

Abstract
Tumor cell proliferation requires sufficient metabolic flux through the pentose phosphate pathway to meet the demand for biosynthetic precursors and to increase protection against oxidative stress which in turn requires an upregulation of substrate flow through glycolysis. This metabolic poise is often coupled with a shift in ATP production from mitochondrial OXPHOS to substrate-level phosphorylation. Despite major advances that were facilitated by using tumor-derived cell lines in research areas spanning from membrane to cytoskeletal biology, this distorted metabolic profile limits their impact as a model in physiology and toxicology. Substitution of glucose with galactose in the cell culture medium has been demonstrated to shift ATP production from substrate-level phosphorylation to mitochondrial OXPHOS. This increase in oxygen utilization is coupled to a global metabolic reorganization with potential impacts on macromolecule biosynthesis and cellular redox homeostasis, but a comprehensive analysis on the effects of sugar substitution in tumor-derived cells is still missing. To address this gap in knowledge we performed transcriptomic and metabolomic analyses on human hepatocellular carcinoma (HepG2) cells adapted to either glucose or galactose as the aldohexose source. We observed a shift toward oxidative metabolism in all primary metabolic pathways at both transcriptomic and metabolomic levels. We also observed a decrease in nicotinamide dinucleotide (NAD(P)) levels and subcellular NAD+-to-NADH ratios in cells cultured with galactose compared with glucose control cells. Our results suggest that galactose reduces both glycolytic and biosynthetic flux and restores a metabolic poise in HepG2 cells that closely reflects the metabolic state observed in primary hepatocytes.
Keywords: galactose, HepG2, mitochondria, NAD, redox state
INTRODUCTION
The glycolytic pathway represents a series of enzymatically catalyzed steps in which glucose is converted to pyruvate. Under aerobic conditions most primary cells catabolize pyruvate to CO2 in the mitochondrion and generate ATP via oxidative phosphorylation (OXPHOS). Certain primary cells can temporarily survive anaerobic conditions by converting pyruvate to lactate to produce ATP by substrate-level phosphorylation (anaerobic glycolysis). Tumor cells often convert glucose to lactate even under normoxic conditions despite the lower ATP yield per glucose molecule compared with complete oxidation during OXPHOS. This process, termed aerobic glycolysis, represents a signature metabolic difference between some tumor and primary cells in which ATP production is shifted from OXPHOS to substrate-level phosphorylation even in presence of supraphysiological oxygen levels (1).
Diminished respiration rates were originally attributed to dysfunctional mitochondria, but many cancer lines have since been shown to possess fully functional mitochondria (2, 3). It is becoming clear that biosynthetic precursors, not ATP, limit proliferation in rapidly dividing cells (4). By employing aerobic glycolysis, tumor cells accelerate the conversion of glucose to pyruvate (glycolytic flux) and simultaneously increase activity in associated biosynthetic pathways (5, 6). For example, the pentose phosphate pathway (PPP) originates with the glycolytic intermediate glucose-6-phosphate (G6P) and throughput activity is elevated in cancer cells employing aerobic glycolysis. The PPP generates biosynthetic precursors such as ribose-5-phosphate for nucleotide synthesis and NADPH for both lipid synthesis and maintenance of cellular redox homeostasis (7, 8). Unfortunately, these wide-ranging metabolic differences observed in tumor-derived cells compared with primary tissue cells are frequently overlooked in both basic and biomedical research. Here we provide evidence that replacing glucose with galactose reestablishes a metabolic profile more reminiscent of a primary cell in a tumor-derived cell line. Beyond improving the physiological relevance of the HepG2 cell culture model, the galactose induced metabolic shift also illuminates potential targets for rational development of novel cancer therapeutics.
In certain immortalized cell types replacement of glucose with galactose shifts metabolic poise toward oxidative phosphorylation (OXPHOS) (9). Reduced lactate production and increased mitochondrial oxygen consumption indicates galactose slows glycolytic flux and increases mitochondrial ATP production (10, 11). Cells grown in presence of galactose instead of glucose as carbohydrate source have been shown to increase expression of OXPHOS proteins and become sensitized to mitochondrial dysfunctions and toxins (9, 12–14). Beyond the effects on rates of OXPHOS and lactate production, the transcriptomic and metabolic changes that occur in tumor-derived cells cultured with galactose have not been sufficiently characterized.
Glucose and galactose are both converted to the glycolytic intermediate, G6P, after transport into the cell via solute carrier 2 A (SLC2A) proteins and/or sodium-dependent hexose transporters (SGLUTs) (15–17). Conversion into G6P is a single enzymatic step when starting with glucose whereas galactose conversion requires four catalyzed reactions of the Leloir pathway (18). The potential ATP yield produced from catabolism of either aldohexose is not significantly different and G6P derived from either sugar can proceed through glycolysis or enter the PPP (19). The increased PPP activity in tumor cells has broad impacts on the cellular redox state by altering nucleotide and NAD(P) synthesis, the cellular NADPH-to-NADP+ ratio, and subsequently homeostasis of the glutathione redox equilibrium (8, 20). Although reduced aerobic glycolysis has been reported in galactose cultured cells, the implications of reduced G6P availability on the PPP and cellular redox state have not been explored.
Our study examines the physiological effects of replacing glucose with galactose in the hepatocellular carcinoma (HepG2) cell culture model. These cells were selected because of their metabolic plasticity and robust use for assessment of hepatoxicity (14, 21, 22). Our study reports the first transcriptomic and metabolomic comparison of a tumor-derived cell cultured with either glucose or galactose as aldohexose source. Metabolic flux decreases in HepG2 cells cultured with galactose for both glycolysis and PPP. Amino acid catabolism increases and nucleotide synthesis decreases resulting in decreased levels of nucleotide phosphates and cellular NAD(H) and NADP(H). Finally, we observe a decrease in the cytosolic and mitochondrial NAD+-to-NADH ratios when cells are shifted to galactose as the primary aldohexose source compared with cells cultured in glucose. These results suggest that substitution of glucose with galactose can restore aspects of carbon metabolism in hepatocellular carcinoma cells reminiscent to the hallmark characteristics found in primary hepatocytes making this commonly used cell model more physiologically relevant.
METHODS
Chemicals
All chemicals used for respirometry and solution preparations were of the highest grade and purchased from Sigma-Aldrich (St. Louis, MO) or Thermo Fischer Scientific (Waltham, MA.)
Cell Culture
Human hepatocellular carcinoma cells (HepG2) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown in 100 mm culture dishes (Corning Company, Corning, NY). For standard culture cells were grown in medium composed of glucose-free Dulbecco’s modified Eagles medium (DMEM) (Thermo Fischer Scientific, Waltham, MA) supplemented with 2 mM glutamine (VWR, West Chester, PA), 1 mM pyruvate (Corning Company, Corning, NY), 10 mM glucose (Thermo Fischer Scientific, Waltham, MA), and 10% dialyzed fetal bovine serum (12–14 kDa) (Atlanta Biologicals Inc., Flowery Branch, GA). For all experimental conditions, a base media was used which consisted of glucose-free DMEM supplemented with 2 mM glutamine and 1 mM pyruvate. For certain experiments 10% non-dialyzed FBS (Atlanta Biologicals Inc., Flowery Branch, GA) was used instead of 10% dialyzed FBS. Both types of FBS were purchased directly from the manufacturer (Atlanta Biologicals Inc., Flowery Branch, GA) and no additional dialysis was performed. Further, depending on the experiment, DMEM containing glutamine, pyruvate, and FBS (nondialyzed or dialyzed) was either used directly (no hexose medium) or was further supplemented with 10 mM glucose (Thermo Fischer Scientific, Waltham, MA (glucose medium) or 10 mM galactose (Thermo Fischer Scientific, Waltham, MA) (galactose medium). All cells were maintained in a humidified atmosphere at 5% CO2 and 95% air at 37°C. Culture medium was renewed every 3–4 days and cells were subcultured every 7 days or before reaching 90% confluency. To subculture, cells were dissociated from the culture plate using 0.25% trypsin and 1 mM EDTA in a balanced salt solution (Thermo Fischer Scientific, Waltham, MA), centrifuged at 200 × g for 5 min, and seeded at 1 × 106 cells per 100 mm cell culture plate.
AlamarBlue Proliferation Assay
The alamarBlue assay (Bio-Rad, Hercules, CA) was used to assess the impact of supplementing galactose medium with either nondialyzed FBS or dialyzed FBS on HepG2 cell proliferation. HepG2 cells were maintained in DMEM supplemented with glucose, glutamine, pyruvate, and 10% nondialyzed FBS before experiments. Cells were plated at 100,000 cells per well in a 24-well plate (Corning Company, Corning, NY) and allowed to attach overnight. After 24 h the cell culture medium was removed, and each well was washed with 500 µL of room temperature PBS. Cells were then grown in DMEM supplemented with glutamine, pyruvate, and either 10% nondialyzed or dialyzed FBS without an added sugar (no hexose) or with media fortified with either 10 mM glucose or 10 mM galactose. Cells were grown in these culture media for an additional 5 days and media were exchanged against fresh media after 3 days. After this incubation period, 1× alamarBlue solution was prepared by diluting the stock solution 10-fold into each of the respective media warmed to 37°C. Each well was rinsed with 500 µL of room temperature PBS and the alamarBlue working solutions were added. A fully oxidized growth control sample was produced by adding 1× alamarBlue to wells without cells. The plate was then placed into a humified 37°C cell culture incubator for 2 h. After 2 h, 3 replicates of 100 µL was removed from each well and transferred to a black clear-bottom 96-well plate (Greiner Bio-one, Frickenhausen, Germany). A SpectraMax I3× (Molecular Devices, San Jose, CA) microplate reader was used to estimate alamarBlue reduction by measuring fluorescent emission at λ = 595 nm after excitation at λ = 565 nm.
