

Abstract
The biguanide metformin is the most commonly used antidiabetic drug. Recent studies show that metformin not only improves chronic inflammation by improving metabolic parameters but also has a direct anti-inflammatory effect. In light of these findings, it is essential to identify the inflammatory pathways targeted by metformin to develop a comprehensive understanding of the mechanisms of action of this drug. Commonly accepted mechanisms of metformin action include AMPK activation and inhibition of mTOR pathways, which are evaluated in multiple diseases. Additionally, metformin’s action on mitochondrial function and cellular homeostasis processes such as autophagy is of particular interest because of the importance of these mechanisms in maintaining cellular health. Both dysregulated mitochondria and failure of the autophagy pathways, the latter of which impair clearance of dysfunctional, damaged, or excess organelles, affect cellular health drastically and can trigger the onset of metabolic and age-related diseases. Immune cells are the fundamental cell types that govern the health of an organism. Thus, dysregulation of autophagy or mitochondrial function in immune cells has a remarkable effect on susceptibility to infections, response to vaccination, tumor onset, and the development of inflammatory and autoimmune conditions. In this study, we summarize the latest research on metformin’s regulation of immune cell mitochondrial function and autophagy as evidence that new clinical trials on metformin with primary outcomes related to the immune system should be considered to treat immune-mediated diseases over the near term.
Keywords: autophagy, complex 1, inflammation, metformin, mitochondria
INTRODUCTION
The biguanide metformin is the first-line pharmacological agent for treating type II diabetes mellitus (T2D) in the United States and worldwide. No drug to date is as versatile and as clinically important as metformin. Absorption of metformin occurs within 6 h of consuming the drug, which has a bioavailability of 50%–60% and is excreted unchanged in the urine with 30% excreted in the feces. The drug is generally well tolerated in people with normal kidney function. The reported side effects are primarily gastrointestinal, with nausea, abdominal pain, and diarrhea in 20%–30% of people treated, and these undesirable effects often resolve spontaneously. More severe complications, such as lactic acidosis, are rarely reported (<1.5/100,000 people/yr). Some of the most notable clinical effects of metformin include glycemic control due to lowering of hepatic glucose output (1), inhibition of autoimmune inflammation by regulating T-cell balance (2–4), reduction of cardiovascular mortality (5), cancer prevention (6), and dementia (7). Metformin is known to reduce inflammation, which makes it an attractive exploratory drug for numerous other pathologies. This review focuses on the effect of metformin on mitochondria and cellular autophagic mechanisms and its relevance to immune cell health and inflammation.
METFORMIN’S ACTION ON MITOCHONDRIAL FUNCTION
Metformin’s use in the United States began in 1995, and for the past quarter century it has good efficacy, safety, and affordability. To date, several molecular mechanisms are regarded as key for metformin’s antidiabetic functions. However, none can be pinpointed as the singular mechanism, which indicates that metformin’s effects are likely combinatorial. The most notable cellular processes that are modulated by metformin are the mitochondrial respiratory chain, AMP-activated protein kinase (AMPK), the AMP:ATP ratio, cyclins, the human epidermal growth factor receptor 2 (HER2) oncoprotein, and cellular homeostatic mechanisms such as autophagy, apoptosis, and redox balance. The cellular mechanisms that are either activated or inhibited by metformin are reported in Table 1.
Table 1.
Effect of metformin on cellular processes
| Cellular Process | Activated or Inhibited by Metformin | Selected References |
|---|---|---|
| AMPK | Activation | Kajiwara et al. (8) |
| AMP:ATP | Activation | El-Mir et al. (9) Rena et al. (10) |
| Autophagy | Activation/inhibition* | Bharath et al. (11) Wang et al. (12) |
| Apoptosis | Activation/inhibition* | Wang et al. (12) |
| Cyclins | Inhibition | Saraei et al. (6) |
| HER2 oncoprotein | Inhibition | Saraei et al. (6) |
| Mitochondrial complex 1 | Inhibition | El-Mir et al. (9) Fontaine (13) |
| mTOR | Inhibition | Dewaele et al. (14) Li et al. (15) |
| ROS | Inhibition/activation* | Kelly et al. (16) Kajiwara et al. (8) |
*Cell/tissue-specific effects are reported.
AMPK, AMP-activated protein kinase; BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3; HER2, human epidermal growth factor receptor 2; LC3, light chain protein 3; mTOR, mechanistic target of rapamycin; PINK1, PTEN-induced putative kinase 1; p62, protein 62/sequestosome 1.
Metformin’s action on respiratory chain complex 1 is consistently reported, especially in cancer biology (17–20). El-Mir et al. (9) were one of the first to report a new mitochondrial complex 1-dependent action of metformin, showing the biguanide inhibited oxygen consumption in a concentration-dependent manner, due to its action on respiratory chain complex 1. Metformin, a hydrophilic compound at physiological pH, does not diffuse into cells. Instead, it enters and exits via transporters such as organic cation transporters (OCT), plasma membrane mono amino transporter (PMAT), and multidrug and toxin extrusion transporters (MATE 1/2), depending on the tissue and organ (21). Although metformin enters cells, it is not shown conclusively to accumulate within the organelles.
