EMBL, Heidelberg; Diamond Light Source, Oxfordshire) equipped with a Gatan Quantum K2 direct electron detector at 103.000x magnification, yielding a pixel size of 0.66 Å on the object scale. Movie stacks were collected in super-resolution mode with SerialEM (Mastronarde, 2005) with the following parameters: defocus range of 0.5-2.5 μm, 40 frames per movie, 20 s exposure time, electron dose of 1.589 e/Å2/s and a cumulative dose of 31.78 e/Å2 per movie.
Computational analysis
High-resolution data collection yielded 5379 movies. The movies were aligned and dose-weighted using MotionCor2 (Zheng et al., 2017) and initial estimation of the contrast transfer function (CTF) was performed with the CTFfind4 package (Mindell and Grigorieff, 2003). Resulting micrographs were manually inspected to exclude images with substantial contaminants (typically lipid/membranes) or grid artifacts. Power spectra were manually inspected to exclude images with astigmatic, weak, or poorly defined spectra. The dataset included 4501 micrographs after these quality control steps (84% of total). Ribosomal particle images were identified using the “swarm” function within e2boxer from the EMAN2 software package (Tang et al., 2007). After the manual removal of artifact particle images, the dataset contained 208206 particle images.
For multiparticle sorting and 3D refinement, the SPHIRE package (Moriya et al., 2017) was used for all steps except for 3D classification, which was performed using a python/SPARX-implementation of the incremental k-means algorithm described previously (Loerke et al., 2010). Therein, two modes for classification exist: (1) refinement, either global or local; and (2) focused classification based on a binary mask, defining a region of interest (ROI) (Penczek et al., 2006). Such a focused mask was derived from 3D variability calculations, which visualizes regions of high heterogeneity with the 3D volume (Behrmann et al., 2015).
However, since heterogeneous regions outside the binary mask can influence the classification, a more sensitive approach was implemented. The “nue” mode, named after the hybrid beast in japanase folklore, creates a hybrid map for each class in a simple procedure: a weighted average of all classes is calculated and used as the “outside.” The ROI within the focused mask is extracted for each class and used as the “inside.” Therefore, the focused mask is transformed into a soft mask by adding a smooth falloff at the edges. For each class, the “outside” map is combined with the respective “inside” map, normalized and filtered, forming the “nue”- map for each class. These “nue” maps are then used as references for focused classification. The “nue” maps only differ within the region of interest, reducing the influence of any peripheral variations. A new set of “nue” maps are calculated at the beginning of each iteration. A similar approach was implemented in Frealign/cisTEM, in which the outside area can be filtered or weighted down in order to reduce its influence during the classification (Grant et al., 2018; Grigorieff, 2016; Zhang et al., 2019).
For the initial refinement, particle images were extracted at a box size of 360 pixels with a pixel size of 1.32 Å/px. All particles were aligned using sxmeridien using a filtered 80S yeast ribosome cryo-EM map as a reference. The refinement yielded a consensus map with sub-nanometer global resolution depicting fragmented densities for the small subunit, tRNAs, eEF2, and Ebp1. In order to separate this dataset into homogeneous sub-states, a hierarchical classification scheme was employed as described previously (Behrmann et al., 2015). Three tiers of sorting were performed, whereby large-scale heterogeneity (e.g., subunit rotation) was classified first, before sorting based on more subtle differences (+/− Ebp1).
In the first tier of sorting, particle images and parameters were decimated to 3.96 Å/px at a box size of 120 px to minimize computational expense and limit the resolution for classification. This yielded a rotated 80S, classical 80S, and an artifact population, achieved by an incremental K-means procedure using global and local refinement.
In the second tier of sorting, rotated and classical populations were separated and treated independently. Particle images were decimated to 2.64 Å/px at a box size of 180 px. Focused classification was performed, since the maps already depicted high-resolution features. A strong signal of 3D variability was detected in the tRNA-binding site and at the eEF2 binding site, and thus focus masks were constructed in order to separate classes with different compositions of tRNA and eEF2. The “rotated”-branch was separated into two classes: (1) +eEF2, and (2) +eEF2 +P/E-tRNA. The “classical”-branch was separated into three classes: (1) +A-tRNA+-P-tRNA, (2) +E-tRNA, and (3) empty 80S. However, within these five classes, the Ebp1 density still appeared fragmented, suggesting further heterogeneity in this region. These findings were confirmed by 3D variability calculations.
In the final sorting tier, particle images were separated into these five classes, and decimated to 2.64 Å/px at a box size of 180 px. A focus mask enclosing the Ebp1 region was defined based on the 3D variability of each of the second-tier classes, and used for sorting into Ebp1-positive and Ebp1-negative classes. The results yielded a nearly equal distribution of Ebp1-positive and Ebp1-negative ribosomes in each sub-state, with an overall Ebp1 · 80S occupancy of 52% in our dataset.
Finally, four of these classes were refined at 1.326 Å/px decimation with box size of 360 px, yielding near-atomic global resolution for all ribosomal complexes, and allowing for the building of an atomic model of the mouse neocortical ribosome 60S·Ebp1. Euler distributions and global Fourier Shell Correlations (FSCs) were calculated, in addition to the local resolutions of these maps with SPHIRE. Local resolution for Ebp1 ranges from 4 Å at the rRNA binding site to 6 Å at the solvent-side periphery. Ebp1-positive and Ebp1-negative maps yielded similar global and local resolutions from a similar particle number, permitting the use of the Ebp1-negative map as an internal control for the structural interpretation.
Cryo-EM maps for the neocortical 80S·Ebp1 complex, including both the rotated state with eEF2 and the classical state with A/A+P/P tRNAs, are deposited in the Worldwide Protein Data Bank (wwPDB; https://www.wwpdb.org/) with accession code EMD-10321.
Model building
Since our focus was the interaction surface of Ebp1 on the mouse neocortical ribosome, we modeled the 60S subunit in complex with Ebp1 from the cryo-EM map. Modeling was performed in density for the rotated sub-state (+) Ebp1, since this map achieved the highest global resolution of 3.1 Å. A 60S model derived from human polysomes (PDB: 5AJ0) (Behrmann et al., 2015) was used as a starting model for the ribosomal proteins, and a rabbit 60S model (PDB: 6GZ5) (Flis et al., 2018) was the starting model for rRNA. A pre-existing crystallographic model of mouse Ebp1 (PDB: 2V6C) (Monie et al., 2007) was utilized to model Ebp1 density, downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) (Berman et al., 2000) website https://www.rcsb.org/. For all models, an initial rigid body docking was performed in UCSF Chimera v1.10.2 (Pettersen et al., 2004) (http://www.rbvi.ucsf.edu/chimera), with subsequent adjustment within the density performed in COOT (Emsley and Cowtan, 2004). Thereafter, the models were globally optimized by real-space refinement in PHENIX (Adams et al., 2010) and validated with MolProbity (Chen et al., 2010). To prevent over-fitting during refinement, the applied weight was optimized by monitoring correlation of the map versus model in half-sets of the cryo-EM map (Brown et al., 2015; Greber et al., 2014; Sprink et al., 2016), using individually-determined weight factors. rRNA stuctures were further refined with ERRASER (Chou et al., 2013). Molecular graphics and analysis for figure preparation was performed with UCSF Chimera v1.10.2 and UCSF ChimeraX v0.9.0 (Goddard et al., 2018) (https://www.cgl.ucsf.edu/chimerax/). Analysis of atomic interactions between Ebp1 residues and ribosomal proteins/rRNA was aided by the CCP4Interface 7.0.073 (Potterton et al., 2003) CONTACT algorithm to compute atomic distances between the Ebp1 crystallographic model (PDB 2V6C) and modeled ribosomal proteins/rRNA as input. Atomic distances deemed significant and highlighted as electrostatic contacts were between 0.93-3.95Å. Electrostatic potential maps were generated for Ebp1 (PDB 2V6C), Metap2 (PDB 1KQ9), and Arx1 (PDB 5APN) in UCSF Chimera v1.10.2 using the APBS (Jurrus et al., 2018) interface and webserver https://nbcr-222.ucsd.edu//pdb2pqr_2.1.1/.
The neocortical 60S·Ebp1 atomic model is deposited in the wwPDB with accession code PDB ID 6SWA.
Binding specificity and affinity analysis of Ebp1 ·ribosmal subunits
40S and 60S subunits were purified from mouse neocortex and rabbit reticulocyte lysate (RRL) essentially as described previously (Pisarev et al., 2007). Briefly, 40 frozen P0 neocortices (40 ODU) were lysed as described above, and ribosomes pelleted through a 1 M sucrose cushion in base buffer (20 mM HEPES, 100 mM KCl, 10 mM MgCl2, 42 U/mL SUPERase-In RNase inhibitor, pH 7.4) in Beckman Coulter Ultra-Clear Tubes (344057) with a SW55 rotor at 50000 rpm for 5.5 hr, 4°C. 80S ribosome pellets were resuspended in base buffer, and subjected to a puromycin reaction as described (Pisarev et al., 2007) to release 40S and 60S subunits. Subunits were separated on a 10%–30% sucrose high-salt gradient (20 mM HEPES, 0.5 M KCl, 10 mM MgCl2, 8 U/mL SUPERase-In RNase inhibitor) prepared as described above, by ultracentrifugation in Beckman Coulter Ultra-Clear Tubes (344060) with a SW40 rotor at 27000 rpm for 12 hr, 4 °C. Subunits were fractionated and collected as described above, and desalted using Amicon Ultra 0.5 mL 100 kDa MWCO spin columns (Millipore/Sigma UFC510024) and reconstituted 1:3 v/v with low salt buffer (20 mM HEPES, 10 mM KCl, 2 mM MgCl2). 40S and 60S subunit concentrations were quantified by NanoDrop Spectrophotometer.
