

Abstract
Spleen tyrosine kinase (Syk) and Bruton’s tyrosine kinase (BTK) play critical roles in platelet physiology, facilitating intracellular immunoreceptor tyrosine-based activation motif (ITAM)-mediated signaling downstream of platelet glycoprotein VI (GPVI) and GPIIb/IIIa receptors. Small molecule tyrosine kinase inhibitors (TKIs) targeting Syk and BTK have been developed as antineoplastic and anti-inflammatory therapeutics and have also gained interest as antiplatelet agents. Here, we investigate the effects of 12 different Syk and BTK inhibitors on GPVI-mediated platelet signaling and function. These inhibitors include four Syk inhibitors, Bay 61-3606, R406 (fostamatinib), entospletinib, TAK-659; four irreversible BTK inhibitors, ibrutinib, acalabrutinib, ONO-4059 (tirabrutinib), AVL-292 (spebrutinib); and four reversible BTK inhibitors, CG-806, BMS-935177, BMS-986195, and fenebrutinib. In vitro, TKIs targeting Syk or BTK reduced platelet adhesion to collagen, dense granule secretion, and alpha granule secretion in response to the GPVI agonist cross-linked collagen-related peptide (CRP-XL). Similarly, these TKIs reduced the percentage of activated integrin αIIbβ3 on the platelet surface in response to CRP-XL, as determined by PAC-1 binding. Although all TKIs tested inhibited phospholipase C γ2 (PLCγ2) phosphorylation following GPVI-mediated activation, other downstream signaling events proximal to phosphoinositide 3-kinase (PI3K) and PKC were differentially affected. In addition, reversible BTK inhibitors had less pronounced effects on GPIIb/IIIa-mediated platelet spreading on fibrinogen and differentially altered the organization of PI3K around microtubules during platelets spreading on fibrinogen. Select TKIs also inhibited platelet aggregate formation on collagen under physiological flow conditions. Together, our results suggest that TKIs targeting Syk or BTK inhibit central platelet functional responses but may differentially affect protein activities and organization in critical systems downstream of Syk and BTK in platelets.
Keywords: BTK, ibrutinib, platelet, Syk, tyrosine kinase
INTRODUCTION
Spleen tyrosine kinase (Syk) and Bruton’s tyrosine kinase (BTK) play central roles in the physiology of immune and hematological cells (1, 2). In platelets, Syk and BTK mediate hemostasis, vascular repair, and a range of inflammatory and pathological responses (3). For example, Syk and BTK transduce signals from collagen receptor glycoprotein VI (GPVI) to drive thrombo-inflammatory platelet responses (4). When activated, GPVI associates with Fc receptor γ-chain (FcRγ) and becomes phosphorylated by Src family kinases (SFKs, namely, Lyn and Fyn) on intracellular immunoreceptor tyrosine-based activation motifs (ITAMs) (5). Phosphorylated ITAMs on GPVI-associated FcRγ then serve as a platform to recruit, phosphorylate, and activate Syk (5). Then, Syk phosphorylates and activates a number of substrates associated with the linker for activation of T cells (LAT) at the LAT signalosome, including BTK, phospholipase C γ2 (PLCγ2), phosphoinositide 3-kinase (PI3K) (4–6), and protein kinase C (PKC) to support platelet hemostatic as well as inflammatory and thrombotic responses (4, 5).
Given their roles in thrombo-inflammatory and other hematological diseases, Syk and BTK have emerged as targets of interest in pathologies driven by platelets and other immune cells (7–9). For instance, small molecule tyrosine kinase inhibitors (TKIs) targeting BTK, such as ibrutinib, have already proved as highly effective, first-line therapeutics used in the treatment of hematologic malignancies and offer promise against a range of immunological and inflammatory disorders (10–13). However, although Syk and BTK are not absolutely required for hemostatic responses of platelets, TKIs with high potency for BTK, such as ibrutinib, are associated with undesirable effects on platelets and bleeding (12). Interestingly, despite key roles as an upstream activator of BTK, inhibition of Syk with broad-spectrum immunomodulatory agents such as fostamatinib are not associated with platelet toxicities and bleeding (14). Overall, several questions remain regarding how Syk and BTK as well as TKIs targeting these kinases affect platelet function.