Some researchers speculate that metformin penetrates and accumulates in the mitochondria (10), inhibiting the respiratory chain activity, whereas others disagree. The mitochondrial inner membrane is not permeable to hydrophilic molecules, and thus metformin would have to enter the mitochondria via a specific transporter. Alternatively, it is suggested that the drug accumulates in the cells and the mitochondria because of the membrane potentials across both the plasma membrane and the mitochondrial inner membrane (22). These conflicting views gave rise to the direct and indirect hypotheses regarding metformin localization in the mitochondria. The direct hypothesis assumes a plasma membrane potential of 35 mV, passive OCT and MATE, and a hypothetical metformin carrier, which would result in metformin accumulation at about a 1,000-fold higher in the mitochondria than the cytosol, at a mitochondrial potential of 180 mV. The indirect hypothesis states that metformin does not enter the mitochondria but stimulates signaling pathways that regulate complex 1 activity (13). Given the contradictory cellular effects induced by metformin in different cell types and under different contexts, we speculate that both the mechanisms might be operational. For example, metformin might accumulate in the mitochondria in specific cell types due to the presence of transporters/carriers. On the other hand, if the cells lack the transporter/carrier, metformin might induce an effect by activating cellular signaling.
Canonical mitochondrial complex 1 inhibitors such as piericidin differ from the biguanides in being highly hydrophobic, uncharged aromatic molecule that competes for ubiquinone-binding sites. Bridges et al. (23) reported that biguanides do not inhibit complex 1 by inhibiting NADH oxidation or preventing intermolecular electron transfer or by competing with ubiquinone. Biguanides are hydrophilic, positively charged molecules that can interact with two separate sites on complex 1. One site perturbs the reactivity of flavin and the other inhibits catalysis. Metformin-mediated inhibition of complex 1 is weak and reversible compared with canonical inhibitors, which can be advantageous because the fast but weak and reversible inhibition is modulated according to cellular needs.
METFORMIN’S REGULATORY EFFECT ON IMMUNE CELL MITOCHONDRIAL ROS, COMPLEX 1, AND AMPK
Immune cells circulate through metabolic tissues and produce inflammatory cytokines that trigger pathologies such as insulin resistance and diabetes (24, 25). Immune cell mitochondrial function declines during pathophysiological conditions such as obesity and aging, raising the possibility that obesity and aging-associated changes in the mitochondria drive inflammation and accelerate pathologies such as type 2 diabetes (T2D), autoimmune disorders, and cancer. Mitochondria regulate immune cell function via mitochondrial ROS (mtROS), mitochondrial DNA (mtDNA), both of which can regulate transcription and cellular signaling, and by altering metabolic pathways that influence the cells. The mitochondria can also activate inflammatory responses by signaling via NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) activation, mitochondrial fission, fusion, mitophagy and by interacting with the endoplasmic reticulum, reviewed earlier (26).
Metformin’s function was evaluated in leukocyte mitochondria in polycystic ovarian syndrome (PCOS), an endocrine disorder that significantly raises the risk for type 2 diabetes (27). The lower leukocyte oxygen consumption rate, membrane potential, and mitochondrial mass in peripheral blood mononuclear cells (PBMCs) from patients with PCOS was restored to control values after metformin treatment. Metformin also decreased reactive oxygen species (ROS), IL-6, and TNFα and increased glutathione levels of leukocytes from PCOS subjects. The investigators observed that HOMA-IR and mitochondrial function biomarkers positively correlated with ROS production and GSH content and negatively correlated with membrane potential at baseline. These differences disappeared after metformin treatment. Assessing the levels of clinically relevant cytokines in PCOS, such as MCP-1 and IL-8 (28, 29), would be essential for defining the anti-inflammatory action of metformin in PCOS.
Metformin alleviated particulate matter (PM)-induced airway inflammation and thrombosis in a mouse model (30). Primary alveolar macrophages obtained from mice exposed to PM and treated with a therapeutic dose of 100 mg/kg/day of metformin had lower NAD+/NADH ratio, ROS, and IL-6 production. Furthermore, the study showed that mitochondrial ROS modulated IL-6 production by activating calcium release-activated channels (CRAC). Metformin caused additional changes in murine macrophages, including reductions in glycolytic and tricarboxylic cycle intermediates and high energy phosphates. Mitochondrial ROS production and calcium levels were also measured in primary human macrophages from healthy volunteers after the cells were treated in vitro with either metformin or Synta-66, a CRAC channel inhibitor. Metformin inhibited mitochondrial ROS and calcium-mediated activation of IL-6, thus recapitulating the findings in murine macrophages. Metformin’s putative mechanism of action was reported to be via the upregulation of unfolded protein response and triggering global cellular stress response pathway.