Recombinant Ebp1 with an N-terminal Histidine tag (Ebp1-His) was cloned into a pET-28a(+) backbone (Novagen #69864-3) and purified as described (Kowalinski et al., 2007). For Ebp1-His binding to mouse neocortex 40S and 60S subunits, 5 nM and 20 nM of Ebp1-His was reconstituted with 100 nM subunit in 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, and incubated for 30 min at 37°C. Samples were pelleted through a 15% sucrose cushion containing 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, 0.04 mM Spermine, 0.5 mM Spermidine in Beckman Coulter 230 μL Thickwall Polypropylene Tubes (343621) with a TLA100 rotor at 35000 rpm for 20 hr at 4°C, separating unbound Ebp1-His from pelleted subunits with bound Ebp1-His. Pellets of subunits-Ebp1-His were resuspended in 20 mM HEPES, 100 mM KCl, 10 mM MgCl2. Binding was assessed by western blot loading supernatant and pellet resuspensions of 40S and 60S samples on the same gel, and probing with Ebp1CT (rabbit, Abcam, ab35424), uL30/Rpl7 (rabbit, Abcam, ab72550), and uS7/Rps5 (mouse, Santa Cruz, sc-390935) antibodies on the same membrane.
Rabbit reticulocyte 40S and 60S subunits were purified and reconstituted to 80S ribosomes as described (Pisarev et al., 2007) from RRL. Binding of 200 nM Ebp1-His to 100 nM rabbit 40S, 60S, and 80S was performed as described above. For dose-response binding of Ebp1-His to 60S rabbit subunits, 100 nM 60S was reconstituted with 1:1 serial dilutions (to 0.5x concentrations) of Ebp1-His from 500 nM to 15.625 nM with 20 mM HEPES, 100 mM KCl, 10 mM MgCl2, 0.04 mM Spermine, 0.5 mM Spermidine. Binding was assessed by pelleting and western blot as described above at each dilution in parallel. This was repeated with a different Ebp1-His dose range between 325 nM to 81.25 nM. Western blot quantification was performed by normalizing Ebp1CT signal to uL30 (Rpl7) signal, subtracting any signal detected in the supernatant, and generating a single dose-response curve including data from both independent experiments. The Ebp1-His concentration demonstrating maximum binding (Ebp1CT/uL30) in each experiment was set to 100%. Curves were fit using the GraphPad Prism, with the best fit achieved by non-linear one site-specific binding with Hill slope accommodation.
Binding dynamics of Ebp1 to the rabbit 60S during mRNA translation were assessed by comparing the following mixtures: (1) 100 nM of rabbit 60S with saturating levels of (350 nM) Ebp1-His; (2) endogenous Ebp1 in RRL (100 nM ribosomes estimated by A260 measurement after ribosome pelleting, 1 A260 ~20 pmol 80S); (3) 350 nM Ebp1-His added to RRL (100 nM ribosomes). All the above mixtures were prepared in parallel, and incubated at 30°C for 30 min, 650 rpm. Mixtures were pelleted through a sucrose cushion as described above to separate unbound versus bound Ebp1, and pellets likewise analyzed by western blot.
Ebp1-Selective Ribosome Profiling (Ebp1 SeRP)
Using our cryo-EM structure of the Ebp1-ribosome complex, we chose the Ebp1CT antibody (Abcam, ab35424) that targets a solvent-exposed epitope (Ebp1 C-terminal domain) for immunoprecipitation, which we validated by western blot in comparison to the band for full-length recombinant Ebp1 (Figure 1F), and by Ebp1 knockdown in Neuro2a cells (Figure S13A). We first confirmed that Ebp1 specifically associates with Neuro2a ribosomes by 10%–50% sucrose density gradient fractionation as described above, with and without 100 mM EDTA in the lysis and gradient buffers to dissociate ribosome subunits (Figures S11B and S11C).
Nuclease titration
Three 15 cm plates of Neuro2a cells were grown in standard DMEM (GIBCO, 31966047) with 10% FBS (GIBCO, 10270106) and 1% penicillin-streptomycin (GIBCO, 15140122) to a confluence of 80% in humidified 37°C, 5% CO2. Cells were washed and detached in 10 mL ice-cold PBS supplemented with 12 mM MgCl2 and 100 mg/mL cycloheximide (Sigma-Aldrich, C7698) and centrifuged for 4 min at 300 xg, 4°C. The cell pellet was lysed in 0.5 mL lysis buffer (20 mM Tris-HCl, pH 8.0, 140 mM KCl, 12 mM MgCl2, 0.5% NP-40, 100 μg/mL cycloheximide, 25 U/mL DNase-I (Roche, 4716728001), 1x Protease Inhibitor cOmplete EDTA-free (Roche, 05056489001), 1 mM PMSF (Roth, 6367)). The lysate was passed five times through a 23 G needle and cleared by centrifugation (2 min at 13000 xg, 4°C). The supernatant was equally divided into six aliquots, and aliquots destined for MNase digest were supplemented with 5 mM CaCl2. Lysate aliquots were then digested with RNase-I (Ambion #AM2294) or MNase (homemade) under the conditions specified in Figure S12A. Nuclease digests were stopped by addition of 10 μL SUPERase-In (Ambion, AM2696) (RNase-I digests) or 10 mM EGTA (MNase digests) and subsequent cooling on ice. Afterward, digested samples were loaded on 5%–45% linear sucrose gradients (5%–45% sucrose, 20 mM Tris-HCl pH 8.0, 140 mM KCl, 12 mM MgCl2, 100 μg/mL cycloheximide) and centrifuged for 2.5 hr, 35000 rpm, at 4°C (Beckman SW 40 Ti Rotor). Polysome profiles were recorded with the Piston Gradient FractionatorTM (Biocomp).
As previously described for mouse tissue lysates (Gerashchenko and Gladyshev, 2017), we observed both monosome and polysome degradation with RNase-I digestion, which may compromise the quality of ribosome immunoprecipitation (IP) for selective ribosome profiling, since we found Ebp1 binds 60S via rRNA helices. In contrast, MNase digestion decreased polysome enrichment with a concomitant increase in monosomes, and was utilized for this experiment.
Purification of Ebp1-ribosome-nascent chain complexes for SeRP
Two 15 cm plates of Neuro2a cells were grown and lysed as above, comprising n = 2 biological replicates, using the lysis buffer supplemented with 5mM CaCl2. After clearing the lysate by centrifugation (2 min at 13000 xg, 4°C), the supernatant was divided for total (200 μL) and IP (400 μL) translatome samples.
Total samples were digested with MNase (300 U / 1 A260 nm) for 30 min at 4°C, under rotation. The reaction was terminated by addition of 10 mM EGTA and ribosomes were purified by sucrose cushion centrifugation (25% sucrose, 20 mM Tris-HCl pH 8.0, 140 mM KCl, 12 mM MgCl2, 100 μg/mL cycloheximide, 1 mM PMSF) for 90 min, 75000 rpm, at 4°C (Beckmann AT2 S120 rotor). Pelleted ribosomes were resuspended in lysis buffer lacking CaCl2 and subjected to phenol-chloroform extraction of RNA.
IP samples were mixed with 100 μL Dynabeads (Life Technologies, 10008D) conjugated with 10 μg anti-Ebp1CT (Abcam, ab35424) and digested using MNase (300U / 1 A260nm) for 30 min at 4°C, under rotation. The reaction was terminated by addition of 10mM EGTA and the beads were washed three times in 400 μL wash buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 12mM MgCl2, 1 mM PMSF, 100 μg/mL cycloheximide, 0.1% NP-40) followed by phenol-chloroform extraction of RNA.
cDNA library preparation for deep sequencing
Gel purification and 3′-dephosphorylation of ribosome-protected footprints from total and IP samples was performed as previously described (Galmozzi et al., 2019). Footprints were then 5′-phosphorylated using T4 PNK (NEB, M0201S) and ATP for 1 hr at 37°C. Deep sequencing libraries were prepared from these RNA fragments using the NEXTflex Small RNA-seq Kit v3 (Bio Scientific, NOVA-5132-06). Deep sequencing was performed on an Illumina Next-Seq 550 system.
Data analysis
Raw reads were processed as described previously (Galmozzi et al., 2019) using standard analysis tools (Cutadapt, Bowtie2, To-pHat2) and python scripts adapted to Mus musculus. In short, trimmed reads between 23 and 37 nucleotides in length were aligned to the mouse transcriptome (NCBI RefSeq mm10) by TopHat2 (Kim et al., 2013), allowing up to two mismatches. Ribosomal A-sites were statistically estimated by (virtually) cutting 11 nucleotides from both 5′ and 3′ read ends (‘center weighting’). RPKM values were calculated using the Plastid CS program (Dunn and Weissman, 2016). Further analyses were performed using customized python scripts. SeRP analyses are based on two independent biological replicates that were highly reproducible. Genes were only considered for analysis if they fulfill the following requirements: i) at least 64 reads in all total and Ebp1-bound translatome datasets; ii) at least 2 RPKM in all total and Ebp1-bound translatome datasets.