Studies over the past decade have determined that a number of Syk and BTK inhibitors have effects on platelets in vivo as well as in vitro (15, 16). However, no studies to date have examined multiple Syk and BTK inhibitors simultaneously to compare as well as to uncover the physiological mechanisms, both the commonalities and disparities, by which these agents interfere with platelet signaling and function. In this study, we investigate the effects of a panel of 12 different clinically relevant Syk and BTK inhibitors on platelet adhesion, granule secretion, activation, aggregation, and protein phosphorylation. Our findings characterize the effects of these inhibitors on GPVI- and GPIIb/IIIa integrin-mediated platelet activation pathways and the interplay of Syk-BTK-PI3K organization to better elucidate how Syk and BTK inhibitors target signaling events central in platelet hemostasis, inflammation, and thrombosis.
MATERIALS AND METHODS
Reagents
Collagen was obtained from Chrono-Log (Havertown, PA), cross-linked collagen-related peptide (CRP-XL) from R. Farndale (Cambridge University, Cambridge, UK), human fibrinogen from Enzyme Research (South Bend, IN), prostacyclin (PGI2) from Cayman Chemical (Ann Arbor, MI), and integrilin from Merck & Co. (Whitehouse Station, NJ). Bovine thrombin, fatty acid-free bovine serum albumin (BSA), and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO) or as previously mentioned (17).
Tyrosine Kinase Inhibitors
Ro 31-8220 and PP2 were obtained from Tocris (Bristol, UK). Bay 61-3606 was from Sigma-Aldrich (St. Louis, MO). R406/fostamatinib, entospletinib, TAK-659, ibrutinib, acalabrutinib, ONO-4059, AVL-292/spebrutinib, BMS-935177, BMS-986195, and fenebrutinib were obtained from Selleck (Houston, TX). CG-806 was from MedChemExpress (Monmouth Junction, NJ).
Antibodies
Primary antisera against phosphorylated Syk Y525 (No. 2711S), AKT T308 (No. 4056S), AKT S473 (No. 4060S), phosphorylated Akt substrates (No. 9614S), PLCγ2 Y1217 (No. 3871S), DAPP1 Y139 (No. 13703S), and phosphorylated PKC substrates (No. 2261) were obtained from Cell Signaling Technology (Danvers, MA). Primary antisera against phosphorylated BTK Y551 (No. MAB7659) was obtained from R&D Systems (Minneapolis, MN). Anti-α tubulin antibody (No. T6199) was obtained from Sigma-Aldrich (St. Louis, MO), and PI 3-kinase p85α antibody (No. sc-376112) was obtained from Santa Cruz Biotechnology (Dallas, TX). APC anti-human CD62P (P-selectin) (No. 304910) was obtained from BioLegend (San Diego, CA), and FITC mouse anti-human PAC-1 (No. 340507) was obtained from BD Biosciences (San Jose, CA). Alexa Fluor secondary antibodies: goat anti-mouse IgG1 488 (No. A-21121), goat anti-mouse IgG (H + L) 488 (No. A-11029), goat anti-mouse IgG2b 546 (No. A-21143), and goat anti-rabbit IgG (H + L) 546 (No. A-21143) were obtained from Thermo Fisher Scientific (Waltham, MA).
Platelet Preparation
Human venous blood was drawn from healthy adult male and female volunteers into 1:10 sodium citrate (3.8% w/v) and warmed 1:10 acid citrate dextrose (ACD, 30°C), as previously described (18–20). Written informed consent was obtained before the blood draw, and procedure was conducted according to a protocol approved by the Institutional Review Board of Oregon Health & Science University. The drawn blood was centrifuged at 200 g for 20 min to isolate and obtain the platelet-rich plasma (PRP). Prostacyclin (0.1 µg/mL) was added to the PRP, and the mixture was centrifuged at 1,000 g for 10 min to isolate and obtain platelets. The collected platelets were resuspended in modified HEPES/Tyrode buffer (129 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2, pH 7.3) and ACD to the desired concentration for experimental use.