High-Resolution Respirometry
Respirometry was used to assess the impact of galactose medium supplemented with either nondialyzed FBS or dialyzed FBS on HepG2 oxygen consumption rates. HepG2 cells were plated at 4 × 106 cells on a 100 mm plate 48 h before respirometry. Cells were plated in media containing 10 mM glucose and 10% nondialyzed FBS and after 24 h cells were washed twice with 10 mL room temperature PBS and the experimental medium was added. Cells were harvested as described above after 24 h at a confluency of ∼70%, resuspended to ∼50 × 106 cells per mL and placed on ice until evaluated (<1 h). Respiration of nonpermeabilized cells was measured at 37°C using 0.75–1 × 106 cells per mL in each chamber of the Oxygraph-2K (OROBOROS Instruments, Innsbruck, Austria). Routine respiration was measured after stabilization of oxygen flux. Leak respiration was determined after introduction of oligomycin (2 µg/mL) followed by determination of maximal uncoupled respiration which was accomplished by successive titrations of carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 0.5 µM steps). Respiration was assessed for HepG2 cells cultured in media and for durations of time as described in the figure legends. For experiments with permeabilized cells, oxygen consumption was measured at 37°C in 2 mL of MiR05 (110 mM sucrose, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 0.5 mM EGTA, 0.1% BSA, 20 mM HEPES-KOH, pH 7.1). To supply mitochondrial substrates, cells were permeabilized by the addition of digitonin dissolved in dimethyl sulfoxide (DMSO) at 10 mg/mL (final concentration 10 μg × 106 cells). This digitonin concentration was sufficient to permeabilize the plasma membrane of HepG2 cells with minimal impact on the integrity of the outer mitochondrial membrane as demonstrated by a lack of increase in oxygen flux after addition of cytochrome c or additional digitonin (data not shown). Electron flow through complex I was stimulated by adding 2 mM malate, 10 mM glutamate, and 5 mM pyruvate. To engage the phosphorylation system, 1 mM ADP was added followed by the addition of 10 mM succinate to supply electrons to the ubiquinone pool via succinate dehydrogenase. Leak respiration in presence of ADP was measured after addition of oligomycin (2 μg/mL). Maximal uncoupled respiration was accomplished by successive titrations of FCCP in 0.5 µM increments. The contribution of complex I to maximal uncoupled respiration was recorded after addition of rotenone (0.5 μM). Non-mitochondrial oxygen consumption was recorded after addition of 2.5 μM of antimycin A. DATLAB software (OROBOROS Instruments, Innsbruck, Austria) was used for data analysis and acquisition.
Microcalorimetry
Heat dissipation of HepG2 cells was measured as previously described (23). Samples were prepared for microcalorimetry following the same protocol described above for respirometry except cells were diluted to ∼2 × 105 cells per mL in pre-warmed culture medium equilibrated with 5% CO2 and 95% air. To measure heat flow, a LKB 2277 thermal activity monitor (Bromma, Sweden) was charged with a 4 mL sealed ampoule filled with 2.5 mL of HepG2 cells at 0.125 × 106 cells per mL. The reference vessel contained 2.5 mL of water and all calorimetric measurements were performed at 37°C. After a 15 min period for thermal equilibration the sample ampoule was lowered to the measuring position and heat flow (µW) was recorded after 30 min.
SDS PAGE and Immunoblotting
Protein was isolated from 5 × 106 cells grown on 60 mm plates (Corning Company, Corning, NY) 48 h after plating using 150 µL of lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na2VO4, 1 μg/mL leupeptin, pH 7.5) (Cell Signaling Technology, Danvers, MA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Samples were sonicated using a 500 W sonicator (QSonica, Newtown, CT) 3 × 5 s on ice in a 1.5 mL microcentrifuge tube at 25% capacity. Cell extracts were next centrifuged for 15 min at 14,000 × g and 4°C. The supernatant was collected, and protein concentrations were determined using the Pierce Coomassie Plus protein assay (Thermo Fischer Scientific, Waltham, MA) with bovine serum albumin as standard in clear 96-well plates (Greiner Bio-one, Frickenhausen, Germany) using a SpectraMax I3× (Molecular Devices, San Jose, CA) microplate reader. The extracts were either stored at −80°C until used, or immediately diluted 1:1 with 2× Laemmli buffer (2% SDS, 25% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue, and 62.5 mM Tris-HCl, pH 6.8). Proteins were denatured at 95°C for 5 min and 15–20 µg of protein was loaded per lane on a 12% polyacrylamide gel. Gels were run using the Mini-PROTEAN 3 Cell system (Bio-Rad, Hercules, CA). After electrophoresis proteins in the gel were electrophoretically transferred in buffer (192 mM glycine, 20% methanol, 0.025% SDS, and 25 mM Tris) onto a nitrocellulose membrane (0.2 µm, Bio-Rad, Hercules, CA) using the Mini Trans-Blot apparatus (Bio-Rad, Hercules, CA). Membranes were stained with Ponceau S (Thermo Fischer Scientific, Waltham, MA) in 0.1% glacial acetic acid to confirm transfer of proteins. The nitrocellulose membrane was then incubated in blocking buffer (5% w/v milk powder, 137 mM NaCl, 0.1% Tween-20, 20 mM Tris-HCl, pH 7.6) for 1 h. The blots were incubated overnight with primary antibody at a dilution of 1:2500 in blocking buffer at 4°C (LDH-A/C 35582, GDH-1/2 12793S, β-actin 8457S, Cell Signaling Technology, Danvers, MA; COX-IV ab16056, Abcam, Cambridge, MA). Appropriate secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:5000 (Anti-Rabbit IgG 7074 Cell Signaling Technology, Danvers, MA; Goat Anti-Rabbit IgG H&L ab205718 Abcam, Cambridge, MA). Biotinylated proteins with known mass were used as molecular weight marker (#81851 Cell Signaling Technology, Danvers, MA). The anti-biotin antibody (#7075 Cell Signaling Technology, Danvers, MA) was applied at a dilution of 1:5000 dilution concurrently with secondary antibodies. Proteins were visualized using LumiGLO (Cell Signaling Technology, Danvers, MA) and Hyperfilm ECL (GE Healthcare, Pittsburgh, PA).
RNA-Seq and Differential Gene Expression Analysis
HepG2 cells were grown for 4 to 5 weeks in either glucose or galactose medium supplemented with 10% dialyzed FBS. Cells grown in galactose proliferate slower than in glucose and to yield equal amounts of RNA during isolation, 3.5 × 105 cells were plated for glucose samples and 7 × 105 cells were plated in galactose medium in 6-well plates. After 24 h the respective medium was exchanged with fresh medium. RNA was isolated after 48 h using the RNAeasy Protect Cell Mini kit and QiaShredder columns according to the manufacturers’ protocols (Qiagen, Hilden, Germany). RNA was stored at −80°C until further preparation of the samples was performed. The Truseq Strated mRNA kit (Illumina, San Diego, CA) was used to prepare mRNA libraries from 1 μg of total RNA. Size, purity and semi quantitation was performed on the Agilent 2100 Bioanalyzer using the Agilent DNA 1000 Kit. Sequencing library quantitation was performed by qPCR using the KAPA Library Quantitation Kit for Illumina. Sequencing was performed on the University of Louisville Center for Genetics and Molecular Medicine’s (CGeMM) Illumina NextSeq 500 using the NextSeq 500/550 75 cycle High Output Kit v2. The sequence reads were directly aligned to the Homo sapiens (hg38) reference genome assembly (hg38.fa) using tophat2 (version 2.0.13) (24). Differentially expressed genes were identified using cuffdiff2 (version 2.2.1) (25, 26).
Metabolomics
HepG2 cells were grown for 4 to 5 weeks in either glucose or galactose medium before sample preparation. Samples were prepared by plating 1.5 × 106 glucose cells or 3.0 × 106 galactose cells on 60 mm plates and the culture medium was exchanged after 24 h (also see RNA isolation). Samples were prepared the following day by quickly washing the plates with 5 mL of ice-cold 150 mM ammonium acetate and liquid nitrogen was poured onto the plates which were immediately wrapped with aluminum foil and stored at −80°C. After four replicates were collected, samples were shipped on dry ice to the University of Michigan Metabolomics facility where the TCA-Plus assay was performed using high performance liquid chromatography-mass spectrometry as previously described (27). Quantitative analysis of metabolite levels was normalized to cellular protein (pmol/μg). Semiquantitative analysis was performed on additional compounds and normalized to protein content in the cell pellet (area/μg). These data along with total protein quantity and concentration are available in Supplemental Tables S2 and S3 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.13522946).
Extracellular Acidification Rates (ECAR)
A XFp Seahorse flux analyzer (Agilent Technologies, Santa Clara, CA) was used to characterize extracellular acidification rates (ECAR) of HepG2 cells. All Seahorse XFp consumables were purchased from Agilent. The day before ECAR measurements, cells were plated in glucose medium in microwell plates at a seeding density of 2 × 104 cells per well. ECAR measurements were taken using the standard Seahorse protocol and cartridge sensors were hydrated overnight with calibrant. On the day of the experiment the medium was removed and replaced with the experimental medium. Cells were placed in an incubator with ambient air at 37°C for 1 h. After incubation, ten measurements were taken in three-minute intervals. The lactate dehydrogenase inhibitor GSK 2837808 A (Torcis, Bristol, United Kingdom) was used at a final concentration of 20 μM. The plate was mixed before and after injections to obtain steady ECAR values.
Raman Microspectroscopy
HepG2 cells were grown for 4 to 5 weeks in either glucose or galactose medium before Raman microspectroscopic experiments. Spatially correlated Raman microspectroscopic measurements were acquired using a customized confocal microscope (Zeiss Corp., Oberkochen, Germany) and Raman spectrometer combination (UHTS 300, WITec Instruments Corp., Ulm, Germany) connected to a CCD camera (Andor Technology, Belfast, Ireland). A 532 nm solid-state laser calibrated to 20 mW was used for excitation and a custom 50× objective for observation (Mitutoyo, Sakado Japan). The changes in the spectral signature from the intracellular space were analyzed using a hyperspectral molecular analysis technique. Spectral arrays were collected using a spatial dimension of 30 μm × 30 μm with a spectral dimension of 90 × 90 pixels (n = 3). Each array of scans was collected using an integration time of 0.5 s. Typical background and substrate subtractions were employed, using WITec Project 5 and Peakfit v4 software. Principal component analysis (PCA) was used to reduce the dimensionality of the collected spectral data arrays into principal components (PCs) and then reconstructed according to a signal-to-noise threshold for selecting PC number. Following noise reduction using PCA, hierarchal cluster analysis was used to construct average spectral signatures for regional components of the cellular hyperspectral arrays based on significant components. Bright-field images were taken to locate desired cells. Following acquisition of hyperspectral Raman array scans, a cluster analysis was performed to yield discreet binned spectral groups. Each cluster produces an averaged Raman spectrum (755 rel. 1/cm is assigned to cytochrome-c, 1012 rel. 1/cm is assigned to NAD+, 1594 rel. 1/cm is assigned to NADH, and the wide band centered at 2940 rel. 1/cm represents CH2 stretching mode, used primarily for self-normalization). The wavenumber assignment for NAD+, NADH, and cytochrome c mentioned here were verified by collection of spectral signatures from pure reagents, respectively.