For metagene enrichment analysis of Ebp1 binding, we aligned open reading frames (ORFs) as indicated and divided the mean RPM value of the IP translatome by the mean RPM value of the total translatome at each codon along the aligned ORFs. Values at each codon were smoothed using a 20 residue rolling average. Analyses were performed using either all genes that were detected in the dataset or by splitting genes into pre-defined subsets based on their cellular localization or protein features. Information about cellular localization and protein features were received from UniProt (https://www.uniprot.org/).
The enrichment profile of L1cam (Figure 6G) was generated by dividing the RPM value of the IP translatome by the RPM value of the total translatome at each codon along that particular ORF. Values at each codon were smoothed using a 10 residue rolling average.
For the metagene profiles of IP and total translatomes (Figure S12C), we aligned ORFs to their start and stop codon, respectively, including 90 nucleotides of adjacent untranslated regions. We then calculated the 1% trimmed mean of RPM values at each codon along the aligned ORFs. Ebp1-selective ribosome profiling data have been deposited in the NIH Gene Expression Omnibus (GEO): GSE157425
Ebp1 knockdown Ribosome Profiling
Ebp1 knockdown in Neuro2a cells
The mouse and human siEbp1 oligos were obtained from the Dharmacon SMARTpool ON-TARGETplus collection (mouse siPa2g4 #18813, L-042883-01-0005; human siPa2g4 #5036; L008860-00-0005) and compared to non-targeting siRNA control (D-001810-10-05). Transfection was performed with Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher, 13778075) according to the manufacturer’s protocol. To confirm robust and specific knockdown with the mouse siEbp1 oligos, Neuro2a cells were treated in parallel with the following conditions in biological duplicate, followed by western blot analysis of total lysates: (1) mock transfection, (2) control siRNA, (3) mouse siEbp1, (4) human siEbp1, and (5) 1:1 mouse + human siEbp1 (Figure S13A).
Ribosome profiling
Neuro2a cultures were prepared in biological triplicate (n = 3), with 3x 10 cm plates per non-targeting siRNA control and knockdown (mouse siEbp1) conditions. Neuro2a cells were grown in standard DMEM (GIBCO, 31966047) with 10% FBS (GIBCO, 10270106) and 1% penicillin-streptomycin (GIBCO, 15140122) in humidified 37°C, 5% CO2. Ribosome profiling was performed as described (Ingolia et al., 2009) with minor modifications. Neuro2a cells were grown to ~90% confluence, medium was removed, and plates placed on dry ice. 400 μl mammalian polysome buffer (20 mMTris-HCl pH 7.4, 150 mM NaCl, 5mM MgCl2, 1 mM DTT, 100 μg/ml cycloheximide, 1% Triton X-100 and 25 U/mLTurboDNase (Thermo Fisher, AM2238]) was added to each plate, which were then placed on ice. Cells were scraped into a slurry, and then passed 10 times through a 26 G needle. After lysate clearance by centrifugation (20000 xg, 10 min, 4°C), 120 μl lysate aliquots were flash frozen and stored at −80°C. One aliquot of cell lysate per replicate was processed for RNA sequencing, with RNA isolated by Trizol LS in combination with the RNA Clean & Concentrator-25 Kit (Zymo Research, R1017), and further processed with the NEBNext Ultra Directional RNA Library Prep Kit (Illumina, E7420L) according to manufacturer’s instructions. Another aliquot of lysate was processed to isolate ribosome-protected fragments by adding 300 U RNase-I (Thermo Fisher, EN0601) for 45 min at room temperature with gentle agitation. Meanwhile, MicroSpin S-400 HR Columns (GE Healthcare, GE27-5140-01) were equilibrated by adding cold mammalian polysome buffer (without DTT, cycloheximide, Triton X-100, TurboDNase) to the columns. Columns were then centrifuged (600 xg, 4 min, 4°C). 100 U SUPERaseIn was added to each sample, mixed, and subsequently the samples were pipetted drop-wise to the columns (100 μl cell lysate per column). Columns were then centrifuged (600 xg, 2 min, 4°C), and flow-through was collected. RNA was isolated using Trizol LS in combination with RNA Clean & Concentrator-25 kit. The ribosome protected fragments were then rRNA depleted with the RiboZero Kit (Illumina, 20037135) according to manufacturer’s protocol, then separated on a 17% denaturing urea-PAGE gel (Carl Roth), with RNA fragments in the range of 27-30 nucleotides excised, defined by markers oligonucleotide references:
Marker-27 nt, rArUrGrUrArCrArCrGrGrArGrUrCrGrArGrCrUrCrArArCrCrCrGrC-P
Marker-30 nt, rArUrGrUrArCrArCrGrGrArGrUrCrGrArGrCrUrCrArArCrCrCrGrCrArArC-P
For sequencing library preparation, the RNA was first ligated to a 3′ adaptor 4N-RA3 (see below), and gel-purified using a 15% denaturing urea-PAGE gel (Carl Roth). Next the 5′ adaptor OR5-4N (see below) was ligated and gel purified. The RNA was reverse transcribed and PCR-amplified by Phusion High-Fidelity DNA polymerase (Thermo Fischer, F-530XL). The cDNA was visualized on a 2.5% agarose gel, a ~150 bp sized fragment was excised and purified by Zymoclean Gel DNA Recovery kit (Zymo Research, D4007/D4008). Next generation sequencing was carried out on a HighSeq 4000 Illumina instrument (1x51+7 cycles).
Oligonucleotides
3′ adaptor 4N-RA3, rApp-NNNNTGGAATTCTCGGGTGCCAAGG-InvdT;
5′ adaptor OR5-4N; rGrUrUrCrArGrArGrUrUrCrUrArCrArGrUrCrCrGrArCrGrArUrCrNrNrNrN;
RT primer RTP; GCCTTGGCACCCGAGAATTCCA
Analysis
Riboseq reads were stripped of adaptor sequences using cutdapt, and contaminants such as transfer RNAs (tRNAs) and rRNA were removed by alignment to a contaminants index via STAR 2.7.0, consisting of nucleotide sequences from known mouse rRNA and tRNA sequences drawn from the gencode annotation. Unaligned reads from this analysis were then aligned to mouse genome version GRCm38 with the STAR v 2.7.0 splice-aware alignment tool allowing for up to 1 mismatch. The star genome index was built using GENCODE M12. Only uniquely aligning reads were used. The RiboseQC pipeline v1.1 (https://github.com/ohlerlab/RiboseQC) was used to confirm 3-nucleotide periodicity of the data and deduce P-site positions from the Riboseq reads. Ribosome P-site count metaplots and fold change were calculated by first normalizing the P-site coverage track for each gene to the gene’s total density, excluding genes with <32 reads or less (low count filtering), and then all genes’ tracks were added together to generate a mean P-site density at each point for each sample. Confidence intervals were calculated by resampling from the 3 replicates for each condition. Ebp1-knockdown ribosome profiling and RNaseq data have been deposited in the NIH Gene Expression Omnibus (GEO): GSE157425
Pulsed SILAC and BONCAT mass spectrometry
Sample preparation
We confirmed robust and specific knockdown with mouse siEbp1 oligos in Neuro2a cells as described above for Ribosome Profiling (Figure S13A). For pulsed stable isotope labeling by amino acids in cell culture (pSILAC) (Schwanhäusser et al., 2009; Selbach et al., 2008) and bioorthogonal noncanonical amino acid tagging (BONCAT) (Dieterich et al., 2006) coupled mass spectrometry (QuaNCAT) (Eichelbaum et al., 2012; Howden et al., 2013), eight 10 cm plates of Neuro2a cells were grown in standard DMEM (GIBCO, 31966047) with 1% FBS (GIBCO #10270106) to a confluence of 50% in humidified 37°C, 5% CO2. Then, mouse siEbp1 versus control siRNA tranfection was performed with Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher, 13778075) according to the manufacturer’s protocol, in four plates each. The next morning, media was changed in each condition to either heavy SILAC (2 plates; Cambridge Isotope Labs, CNLM-539, CNLM-291) or medium SILAC (2 plates; Cambridge Isotope Labs, CLM-2265, DLM-2640) prepared with DMEM (Pan-Biotech, P04-02505), 1% dialyzed FBS (PAN-Biotech, P30-2102), GlutaMAX (Thermo Fisher, 35050-038), and Penicillin-Streptomycin (Thermo Fisher, 15140-122) along with repeated application of the siRNAs. Thus, throughout the course of Ebp1 versus control knockdown, all newly made proteins were labeled with either heavy or light SILAC (pSILAC), i.e., “label swap” biological replicates (n = 2). After 48 hr, SILAC media and siRNAs were refreshed. After another 24 hr, one heavy SILAC and one medium SILAC plate from each condition were pulsed with 1 mM L-azidohomoalaine (AHA; Anaspec, AS-63669) for four hours in the corresponding SILAC media prepared with methionine-free DMEM (Sigma-Aldrich, D0422) and 1% dialyzed FBS, labeling all newly made proteins during this acute interval with AHA in addition to the original SILAC label. Thus, acutely synthesized proteins at the point of maximal Ebp1 knockdown were labeled with both SILAC and AHA in parallel (pSILAC-AHA).