In another study, metformin decreased the production of IL-1β, but had no effect on TNFα in response to LPS stimulation in macrophages. Metformin enhanced IL-10 production, reduced mitochondrial ROS, and inhibited NADH:ubiquinone oxidoreductase in macrophages. In this experimental context, the activity of metformin was independent of both AMPKα1 or AMPKβ1 (16). Although this study showed that metformin inhibited mitochondrial ROS (mROS) in macrophages, Kajiwara et al. (8) showed that metformin treatment enhanced mitochondrial ROS and AMPK phosphorylation in a murine macrophage model of Legionella pneumophila infection. L. pneumophila is highly resistant to killing by macrophages and monocytes. Metformin treatment showed promising results in killing the intracellular pathogen within 12–48 h postinfection. L pneumophila-induced secretion of IL-12P35, IFNγ, and TNFα production was prevented by metformin treatment along with reductions in bacterial number.
ROS plays a significant role in the clearance of bacterial pathogens. Metformin increased ROS production within 6 h in L. pneumophila-infected RAW cells, and further ROS exaggeration was observed within 24 h postinfection. Metformin treatment alone induced mROS, whereas cytoplasmic ROS (cROS) levels were unaltered by metformin irrespective of the bacterial infection. Interestingly, GSH treatment of bone marrow-derived macrophages (BMDMs) prevented metformin-induced inhibition of bacterial growth. Taken together, these results indicate that bactericidal effects of metformin are mediated via its ability to generate ROS in the mitochondria. It remains unclear how metformin promotes and alleviates ROS in the same cell type under different experimental contexts, but it may depend on the nature of the challenge, the receptors activated, and intracellular signaling events the stimuli generate. Similar to its effect on ROS, metformin’s action on AMPK varies. Metformin acts via AMPK in certain context, whereas it is AMPK-independent in others. We reported that metformin’s ability to alleviate aging-induced Th17 proinflammatory cytokine production was independent of AMPK (11).
Atherosclerosis progression in diabetic patients is significantly influenced by inflammation downstream of advanced glycation end products (AGEs) (31). Proinflammatory cytokines IL-1β, IL-6, TNFα, and anti-inflammatory IL-10 production were assessed in bone marrow-derived macrophages (BMDMs) from mice in the presence of AGE activators and metformin. Metformin at a modest dose of 0.25–2 µM inhibited the AGE-induced expression of proinflammatory cytokines, reduced p-p65/p65 ratio, prevented p65 nuclear translocation, and boosted the anti-inflammatory IL-10 expression. Metformin-induced activation of AMPK was inhibitory to NF-κB pathways that were promoted by AGEs. Metformin’s inhibition of the NF-κB pathway was negated in the presence of AMPK inhibitor compound C. The significance of metformin’s influence on AGEs in human macrophages needs further evaluation and can be of therapeutic importance in preventing cardiovascular inflammation.
METFORMIN AND MITOPHAGY
Mitochondrial health plays a critical role in obesity-associated inflammation. Thus the general paradigm that mitochondrial recycling, or mitophagy, is compromised in inflammatory disease raises the possibility that mitophagy defects impact chronic inflammation (Fig. 1). Mitophagy is triggered by cellular and mitochondrial stressors and the presence of superfluous, damaged, or dysfunctional mitochondria, all of which are potent signals for mitophagy initiation. The phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)/Parkin pathway is shown to be vital in removing damaged mitochondria in all cell types, including CD4+ T cells (11). The E3 ubiquitin ligase, Parkin, translocates from the cytosol to the mitochondria if the membrane potential of the mitochondria drops. PINK1 stabilizes and accumulates on the mitochondria, after which Parkin is recruited to the mitochondria. Parkin ubiquitinylates many mitochondrial outer membrane proteins, followed by adaptor protein-mediated recruitment of dysfunctional mitochondria into the autophagosome. This recruitment culminates in the fusion of the autophagosome with the lysosome, resulting in the destruction and recycling of the autophagolysosome contents, including the mitochondria. Many different proteins regulate mitophagy, including PINK/Parkin, B-cell lymphoma 2 (BCL2) interacting protein 3 (BNIP3), NIX-BCL2/adenovirus E1B 19 kDa interacting protein 3-like (BNIP3L), mitofusin 2. Although such details have not been confirmed in immune cells, impaired mitophagy has been attributed to inflammasome activation and plays an important role in health and disease (32). Peripheral blood mononuclear cells from drug naïve patients with T2DM on metformin intervention had improved mitochondrial oxidative stress and HbA1c compared with placebo. Metformin intervention group also had increased protein markers of mitophagy and PINK1 expression, which positively correlated with HOMA-β indices. Electron microscopy revealed that metformin treatment of the mononuclear cells reduced abnormalities in mitochondrial morphology via enhancing mitophagy of abnormal cells; this effect was independent of metformin’s glucose lowering effect (33). This study further showed that NLRP3 was augmented by metformin, leading to caspase 1-mediated pyroptosis of chronically activated macrophages that coincided with a decrease in chronic inflammation. These results contradicted the demonstration that metformin inhibited NLRP3, caspase 1, and IL-1β in previously drug naïve T2D subjects (34). Other than the differences in the participants’ ethnicity, the study participants in one study were advised for lifestyle modifications. It is difficult to predict if these differences resulted in contradictory findings regarding metformin’s effect on NLRP3 activation. Such contradictory findings warrant clinical trials to sort out metformin’s mechanisms and its action in humans. Although uncontrolled pyroptosis is detrimental, controlled pyroptosis limits the extent of inflammation and preserves homeostasis.