Static Adhesion Assays
For platelet spreading experiments on fibrinogen or collagen, 12-mm no. 1.5 glass coverslips (Fisher Scientific) were coated with human fibrinogen (100 µg/mL) or soluble collagen (50 µg/mL), respectively. The coated surfaces were then treated and blocked with filtered, denatured fatty acid-free BSA (5 mg/mL). The selected Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) were added to platelets (2 × 107/mL) in solution for 10 min. The inhibitor-treated platelets were then seeded onto the immobilized fibrinogen- or collagen-coated coverslip surfaces and incubated at 37°C for 45 min and 30 min, respectively. The glass coverslips were then washed three times with PBS to remove nonadherent platelets. Adherent platelets were fixed in 4% paraformaldehyde (PFA) for 10 min and washed three times with PBS. Coverslips were mounted on precleaned microscope slides (25 × 75 × 1 mm) using Fluoromount G (Southern Biotech). Platelets are imaged using Kohler-illuminated Nomarski differential interference contrast (DIC) optics with a Zeiss ×63 oil immersion 1.40 numerical aperture (NA) plan-apochromat lens on a Zeiss Axio Imager M2 microscope using Slidebook 5.5 image acquisition software (Intelligent Imaging Innovations, Denver, CO), as previously described (19). Three images for each treatment condition were taken. Individual platelet surface areas, total surface area covered by platelets, and average number of platelets per imaged field adherent on collagen were measured and analyzed using Image J (NIH). Similarly, the average platelet surface area of platelets spread on fibrinogen were measured and calculated using Image J (NIH). Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test on GraphPad PRISM, where a P value < 0.05 was considered significant.
Fluorescence Microscopy and Quantification
Washed, purified platelets were prepared to a concentration of 2 × 107/mL, incubated on fibrinogen-coated glass coverslips, and fixed using 4% PFA, as described in Static Adhesion Assays method section. To fluorescently visualize platelet tubulin, PI3K, and PKC, the adherent platelets were first permeabilized with blocking solution (1% BSA and 1% SDS in PBS, 1:100) and then stained with anti-α-tubulin, anti-p85 PI3K, and anti-PKC primary antibodies in blocking buffer overnight at 4°C. Coverslips were washed with PBS, and adherent platelets were stained using Alexa Fluor secondary antibodies (1:500) and mounted on precleaned microscope slides (25 × 75 × 1 mm) using Fluoromount G (Southern Biotech). Adherent platelets were imaged using a Zeiss Axio Imager M2 microscope, as described above. Three images for each treatment condition were taken. Colocalization analyses of α-tubulin with either p85 PI3K or PKC were performed using a custom FIJI script (SciJava, NIH) incorporating Just Another Co-localization Plugin (JACoP, ver. 2.1.1) (21). Images were preprocessed by smoothing using a median filter with radius = 2 pixels (px) to remove noise outliers and background subtracted with rolling radius = 50 px. Each image was then automatically quantified using the Mander’s colocalization method within JACoP. The Mander’s overlap coefficient (MOC) is reported as the percentage of overlap of tubulin signal in channel 1 with PI3K or PKC signal in channel 2, as previously described (22, 23). Each image was then automatically quantified using the Pearson’s correlation method within a custom MATLAB script. The Pearson’s correlation coefficient (PCC) is reported as the degree of fluorescence intensity correlation between the two channels, as previously described (20, 24).
Platelet Secretion Assay
Washed, purified platelets were prepared to a concentration of 2 × 108/mL. Platelets were incubated with the selected panel of Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) for 10 min in a clear flat bottom 96-well plate (Corning Costar, Tewksbury, MA) and then stimulated with CRP-XL (10 µg/mL) or thrombin (1 U/mL). The 96-well plate was placed in an Infinite M200 spectrophotometer (TECAN, Mannderdorf, Switzerland) and shook for 20 s. Detection reagent Chrono-Lume (Chrono-Log) was added to each well to detect for ATP released, measured as the light output generated by an ATP-luciferin-luciferase reaction. An automated protocol is carried out to shake for 10 s, and sample luminescence was measured and recorded at 30-s intervals for 5 min. Four kinetic profiles were recorded for each treatment condition, and the average luminescence measured at 4 min was calculated and normalized by the average luminescence of untreated platelets stimulated with CRP-XL or thrombin. Statistical analysis was performed using a one-way ANOVA test on GraphPad PRISM, where a P value < 0.05 was considered significant.