Media was then gently aspirated from each plate, followed by washing with ice-cold PBS, then scraping cells into 1 mL ice-cold PBS. Samples were lysed by the addition of 50 mM TrispH 8, 150 mM NaCl, 1% IGEPAL CA-630 detergent (Sigma, I8896), and 0.5% Sodium Deoxycholate, followed by 5 min of boiling, then lysate clarification by centrifugation at 16000 xg 4°C for 30 min. 10% of each sample was frozen for western blot confirmation of Ebp1 knockdown. The remaining 90% of samples were then mixed 1:1 as per the following:
Control+Heavy SILAC: siEbp1+Medium SILAC
Control+Medium SILAC: siEbp1+Heavy SILAC
Control+Heavy SILAC-AHA: siEbp1+Medium SILAC-AHA
Control+Medium SILAC-AHA: siEbp1+Heavy SILAC-AHA
Mixtures (1) and (2) were combined with nine volumes of ice-cold ethanol, and frozen at −80°C for downstream MS analysis. Mixtures (3) and (4) were subjected to AHA-enrichment.
AHA-enrichment
In preparation for on-bead digestion, azide-containing proteins were enriched from Neuro2a cell lysates using alkyne-agarose beads (Click-Chemistry Tools, 1033). Alkyne-agarose beads were rinsed 2 times in pure injection grade water (AMPUWA) before use. To facilitate azide-alkyne binding, a 4x-concentrated “click-solution” was prepared in pure water: 0.8 mM Tris(3-hydroxypropyltriazolylmethyl) amine (THPTA, Sigma-Aldrich, 762342), 80 mM Sodium L-ascorbate (Sigma-Aldrich, A7631), and 0.8mM Copper(II) sulfate pentahydrate (Sigma-Aldrich, 209198). 200 μl of 4x-click-solution and 200 μl of alkyne-agarose beads were first mixed before addition to 400 μl of Neuro2a lysate. To prevent protease degradation of peptides, Protease Inhibitor Cocktail Set III, EDTA-Free (Calbiochem/Sigma-Aldrich, 539134) was added to the final solution (1:50 dilution). To allow time for the click reaction to proceed, the click-bead-lysate mix was briefly vortexed (~8000 xg for 5 s) before being placed on an orbital shaker maintained in the dark at room temperature. Following 3.5 hr of incubation, alkyne-agarose beads were briefly centrifuged at 3000 xg for 2 min, and resuspended in agarose wash buffer (100 mM Tris, 1% SDS, 250 mM NaCl, 5 mM EDTA, pH 8.0) containing dithiothreitol (DTT, 10 mM). To break disulfide bonds, alkyne-agarose beads were incubated with DTT-solution for 20 min at room temperature, then 10 min at 70°C, at 1000 rpm in a thermomixer (Eppendorf). Following DTT treatment, alkyne-agarose beads were resuspended in agarose wash buffer containing 40 mM Iodoacetamide (IAA). For the alkylation of free thiol groups, alkyne-agarose beads were incubated with IAA for 45 min on an orbital shaker maintained in the dark at room temperature. Following incubation, alkyne-agarose beads were washed using a bench top centrifuge (Roth) and 2 mL centrifuge columns (Pierce) with the following solutions, 10 times each: (1) agarose wash buffer, (2) 8 M Urea in 100 mM Tris, and (3) 70% acetonitrile solution (100 mM ammonium bicarbonate buffer; ABC). Following washing, beads were then resuspended in 35% acetonitrile (50 mM ABC buffer) before centrifugation at 3000 xg for 2 min to form a bead-pellet. The resulting supernatant was removed and the tube containing the pellet was frozen on liquid nitrogen before storage at −20°C until on-bead digestion.
Mass spectrometry analysis
Proteins from cell lysates were precipitated in 90% ethanol solution at −20°C followed by 30 min centrifugation at 20000 xg at 4°C. Protein pellets and AHA-clicked beads were resuspended in 2 M urea, 6 M Thiourea, 0.1 M Tris pH 8 solution. Proteins were reduced and alkylated with 10 mM DTT and 55 mM iodoacetamide at room temperature, respectively. For lysis, proteins were incubated with lysyl endopeptidase (LysC) (Wako) at a protein-to-LysC ratio of 100:1 (w/w) at room temperature for 3 hr. Three volumes of 50 mM ammonium bicarbonate solution were added, and proteins were further digested with trypsin (Promega) at a protein-to-trypsin ratio of 100:1 (w/w) under constant agitation at room temperature for 16 hr. Peptides were desalted with C18 Stage Tips prior to LC-MS/MS analysis. Peptide concentration was measured based on 280 nm UV light absorbance.
Reversed-phase liquid chromatography was performed employing an EASY nLC II (Thermo Fisher) using self-made C18 microcolumns (75 μm ID, packed with ReproSil-Pur C18-AQ 1.9 μm resin, Dr. Maisch, Germany) connected on-line to the electrospray ion source (Proxeon, Denmark) of a Q Exactive HF-X mass spectrometer (Thermo Fisher). Peptides were eluted at a flow rate of 250 nL/min over 1 or 2 hr with a 9% to 55.2% acetonitrile gradient in 0.1% formic acid. Settings for mass spectrometry analysis were as follows: one full scan (resolution, 60,000; m/z, 350-1,800) followed by top 20 MS/MS scans using higher-energy collisional dissociation (resolution, 15,000; AGC target, 1e5; max. injection time, 22 ms; isolation width, 1.3 m/z; normalized collision energy, 26). The Q Exactive HF-X instrument was operated in data dependent mode with a full scan in the Orbitrap followed by up to 20 consecutive MS/MS scans. Ions with an unassigned charge state, singly charged ions, and ions with charge state higher than six were rejected. Former target ions selected for MS/MS were dynamically excluded for 20 or 30 s.
All raw files were analyzed with MaxQuant software (v1.6.0.1) with default parameters, and with match between runs and requantify options on. Search parameters included two missed cleavage sites, cysteine carbamidomethyl fixed modification, and variable modifications including methionine oxidation, protein N-terminal acetylation and deamidation of glutamine and asparagine. Peptide mass tolerance was 4.5 ppm and the MS/MS tolerance was 20 ppm. Database search was performed with Andromeda against UniProt/Swiss-Prot mouse database (downloaded on January 2019) with common serum and enzyme contaminant sequences. False discovery rate (FDR) was set to 1% at peptide spectrum match (PSM) and protein levels. Minimum peptide count required for protein quantification was set to two. Potential contaminants, reverse database hits and peptides only identified by modification were excluded from analysis. MaxQuant normalized SILAC ratios were used for quantitative data analysis.
We tested for miRNA-like off-target effects using seeds based on the siRNA sequences in the mouse siEbp1 (siPa2g4) knockdown pool with the cWord software (Rasmussen et al., 2013). All proteins from our dataset were ranked according to their change ratio (mean H/M ratio in Forward and Reverse experiments; lowest to highest fold change). UniProt IDs were converted to Ensembl IDs and searched against mouse 3′UTR using cWords (http://servers.binf.ku.dk/cwords/) (Rasmussen et al., 2013). None of the possible 7-mers from these seeds showed specific enrichment in either AHA or pSILAC datasets. Significance was assessed at > 1.25, 1.5, and 2-fold change in siEbp1 conditions compared to control in both replicates. Gene ontology (GO) pathway analysis was performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009) for proteins with > 2-fold change from control in siEbp1 conditions (against all quantified proteins). Importantly, peptides corresponding to Ebp1 measured in siEbp1 samples did not meet the minimum requirements for quantification, confirming robust knockdown, and therefore show no fold change ratio. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository: PXD014740
In utero electroporation and morphology analysis
The mouse shEbp1 plasmid was obtained from the Sigma MISSION collection (shPa2g4; oligo name: TRCN0000236756; RefSeq NM_011119) in bacterial glycerol stock format, and amplified according to the manufacturer’s protocol, followed by plasmid purification with the Nucleobond Xtra Midi Kit (Macherey & Nagel, 740410.100). The non-targeting scrambled shRNA control was generated as described in a prior study (Ambrozkiewicz et al., 2018). The Ebp1 overexpression plasmid was generated by insert amplification from the Clone IRAVp968A0190D I.M.A.G.E. Fully Sequenced cDNA (Source BioScience) with primers forward 5′-gtctcatcattttggcaaagATGTACCCATACGATGTTCCAGATTACGCTTCGGGCGAAGACGAG-3′ and reverse 5′-cggccgcgatatcctcgaggTCAGTCCCCAGCTCCATTC-3′, followed by cloning into the pCAG-IRES-GFP backbone (Ambrozkiewicz et al., 2018) with the restriction enzyme EcoRI (NEB). Co-electroporation of the pCAG-IRES-GFP plasmid was used as a transfection reporter and to trace cell morphology.