Figure 1.
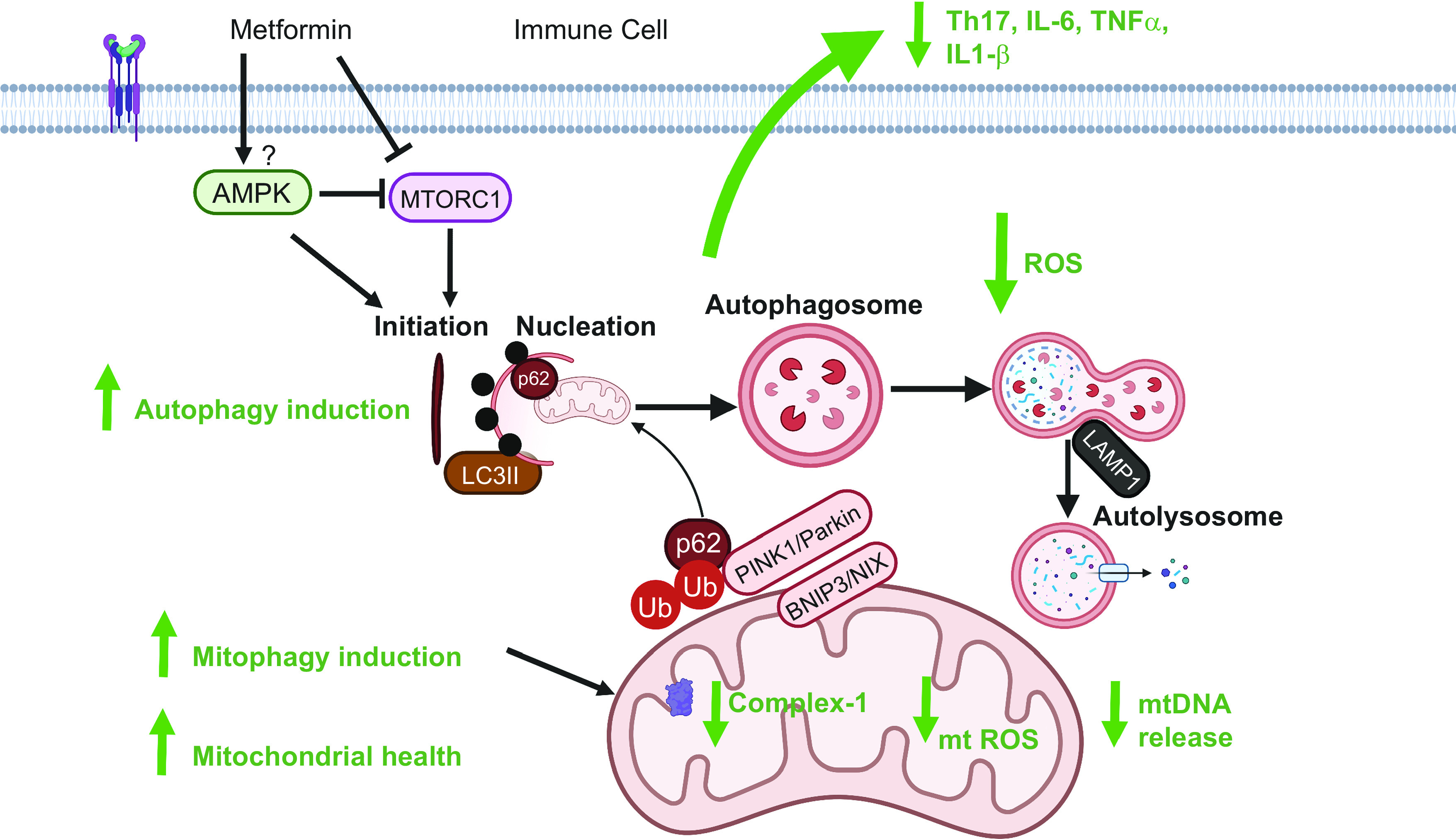
Model: metformin’s regulation of mitochondrial respiratory complex activity and homeostatic processes such as autophagy and mitophagy prevent cellular and mitochondrial reactive oxygen species (ROS) generation and release of mitochondrial DNA, resulting in lower proinflammatory cytokine production and improved cellular health (figure created with Biorender.com). AMPK, AMP-activated protein kinase; BNIP3, BCL2/adenovirus E1B 19 kDa interacting protein 3; LAMP1, lysosomal associated membrane protein 1; LC3, light chain protein 3; mTOR, mechanistic target of rapamycin; PINK1, PTEN-induced putative kinase 1; p62, protein 62/sequestosome 1.