Flow Cytometry Analysis
Washed, purified platelets were prepared to a concentration of 2 × 107/mL and incubated with the selected panel of Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) for 10 min. FITC PAC-1 (3:100) and APC CD62P (3:100) were added to stain for activated integrin αIIbβ3 and P-selectin, respectively. Each platelet mixture was stimulated with CRP-XL (10 µg/mL) and incubated for 20 min. Untreated platelets (vehicle, 0.1% DMSO) unstimulated and stimulated with CRP-XL (10 µg/mL) served as the negative and positive controls, respectively. Each sample was analyzed using flow cytometry on a BD FACSCantoII flow cytometer. Platelet populations were identified and gated by logarithmic signaling amplification for forward and side scatter as well as CD41+ staining, as previously described (25). Statistical analysis was performed using a one-way ANOVA test on GraphPad PRISM, where a P value < 0.05 was considered significant.
Western Blotting
Washed, purified platelets were prepared to a concentration of 1 × 109/mL and incubated with glycoprotein (GP) IIb/IIIa inhibitor integrilin (20 µg/mL). The platelets were then incubated with the selected panel of Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) for 10 min. CRP-XL (10 µg/mL) was added to stimulate each platelet mixture and then incubated in a 25°C water bath for 10 min. Laemmli Sample Buffer was added at a 1:1 volume ratio in dithiothreitol (DTT, 200 mM). The platelet samples were separated by SDS-PAGE and then transferred to nitrocellulose for western blotting. The platelet samples were separated by SDS-PAGE and then transferred to nitrocellulose membranes, which were then blocked in 5% milk + PBS before antibody staining for Western blot. Antibody staining was performed in 5% BSA + PBS. Each platelet/inhibitor sample was blotted and stained using the antibodies for phospho-Syk (Tyr525/526), phospho-BTK (Y551) antibody, phospho-DAPP1 (Tyr139), phospho-PLCγ2 (Tyr1217), phospho-Akt (Thr308), phospho-Akt (Ser473), phospho-Akt substrate, phospho-(Ser) PKC substrate, and α tubulin, as previously described (26).
Flow Adhesion and Analysis
Glass capillary tubes (0.2 × 2 × 200 mm; Vitrocom, Mountain Lakes, NJ) were coated with fibrillar type I collagen (100 µg/mL) for 1 h at room temperature, as previously described (27). Glass capillaries were then washed with PBS and blocked with 5 mg/mL denatured BSA for 1 h at room temperature before connecting to a syringe pump system. Human venous blood was drawn into syringe containing sodium citrate (3.8% w/v) in 1:9 sodium citrate:whole blood and pretreated with a subset of inhibitors, including entospletinib, ibrutinib, and fenebrutinib (10 µM), before perfusion through the glass capillary tube at a venous shear rate of 300 s−1 for 10 min. After 10 min of whole blood perfusion, glass capillary tubes were washed with PBS, fixed with 4% paraformaldehyde, and sealed with Fluoromount G. Z-stack images of platelet aggregates in three random fields of view were captured using a ×63 Zeiss Axio Imager M2 microscope with Slidebook 5.5 image acquisition software, as previously described (28, 29). The surface area and volume of each platelet aggregate were determined using a custom MATLAB script (28, 29). Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test on GraphPad PRISM, where a P value < 0.05 was considered significant.