E12 In utero electroporation (IUE) of control, shEbp1, and shEbp1+oeEbp1 conditions along with CAG-GFP reporter followed by analysis at E16 with confocal imaging, morphology tracing, and Sholl analysis was performed as described (Ambrozkiewicz et al., 2018). Briefly, GFP labeling of electroporated neurons in confocal images was analyzed by morphology tracing with the Neurite Tracer plugin (Longair et al., 2011) by a blinded investigator, followed by the Sholl analysis (Ferreira et al., 2014) plugin run in FIJI with 1 μm radius of concentric circles, plotting the average intersections over distance from the soma and average total summed intersections in each condition. Significance was assessed by one-way ANOVA followed by Bonferroni corrected post hoc testing in GraphPad Prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mass spectrometry
Protein quantification across samples was performed using the label-free quantification (LFQ) algorithm (Cox et al., 2014). A minimum peptide count required for LFQ protein quantification was set to two. Only proteins quantified in at least two out of the three biological replicates in input, 80S, and polysome samples, and two out of two (label swap) biological replicates in pSILAC/AHA samples, were considered for further analyses. LFQ intensities were log2-transformed and imputation for missing values was performed in Perseus software (Tyanova et al., 2016) based on a simulated normal distribution to represent low abundance values below the noise level (generated at 1.8 standard deviations of the total intensity distribution, subtracted from the mean, and a width of 0.3 standard deviations). For stoichiometry calculations, the IBAQ algorithm (Schwanhäusser et al., 2011) was used to quantify within-sample abundance. False discovery rate (FDR) was set to 1% at peptide spectrum match (PSM) and protein levels. Minimum peptide count required for protein quantification was set to two. Potential contaminants, reverse database hits and peptides only identified by modification were excluded from analysis.
For input, 80S, and polysome MS performed in biological triplicate, Ebp1 and the median protein abundance within protein groups (Rpl, Rps, translation-associated) were tested for significantly changing levels across developmental stages by one-way ANOVA with Bonferroni corrected post hoc testing. For pSILAC/AHA MS performed in biological duplicate with SILAC label swap, MaxQuant normalized SILAC ratios were used for quantitative data analysis. All proteins from our dataset were ranked according to their change ratio (mean H/M ratio in Forward and Reverse experiments; lowest to highest fold change). Significance was assessed at > 1.25, 1.5, and 2-fold change in both replicates.
RNaseq and Ribosome Profiling
For RNaseq data, significantly changing levels over time of Ebp1, or the median value of Rpl, Rps, and translation-associated gene groups, was assessed by one-way ANOVA followed by Bonferroni corrected post hoc testing versus E12.5. For Ebp1-knockdown ribosome profiling, to assess differences in enrichment at the start codon, start +1 codon, stop codon, and stop −1 codon, we calculated densities at each position per gene, per sample, by dividing p-site counts at that position by the total number for that gene. We then averaged these for each gene of interest to derive mean densities per condition (control, knockdown), and used these mean densities to calculate a ratio between conditions. Genes for which the density at either condition, in either position, was 0 were excluded, and a Student’s t test was used to test the hypothesis that the mean of the distribution of these ratios was greater or less than 1.
Sucrose density gradient ultracentrifugation fractionation curves
A260 values measured by PicoLogger recorder and software were summed corresponding to 40-60S, 80S, and polysome peaks of the gradient. Significance was tested by one-way ANOVA with Dunnett’s post hoc test versus E12.5, performed in GraphPad Prism.
Quantitative western blot
Western blot band signal intensity measured in duplicate membranes was quantified using GE Amersham Imager 600 software, with significance testing by two-tailed unpaired t test (≤2 comparisons), versus E12.5 with GraphPad Prism.
Sholl analysis
Significance of total summed intersections was assessed by one-way ANOVA followed by Bonferroni corrected post hoc testing in GraphPad Prism.
Supplementary Material
Highlights.
Near-atomic resolution structure of translating ribosomes in the nervous system
Ebp1 binds the 60S peptide tunnel exit during active protein synthesis
Ebp1 regulates start codon initiation and N-terminal peptide elongation
Ebp1 is abundant in early-born neocortex neurons and regulates their morphology
ACKNOWLEDGMENTS
We apologize to the authors of key papers who we could not cite due to space limitations. We are particularly grateful to Rainer Nikolay, Anett Unbehaun, Tatyana Budkevich, Justus Loerke, and Dennis Kwiatkowski for fruitful scientific discussions and technical support. We thank Ludovic Telley and the Denis Jabaudon lab for technical support with the scRNA-seq data. M.L.K. would like to thank James Millonig and Daniel Mehan of the Rutgers-RWJMS-Princeton Universities MD/PhD Physician-Scientist Program. M.L.K. was supported by an EMBO long-term postdoctoral fellowship (190-2016), Alexander von Humboldt Foundation postdoctoral fellowship, and a NIH NRSA F30 MD/PhD fellowship (1F30MH106220). The study was further supported by funding from the Deutsche Forschungsgemeinschaft (DFG; SFB-740 and SFB-958 to C.M.T.S. and SFB-740 to T.M.). High-resolution cryo-EM data collection was performed with support from an iNEXT cryo-EM instrumentation grant (PID:2227; EMBL Heidelberg, special thanks to Wim Hagen and Felix Weis), and an Instruct Structural Biology pilot R&D grant (APPID: 2016-232; Diamond Light Source Oxfordshire, special thanks to Jason van Rooyen) awarded to M.L.K. Funding from a NeuroCure/Charité Cluster of Excellence innovation project grant awarded to M.L.K. and C.M.T.S. further supported this work. This work was also supported by funding fromthe German Research Foundation (grant EXC257 to I.V.) and the European Research Council (advanced grant 743118 to Bernd Bukau, ZMBH-Heidelberg). Computational resources were supported by the North German Supercomputing Alliance (Norddeutscher Verbund zur Förderung des Hochund Höchstleistungsrechnens). In utero electroporation experiments were funded by Russian Science Foundation (grant 19-14-00345).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.037.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Miettinen and Björklund (2015); Xia et al. (2001).
DATA AVAILABILITY
Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository: PXD014740, PXD014841
Neocortex developmental RNaseq, Ebp1-selective ribosome profiling, Ebp1-knockdown ribosome profiling, and Ebp1-knockdown RNaseq data have been deposited in the NIH Gene Expression Omnibus (GEO): GEO: GSE157425.
Cryo-EM maps for the neocortical 80S·Ebp1 complex, including both the rotated state with eEF2 and the classical state with A/A+P/P tRNAs, have been deposited in the wwPDB with accession code EMD-10321.
The neocortical 60S·Ebp1 atomic model has been deposited in the wwPDB with accession code PDB ID 6SWA.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrozkiewicz MC, Schwark M, Kishimoto-Suga M, Borisova E, Hori K, Salazar-Lázaro A, Rusanova A, Altas B, Piepkorn L, Bessa P, et al. (2018). Polarity acquisition in cortical neurons is driven by synergistic action of Sox9-regulated Wwp1 and Wwp2 E3 ubiquitin ligases and intronic miR-140. Neuron 100, 1097–1115.e15. [DOI] [PubMed] [Google Scholar]
- Archer SK, Shirokikh NE, Beilharz TH, and Preiss T (2016). Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 535, 570–574. [DOI] [PubMed] [Google Scholar]
- Behrmann E, Loerke J, Budkevich TV, Yamamoto K, Schmidt A, Penczek PA, Vos MR, Bürger J, Mielke T, Scheerer P, and Spahn CM (2015). Structural snapshots of actively translating human ribosomes. Cell 161, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, LaVoie MJ, Petsko GA, and Ringe D (2011). Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Biol 3, a007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradatsch B, Katahira J, Kowalinski E, Bange G, Yao W, Sekimoto T, Baumgärtel V, Boese G, Bassler J, Wild K, et al. (2007). Arx1 functions as an unorthodox nuclear export receptor for the 60S preribosomal subunit. Mol. Cell 27, 767–779. [DOI] [PubMed] [Google Scholar]
- Brown A, Shao S, Murray J, Hegde RS, and Ramakrishnan V (2015). Structural basis for stop codon recognition in eukaryotes. Nature 524, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CR, Bhaduri A, Mostajo-Radji MA, Keefe MG, and Nowakowski TJ (2019). Development and arealization of the cerebral cortex. Neuron 103, 980–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, and Reilein A (2000). Leginon: an automated system for acquisition of images from vitreous ice specimens. J. Struct. Biol 132, 33–45. [DOI] [PubMed] [Google Scholar]