METFORMIN AND GLOBAL AUTOPHAGY IN IMMUNE CELLS
Understanding the regulation of immune cell autophagy is vital for combating many illnesses driven by underlying inflammation. Metformin is a top candidate based on its ability to regulate global autophagy in immune cells (11). Autophagy induction and immunomodulatory effects of metformin were evaluated in macrophages and in a mouse model of allergic contact dermatitis (12). Lipopolysaccharide-induced expression of nitric oxide (NO) and mRNA expression of inflammatory cytokines, TNFα, IL-6, and IL-1β were reduced in a dose-dependent manner along with activation of autophagy within 24 h of metformin treatment in macrophage cell line and in bone marrow-derived primary macrophages (BMDMs) (12). In addition to inhibiting proinflammatory signaling pathways such as mitogen-activated protein kinase (MAPK) and NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3), metformin inhibited protein kinase3/mechanistic target of rapamycin (Akt/mTOR) axis. Inhibition of Akt/mTOR is known to activate autophagy (14, 15). Metformin increased autophagy-related proteins microtubule-associated protein 1 light chain protein 3 (LC3), autophagy protein 5 (Atg5), and decreased protein 62/sequestosome 1 (p62) and small RNA negative regulator of autophagy, miR-30a. In a mouse model of dinitrofluorobenzene (DNFB)-induced allergic dermatitis, metformin treatment (100 and 250 mg/kg) activated autophagy and decreased the number of infiltrating (F4/80) macrophages as well as the mRNA levels of MCP-1, but increased IL-10. DNFB-induced allergic dermatitis causes severe inflammatory response, which was marked by macrophage infiltration, epidermal hyperkeratosis, and edema, all of which were prevented in metformin-treated animals. The authors make a correlative observation that the functional improvements in allergic dermatitis are due to metformin’s ability to activate autophagy and inhibit the inflammatory signaling pathways such as MAPK. Kim et al. (35) reported that metformin inhibited proinflammatory responses and the accumulation of lipid droplets within cells by increasing autophagic flux in human vascular endothelial cell culture models. Ha et al. (36) reported metformin’s effect on microglial cells treated with inflammation inducers LPS and S100 calcium-binding protein A8 (S100A8). Metformin attenuated proinflammatory cytokines, suppressed phosphorylation of ERK, NLRP3, caspase-1, and activated autophagy. Yan et al. (37) reported the correlative association between metformin’s ability to upregulate autophagy and downregulate proinflammatory responses in an experimental arthritis model. The authors observed that autophagy was severely impaired in K/BxN serum-transfer model of rheumatoid arthritis (RA). Induction of autophagy by metformin through AMPK activation and mTOR inhibition resulted in the mitigation of inflammation in experimental arthritis (37). Nakahira et al. (38) demonstrated a direct relationship between autophagy and innate immune cell inflammation. Depletion of key autophagy proteins such as LC3 and Beclin resulted in caspase-1 activation and secretion of IL-1β and IL-18. Interestingly, in addition to the accumulation of dysfunctional mitochondria, cytosolic translocation of mitochondrial DNA occurred due to exaggerated mtROS, and NLR family pyrin domain containing 3 (NALP3) activation in macrophages stimulated with LPS and ATP. The presence of mitochondrial DNA in the cytosol promoted the secretion of IL-1β and IL-18, the two cytokines that require NALP3 processing before secretion. Additionally, mice deficient in autophagy protein LC3B were susceptible to LPS-induced mortality and produced caspase-1-dependent cytokines in sepsis models. We speculate that the mortality of autophagy-deficient mice upon LPS challenge could be due to the dysregulation of p21-LC3B interaction. Protein 21 (p21) is an essential protein for the regulation of cardiovascular physiology and inflammatory responses. p21 interacts with LC3B to promote autophagy to improve cardiac function during sepsis, as demonstrated in a mouse model (39). Thus the deficiency of LC3B in mice could disrupt this axis and can promote cardiac dysfunction and death.
Our group showed that a physiological dose of metformin (100 µM) prevented Th17 inflammation through an autophagy-dependent mechanism in CD4+ T cells from older adults (11). T cells from older subjects had higher oxygen consumption rate (OCR), which was lowered by metformin, surprisingly irrespective of AMPKα silencing. Metformin instead increased autophagy and decreased ROS in activated T cells from older subjects. Complementary work in T cells from younger subjects showed that autophagy inhibition, achieved by siRNA targeting the autophagy protein Atg3, recapitulated the respiratory profiles and the inflammatory profiles of T cells from older adults. Metformin failed to change inflammatory cytokine production in autophagy-deficient T cells from younger subjects, thus supporting the conclusion that metformin counters age-related inflammation by increasing autophagy. Kelly et al. (16) showed that metformin action could be independent of both a1 and b1 subunits of AMPK in macrophages. It is important to note that metformin could act through AMPK independent but regulated in development and DNA damage responses 1 (REDD1)-dependent mechanisms. REDD1 is a negative regulator of mTOR, a possible mechanism that must be experimentally validated.