Statistical Analysis
Data were analyzed using an unpaired two-tailed t-test, one-way analysis of variance (ANOVA) with multiple comparisons (Holm-Sidak method), or two-way ANOVA with multiple comparisons (Holm-Sidak method). Analyses were performed using GraphPad Prism 8.4 software (GraphPad Software Inc., San Diego, CA).
RESULTS
A Hexose Source Is Required for Hepg2 Proliferation
The alamarBlue assay was used to investigate the effects of sugar and FBS supplements to DMEM on the resazurin reduction rate of HepG2 cells which indirectly indicates metabolic activity and cell proliferation. Cells were cultured for 5 d in DMEM supplemented with 2 mM glutamine and 1 mM pyruvate and either 10% nondialyzed or dialyzed FBS (12–14 kDa). This medium was either used without additional supplementation (no hexose) or was further supplemented with 10 mM glucose or galactose. The resazurin reduction rate of HepG2 cells grown in presence of glucose and nondialyzed FBS was chosen as the control condition. Resazurin reduction rates for cells grown in media supplemented with nondialyzed FBS, nondialyzed FBS + galactose, and dialyzed FBS + 10 mM glucose were not statistically different from control (Fig. 1A). Reduction rates decreased by 47.5 ± 14% (SE) (n = 5) for cells grown in DMEM supplemented with dialyzed FBS + 10 mM galactose, however the results were also not statistically significant (P = 0.051). In contrast, cells did not remain viable if neither aldohexose was added to DMEM supplemented with glutamine, pyruvate, and dialyzed FBS. After 5 d no intact cells were observed and the resazurin reduction decreased by 96 ± 12% (SE) (n = 5).
Figure 1.
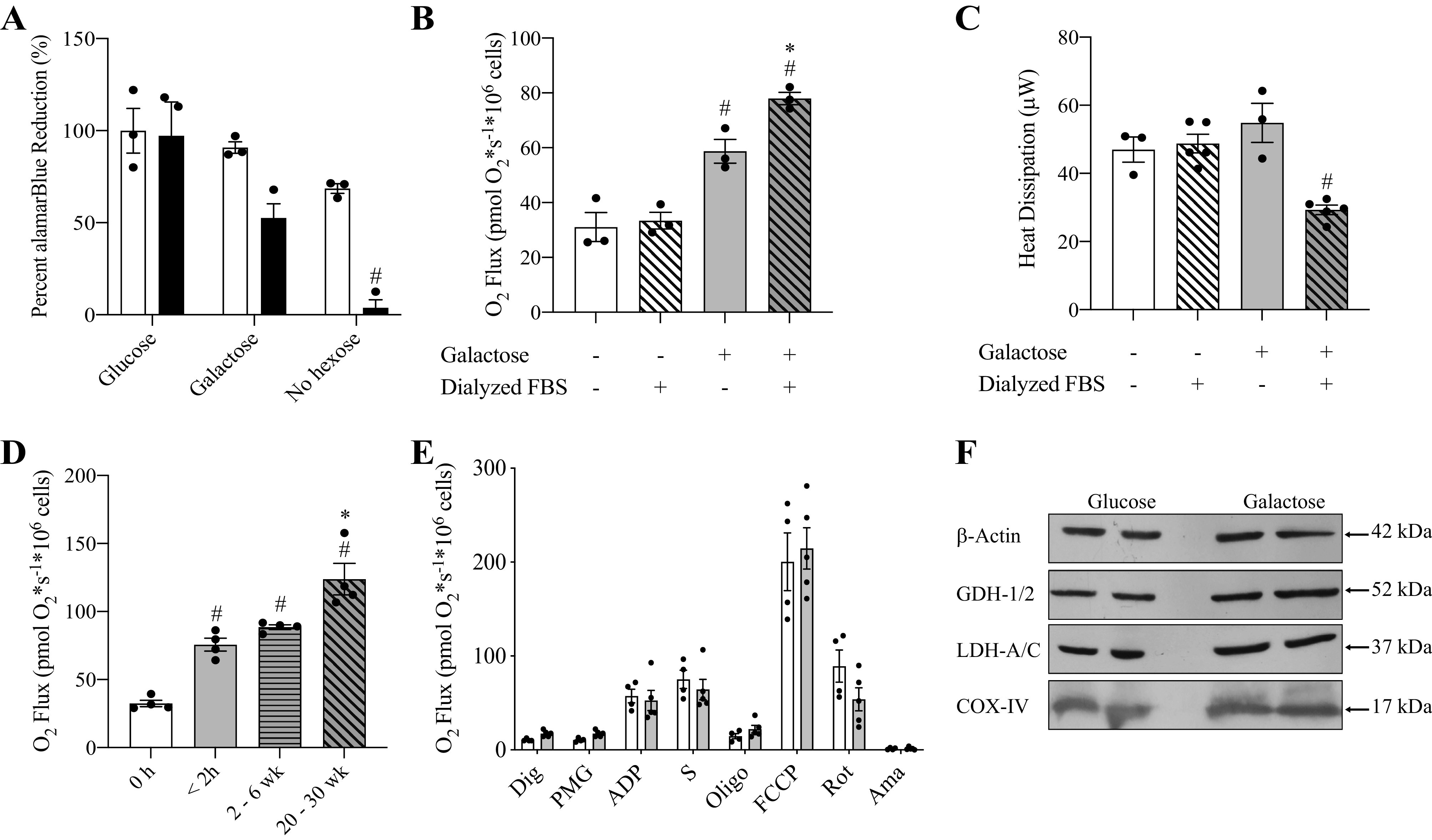
Time-dependent increase in mitochondrial respiration of HepG2 cells cultured with galactose medium. A: alamarBlue reduction by HepG2 cells cultured 24 h with DMEM supplemented with either nondialyzed (white bars) or dialyzed FBS (black bars). Certain media were further supplemented with glucose or galactose. #Indicates statistically significant differences in conditions compared with control (glucose + nondialyzed FBS). (Two-way ANOVA, P < 0.05, n = 5, average ± SEM). B: respiration rates of HepG2 cells cultured in glucose medium (white bars) or galactose medium (gray bars) with either 10% nondialyzed FBS (solid) or 10% dialyzed FBS (slanted lines) for 24 h. #Indicates statistically significant differences in conditions compared with control (glucose + nondialyzed FBS). *Indicates statistically significant differences between galactose conditions. (One-way ANOVA, P < 0.05, n = 3, average ± SEM). C: heat dissipation of HepG2 cells cultured in glucose medium (white bars) or galactose medium (gray bars) with either 10% nondialyzed FBS (solid) or 10% dialyzed FBS (slanted lines) for 24 h. #Indicates statistically significant differences in conditions compared with control (glucose + nondialyzed FBS) (One-way ANOVA, P < 0.05, n = 3–5, average ± SEM). D: respiration rates of HepG2 cells cultured for increasing time in galactose medium with 10% dialyzed FBS. Cells cultured in glucose medium with dialyzed FBS (white bars) served as the control. HepG2 cells were cultured with galactose for 2 h (solid gray bar), 2-6 weeks (horizontal gray bar), or 20-30 weeks (slanted gray bar). #Indicates statistically significant differences in conditions compared with control (glucose + dialyzed FBS). *Indicates statistically significant differences in cells for cultured 20-30 weeks in galactose compared with shorter galactose exposures. (One-way ANOVA, P < 0.05, n = 4, average ± SEM). E: oxygen consumption of permeabilized HepG2 cells cultured in glucose or galactose medium (4-5 weeks) (unpaired t-test, P < 0.05, n = 4-5, average ± SEM). F: Western blot analysis of GDH-1/2, LDH-A/C, and COX-IV proteins levels in HepG2 cells cultured 4 weeks in galactose medium compared with cells cultured in glucose medium. β−actin served as loading control.
Residual Glucose in Non-dialyzed FBS Containing Medium Inhibits the Galactose Effect on Respiration
Mitochondrial respiration was assessed for HepG2 cells cultured for 24 h in the same media used in the alamarBlue experiments without addition of the indicator dye. Cells cultured in medium containing 10 mM glucose and nondialyzed FBS served as control (Fig. 1B). Routine respiration, uncoupled respiration, and LEAK respiration was identical for cells cultured with glucose medium supplemented with dialyzed FBS compared with control (Supplemental Fig. S2, A and B). However, replacement of glucose with galactose resulted in an FBS-dependent increase in routine respiration. Compared with control, oxygen consumption was 89 ± 18% (SE) (n = 3) higher for galactose treated cells if cultured with nondialyzed FBS and 150 ± 17% (SE) (n = 3) higher when dialyzed FBS was employed. Despite increased routine respiration, no significant changes were observed for maximal uncoupled respiration or LEAK respiration compared with glucose controls (Supplemental Fig. S2, A and B). Microcalorimetry was performed to assess heat production for HepG2 cells cultured 24 h in glucose or galactose medium supplemented with nondialyzed or dialyzed FBS. Cells cultured with galactose medium and dialyzed FBS had a significantly lower heat output compared with all other conditions (Fig. 1C). Based on these results, all further experiments were performed in DMEM supplemented with glutamine, pyruvate, dialyzed FBS and either glucose or galactose.
The Galactose Effect on Mitochondrial Oxygen Consumption Depends on Length of Culture
HepG2 cells were shifted from glucose medium (DMEM + glutamine + pyruvate + glucose + dialyzed FBS) to galactose medium (DMEM + glutamine + pyruvate + galactose + dialyzed FBS) for 2 h, 2–6 weeks, and 20–30 weeks before respirometry and the effect on routine, maximal uncoupled, and LEAK respiration was measured (Fig. 1D). HepG2 cells exposed to galactose medium for 2 h significantly increased routine respiration compared with glucose control by 132 ± 10% (SE) (n = 4) and this increase stayed constant over to 2–6 weeks of culture in this medium (Fig. 1D). In contrast, culturing HepG2 cells for 20–30 weeks in galactose medium resulted in a 281 ± 11% (SE) (n = 4) increase in respiration compare to control and oxygen flux was significantly higher compared with all other treatment groups (Fig. 1D). Further, both maximal and LEAK respirations were significantly elevated in the 20–30 week galactose treatment group compared with control (Supplemental Fig. S2, C and D). Respiration rates were not altered in HepG2 cells cultured with glucose for 20–30 weeks compared with the lower passage cells (4–12 weeks) cultured in glucose used for control data (Supplemental Fig. S2, E–G). Next, parts of the OXPHOS machinery were investigated in permeabilized HepG2 cells after cultured for 4–5 weeks in galactose medium (Fig. 1E). No significant differences were observed between glucose and galactose conditions for either complex I (pyruvate, malate, and glutamate) or complex I + II (pyruvate, malate, glutamate, and succinate) stimulated oxygen flux including LEAK and maximal uncoupled respiration rates. Finally, immunoblotting was performed on lactate dehydrogenase (LDH-A/C) and two mitochondrial proteins (COX-IV and GDH-1/2). No differences in expression levels occurred for these proteins during cultured for 4–5 weeks in galactose medium compared with glucose control (Fig. 1F).