- Chartron JW, Hunt KCL, and Frydman J (2016). Cotranslational signal-independent SRP preloading during membrane targeting. Nature 536, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau KF, Shannon ML, Fame RM, Fonseca E, Mullan H, Johnson MB, Sendamarai AK, Springel MW, Laurent B, and Lehtinen MK (2018). Downregulation of ribosome biogenesis during early forebrain development. eLife 7, e36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, and Das R (2013). Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat. Methods 10, 74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, and Ghosh A (2016). Specification of synaptic connectivity by cell surface interactions. Nat. Rev. Neurosci 17, 22–35. [DOI] [PubMed] [Google Scholar]
- DeBoer EM, Kraushar ML, Hart RP, and Rasin M-R (2013). Post-transcriptional regulatory elements and spatiotemporal specification of neocortical stem cells and projection neurons. Neuroscience 248, 499–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G, et al. (2018). mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell 174, 338–349.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, Gamerdinger M, and Kreft SG (2019). Chaperone interactions at the ribosome. Cold Spring Harb. Perspect. Biol 11, 033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, and Schuman EM (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, and Weissman JS (2016). Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelbaum K, Winter M, Berriel Diaz M, Herzig S, and Krijgsveld J (2012). Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 30, 984–990. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Evangelopoulos ME, Weis J, and Krüttgen A (2005). Signalling pathways leading to neuroblastoma differentiation after serum withdrawal: HDL blocks neuroblastoma differentiation by inhibition of EGFR. Oncogene 24, 3309–3318. [DOI] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjöström PJ, and van Meyel DJ (2014). Neuronal morphometry directly from bitmap images. Nat. Methods 11, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flis J, Holm M, Rundlet EJ, Loerke J, Hilal T, Dabrowski M, Bürger J, Mielke T, Blanchard SC, Spahn CMT, and Budkevich TV (2018). tRNA translocation by the eukaryotic 80S ribosome and the impact of GTP hydrolysis. Cell Rep. 25, 2676–2688.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, and Leith A (1996). SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol 116, 190–199. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47 (D1), D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmozzi CV, Merker D, Friedrich UA, Döring K, and Kramer G (2019). Selective ribosome profiling to study interactions of translating ribosomes in yeast. Nat. Protoc 14, 2279–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M, Hanebuth MA, Frickey T, and Deuerling E (2015). The principle of antagonism ensures protein targeting specificity at the endoplasmic reticulum. Science 348, 201–207. [DOI] [PubMed] [Google Scholar]
- Gerashchenko MV, and Gladyshev VN (2017). Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 45, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, and Ban N (2014). The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515, 283–286. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Gerhardy S, Leitner A, Leibundgut M, Salem M, Boehringer D, Leulliot N, Aebersold R, Panse VG, and Ban N (2016). Insertion of the biogenesis factor rei1 probes the ribosomal tunnel during 60S maturation. Cell 164, 91–102. [DOI] [PubMed] [Google Scholar]
- Grigorieff N (2016). Frealign: an exploratory tool for single-particle cryo-EM. Methods Enzymol. 579, 191–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A-S, Donlin-Asp PG, Leitch B, Herzog E, and Schuman EM (2019). Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 364, 363184. [DOI] [PubMed] [Google Scholar]
- Holt CE, Martin KC, and Schuman EM (2019). Local translation in neurons: visualization and function. Nat. Struct. Mol. Biol 26, 557–566. [DOI] [PubMed] [Google Scholar]
- Howden AJM, Geoghegan V, Katsch K, Efstathiou G, Bhushan B, Boutureira O, Thomas B, Trudgian DC, Kessler BM, Dieterich DC, et al. (2013). QuaNCAT: quantitating proteome dynamics in primary cells. Nat. Methods 10, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Imami K, Milek M, Bogdanow B, Yasuda T, Kastelic N, Zauber H, Ishihama Y, Landthaler M, and Selbach M (2018). Phosphorylation of the ribosomal protein RPL12/uL11 affects translation during mitosis. Mol. Cell 72, 84–98.e9. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Rappsilber J, Andersen JS, and Mann M (2002). Microcolumns with self-assembled particle frits for proteomics. J. Chromatogr. A 979, 233–239. [DOI] [PubMed] [Google Scholar]
- Jan CH, Williams CC, and Weissman JS (2014). Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraj GG, Hipp MS, and Hartl FU (2020). Functional modules of the proteostasis network. Cold Spring Harb. Perspect. Biol 12, 033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Gkogkas CG, Sonenberg N, and Holt CE (2014). Remote control of gene function by local translation. Cell 157, 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, et al. (2018). Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, Monaghan CE, and Ackerman SL (2017). Regulation of mRNA translation in neurons: a matter of life and death. Neuron 96, 616–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr AG, Schmidt C, Tesina P, Berninghausen O, Becker T, Beatrix B, and Beckmann R (2019). Ribosome-NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nat. Struct. Mol. Biol 26, 35–39. [DOI] [PubMed] [Google Scholar]
- Ko HR, Hwang I, Ahn SY, Chang YS, Park WS, and Ahn JY (2017). Neuron-specific expression of p48 Ebp1 during murine brain development and its contribution to CNS axon regeneration. BMB Rep. 50, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Jomaa A, Lee JH, Chandrasekar S, Boehringer D, Shan SO, and Ban N (2018). Structure of a prehandover mammalian ribosomal SRP-SRP receptor targeting complex. Science 360, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E, Bange G, Bradatsch B, Hurt E, Wild K, and Sinning I (2007). The crystal structure of Ebp1 reveals a methionine aminopeptidase fold as binding platform for multiple interactions. FEBS Lett. 581, 4450–4454. [DOI] [PubMed] [Google Scholar]
- Kramer G, Shiber A, and Bukau B (2019). Mechanisms of cotranslational maturation of newly synthesized proteins. Annu. Rev. Biochem 88, 337–364. [DOI] [PubMed] [Google Scholar]
- Kraushar ML, Viljetic B, Wijeratne HRS, Thompson K, Jiao X, Pike JW, Medvedeva V, Groszer M, Kiledjian M, Hart RP, and Rasin MR (2015). Thalamic WNT3 secretion spatiotemporally regulates the neocortical ribosome signature and mRNA translation to specify neocortical cell subtypes. J. Neurosci 35, 10911–10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushar ML, Popovitchenko T, Volk NL, and Rasin M-R (2016). The frontier of RNA metamorphosis and ribosome signature in neocortical development. Int. J. Dev. Neurosci 55, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon IS, and Ahn JY (2011). p48 Ebp1 acts as a downstream mediator of Trk signaling in neurons, contributing neuronal differentiation. Neurochem. Int 58, 215–223. [DOI] [PubMed] [Google Scholar]
- Kraushar ML, Thompson K, Wijeratne HRS, Viljetic B, Sakers K, Marson JW, Kontoyiannis DL, Buyske S, Hart RP, and Rasin M-R (2014). Temporally defined neocortical translation and polysome assembly are determined by the RNA-binding protein Hu antigen R. Proc. Natl. Acad. Sci. USA 111, E3815–E3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Belgard TG, Hawrylycz M, and Molnár Z (2017). Transcriptomic perspectives on neocortical structure, development, evolution, and disease. Annu. Rev. Neurosci 40, 629–652. [DOI] [PubMed] [Google Scholar]
- Lin Z, Gasic I, Chandrasekaran V, Peters N, Shao S, Mitchison TJ, and Hegde RS (2020). TTC5 mediates autoregulation of tubulin via mRNA degradation. Science 367, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Ahn J-Y, Liu X, and Ye K (2006). Ebp1 isoforms distinctively regulate cell survival and differentiation. Proc. Natl. Acad. Sci. USA 103, 10917–10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerke J, Giesebrecht J, and Spahn CMT (2010). Multiparticle cryo-EM of ribosomes. Methods Enzymol. 483, 161–177. [DOI] [PubMed] [Google Scholar]
- Longair MH, Baker DA, and Armstrong JD (2011). Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27, 2453–2454. [DOI] [PubMed] [Google Scholar]