RESULTS
Effects of TKIs Targeting Syk and BTK on Platelet Adhesion to Collagen
We first examined the effects of 12 Syk and BTK inhibitors (Supplemental Table S1; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.13640606) on GPVI-mediated platelet adhesion to immobilized collagen. Washed platelets were prepared from citrate-anticoagulated whole blood collected from a pool of healthy adult human donors. Purified platelets (2 × 107/mL) were treated with the selected Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) in solution, before incubation on collagen-coated coverslips. As seen in Fig. 1A, platelets readily adhered to collagen-coated surfaces. Following 30-min incubation, 104 ± 5 platelets readily adhered onto collagen-coated glass in an imaged area of 14,587 µm2 under control conditions (Fig. 1A), with an average platelet surface area of 41.2 ± 1.54 µm2 per platelet (Fig. 1C) and a total surface area of 2,827.3 ± 232.6 µm2 covered by all platelets in each imaged field of view (Fig. 1D). Preincubation of platelets with the Syk and BTK inhibitors significantly decreased the number of platelets adherent to collagen and the average platelet surface area relative to control. The Syk inhibitor Bay 61-3606 reduced the number of platelets adherent to collagen by 31% ± 2.9% and average platelet surface area by 33.3% ± 3.1% similar to other Syk inhibitors, including R406 (27% ± 3.8% reduction in number, 28% ± 7.6% reduction in surface area) and entospletinib (34% ± 2.9% reduction in number, 21.6% ± 1.9% reduction in surface area); TAK-659 (70% ± 7.5% reduction in number, 91.6% ± 11.5% reduction in surface area) had more pronounced effects (Fig. 1, B and C). In an analogous manner, the BTK inhibitor, ibrutinib (52% ± 5.8% reduction in number, 33.5% ± 10% reduction in surface area) reduced platelet adhesion to collagen more than the other BTK inhibitors tested (Fig. 1, B and C). Similarly, preincubation of platelets with the Syk and BTK inhibitors significantly decreased the total surface area covered by the adherent platelets on collagen (Fig. 1D). Inhibitors of platelet GPVI targeting SFKs (PP2) and protein kinase C (Ro 31-8220) served as controls and likewise inhibited platelet adhesion to collagen. Together, these results demonstrate that different inhibitors targeting Syk and BTK signaling similarly inhibit the adhesion of platelets to collagen in vitro.
Figure 1.
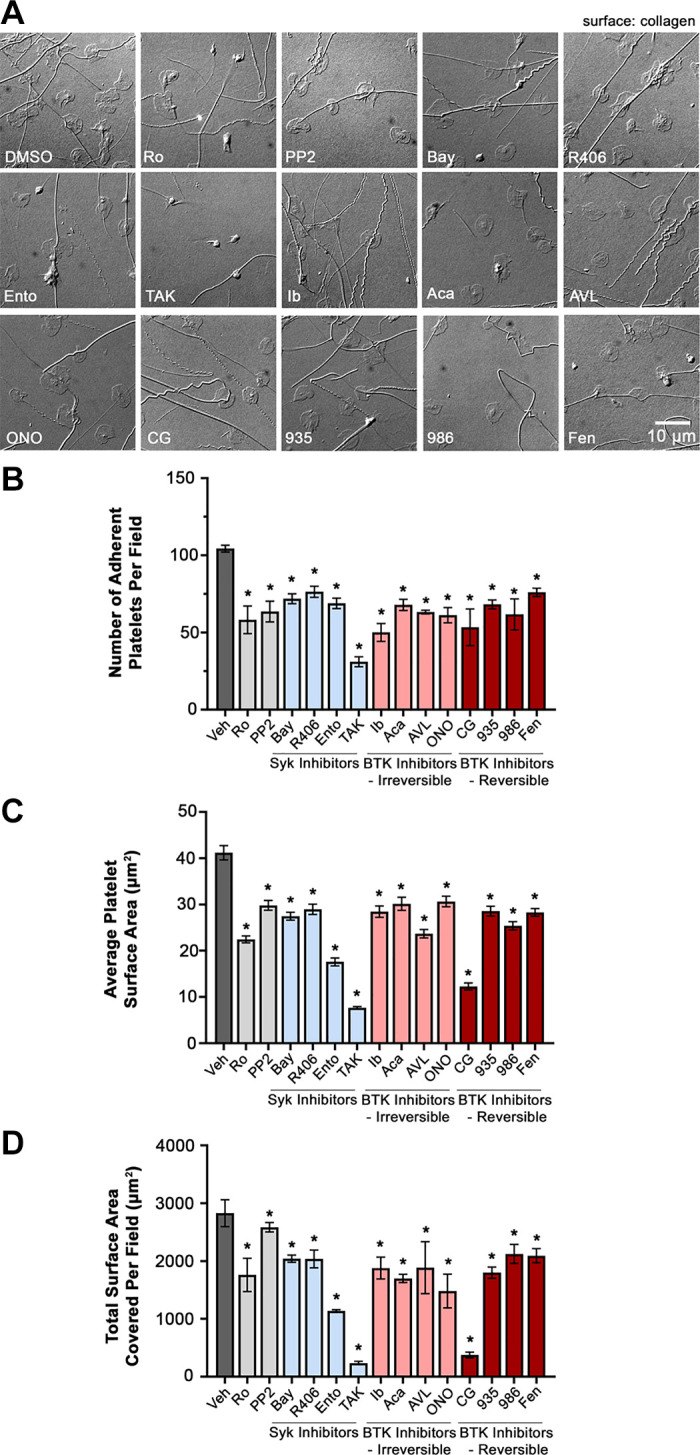
Effects of Syk and BTK inhibitors on platelet adhesion to collagen. A: replicate samples (n = 3) of washed human platelets (2 × 107/mL) were treated with the selected Syk and BTK inhibitors (10 µM) or vehicle (0.1% DMSO) in solution for 10 min and incubated on immobilized collagen-coated (100 µg/mL) glass coverslips for 30 min at 37°C. After fixation, coverslips were mounted on microscope slides (25 × 75 × 1 mm). Adherent platelets were visualized using Nomarski differential interference contrast (DIC) microscopy at ×63,000 magnification. Images representative of each replicate sample is shown. Scale bar = 10 µm. B: three images of platelets under each TKIs treatment condition were captured, and the number of platelets adherent to collagen per field of view (14,587 µm2) were counted. The average surface area of platelets (C) and the total surface area covered per imaged field of view (D) were determined using Image J. Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM, where a P value < 0.05 was considered significant, indicated by *. AVL, AVL-292 (spebrutinib); Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; CG, CG-806; Fen, fenebrutinib; ONO, ONO-4059 (tirabrutinib); R406, fostamatinib; Syk, spleen tyrosine kinase; Veh, vehicle; 935, BMS-935177; 986, BMS-986195; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220; Aca, acalabrutinib.