Reduction in Flux through Primary Metabolic Pathways Is Reflected in Gene Expression and Metabolite Levels
RNA sequencing and targeted metabolomics were performed on HepG2 cells cultured in galactose medium for 4–5 weeks and results were compared with glucose control cells. A comprehensive list of genes and metabolites analyzed are available in supplementary material (Supplemental Tables S1 and S3). Replacement of glucose with galactose altered expression of transcripts from the SLC2A gene family which codes for GLUT transporters. HepG2 cells cultured in galactose had elevated expression of SLC2A2 and SLC2A4 and decreased expression of SLC2A1 and SLC2A3 compared with glucose control cells (Table 1). Further, cells cultured with galactose had elevated expression of two genes involved in the Leloir pathway, GALM and GALT. However, intracellular levels of UDP-glucose, a metabolite in the Leloir pathway, was not impacted by the hexose used in the culture medium (Fig. 2).
Table 1.
Differentially expressed metabolic genes in HepG2 cells cultured with galactose
| Biological Process | Gene Symbol | Description | Fold Change | P Value |
|---|---|---|---|---|
| Galactose metabolism | GALTGALM | Galactose-1-phosphate uridylyltransferaseGalactose mutarotase (aldose 1-epimerase) | 1.61.3 | 0.0010.033 |
| Hexose Transport | SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | −2.0 | 5.00E−05 |
| SLC2A2 | Solute carrier family 2 (facilitated glucose transporter), member 2 | 1.5 | 0.036 | |
| SLC2A3 | Solute carrier family 2 (facilitated glucose transporter), member 3 | −2.0 | 5.00E−0.5 | |
| SLC2A4 | Solute carrier family 2 (facilitated glucose transporter), member 4 | 1.5 | 0.002 | |
| Glycolysis | ALDOB | Aldolase B, fructose-bisphosphate | 8.5 | 5.00E−0.5 |
| Mitochondrial Pyruvate Carriers | MPC1 | Mitochondrial pyruvate carrier 1 | 1.3 | 0.012 |
| MPC2 | Mitochondrial pyruvate carrier 2 | 1.3 | 0.043 | |
| Gluconeogenesis | PCK1 | Phosphoenolpyruvate carboxykinase 1 (soluble) | 11 | 5.00E−05 |
| TCA Cycle | IDH1 | Isocitrate dehydrogenase 1 (NADP+), soluble | 1.2 | 0.020 |
| IDH2 | Isocitrate dehydrogenase 2 (NADP+), mitochondrial | 1.3 | 0.004 | |
| SUCLG2 | Succinate-CoA ligase, GDP-forming, beta subunit | 1.3 | 0.005 | |
| MDH1 | Malate dehydrogenase 1, NAD (soluble) | 1.2 | 0.023 | |
| Pentose Phosphate Pathway | G6PD | Glucose-6-phosphate dehydrogenase | −1.3 | 0.010 |
| RPIA | Ribose 5-phosphate isomerase A | −1.2 | 0.026 | |
| TKT | Transketolase | −1.2 | 0.028 | |
| Amino Acid Metabolism | HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | 1.2 | 0.008 |
| MUT | Methylmalonyl Coa mutase | 1.3 | 0.002 | |
| HIBADH | 3-hydroxyisobutyrate dehydrogenase | 1.2 | 0.034 | |
| SHMT2 | Serine hydroxymethyltransferase 2 | −1.3 | 0.004 | |
| AOX1 | Aldehyde oxidase 1 | 1.7 | 0.002 | |
| ALDH1A1 | Aldehyde dehydrogenase 1 family, member A1 | 1.3 | 0.006 | |
| ALDH1B1 | Aldehyde dehydrogenase 1 family, member B1 | 1.2 | 0.012 | |
| PAH | Phenylalanine hydroxylase | 1.4 | 0.008 | |
| TAT | Tyrosine aminotransferase | 1.4 | 0.034 | |
| HPD | 4-Hydroxyphenylpyruvate dioxygenase | 1.9 | 5.00E−05 | |
| AGXT | Alanine-glyoxylate aminotransferase | 3.2 | 5.00E−05 | |
| KYNU | Kynureninase | 1.5 | 5.00E−05 | |
| ARG1 | Arginase 1 | 5.4 | 0.002 | |
| HAL | Histidine ammonia-lyase | 3 | 0.002 |
A P value cutoff ≤ 0.05, q-value cutoff ≤ 1 with |FC| ≥ 1 was used to determine differential expression (n = 3). Complete list of genes analyzed for differential expression and additional details of analysis are included in Supplemental Table S1.
Figure 2.
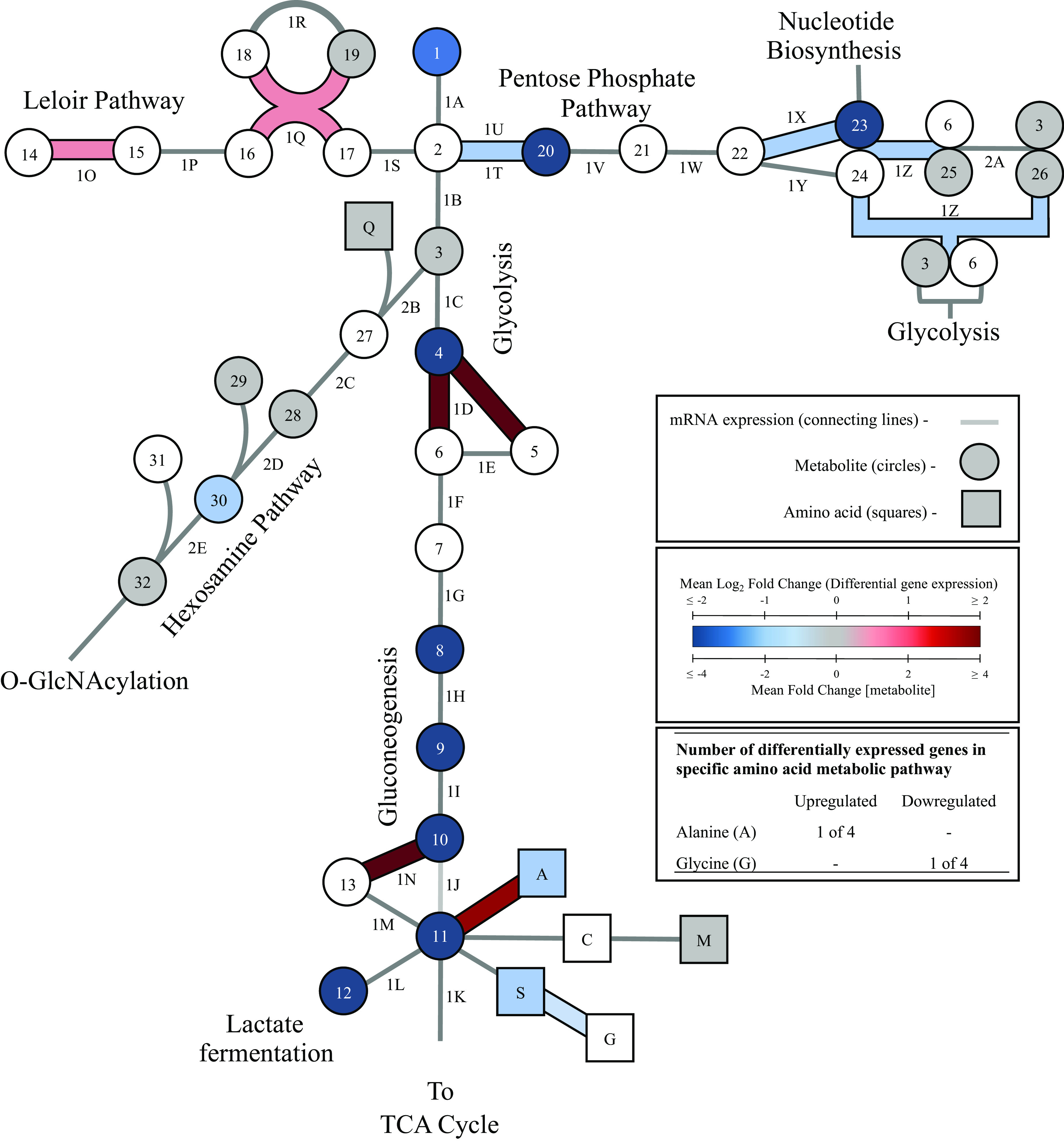
Transcriptomic and metabolomic profiling of the glycolytic, gluconeogenic, PPP, hexosamine, and Leloir pathways for HepG2 cells cultured in galactose medium. HepG2 cells were cultured for 4-5 weeks in glucose or galactose medium and glucose conditions were used as the control. Metabolites in glycolytic, gluconeogenic, Leloir, and pentose phosphate pathways are represented with circles and amino acids levels are represented with squares along with a one letter amino acid code. S, succinate; A, alanine; W, tryptophan; G, glycine; T, threonine. Blue symbols represent metabolites with decreased levels, red represents increased levels, gray represents no change, and white indicates the metabolite level was not analyzed (unpaired t-test, P < 0.05, n = 4, average ± SEM). Differentially expressed genes were assessed under the same conditions for all enzymes involved in the metabolic diagram. The same color-code used for metabolite concentration is used to represent changes in expression and differentially expressed genes are bolded. The represented catabolic pathways for certain amino acids are simplified to one reaction and do not represent all enzymatic steps and if differential expression was observed for one or more gene in a catabolic pathway, the single step reflected in the figure is bolded and colored to indicate this. A P-value cutoff ≤ 0.05, q-value cutoff ≤ 1 with |FC| ≥ 1 was used to determine differential expression (n = 3).
In respect to gene products involved in glycolysis, the only differentially expressed gene was ALDOB (Fig. 2). This gene codes for liver-specific aldolase B and expression increased in galactose cultured cells by 8.5-fold compared with glucose control. Further, galactose exposure reduced the levels of several glycolytic metabolites (glucose, fructose-1,6-bisphosphate, 3-phosphoglycerate/2-phosphoglycerate, phosphoenolpyruvate, and pyruvate (Fig. 2)). Intracellular lactate concentration decreased by 83-fold in response to galactose even though mRNA and protein expression levels for LDH were unchanged in galactose versus glucose treated cells. The mRNA levels for phosphoenolpyruvate carboxykinase (PEPCK), which generates phosphoenolpyruvate in gluconeogenesis, significantly increased by 11-fold in galactose treated cells.