- Martínez G, Khatiwada S, Costa-Mattioli M, and Hetz C (2018). ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. 41, 610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Miettinen TP, and Björklund M (2015). Modified ribosome profiling reveals high abundance of ribosome protected mRNA fragments derived from 3′ untranslated regions. Nucleic Acids Res. 43, 1019–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EW, and Green R (2017). Ribosomopathies: there’s strength in numbers. Science 358, eaan2755. [DOI] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JRL, and Macklis JD (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Monie TP, Perrin AJ, Birtley JR, Sweeney TR, Karakasiliotis I, Chaudhry Y, Roberts LO, Matthews S, Goodfellow IG, and Curry S (2007). Structural insights into the transcriptional and translational roles of Ebp1. EMBO J. 26, 3936–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya T, Saur M, Stabrin M, Merino F, Voicu H, Huang Z, Penczek PA, Raunser S, and Gatsogiannis C (2017). High-resolution single particle analysis from electron cryo-microscopy images using SPHIRE. J. Vis. Exp 123, e55448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DQ, Hoang DH, Nguyen Vo TT, Huynh V, Ghoda L, Marcucci G, and Nguyen LXT (2018). The role of ErbB3 binding protein 1 in cancer: Friend or foe? J. Cell. Physiol 233, 9110–9120. [DOI] [PubMed] [Google Scholar]
- Nonato MC, Widom J, and Clardy J (2006). Human methionine aminopeptidase type 2 in complex with L- and D-methionine. Bioorg. Med. Chem. Lett 16, 2580–2583. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, and Steinmetz LM (2015). Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161, 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczek PA, Frank J, and Spahn CMT (2006). A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J. Struct. Biol 154, 184–194. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pilipenko EV, Pestova TV, Kolupaeva VG, Khitrina EV, Poperechnaya AN, Agol VI, and Hellen CUT (2000). A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 14, 2028–2045. [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Unbehaun A, Hellen CUT, and Pestova TV (2007). Assembly and analysis of eukaryotic translation initiation complexes. Methods Enzymol. 430, 147–177. [DOI] [PubMed] [Google Scholar]
- Potterton E, Briggs P, Turkenburg M, and Dodson E (2003). A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, and Tomancak P (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rakwalska M, and Rospert S (2004). The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol. Cell. Biol 24, 9186–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Rasmussen SH, Jacobsen A, and Krogh A (2013). cWords - systematic microRNA regulatory motif discovery from mRNA expression data. Silence 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ES (1963). The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol 17, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibich D, Gloge F, Pöhner I, Björkholm P, Wade RC, von Heijne G, Bukau B, and Kramer G (2016). Global profiling of SRP interaction with nascent polypeptides. Nature 536, 219–223. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Gossen M, Dittmar G, and Selbach M (2009). Global analysis of cellular protein translation by pulsed SILAC. Proteomics 9, 205–209. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, and Rajewsky N (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63. [DOI] [PubMed] [Google Scholar]
- Shao S, Brown A, Santhanam B, and Hegde RS (2015). Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Gamerdinger M, Chan R, Gense K, Martin EM, Sachs N, Knight PD, Schlömer R, Calabrese AN, Stewart KL, et al. (2019). Dual role of ribosome-binding domain of NAC as a potent suppressor of protein aggregation and aging-related proteinopathies. Mol. Cell 74, 729–741.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, and Barna M (2015). Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu. Rev. Cell Dev. Biol 31, 31–54. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbereis JC, Pochareddy S, Zhu Y, Li M, and Sestan N (2016). The cellular and molecular landscapes of the developing human central nervous system. Neuron 89, 248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinturel F, Gerber A, Mauvoisin D, Wang J, Gatfield D, Stubblefield JJ, Green CB, Gachon F, and Schibler U (2017). Diurnal oscillations in liver mass and cell size accompany ribosome assembly cycles. Cell 169, 651–663.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS, and Costa-Mattioli M (2019). Translational control in the brain in health and disease. Cold Spring Harb. Perspect. Biol 11, 032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprink T, Ramrath DJF, Yamamoto H, Yamamoto K, Loerke J, Ismer J, Hildebrand PW, Scheerer P, Bürger J, Mielke T, and Spahn CM (2016). Structures of ribosome-bound initiation factor 2 reveal the mechanism of subunit association. Sci. Adv 2, e1501502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squatrito M, Mancino M, Sala L, and Draetta GF (2006). Ebp1 is a dsRNA-binding protein associated with ribosomes that modulates eIF2alpha phosphorylation. Biochem. Biophys. Res. Commun 344, 859–868. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Teixeira FK, and Lehmann R (2019). Translational control during developmental transitions. Cold Spring Harb. Perspect. Biol 11, a032987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Agirman G, Prados J, Amberg N, Fièvre S, Oberst P, Bartolini G, Vitali I, Cadilhac C, Hippenmeyer S, et al. (2019). Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, eaav2522. [DOI] [PubMed] [Google Scholar]
- Turko P, Groberman K, Browa F, Cobb S, and Vida I (2019). Differential dependence of GABAergic and glutamatergic neurons on glia for the establishment of synaptic transmission. Cereb. Cortex 29, 1230–1243. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Fernández IS, Scheres SHW, and Hegde RS (2014). Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 157, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild K, Aleksić M, Lapouge K, Juaire KD, Flemming D, Pfeffer S, and Sinning I (2020). MetAP-like Ebp1 occupies the human ribosomal tunnel exit and recruits flexible rRNA expansion segments. Nat. Commun 11, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Cheng A, Lessor T, Zhang Y, and Hamburger AW (2001). Ebp1, an ErbB-3 binding protein, interacts with Rb and affects Rb transcriptional regulation. J. Cell. Physiol 187, 209–217. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Borrett MJ, Innes BT, Voronova A, Ketela T, Kaplan DR, Bader GD, and Miller FD (2017). Developmental emergence of adult neural stem cells as revealed by single-cell transcriptional profiling. Cell Rep. 21, 3970–3986. [DOI] [PubMed] [Google Scholar]
- Zappulo A, van den Bruck D, Ciolli Mattioli C, Franke V, Imami K, McShane E, Moreno-Estelles M, Calviello L, Filipchyk A, Peguero-Sanchez E, et al. (2017). RNA localization is a key determinant of neurite-enriched proteome. Nat. Commun 8, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Cantara W, Jeon Y, Musier-Forsyth K, Grigorieff N, and Lyumkis D (2019). Analysis of discrete local variability and structural covariance in macromolecular assemblies using cryo-EM and focused classification. Ultramicroscopy 203, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Yuan Y, Zhu J, Li N, Chen C, Wu S, Yu L, Lei J, and Gao N (2014). Structural basis for interaction of a cotranslational chaperone with the eukaryotic ribosome. Nat. Struct. Mol. Biol 21, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for code generated during this study, i.e., not otherwise referenced in the Method Details and Key Resources Table, may be directed to and will be fulfilled by the Lead Contact, christian.spahn@charite.de (C.M.T.S.). Data have been deposited in publicly available repositories as indicated in the Method Details and Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Ebp1CT | rabbit, Abcam | ab35424; RRID:AB_732061 |
| anti-Ebp1NT | rabbit, Millipore | ABE43; RRID:AB_10616223 |
| anti-eEF2 | rabbit, Cell Signaling | 2332S; RRID:AB_10693546 |
| anti-Gapdh | mouse, Millipore | MAB374; RRID:AB_2107445 |
| anti-GFP | chicken, Abcam | ab13970; RRID:AB_300798 |
| anti-Map2 | chicken, Millipore | AB5543; RRID:AB_571049 |
| anti-Nestin | mouse, Millipore | MAB353; RRID:AB_94911 |
| anti-Rpl7 (uL30) | rabbit, Abcam | ab72550; RRID:AB_1270391 |
| anti-Rps5 (uS7) | mouse, Santa Cruz | sc-390935; RRID:AB_2713966 |
| Gold-conjugated-anti-rabbit IgG | goat, Nanoprobes | 2003; RRID:AB_2687591 |
| HRP-anti-rabbit-Light Chain | mouse, Dianova | 211-032-171; RRID:AB_2339149 |
| HRP-anti-mouse-Heavy Chain | goat, Millipore | 71045; RRID:AB_11211441 |
| 488-anti-chicken | donkey, Jackson ImmunoResearch | 703-545-155; RRID:AB_2340375 |
| 488-anti-rabbit | donkey, Jackson ImmunoResearch | 711-545-152; RRID:AB_2313584 |
| 594-anti-mouse | donkey, Jackson ImmunoResearch | 715-585-150; RRID:AB_2340854 |
| 647-anti-chicken | donkey, Jackson ImmunoResearch | 703-605-155; RRID:AB_2340379 |
| Recombinant DNA | ||
| Control siRNA (non-targeting) | Dharmacon | D-001810-10-05 |