Effects of Syk and BTK Inhibitors on GPVI-Mediated Granule Secretion and Integrin Activation
Following GPVI activation, platelets secrete adenosine diphosphate (ADP) from dense granules as an autocrine and paracrine activator of platelet purinergic receptors (P2Y1, P2Y12) to progress platelet activation and hemostatic plug formation. We next assessed for platelet dense granule secretion to determine the effects of Syk and BTK inhibitors on GPVI-mediated platelet activation. Following stimulation of platelets with the GPVI-specific agonist CRP-XL in the presence of Chrono-Lume reagent, luminescence was measured over a time course to record the kinetic profile of ATP release as a marker of dense granule secretion. As seen in Fig. 2A, platelets stimulated with CRP-XL secreted dense granule contents (normalized as 1.0 luminescence intensity). Preincubation of platelets with Syk and BTK inhibitors before stimulation with CRP-XL significantly decreased dense granule release to baseline levels, with the exception of CG-806, which had a less pronounced although still significant effect on platelet ATP release (Fig. 2A). In comparison, preincubation of platelets with Syk and BTK inhibitors before stimulation with thrombin did not significantly decrease dense granule secretion (Fig. 2B), with the exception of the Syk inhibitor entospletinib, which appeared to interfere with assay luciferase activity for reasons to be determined.(30) Together, these results suggests that Syk and BTK inhibitors tested selectively block GPVI-mediated dense granule secretion without effects on PAR-mediated platelet activation in response to thrombin.
Figure 2.

Effects of Syk and BTK inhibitors on platelet-dense granule secretion. Replicate samples (n = 3) of washed human platelets (2 × 108/mL) were treated with the selected Syk and BTK inhibitors or vehicle (0.1% DMSO) and then stimulated with CRP-XL (A; 10 μg/mL) or thrombin (B; 1 Unit/mL). Platelet samples were monitored using ChromoLume (luciferase enzyme) to measure and record the luminescence kinetics of the dense granule ATP release profile of platelets, at 30-s intervals over the course of 5 min. Four kinetic profiles were recorded per platelet sample (n = 3). The average luminescence measured at 240 s was calculated and normalized by the average luminescence of the untreated platelets stimulated with CRP-XL or thrombin (+CRP-XL/−inhibitor or +thrombin/−inhibitor). Statistical analysis was performed using a one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM. Statistical significance is indicated by two asterisks (**) for a P value < 0.001. AVL, AVL-292 (spebrutinib); Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; CG, CG-806; Fen, fenebrutinib; ONO, ONO-4059 (tirabrutinib); R406, fostamatinib; Syk, spleen tyrosine kinase; 935, BMS-935177; 986, BMS-986195; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220; Aca, acalabrutinib; CRP-XL, cross-linked collagen-related peptide.