Next, changes to the pentose phosphate pathway (PPP) were assessed in cells cultured with galactose compared with glucose. Expression levels of three genes (G6PD, RPIA, and TKT) decreased significantly and cells cultured with galactose had significantly lower levels of ribose-5-phosphate (−5.4-fold) and 6-phosphogluconate (−8.9-fold) (Fig. 2). No significant differences were detected in the levels of additional PPP intermediates including fructose-6-phosphate, erythose-4-phosphate, and seduheptulose-7-phosphate. Further, differential gene expression was assessed for the mitochondrial pyruvate transporter (MPC) which consists of two subunits, MPC1 and MPC2 (28). Expression levels increased for MPC1 and decreased for MPC2 in galactose cultured HepG2 cells compared with glucose control (Table 1). No significant changes were detected in gene expression for components of the pyruvate dehydrogenase (PDH) complex. Compared with control, HepG2 cells cultured in galactose medium had significantly increased expression of three genes encoding enzymes of the TCA cycle (IDH1/2, SUCLG2, and MDH1) (Fig. 3). No differences were detected in acetyl-CoA or α-ketoglutarate levels compared with control, but decreased levels of citrate (−22.9-fold), succinate (−3.9-fold), fumarate (−7.9-fold), and malate (−9-fold) were observed (Fig. 3) in galactose cultured cells.
Figure 3.
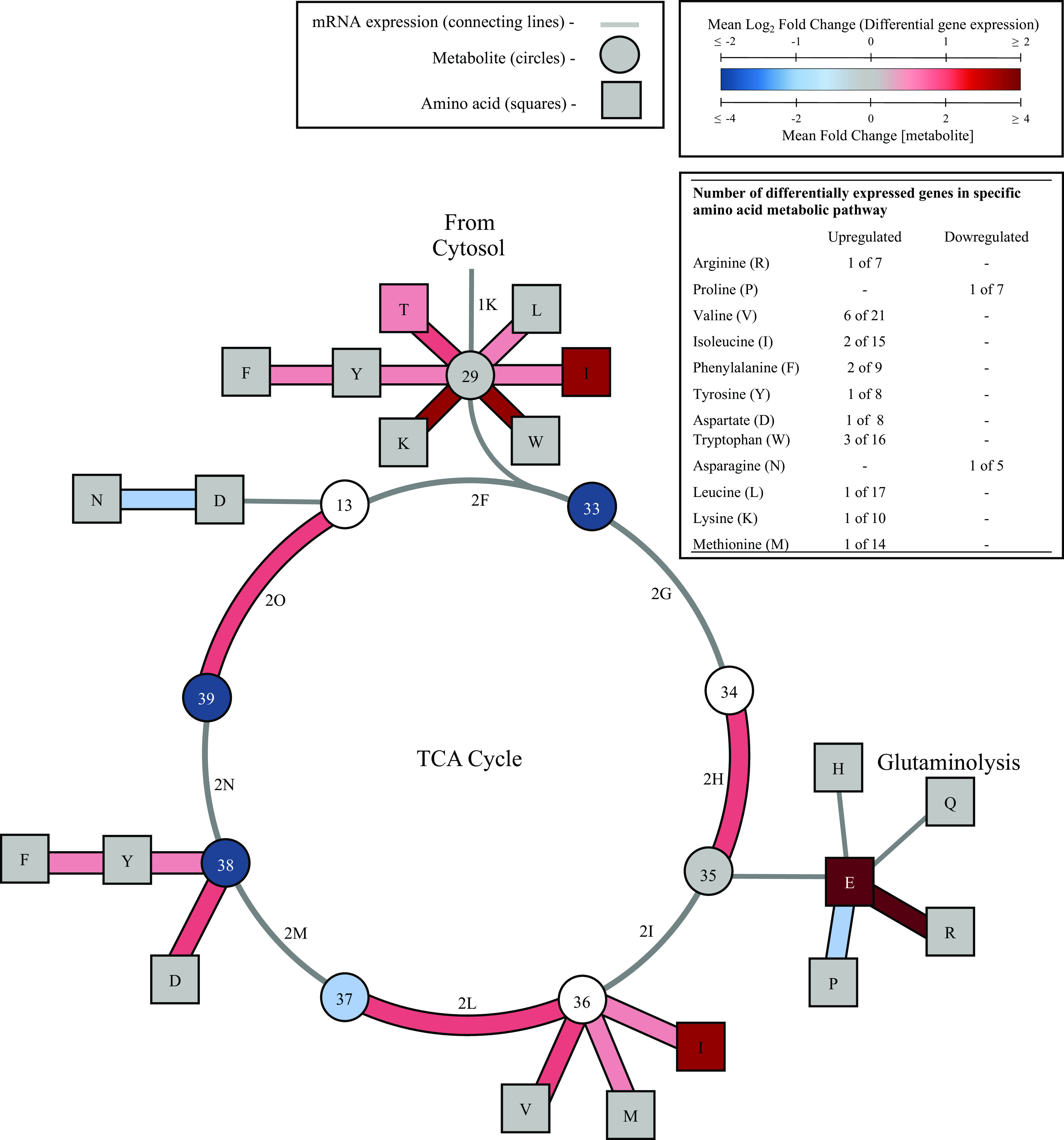
Transcriptomic and metabolomic profiling of the TCA cycle and amino acid metabolism for HepG2 cells cultured in galactose medium. HepG2 cells were cultured 4-5 weeks in glucose or galactose medium and glucose conditions were used as the control. Metabolites in the TCA cycle are represented with circles and amino acids levels are represented with squares along with a one letter amino acid code. L, leucine; I, isoleucine; Y, tyrosine; W, tryptophan; N, asparagine; D, aspartate; F, phenylalanine; V, valine; M, methionine; T, threonine; H, histidine; Q, glutamine; R, arginine; P, proline; E, glutamate. Blue symbols represent metabolites with decreased levels, red represents increased levels, gray represents no change, and white indicates the metabolite level was not analyzed (unpaired t-test, P < 0.05, n = 4, average ± SEM). Differentially expressed genes were assessed under the same conditions for all enzymes involved in the metabolic diagram according to KEGG pathways. The same color-code used for metabolite concentration is used for gene expression data and differentially expressed genes are bolded. Represented catabolic pathways for certain amino acids are simplified to one reaction and do not represent all enzymatic steps. If differential expression was observed for one or more gene in a catabolic pathway, the single step reflected in the figure is bolded and colored to indicate differential expression. A P-value cutoff ≤ 0.05, q-value cutoff ≤ 1 with |FC| ≥ 1 was used to determine differential expression (n = 3).
Changes to gene expression and metabolite levels were then assessed for the hexosamine pathway which branches from glycolysis at fructose-6-phosphate (Fig. 2). No genes were differentially expressed between treatment groups, and n-acetyl-glucosamine-1-phosphate was the only assessed metabolite with altered levels in this pathway (−1.7-fold). Levels of fructose-6-phosphate, glutamine, acetyl-CoA, N-acetyl-glucosamine-6-phosphate, and UDP-GlcNAc did not change compared with glucose control cells.
Galactose Elevates Amino Acid Catabolism
Replacement of glucose with galactose altered amino acid metabolism in HepG2 cells. Levels were assessed for 18 of the 20 standard amino acids and gene expression changes were monitored in amino acid metabolic pathways. The KEGG database (https://www.genome.jp/kegg/) was used for construction of reference pathways and to analyze gene expression patterns (Table 1). Metabolite levels were assessed for 4 of the 6 amino acids known to be metabolized to pyruvate. Alanine levels decreased (−2.4-fold) and expression increased for AGXT which codes for an enzyme that converts alanine to pyruvate (Fig. 2). Glycine and serine are metabolized to pyruvate through a shared pathway. Glycine levels were not measured, however expression of SHMT2 increased which codes for an enzyme that catalyzes a reversible conversion of serine to glycine and serine levels decreased (−2.8-fold) compared with glucose control cells. Both methionine and cysteine are also metabolized to pyruvate. However, methionine levels did not change, and cysteine levels were not measured. No genes involved in conversion of methionine and cysteine to pyruvate were significantly altered in galactose cultured cells compared with glucose control.
Galactose exposure increased gene expression in several pathways resulting in amino acid catabolism to a TCA cycle intermediate (Fig. 3). Further, amino acids levels significantly increased for threonine, glutamate, and isoleucine. Glutamate levels increased by 398-fold demonstrating an increase in glutaminolysis (Fig. 3) (29). Catabolic pathways containing one or more upregulated genes include pathways for catabolism of threonine, leucine, isoleucine, tryptophan, lysine, phenylalanine, tyrosine, arginine, valine, and aspartate. Expression decreased for two genes involved in amino acid synthesis; ASNS and PYRC2 which code for enzymes involved in asparagine and proline synthesis, respectively.
Galactose Decreases Nucleotide and NAD Synthesis
Galactose decreased expression of genes involved in nucleotide synthesis (RRM2, TK1, PRPS1, PRPS2, PAICS, and GMPS) and increased expression of genes involved in pyrimidine degradation (DYPD and DYSP) (Table 2). The experimental approach employed in this study did not resolve nucleotide phosphate ratios. Instead, data was combined for each nucleotide phosphate level to evaluate total nucleotide phosphate pool. Several nucleotide phosphate pools were significantly lower in galactose cultured cells compared with control. Total adenosine phosphate levels were impacted by the culture conditions with cells having a >17-fold decrease in the total adenylate pool ([ATP]+[ADP]+[AMP]) (Fig. 4). Galactose increased the level of the only nucleoside assessed, deoxyuridine. Inosine, hypoxanthine, and xanthine levels were assessed as markers for nucleotide catabolism. Cells cultured with galactose had elevated inosine levels, but hypoxanthine and xanthine levels were not altered compared with glucose control cells.
Table 2.