REGULATION OF Th17/Treg BALANCE
In addition to glucose-lowering effects, action on mitochondrial function, cell metabolism, and homeostatic mechanisms such as autophagy, metformin influences inflammation by regulating the Th17/Treg balance. Research shows that inflammatory conditions such as rheumatoid arthritis (RA) are associated with metabolic dysregulation and are often refractory to RA therapeutics, thus posing a challenge in managing this disease. RA is a condition where there is an imbalance in T effector cells (Th1/Th17) versus T regulatory cells (Tregs), and Th17 inflammation is reported in RA (40). Cells cultured in vitro in Th17 polarizing conditions and treated with metformin experienced a reduction in the number of Th17 cells and an increase in Treg cells, thus showing the immunomodulatory effects of metformin (2). Metformin’s ability to alleviate collagen-induced arthritis (CIA) by reciprocally regulating the Th17/Treg axis in joints of mice is promising and shows that metformin can be a potential therapeutic drug for treating RA (2).
It is important to note that metabolism controls the balance of Th17/Treg cells (41). AMPK and mTOR are metabolic sensors that can control the balance between pro- and anti-inflammatory cells. Th17 cells rely on glycolytic fuel utilization, whereas the Tregs depend on fatty acid oxidation. The ability of AMPK to regulate metabolism will influence the type of cells that differentiate. Similarly, mTOR activation and the subsequent induction of hypoxia-inducible factor 1-α (HIF1α) promotes glucose import and glycolysis at the transcriptional and translational level. The absence of induction of HIF1α results in the dramatic reduction of Th17 cells. Metformin activated AMPK, inhibited mTOR, and regulated Th17/Treg ratio in a collagen-induced arthritis (CIA) rat model. Metformin also reduced the higher amounts of proinflammatory cytokines TNFα, IL-1β, IL-6, and IL-17 in serum of rats with CIA and reduced the number of splenic CD4+/RORγt+/IL-17+ T cells (Th17s) in a dose-dependent manner. Regulatory T cells (CD4+/CD25+/FOXP3+) increased in a positive association with metformin dose (42). Similarly, metformin alleviated autoimmune insulitis by regulating the T effector Teff/Treg balance in NOD mice, a model of type 1 diabetes. Female NOD mice were treated with metformin or vehicle starting 4-wk of age. The mice developed insulitis by 12-wk of age, but the mice on metformin had many functional β cells, than mice on vehicle treatment. Metformin significantly decreased the number of splenic pro-inflammatory IFN-γ+ as well as IL17+ CD4 T cells of NOD mice and significantly increased the regulatory IL-10+ and Foxp3+ CD4-T cells along with mitigation of autoimmune insulitis (43). The imbalance in Teff/Treg ratios reported in CIA and type 1 diabetes is recapitulated in multiple diseases, including type 2 diabetes, obesity, aging, and rheumatoid arthritis. In all of these diseases, inflammation is a dominant driver of comorbidities such as cancer and Alzheimer’s disease (44, 45) and may worsen the course of dementia (46).
Metformin at 10–50 mg/kg/day, alters adipose tissue composition by reducing white adipose tissue (WAT)-associated genes and upregulating brown adipose tissue (BAT)-associated genes in a mouse model of obesity. Fat droplet accumulation in the liver was significantly reduced in the metformin-treated group and was secondary to fibroblast growth factor (FGF) 21 production. Metformin treatment also reduced CD4+/IL-17+ (Th17) cells and increased CD4+/Foxp3+ (Treg) cells in spleens of obese mice (47). Collectively these data reinforce metformin’s anti-inflammatory effects and its ability to regulate Th17/Treg axis.
In conclusion, metformin’s potent effect on mitochondrial function, autophagy, and immune modulation significantly impacts organismal inflammatory status and health. The versatile nature of this antidiabetic drug is evident by its effects on several cellular processes and its relevance to human health, independent of its role in blood glucose control.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK108056 (to B.S.N.), National Institute on Aging Grant R56AG069685 (to B.S.N.), and the College of Health Sciences, Faculty Development Grant (to L.P.B.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.P.B. prepared figures; L.P.B. drafted manuscript; L.P.B. and B.S.N. edited and revised manuscript; L.P.B. and B.S.N. approved final version of manuscript.