| Homo sapiens siPa2g4 siRNA | Dharmacon | SMARTpool ON-TARGETplus #5036, #L008860-00-0005 |
| Luciferase reporter pSPUTK-luc+ | Rakwalska and Rospert, 2004 | |
| Mus musculus Pa2g4 cDNA | Source BioScience | IRAVp968A0190D |
| Mus musculus shPa2g4 shRNA | Sigma Mission | TRCN0000236756, RefSeq NM_011119 |
| Mus musculus siPa2g4 siRNA | Dharmacon | SMARTpool ON-TARGETplus #18813, #L-042883-01-0005 |
| pCAGIG (pCAG-IRES-GFP) | Ambrozkiewicz et al., 2018 | |
| pET-28a(+) | Novagen | 69864-3 |
| pSuper-Neo-GFP | OligoEngine | VEC-pBS-0006 |
| pSuper-Neo-GFP-sh-Scramble | Ambrozkiewicz et al., 2018 | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acetonitrile | CHEMSOLUTE | 2697 |
| Acetonitrile (Alkyne-agarose enrichment) | Sigma-Aldrich | 271004 |
| Acetylated Bovine Serum Albumin (BSA-c) | Aurion | 900.022 |
| Agarose | Sigma-Aldrich | A9539 |
| Alkyne-agarose beads | Click-Chemistry Tools | 1033 |
| Ammonium bicarbonate (ABC) | Sigma-Aldrich | 9830 |
| B27 | Thermo Fisher | 17504044 |
| Bovine serum albumin | Sigma-Aldrich | A3294 |
| Copper(II) sulfate pentahydrate | Sigma-Aldrich | 209198 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| DAPI (Nuc Blue, Molecular Probes) | Invitrogen | R37606 |
| Dithiothreitol (DTT) | Sigma-Aldrich/Roche | DTT-RO |
| Dithiothreitol (DTT) (Alkyne agarose enrichment) | BioMol | 40010.25 |
| DMEM | GIBCO | 31966047 |
| DMEM - methionine free | Sigma-Aldrich | D0422 |
| DNase-I | Roche | 4716728001 |
| Ebp1 recombinant protein | mouse, this paper | |
| EcoRI restriction enzyme | New England Biolabs | R0101 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | E-5143 |
| Ethylene glycol bis(β-aminoethylether) tetraacetic acid (EGTA) | Roth | 3054 |
| Epoxy embedding medium Epon 812 | Sigma-Aldrich | 45345 |
| Ethanol | J.T. Baker | 8025 |
| Fetal Bovine Serum | GIBCO | 10270106 |
| Fetal Bovine Serum - dialyzed | PAN-Biotech | P30-2102 |
| Fluoromount-G | Southern Biotech | 0100-01 |
| Formic acid | Sigma-Aldrich | 33015 |
| Glutamax | Thermo Fisher | 35050-038 |
| Glutaraldehyde | Sigma-Aldrich | G5882 |
| HEPES | Sigma-Aldrich | 391338 |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 |
| Iodoacetamide (IAA) | Sigma-Aldrich | I6125 |
| KCl | Roth | 6781.1 |
| L-Arginine:HCl (13C6, 99%; 15N4, 99%) (Arg-10) | Cambridge Isotope Labs | CNLM-539 |
| L-Arginine:HCl (13C6, 99%) (Arg-6) | Cambridge Isotope Labs | CLM-2265 |
| L-azidohomoalaine (AHA) | Anaspec | AS-63669 |
| L-Lysine:2HCl (13C6, 99%; 15N2, 99%) (Lys-8) | Cambridge Isotope Labs | CNLM-291 |
| L-Lysine:2HCl (4,4,5,5-D4, 96-98%) (Lys-4) | Cambridge Isotope Labs | DLM-2640 |
| Lead citrate | Fluka | GA10655 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher | 13778075 |
| Liquid ethane, grade 3.5 | Linde GmbH | |
| Lysyl endopeptidase (LysC) | Wako | 12505061 |
| Methanol | Merck Millipore | 1.06009.2511 |
| MgCl2 | Ambion | AM9530G |
| Nanogold silver enhancement | Nanoprobes | |
| Neurobasal medium | Thermo Fisher | 21103049 |
| Neurobasal custom medium (-met / -arg/ -lys) | GIBCO | 041-96642M |
| Normal goat serum | PAN-Biotech | P30-1002 |
| Osmium tetroxide (OsO4) | Polysciences | 0972A |
| Papain | Sigma-Aldrich | P4762 |
| Paraformaldehye (PFA) | Sigma-Aldrich | P6148 |
| Penicillin-Streptomycin | Thermo Fisher | 15140-122 |
| Phusion High-Fidelity DNA polymerase | Thermo Fischer | F-530XL |
| Phenylmethyl sulphonyl fluoride (PMSF) | Roth | 6367 |
| Poly-L-Lysine | Sigma-Aldrich | P1399 |
| Protease Inhibitor Cocktail Set III, EDTA-Free | Calbiochem/Sigma-Aldrich | 539134 |
| Protease Inhibitor cOmplete EDTA-free | Roche | 5056489001 |
| Rabbit reticulocyte lysate nuclease-treated | Promega | L4960 |
| ReproSil-Pur C18-AQ 3-μm resin | Dr. Maisch GmbH | r13.aq |
| RNasin Plus RNase inhibitor | Promega | N2615 |
| RNase-I | Thermo Fisher | EN0601 |
| SeeBlue Plus2 Prestained Protein Ladder | Thermo Fisher | LC5925 |
| SILAC-DMEM | PAN-Biotech | P04-02505 |
| Sodium borohydride (NaBH4) | Sigma-Aldrich | 452882 |
| Sodium deoxycholate | Sigma-Aldrich | D6750 |
| Sodium dodecyl sulfate (SDS) | Roth | 2326.1 |
| Sodium L-ascorbate | Sigma-Aldrich | A7631 |
| Spermidine●3HCl | Sigma-Aldrich | S2501 |
| Spermine●4HCl | Sigma-Aldrich | S2876 |
| Sucrose | Sigma-Aldrich | S0389 |
| SUPERase-In RNase inhibitor | ThermoFisher | AM2694 |
| T4 PNK | New England Biolabs | M0201S |
| Tris-HCl | Roth | 9090.3 |
| Tris(3-hydroxypropyltriazolylmethyl) amine (THPTA) | Sigma-Aldrich | 762342 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| TRIzol-LS | Invitrogen | 10296010 |
| Trypsin | Promega | V511A |
| TurboDNase | Thermo Fisher | AM2238 |
| Tween | Sigma-Aldrich | P9416 |
| Uranyl acetate | Merck | 1.08473.0100 |
| Urea | Sigma-Aldrich | 51459 |
| Vectashield Antifade Mounting Medium | Vector Laboratories | H-1000 |
| Critical Commercial Assays | ||
| Amersham ECL Prime | GE Healthcare | RPN2232 |
| Dynabeads | Life Technologies | 10008D |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | New England BioLabs | E7420L |
| NEXTflex Small RNA-seq Kit v3 | Bio Scientific | NOVA-5132-06 |
| RNA Clean & Concentrator-25 Kit | Zymo Research | R1017 |
| RiboZero Kit | Illumina | 20037135 |
| TruSeq Stranded mRNA Kit | Illumina | 20020594 |
| Zymoclean Gel DNA Recovery Kit | Zymo Research | D4007/D4008 |
| Deposited Data | ||
| Neocortex total lysate, 80S, polysome mass spectrometry | this paper | ProteomeXchange PXD014841 |
| Neuro2a pSILAC/AHA mass spectrometry | this paper | ProteomeXchange PXD014740 |
| Neocortex total lysate RNA sequencing | this paper | NIH GEO: GSE157425 |
| Cryo-EM maps of the P0 neocortical ribosome | this paper | Worldwide Protein Data Bank EMD-10321 |
| Atomic model of the P0 neocortical 60S●Ebp1 complex | this paper | Worldwide Protein Data Bank PDB: 6SWA |
| Ebp1-selective Ribosome Profiling | this paper | NIH GEO: GSE157425 |
| Ebp1-knockdown Ribosome Profiling | this paper | NIH GEO: GSE157425 |
| Ebp1-knockdown RNaseq | this paper | NIH GEO: GSE157425 |
| Experimental Models: Cell Lines | ||
| Neuro2a | Thermo Fisher | RRID: CVCL_0470 |
| Experimental Models: Organisms/Strains | ||
| CD1 WT mice | Charles River | N/A |
| Nex:Cre;Ai9 mice | Turko et al., 2019 | N/A |
| NMRI WT mice | Charles River and Janvier Labs | N/A |
| Software and Algorithms | ||
| Andromeda | Cox et al., 2011 | N/A |
| APBS | Jurrus et al., 2018 | N/A |
| CCP4Interface CONTACT | Potterton et al., 2003 | N/A |
| CLUSTAL Omega MSA (1.2.4) | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| COOT | Emsley and Cowtan, 2004 | N/A |
| CTFfind4 | Mindell and Grigorieff, 2003 | N/A |
| DAVID | Huang et al., 2009 | N/A |
| EMAN2 | Tang et al., 2007 | N/A |
| EPU | FEI Company | N/A |
| ERRASER | Chou et al., 2013 | N/A |
| FIJI | Schindelin et al., 2012 | https://fiji.sc/ |
| GraphPad Prism 7 | GraphPad Software Inc | https://www.graphpad.com/ |
| IBAQ | Schwanhäusser et al., 2011 | N/A |
| Illustrator | Adobe Creative Cloud | N/A |
| Image stitching plugin (FIJI) | Preibisch et al., 2009 | N/A |
| Leginon | Carragher et al., 2000; Suloway et al., 2005 | N/A |
| LFQ | Cox et al., 2014 | N/A |
| MaxQuant | Cox and Mann, 2008 | N/A |
| MolProbity | Chen et al., 2010 | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus | N/A |
| MotionCor2 | Zheng et al., 2017 | N/A |
| Neurite Tracer plugin (FIJI) | Longair et al., 2011 | N/A |
| Perseus | Tyanova et al., 2016 | N/A |
| PHENIX | Adams et al., 2010 | N/A |
| Photoshop | Adobe Creative Cloud | N/A |
| Plastid CS | Dunn and Weissman, 2016 | N/A |
| RiboseQC v1.1 | https://github.com/ohlerlab/RiboseQC | N/A |
| Sholl analysis plugin (FIJI) | Ferreira et al., 2014 | N/A |
| SPHIRE/SPARX | Moriya et al., 2017 | N/A |
| SPIDER | Frank et al., 1996 | N/A |
| STAR | Dobin et al., 2013 | N/A |
| TopHat2 | Kim et al., 2013 | N/A |
| UCSF Chimera | Pettersen et al., 2004 | N/A |
| UCSF ChimeraX | Goddard et al., 2018 | N/A |
| Primers | ||
| Ebp1-His forward (recombinant protein) | Eurofins | 5′AATTCCATGGGCCACCATCACCATCA CCATTCGGGCGAGGACGAGCAAC3′ |
| Ebp1-His reverse (recombinant protein) | Eurofins | 5′TTAAGGATCCTTAGTCCCCAGCTTCA TTTTCTTC3′ |
| Ebp1-HA forward (overexpression plasmid) | Eurofins | 5′gtctcatcattttggcaaagATGTACCCATA CGATGTTCCAGATTACGCTTCGGG CGAAGACGAG3′ |
| Ebp1-HA reverse (overexpression plasmid) | Eurofins | 5′cggccgcgatatcctcgaggTCAGTCCCC AGCTCCATTC3′ |
Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository: PXD014740, PXD014841
Neocortex developmental RNaseq, Ebp1-selective ribosome profiling, Ebp1-knockdown ribosome profiling, and Ebp1-knockdown RNaseq data have been deposited in the NIH Gene Expression Omnibus (GEO): GEO: GSE157425.
Cryo-EM maps for the neocortical 80S·Ebp1 complex, including both the rotated state with eEF2 and the classical state with A/A+P/P tRNAs, have been deposited in the wwPDB with accession code EMD-10321.
The neocortical 60S·Ebp1 atomic model has been deposited in the wwPDB with accession code PDB ID 6SWA.