Differentially expressed genes involved in nucleotide and NAD metabolism.
| Biological Process | Gene Symbol | Description | Fold Change | P Value |
|---|---|---|---|---|
| Nucleotide synthesis | RRM2 | Ribonucleotide reductase M2 | −1.3 | 0.018 |
| TK1 | Thymidine kinase 1, soluble | −1.3 | 0.001 | |
| PRPS1 | Phosphoribosyl pyrophosphate synthetase 1 | −1.2 | 0.008 | |
| PRPS2 | Phosphoribosyl pyrophosphate synthetase 2 | −1.3 | 0.004 | |
| PAICS | Phosphoribosylaminoimidazole carboxylase, | −1.3 | 0.031 | |
| GMPS | Guanine monophosphate synthase | −1.3 | 0.013 | |
| Nucleotide degradation | DYPD | Dihydropyrimidine dehydrogenase | 2.1 | 5.00E-05 |
| DYPS | Dihydropyrimidinase | 10.8 | 5.00E-05 | |
| NAD Synthesis | NMNAT3 | Nicotinamide Nucleotide adenylyltransferase 3 | 1.5 | 0.047 |
| AFMID | Arylformamidase | 1.3 | 0.017 | |
| QPRT | Quinolinate phosphoribosyltransferase | 1.3 | 0.017 |
A P-value cutoff ≤ 0.05, q-value cutoff ≤ 1 with |FC| ≥ 1 was used to determine differential expression (n = 3). complete list of genes analyzed for differential expression and additional details of analysis are included in Supplemental Table S1.
Figure 4.

Galactose impacts nucleotide levels in HepG2 cells. Metabolite level of nucleotide phosphates and related metabolites in HepG2 cells cultured in glucose or galactose medium for 4-5 weeks. Represented metabolites include ATP+ADP+AMP (A), GTP+GDP+GMP (B), UTP+UDP+UMP (C), CDP+CMP (D), IMP (E), deoxyuridine (F), inosine (G), hypoxanthine (H), and xanthine (I). #Indicates statistically significant differences between cells cultured with galactose compared with glucose (unpaired t-test, P < 0.05, n = 4, average ± SEM).
Culturing HepG2 cells with galactose impacted nicotinamide adenine dinucleotide (NAD) synthesis which can occur through the Preiss-Handler, salvage, or de novo biosynthetic pathways (30). Galactose treatment increased mRNA expression for three genes involved in the Preiss-Handler and de novo biosynthetic pathways and no downregulated genes were observed compared with control (Table 2). Further, cells cultured with galactose had ∼ 10-fold lower levels of NAD+, NADH, NADP+, and NADPH, but the cellular bulk NAD+-to-NADH and NADP+-to-NADPH ratios were not altered (Fig. 5). Neither the redox ratio or metabolite levels of oxidized glutathione (GSSG) and reduced glutathione (GSH) were altered by galactose treatment.
Figure 5.

Galactose decreases NAD(P) levels in HepG2 cells. Metabolite level of selected redox couples in HepG2 cells cultured in glucose or galactose medium for 4-5 weeks. Represented metabolites include NAD+ (A), NADH (B), NADP+ (D), NADPH (E), GSSG (G), and GSH (H). The redox ratio (oxidized/reduced) represents the redox poise for each couple NAD+/NADH (C), NADP+/NADPH (F), and GSSG/GSH (I). #Indicates statistically significant differences between cells cultured with galactose compared with glucose (Unpaired t-test, P < 0.05, n = 4, average ± SEM).
Galactose Lowers Cytoplasmic and Mitochondrial NAD+/NADH Ratios
Metabolomics revealed a non-significant trend toward a lower NAD+-to-NADH redox ratio in galactose-cultured cells but because these bulk measurements did not allow for subcellular resolution a novel Raman microspectroscopic technique was developed. Compartment specific measurements were developed by first scanning pure NAD compounds and then identifying these signatures in HepG2 cells through confocal Raman spectroscopy to independently monitor the cytoplasmic and mitochondrial NAD+-to-NADH redox ratios (Fig. 6A–C). Cytochrome-c, served as a marker to identify mitochondrial regions and allow for comparison between ratios measured in the mitochondrial and cytoplasmic compartments. The accuracy of this technique was tested by introducing a lactate dehydrogenase inhibitor, GSK 2837808 A (20 µM), to prevent cytoplasmic oxidation of NADH to NAD+ while extracellular acidification rates (ECAR) were monitored to confirm LDH inhibition. Before LDH inhibition, cells cultured with galactose had a 169 ± 8.2% (SE) (n = 8) lower rate of ECAR compared with glucose control. LDH inhibition significantly lowered ECAR in glucose and galactose cultured cells by 145 ± 10% (SE) (n = 8) and 37 ± 8% (SE) (n = 8), respectively and resulted in a 51 ± 10% (SE) (n = 8) difference of ECAR in presence of the LDH inhibitor (Fig. 6D). Raman spectroscopy measured that the cytoplasmic NAD+-to-NADH redox ratio for HepG2 cells cultured with galactose was 26 ± 6% (SE) (n = 6) lower in absence of the LDH inhibitor (Fig. 6E). After LDH inhibition the cytoplasmic NAD+-to-NADH redox ratio decreased by 43 ± 7% (SE) (n = 6) in glucose and 32 ± 4% (SE) (n = 6) in galactose cultured cells. Despite the ratios decreasing for both conditions after addition of the LDH inhibitor, galactose-treated cells still had a significantly lower redox ratio compared with glucose cultured cells. Further, the mitochondrial NAD+-to-NADH ratio was assessed in glucose and galactose-cultured cells through colocalization with cytochrome-c. The mitochondrial ratio was lower for both conditions compared with cytoplasmic ratios. Further, cells cultured with galactose had a 20 ± 6% (SE) (n = 6) lower mitochondrial NAD+-to-NADH ratio compared with glucose control cells.
Figure 6.

Galactose lowers cytoplasmic and mitochondrial NAD+-to-NADH ratios. Acquisition of Raman spectra of pure NAD+, NADH, NADP+, and NADPH compounds and identification in HepG2 cells. The fingerprint region of each of each of the compounds were used to spatially correlated with the intracellular distribution of the compounds. A: representative Raman spectra of a cluster average. B: intensity of 755 rel. 1/cm peak, representing cytochrome C, increasing as function of cluster degree. C: cascade graph of Raman spectra for four related NAD compounds. D: extracellular acidification of HepG2 cells cultured with glucose or galactose in presence or absence of the LDH inhibitor, GSK 2837808 A (20 µM). #Indicates statistically significant differences compared with glucose control. *Indicates statistically significant between glucose and galactose condition in presence of LDH inhibitor (One-way ANOVA, P < 0.05, n = 8, average ± SEM). E: cytoplasmic NAD+/NADH ratio obtained from Raman spectra for HepG2 cells cultured with glucose or galactose in presence or absence of GSK 2837808 A (20 µM). #Indicates statistically significant differences compared with glucose control. *Indicates statistically significant between glucose and galactose condition in presence of LDH inhibitor. (One-way ANOVA, P < 0.05, n = 6, average ± SEM). F: mitochondrial NAD+/NADH obtained from Raman spectra for HepG2 cells cultured in glucose or galactose. #Indicates statistically significant differences compared with glucose control. (Unpaired t-test, P < 0.05, n = 6, average ± SEM).
DISCUSSION
Here we demonstrate that the metabolic poise in tumor-derived HepG2 cells can be shifted to reflect conditions reminiscent of a primary hepatocyte by replacing glucose with galactose in the culture medium. Although cells grown with galactose have been previously shown to increase OXPHOS activity the impact on the central metabolic pathways in tumor-derived cells has not been sufficiently investigated (31, 32). To address this gap in knowledge we employed RNAseq and metabolomics on HepG2 cells adapted to DMEM supplemented with dialyzed FBS, glutamine, pyruvate, and either glucose or galactose. To investigate a potential involvement of the cellular redox state in metabolic poise we developed a novel Raman spectroscopic technique to monitor subcellular NAD+-to-NADH ratios. These approaches allowed us to identify hexose-dependent differences in the central metabolic pathways of tumor-derived cells.
OXPHOS Is Increased in HepG2 Cells If Galactose Medium Is Supplemented with Dialyzed FBS
HepG2 cells cultured with galactose do not achieve sufficient glycolytic flux to sustain ATP production from substrate-level phosphorylation and cells are forced to shift energy production to OXPHOS in the mitochondrion. We demonstrate this shift can be more robustly accomplished in HepG2 cells if galactose culture medium is supplemented with dialyzed FBS opposed to standard nondialyzed FBS. Mitochondrial oxygen consumption, alamarBlue reduction, lactate production, and heat dissipation data all support this conclusion. These differences are likely attributed to residual glucose concentrations reaching 1–2 mM if culture medium is supplemented with 10% FBS (33, 34). These glucose concentrations are sufficient to accelerate lactate production and inhibit respiration. We also observed increased oxygen consumption in galactose despite a decrease in resazurin reduction. These results support the observation that resazurin reduction occurs in the cytosol opposed to the mitochondrion (35).
Galactose Is a Necessary Source of Carbon for HepG2 Proliferation
We found that HepG2 cells retain the capacity to proliferate in galactose medium supplemented with 10% dialyzed FBS but did not survive in absence of both aldohexoses in the culture medium. These results conflicted with the observation that galactose was unable to support an oxidative shift in murine-derived myoblasts (C2C12) cells (36). Using C2C12 cells we also found that galactose cannot sustain proliferation in this line which contrasts with HepG2 cells (Supplemental Fig. S1). These results suggest that replacing glucose with galactose to increase OXPHOS activity is limited to specific cell types which may be determined by the expression profile of genes involved in carbon metabolism. Further, our results demonstrate that galactose serves as a necessary source of carbon for HepG2 survival in absence of glucose. Previous claims suggested increased OXPHOS rates occur because galactose catabolism yields no net ATP and is energetically inaccessible (14). No net ATP would be generated if the two ATP equivalents produced during each passage of carbon from galactose to pyruvate were lost because of the synthesis of UDP-glucose before each cycle of the Leloir pathway. However, once produced the nucleotide sugar participates in multiple cycles through the Leloir pathway and offsets the energetic cost incurred during UDP-glucose synthesis (19). Despite an equivalent glycolytic ATP yield when glucose or galactose is metabolized, cells cultured with galactose likely derive most ATP from the catabolism of pyruvate and glutamine (29). We hypothesize galactose to be an important carbon contributor to the PPP and radioactive isotope tracing would likely reveal that the PPP, not the TCA cycle, is the primary metabolic pathway sustained by galactose metabolism.