REFERENCES
- 1.Song R. Mechanism of metformin: a tale of two sites. Diabetes Care 39: 187–189, 2016. doi: 10.2337/dci15-0013. [DOI] [PubMed] [Google Scholar]
- 2.Son H-J, Lee J, Lee S-Y, Kim E-K, Park M-J, Kim K-W, Park S-H, Cho M-L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm 2014: 973986, 2014. doi: 10.1155/2014/973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AJ. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 58: 1302–1311, 2009. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ursini F, Russo E, Pellino G, D’Angelo S, Chiaravalloti A, De Sarro G, Manfredini R, De Giorgio R. Metformin and autoimmunity: a “new deal” of an old drug. Front Immunol 9: 1236, 2018. doi: 10.3389/fimmu.2018.01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han Y, Xie H, Liu Y, Gao P, Yang X, Shen Z. Effect of metformin on all-cause and cardiovascular mortality in patients with coronary artery diseases: a systematic review and an updated meta-analysis. Cardiovasc Diabetol 18: 96, 2019. doi: 10.1186/s12933-019-0900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saraei P, Asadi I, Kakar MA, Moradi-Kor N. The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manag Res 11: 3295–3313, 2019. doi: 10.2147/CMAR.S200059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell JM, Stephenson MD, De Courten B, Chapman I, Bellman SM, Aromataris E. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimers Dis 65: 1225–1236, 2018. doi: 10.3233/JAD-180263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kajiwara C, Kusaka Y, Kimura S, Yamaguchi T, Nanjo Y, Ishii Y, Udono H, Standiford TJ, Tateda K. Metformin mediates protection against Legionella pneumonia through activation of AMPK and mitochondrial reactive oxygen species. J Immunol 200: 623–631, 2018. doi: 10.4049/jimmunol.1700474. [DOI] [PubMed] [Google Scholar]
- 9.El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275: 223–228, 2000. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 10.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 60: 1577–1585, 2017. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, Jiang K, Liu R, Guo Z, Deeney J, Apovian CM, Snyder-Cappione J, Hawk GS, Fleeman RM, Pihl RMF, Thompson K, Belkina AC, Cui L, Proctor EA, Kern PA, Nikolajczyk BS. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab 32: 44–55.e6, 2020. doi: 10.1016/j.cmet.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang M, Qu S, Ma J, Wang X, Yang Y. Metformin suppresses LPS-induced inflammatory responses in macrophage and ameliorates allergic contact dermatitis in mice via autophagy. Biol Pharm Bull 43: 129–137, 2020. doi: 10.1248/bpb.b19-00689. [DOI] [PubMed] [Google Scholar]
- 13.Fontaine E. Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol (Lausanne) 9: 753, 2018. doi: 10.3389/fendo.2018.00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 6: 838–854, 2010. doi: 10.4161/auto.6.7.12113. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Hu X, Wang J, Xu W, Yi C, Ma R, Jiang H. Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway contributes to the protection of hesperidin against myocardial ischemia/reperfusion injury. Int J Mol Med 42: 1917–1924, 2018. doi: 10.3892/ijmm.2018.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly B, Tannahill GM, Murphy MP, O'Neill LAJ. Metformin inhibits the production of reactive oxygen species from NADH: ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem 290: 20348–20359, 2015. doi: 10.1074/jbc.M115.662114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3: e02242, 2014. doi: 10.7554/elife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luengo A, Sullivan LB, Heiden MGV. Understanding the complex-I-ty of metformin action: limiting mitochondrial respiration to improve cancer therapy. BMC Biol 12: 82, 2014. doi: 10.1186/s12915-014-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrzejewski S, Gravel S-P, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab 2: 12, 2014. doi: 10.1186/2049-3002-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron AR, Logie L, Patel K, Erhardt S, Bacon S, Middleton P, Harthill J, Forteath C, Coats JT, Kerr C, Curry H, Stewart D, Sakamoto K, Repiscak P, Paterson MJ, Hassinen I, McDougall G, Rena G. Metformin selectively targets redox control of complex I energy transduction. Redox Biol 14: 187–197, 2018. doi: 10.1016/j.redox.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang X, Giacomini KM. Transporters involved in metformin pharmacokinetics and treatment response. J Pharm Sci 106: 2245–2250, 2017. doi: 10.1016/j.xphs.2017.04.078. [DOI] [PubMed] [Google Scholar]
- 22.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348: 607–614, 2000. doi: 10.1042/bj3480607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462: 475–487, 2014. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wouters K, Gaens K, Bijnen M, Verboven K, Jocken J, Wetzels S, Wijnands E, Hansen D, van Greevenbroek M, Duijvestijn A, Biessen EAL, Blaak EE, Stehouwer CDA, Schalkwijk CG. Circulating classical monocytes are associated with CD11c+ macrophages in human visceral adipose tissue. Sci Rep 7: 42665, 2017. doi: 10.1038/srep42665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Travers RL, Motta AC, Betts JA, Bouloumié A, Thompson D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes (Lond) 39: 762–769, 2015. doi: 10.1038/ijo.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, Tan M. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol 9: 1605, 2018. doi: 10.3389/fimmu.2018.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Victor VM, Rovira-Llopis S, Bañuls C, Diaz-Morales N, Castelló R, Falcón R, Gomez M, Rocha M, Hernandez-Mijares A. Effects of metformin on mitochondrial function of leukocytes from polycystic ovary syndrome patients with insulin resistance. Eur J Endocrinol 173: 683–691, 2015. doi: 10.1530/EJE-15-0572. [DOI] [PubMed] [Google Scholar]