Prolonged Galactose Exposure Increases Routine Respiration
HepG2 cells were grown up to 30 weeks with galactose as the only aldohexose available in the cell culture medium and longer galactose exposure resulted in higher routine respiration rates. We elected to investigate global changes to metabolism in cells adapted to galactose medium for approximately one month. Although longer adaptations to galactose medium would likely induce more robust changes to global metabolism, the one-month time frame permitted sufficient time for metabolic reorganization and reduced the risk for genetic mutations that could accumulate from multiple months of culture. No differences were observed after four weeks in respiration rates of permeabilized cells or in the expression levels of the mitochondrial proteins GDH-1/2 and COX-IV suggesting mitochondrial biogenesis did not change in response to the galactose medium. A previous study observed increased mitochondrial volume and changes to mitochondrial morphology in galactose grown Hela cells (9). Although our current study did not observe altered expression of transcription factors involved in coordinating mitochondrial biogenesis, we did not assess mitochondria ultrastructure. Therefore, we cannot exclude the occurrence of some mitochondrial alterations after one month of HepG2 culture with galactose which may have contributed to the increased respiration rates.
Metabolite-Induced Regulation of the Crabtree Effect
The Crabtree effect has been observed in both cancerous and noncancerous cell types (37–39). Despite decades of research we still do not have a satisfying explanation to account for the inhibition of mitochondrial respiration by glucose. One hypothesis suggests gradual structural changes such as oxidative damage to membranes or changes to subcellular morphology are responsible for inducing the Crabtree effect (40, 41). A conflicting hypothesis links respiration inhibition to a rapid chemical/molecular trigger such as an acute drop in intracellular pH, redistribution of subcellular Ca2+, or post-translational modification of an enzyme (42). We observed that the Crabtree effect can be acutely induced through titration of millimolar glucose concentrations into HepG2 cells adapted to galactose medium. Within 30 seconds of glucose addition, respiration decreased by nearly 70% (Supplemental Fig. S3). Further, acute inhibition of LDH results in a sudden rise in oxygen consumption for HepG2 cells grown in glucose medium. These acute, bidirectional changes suggest that rapid molecular change regulates mitochondrial oxygen consumption. Although an appropriate expression profile of glycolytic enzymes is required, glucose availability appears to activate the molecular trigger responsible for diminishing respiration in HepG2 cells.
Galactose Changes Metabolite Levels in All Primary Metabolic Pathways
Increased oxygen consumption in presence of galactose is attributed to diminished glycolytic flux. Although this metabolic shift has been previously characterized, additional changes beyond glycolysis and the TCA cycle have not been thoroughly investigated (43). We observed HepG2 cells cultured in galactose had elevated expression of SLC2A2 and decreased expression of SLC2A3. Both genes code for sugar transporters with affinities for galactose of Km-value = 92 mM for GLUT2 (SLC2A2), the primary expressed glucose transporter in liver, and Km-value = 8.5 mM for GLUT3 (SLC2A3) (15, 44). Despite increased expression of GLUT2, the low affinity for galactose likely restricted HepG2 cells from a substantial increase in galactose uptake. Assuming a Km-value of 92 mM, GLUT2’s affinity for galactose suggests that increasing galactose concentrations from 10 mM to 30 mM would increase transport velocity only from 10% to 17% of Vmax. Meanwhile, GLUT3 would approach Vmax in presence of 30 mM galactose. Interestingly, HepG2 cells cultured with galactose had reduced expression of GLUT3 which also likely limited galactose uptake. One may suspect that cells with high constitutive GLUT3 expression may exhibit some aerobic glycolysis if galactose levels are raised from 10 mM to 30 mM if galactose metabolism is not bottlenecked in the Leloir pathway. Unfortunately, quantification of GLUT2 and GLUT3 protein expression was out of the scope of the current study, and future work to investigate which transporter contributed most to galactose transport would be constructive.
Considering aerobic glycolysis was reduced in HepG2 cells cultured with galactose it was not surprising to observe changes to gene expression and metabolite levels for the TCA cycle, glycolytic, and PPP pathways in these cells compared with glucose controls. Of the 7 glycolytic intermediates measured in this study, 6 were significantly reduced by the galactose treatment. Glucose and fructose-6-phosphate decreased by an average of 2.8-fold. Meanwhile, metabolites downstream of fructose-6-phosphate decreased on average by 13.2-fold. These data coupled with increased ALDOB expression suggests galactose transport/metabolism was particularly limiting in the maintenance of metabolite pools in the latter portion of the glycolytic pathway. Reduced levels of TCA cycle metabolites and lactate also support this observation. Interestingly, acetyl-CoA levels were not different in glucose and galactose cultured cells. It is unclear what percentage of TCA cycle intermediates were derived directly from the supplemented pyruvate in the cell culture medium compared with derived from galactose. We suspect a significant contribution to the TCA cycle from supplemented pyruvate considering the drop in glycolytic intermediates. These data broadly suggest that supplemented galactose may disproportionately support the PPP compared with other pathways. Meanwhile, the majority of added pyruvate is likely directed to the TCA cycle. Additional studies are needed to assess the relationship between each substrate’s concentration and specific metabolic pathway activity. Finally, our metabolomics approach did not allow to quantify intracellular levels of the metabolites of the Leloir pathway (galactose, galactose-1-phosphate, UDP-galactose, and glucose-1-phosphate) beyond UDP-glucose. Measuring these metabolites was beyond the scope of this study, however their quantification in glucose and galactose cultured cells would provide key insights into potential bottlenecks in galactose metabolism.
We suspect the observed changes in gene expression may be downstream responses to altered activity of metabolic sensors such as AMPK and/or the mTORC1 complex. For example, glycolytic flux regulates mTORC1 activity which in turn regulates expression of genes involved in both anabolic and catabolic pathways (45). A recent study identified that levels of the glycolytic intermediate, DHAP, link glycolysis with mTORC1 activity (46). Although we did not measure DHAP levels, glycolytic metabolite levels upstream and downstream of DHAP were significantly reduced in galactose cultured cells and expression of ALDOB increased. Further, glycerol-3-phosphate levels were lower suggesting reduced triglyceride synthesis. These changes suggest that lower metabolite levels in HepG2 cells cultured with galactose may regulate an energy sensor such as mTORC1 and result in global changes to expression profiles of central metabolic pathways.
Chronic Galactose Exposure Reduces Dinucleotide and Nucleotide Phosphate Levels
NAD(P) participate in hundreds of biochemical reactions making these molecules key regulators of cellular metabolism. The liver serves as the primary site of NAD synthesis and likely performs more NAD-linked chemical reactions than any other tissue (47). Several pathways in the liver contribute to NAD synthesis, however precursors are limited for cultured cells (48, 49). Regardless, NAD synthesis requires phosphoribosyl pyrophosphate (PRPP), a metabolite downstream of the PPP, for generation of adenosine. Both RNAseq and metabolomics data show that HepG2 cells cultured with galactose have reduced levels of nucleotide phosphates, NAD(H), and NADP(H). We suspect slow kinetics of both galactose transport and metabolism reduce PPP metabolic flux and downstream biosynthetic processes such as nucleotide and NAD synthesis. Disruption of heightened NAD synthesis in cancer cells is emerging as a novel therapeutic option. This field has been primarily approached through pharmacological inhibition of enzymes involved in NAD synthesis (50, 51). Here our results demonstrate that this process can also be disrupted by altering carbohydrate availability. Unfortunately, the significance of the 10-fold drop in NAD levels observed in this study is unknown. NAD levels have been widely reported in mouse and rat primary hepatocytes; however, data is lacking for primary human hepatocytes. Therefore, is it difficult to estimate differences in magnitude between glucose and galactose cultured HepG2 cells with primary human hepatocytes.
Galactose Treatment Reduces Total NAD(P) Levels but not their Oxidation State
The NAD+-to-NADH ratio regulates flux through a wide range of metabolic pathways (52). High NAD+-to-NADH ratios likely facilitates proliferation by sustaining flux through oxidative biosynthetic pathways (52–56). Meanwhile, NADP+-to-NADPH ratios remain low for participation in reductive biosynthetic pathways and maintenance the cellular glutathione redox state. Despite NAD(P) and NAD(P)H levels decreasing by ∼ 10 -fold in galactose cultured HepG2 cells, total cellular NAD+-to-NADH and NADP+-to-NADPH ratios remain unchanged. This suggests that both redox ratios are relatively resistant to change to sustain metabolic flux despite a decrease in total coenzyme concentrations. One limitation of standard metabolomics is that it reports total cellular NAD levels, however redox ratios, NAD synthesis rates, and NAD concentration vary depending on subcellular localization (57). For example, mitochondrial NAD levels remain relatively unaffected after depletion of cytoplasmic NAD levels and concentrations are ∼2–10-fold higher compared with the cytoplasm (58–61). Further, the mitochondrial NAD+-to-NADH ratio has been reported to range from 2 to 8 whereas cytoplasmic ratios range from 0.1 to 600 (62–64).
Galactose Decreases Cytosolic and Mitochondrial NAD+-to-NADH Ratios
Using a novel Raman spectroscopic technique, we observed lower mitochondrial NAD+-to-NADH ratios compared with ratios observed in the cytoplasm for HepG2 cells cultured in both glucose and galactose. Further, both the cytoplasmic and mitochondrial NAD+-to-NADH ratios were lower in cells cultured with galactose compared with glucose control cells. This indicates a metabolic shift occurs in both subcellular compartments in response to the aldohexose source. The cytoplasmic NAD+-to-NADH ratio is primarily regulated by the conversion of pyruvate to lactate catalyzed by LDH. The LDH reaction rate is ∼2 orders of magnitude faster than overall flux through glycolysis which results in rapid regeneration of NAD+ in presence of high pyruvate concentrations (63). The increased NAD+ availability may then allow to sustain glycolysis for ATP generation as well as metabolic flux through oxidative biosynthetic pathways (53). Unfortunately, both approaches in this study failed to distinguish [NAD]free versus [NAD]bound, the former of which largely regulates redox potentials of subcellular compartments. The intracellular lactate/pyruvate (L/P) ratio is widely used for determination of the free cytoplasmic NAD+-to-NADH ratio. In this study we observed a 4-fold decrease in the L/P ratio for galactose cultured HepG2 cells. Unfortunately, the steps required to determine equilibrium status of this reaction were outside the scope of our current study and the free cytosolic NAD+-to-NADH ratios were not quantified (65). Despite this limitation we hypothesize that the robust metabolic shift accomplished by culturing HepG2 cells with galactose may lower the free compartmental NAD+-to-NADH ratios. In addition to free NAD+-to-NADH ratios, additional studies are required to examine the relationship between substrate availability in tumor-derived cells and compartmental free NADP+-to-NADPH ratios.