- 28.Glintborg D, Andersen M, Richelsen B, Bruun JM. Plasma monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α are increased in patients with polycystic ovary syndrome (PCOS) and associated with adiposity, but unaffected by pioglitazone treatment. Clin Endocrinol (Oxf) 71: 652–658, 2009. doi: 10.1111/j.1365-2265.2009.03523.x. [DOI] [PubMed] [Google Scholar]
- 29.Adams J, Liu Z, Ren YA, Wun W-S, Zhou W, Kenigsberg S, Librach C, Valdes C, Gibbons W, Richards J. Enhanced inflammatory transcriptome in the granulosa cells of women with polycystic ovarian syndrome. J Clin Endocrinol Metab 101: 3459–3468, 2016. doi: 10.1210/jc.2015-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soberanes S, Misharin AV, Jairaman A, Morales-Nebreda L, McQuattie-Pimentel AC, Cho T, Hamanaka RB, Meliton AY, Reyfman PA, Walter JM, Chen C-I, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Antalek M, Abdala-Valencia H, Chiarella SE, Sun KA, Woods PS, Ghio AJ, Jain M, Perlman H, Ridge KM, Morimoto RI, Sznajder JI, Balch WE, Bhorade SM, Bharat A, Prakriya M, Chandel NS, Mutlu GM, Scott Budinger GR. Metformin targets mitochondrial electron transport to reduce air-pollution-induced thrombosis. Cell Metab 29: 335–347.e5, 2019. [Erratum in Cell Metab 29: 503, 2019]. doi: 10.1016/j.cmet.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z, Tang Y, Jin X, Chen C, Lu Y, Liu L, Shen C. Metformin inhibits advanced glycation end products-induced inflammatory response in murine macrophages partly through AMPK activation and RAGE/NFκB pathway suppression. J Diabetes Res 2016: 4847812, 2016. doi: 10.1155/2016/4847812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuk J-M, Silwal P, Jo E-K. Inflammasome and mitophagy connection in health and disease. Int J Mol Sci 21: 4714, 2020. doi: 10.3390/ijms21134714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhansali S, Bhansali A, Dutta P, Walia R, Dhawan V. Metformin upregulates mitophagy in patients with T2DM: a randomized placebo-controlled study. J Cell Mol Med 24: 2832–2846, 2020. doi: 10.1111/jcmm.14834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62: 194–204, 2013. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H-S, Ren G, Kim T, Bhatnagar S, Yang Q, Bahk YY, Kim JA. Metformin reduces saturated fatty acid-induced lipid accumulation and inflammatory response by restoration of autophagic flux in endothelial cells. Sci Rep 10: 13523, 2020. doi: 10.1038/s41598-020-70347-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha J-S, Yeom Y-S, Jang J-H, Kim Y-H, Im JI, Kim IS, Yang SJ. Anti-inflammatory effects of metformin on neuro-inflammation and NLRP3 Inflammasome activation in BV-2 microglial cells. BSL 25: 92–98, 2019. doi: 10.15616/BSL.2019.25.1.92. [DOI] [Google Scholar]
- 37.Yan H, Zhou H-F, Hu Y, Pham CTN. Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation. J Rheum Dis Treat 1: 5, 2015. doi: 10.23937/2469-5726/1510005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AMK. Autophagy proteins regulate innate immune response by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang S, Xu M, Liu L, Yang J, Wang H, Wan C, Deng W, Tang Q. Autophagy is involved in the protective effect of p21 on LPS-induced cardiac dysfunction. Cell Death Dis 11: 554, 2020. doi: 10.1038/s41419-020-02765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niu Q, Cai B, Huang Z-C, Shi Y-Y, Wang L-I. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int 32: 2731–2736, 2012. doi: 10.1007/s00296-011-1984-x. [DOI] [PubMed] [Google Scholar]
- 41.Sun L, Fu J, Zhou Y. Metabolism controls the balance of Th17/T-regulatory cells. Front Immunol 8: 1632, 2017. doi: 10.3389/fimmu.2017.01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang M, Ding Y, Wang Y, Gu J, Zhang B, Wang H. Metformin regulates of Th17/treg cell balance and reduces hyperplastic synovium via activating AMPK and inhibiting mTOR in a collagen-induced arthritis rat model. Int J Clin Exp Med 10: 11479–11487, 2017. [Google Scholar]
- 43.Duan W, Ding Y, Yu X, Ma D, Yang B, Li Y, Huang Li, Chen Z, Zheng J, Yang C. Metformin mitigates autoimmune insulitis by inhibiting Th1 and Th17 responses while promoting Treg production. Am J Transl Res 11: 2393–2402, 2019. [PMC free article] [PubMed] [Google Scholar]
- 44.Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51: 27–41, 2019. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4: 575–590, 2018. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enciu A-M, Popescu BO. Is there a causal link between inflammation and dementia? Biomed Res Int 2013: 316495, 2013. doi: 10.1155/2013/316495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim EK, Lee SH, Jhun JY, Byun JK, Jeong JH, Lee S-Y, Kim JK, Choi JY, Cho M-L. Metformin prevents fatty liver and improves balance of white/brown adipose in an obesity mouse model by inducing FGF21. Mediators Inflamm 2016: 5813030, 2016. doi: 10.1155/2016/5813030. [DOI] [PMC free article] [PubMed] [Google Scholar]