Conclusions
In summary, extensive changes to gene expression patterns and increased nutrient uptake is required to divert metabolites into biosynthetic pathways for proliferation of malignant cells. This change is often coupled with a shift in ATP production from OXPHOS to substrate-level phosphorylation even in absence of mitochondrial dysfunction. The resulting shift increasingly desensitizes cells to the impacts of mitochondrial toxins and subsequent dysfunction (12). This metabolic poise should be more widely considered when using tumor-derived cells to investigate processes apt for translation to nondividing somatic cells. Fortunately, for certain tumor-derived cell lines replacing glucose with galactose represents a convenient approach to shift cells toward a metabolic poise that closer reflects a primary cell. This shift also illuminates the metabolic activities that sustain rapid proliferation in malignant cells which in turn could inform the development of novel therapeutics for cancer treatment.
SUPPLEMENTAL DATA
Supplemental Tables S1, S2, and S3: https://doi.org/10.6084/m9.figshare.13522946.
Supplemental Figs. S1, S2, and S3: https://doi.org/10.6084/m9.figshare.13522946.
GRANTS
This work was funded by National Science Foundation Grant CHE-160944 to M.A.M, M.K., and N.C. Sequencing and bioinformatics support for this work was initially provided by National Institutes of Health (NIH) National Institute of General Medical Sciences Grants P20GM103436 (Nigel Cooper, PI) and P20GM106396 (Donald Miller, PI). These grants then supported a KBRIN-NGS pilot project award (KBRIN0075) to M.A.M. Further, part of this work was supported by a University of Louisville Graduate Network in Arts & Sciences (GNAS) grant to R.A.S.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.C. and M.A.M. conceived and designed research; R.A.S. and J.S. performed experiments; R.A.S. and J.S. analyzed data; R.A.S., J.S., M.E.K., N.C., and M.A.M. interpreted results of experiments; R.A.S. and J.S. prepared figures; R.A.S. drafted manuscript; M.E.K., N.C., and M.A.M. edited and revised manuscript; R.A.S., J.S., M.E.K., N.C., and M.A.M. approved final version of manuscript.
References
- 1.Warburg O. On the origin of cancer cells. Science 123: 309–314, 1956. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J 274: 1393–1418, 2007. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 3.Warburg O. On respiratory impairment in cancer cells. Science 124: 269–270, 1956. [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20, 2008. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464, 2011. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 7.Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 5: 592–602, 2014. doi: 10.1007/s13238-014-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 39: 347–354, 2014. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64: 985–993, 2004. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 10.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol 28: 249–255, 2010. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimm D, Altamirano L, Paudel S, Welker L, Konkle ME, Chakraborty N, Menze MA. Modulation of cellular energetics by galactose and pioglitazone. Cell Tissue Res 369: 641–646, 2017. doi: 10.1007/s00441-017-2657-1. [DOI] [PubMed] [Google Scholar]
- 12.Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, Mootha VK. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab 24: 875–885, 2016. doi: 10.1016/j.cmet.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bayona-Bafaluy MP, Sanchez-Cabo F, Fernandez-Silva P, Perez-Martos A, Enriquez JA. A genome-wide shRNA screen for new OxPhos related genes. Mitochondrion 11: 467–475, 2011. doi: 10.1016/j.mito.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97: 539–547, 2007. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 15.Augustin R, Mayoux , E. Mammalian solute carrier families SLC2 and SLC5: facilitative and active transport of hexoses and polyols. In: Molecular Life Sciences: An Encyclopedic Reference, edited by Wells RD, Bond JS, Klinman J.. Masters BSS. New York, Springer, 2018, p. 627–651. [Google Scholar]
- 16.Ghezzi C, Loo DDF, Wright EM. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61: 2087–2097, 2018. doi: 10.1007/s00125-018-4656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol 1: 140–151, 2013. [Erratum in Lancet Diabetes Endocrinol 3: e3, 2015]. doi: 10.1016/S2213-8587(13)70050-0. [DOI] [PubMed] [Google Scholar]
- 18.Holden HM, Rayment I, Thoden JB. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 278: 43885–43888, 2003. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 19.Frey PA. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 10: 461–470, 1996. [PubMed] [Google Scholar]
- 20.Boros LG, Puigjaner J, Cascante M, Lee WN, Brandes JL, Bassilian S, Yusuf FI, Williams RD, Muscarella P, Melvin WS, Schirmer WJ. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res 57: 4242–4248, 1997. [PubMed] [Google Scholar]
- 21.Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I. In vitro models for liver toxicity testing. Toxicol Res (Camb) 2: 23–39, 2013. doi: 10.1039/C2TX20051A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tolosa L, Gómez-Lechón MJ, Pérez-Cataldo G, Castell JV, Donato MT. HepG2 cells simultaneously expressing five P450 enzymes for the screening of hepatotoxicity: identification of bioactivable drugs and the potential mechanism of toxicity involved. Arch Toxicol 87: 1115–1127, 2013. doi: 10.1007/s00204-013-1012-x. [DOI] [PubMed] [Google Scholar]
- 23.Menze MA, Chakraborty N, Clavenna M, Banerjee M, Liu XH, Toner M, Hand SC. Metabolic preconditioning of cells with AICAR-riboside: improved cryopreservation and cell-type specific impacts on energetics and proliferation. Cryobiology 61: 79–88, 2010. doi: 10.1016/j.cryobiol.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53, 2013. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578, 2012. [Erratum in Nat Protoc 9: 2513, 2014]. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz MA, Burant CF, Kennedy RT. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal Chem 83: 3406–3414, 2011. doi: 10.1021/ac103313x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bender T, Martinou JC. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim Biophys Acta 1863: 2436–2442, 2016. doi: 10.1016/j.bbamcr.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 29.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254: 2669–2676, 1979. doi: 10.1016/S0021-9258(17)30124-2. [DOI] [PubMed] [Google Scholar]
- 30.Cantó C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53, 2015. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, Harper ME. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS One 6: e28536, 2011. [Errata in PLoS One 7: 2012]. doi: 10.1371/journal.pone.0028536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dott W, Mistry P, Wright J, Cain K, Herbert KE. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol 2: 224–233, 2014. doi: 10.1016/j.redox.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gstraunthaler G. Alternatives to the use of fetal bovine serum: Serum-free cell culture. Altex-Altern Tierexp 20: 275–281, 2003. [PubMed] [Google Scholar]
- 34.Price PJ, Gregory EA. Relationship between in vitro growth promotion and biophysical and biochemical properties of the serum supplement. In Vitro 18: 576–584, 1982. doi: 10.1007/BF02810081. [DOI] [PubMed] [Google Scholar]
- 35.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 267: 5421–5426, 2000. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 36.Elkalaf M, Anděl M, Trnka J. Low glucose but not galactose enhances oxidative mitochondrial metabolism in C2C12 myoblasts and myotubes. PLoS One 8: e70772, 2013. doi: 10.1371/journal.pone.0070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta 1807: 568–576, 2011. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 38.Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem 212: 95–99, 1993. doi: 10.1111/j.1432-1033.1993.tb17637.x. [DOI] [PubMed] [Google Scholar]
- 39.Seshagiri PB, Bavister BD. Glucose and phosphate inhibit respiration and oxidative metabolism in cultured hamster eight-cell embryos: evidence for the "crabtree effect". Mol Reprod Dev 30: 105–111, 1991. doi: 10.1002/mrd.1080300206. [DOI] [PubMed] [Google Scholar]
- 40.Golshani-Hebroni SG, Bessman SP. Hexokinase binding to mitochondria: A basis for proliferative energy metabolism. J Bioenerg Biomembr 29: 331–338, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Yang XL, Borg LAK, Eriksson UJ. Altered metabolism and superoxide generation in neural tissue of rat embryos exposed to high glucose. Am J Physiol Endocrinol Metab 272: E173–E180, 1997. doi: 10.1152/ajpendo.1997.272.1.E173. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Enriquez S, Juarez O, Rodriguez-Zavala JS, Moreno-Sanchez R. Multisite control of the Crabtree effect in ascites hepatoma cells. Eur J Biochem 268: 2512–2519, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Kase ET, Nikolić N, Bakke SS, Bogen KK, Aas V, Thoresen GH, Rustan AC. Remodeling of oxidative energy metabolism by galactose improves glucose handling and metabolic switching in human skeletal muscle cells. PLoS One 8: e59972, 2013. doi: 10.1371/journal.pone.0059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34: 121–138, 2013. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183, 2010. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orozco JM, Krawczyk PA, Scaria SM, Cangelosi AL, Chan SH, Kunchok T, Lewis CA, Sabatini DM. Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat Metab 2: 893–901, 2020. doi: 10.1038/s42255-020-0250-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodman RP, Calvo SE, Mootha VK. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J Biol Chem 293: 7508–7516, 2018. doi: 10.1074/jbc.TM117.000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 28: 115–130, 2008. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Su X, Quinn WJ , 3rd,Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, Migaud M, Mitchison TJ, Baur JA, Rabinowitz JD. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab 27: 1067–1080.e5, 2018. doi: 10.1016/j.cmet.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, Chini EN. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res 20: 120–130, 2014. doi: 10.1158/1078-0432.CCR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat Struct Mol Biol 13: 582–588, 2006. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 52.Hosios AM, Vander Heiden MG. The redox requirements of proliferating mammalian cells. J Biol Chem 293: 7490–7498, 2018. doi: 10.1074/jbc.TM117.000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diehl FF, Lewis CA, Fiske BP, Vander Heiden MG. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab 1: 861–867, 2019. doi: 10.1038/s42255-019-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, Vander Heiden MG. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab 24: 716–727, 2016. doi: 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol 24: 1161–1180, 2017. doi: 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352: 231–235, 2016. doi: 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352: 1474–1477, 2016. doi: 10.1126/science.aad5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alano CC, Tran A, Tao R, Ying WH, Karliner JS, Swanson RA. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res 85: 3378–3385, 2007. doi: 10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- 59.Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem 285: 34106–34114, 2010. doi: 10.1074/jbc.M110.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tischler ME, Friedrichs D, Coll K, Williamson JR. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch Biochem Biophys 184: 222–236, 1977. doi: 10.1016/0003-9861(77)90346-0. [DOI] [PubMed] [Google Scholar]
- 61.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130: 1095–1107, 2007. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol 15: 241–246, 2003. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 63.Sun F, Dai C, Xie J, Hu X. Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLoS One 7: e34525, 2012. doi: 10.1371/journal.pone.0034525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103: 514–527, 1967. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelly RA, Leedale J, Harrell A, Beard DA, Randle LE, Chadwick AE, Webb SD. Modelling the impact of changes in the extracellular environment on the cytosolic free NAD+/NADH ratio during cell culture. PLoS One 13: e0207803, 2018. doi: 10.1371/journal.pone.0207803. [DOI] [PMC free article] [PubMed] [Google Scholar]