Complementary to ADP secretion from dense granules, activated platelets also mobilize proteins through α-granule secretion, including P-selectin (CD62P). We therefore quantified P-selectin expression via flow cytometry as another marker of GPVI-mediated platelet activation. As seen in Fig. 3A and Supplemental Fig. S1, stimulation with the GPVI-agonist, CRP-XL, upregulated the percentage of platelets with surface P-selectin from 2.05 ± 0.1% to 19.2 ± 3.4%. Preincubation of platelets with the majority of Syk and BTK inhibitors before stimulation with CRP-XL significantly decreased platelet surface P-selectin levels relative to control. For instance, the Syk inhibitors, Bay 61-3606 (1.97 ± 0.5%), R406 (3.73 ± 0.5%), entospletinib (2.02 ± 0.5%), and TAK-659 (1.63 ± 0.3%), all significantly reduced the percentage of platelets with P-selectin surface expression following stimulation with CRP-XL. Similarly, BTK inhibitors, including ibrutinib (1.98 ± 0.1%), acalabrutinib (2.17 ± 0.2%), BMS-986195 (1.69 ± 0.1%), and fenebrutinib (1.37 ± 0.1%), all significantly reduced the percentage platelets with surface P-selectin in response to CRP-XL stimulation. Pretreatment of platelets with the BTK inhibitor CG-806 (19.7 ± 5.7%) did not significantly decrease the exposure of platelet P-selectin following CRP-XL stimulation (Fig. 3A).
Figure 3.

Effects of Syk and BTK inhibitors on platelet α-granule secretion (A) and integrin activation (B). Replicate samples (n = 3) of washed human platelets (2 × 108/mL) were treated with the selected Syk and BTK inhibitors or with vehicle (0.1% DMSO), stimulated with CRP-XL (10 µg/mL), and stained with APC-CD62P and FITC-PAC1 to monitor for platelet surface expression of P-selectin and integrin activation, respectively, using flow cytometry. Thresholds for platelet surface integrin and P-selectin expression were set based on the negative control (−CRP-XL/−inhibitor), such that percentage of expression is 0.5%. Representative FACS traces are depicted in Supplemental Fig. S2. Statistical analysis was performed using one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM. Statistical significance is indicated by one asterisk (*) for a P value < 0.001. AVL, AVL-292 (spebrutinib); Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; CG, CG-806; Fen, fenebrutinib; ONO, ONO-4059 (tirabrutinib); R406, fostamatinib; Syk, spleen tyrosine kinase; 935, BMS-935177; 986, BMS-986195; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220; Aca, acalabrutinib; CRP-XL, cross-linked collagen-related peptide.
As platelets activate in response to GPVI stimulation and feedback from ADP secretion and other factors, downstream pathways promote the “inside-out” activation of the platelet surface integrin αIIbβ3 to support fibrinogen binding and platelet-platelet aggregation. Resting and CRP-XL stimulated platelets were incubated in solution with FITC-conjugated PAC-1 antibody, which recognizes specifically open active conformation of human integrin αIIbβ3 to monitor platelet surface integrin αIIbβ3 activation using flow cytometry. As seen in Fig. 3B, the percentage of platelets binding PAC-1 increased in response to CRP-XL stimulation from 2.03 ± 0.03% to 43.3 ± 8.8%. Preincubation of platelets with the majority of Syk and BTK inhibitors before stimulation with CRP-XL significantly decreased platelet PAC-1 binding in response to CRP-XL. However, like P-selectin surface exposure, the BTK inhibitor CG-806 (33.5% ± 11.0%) did not significantly decrease platelet PAC-1 binding in response to CRP-XL (Fig. 3B).
Effects of Syk and BTK Inhibitors on PKC and PI3K/Akt Proximal Signaling Events
Following activation of GPVI, a Syk-dependent signaling cascade leads to formation of the LAT signalosome and activation of PLCγ2, BTK, PKC, PI3K/Akt, and other effectors to mediate platelet adhesion, secretion, and other responses. To determine the effects of the Syk and BTK inhibitors on the Syk-BTK-PI3K signaling axis, we next examined the effects on intracellular phosphorylation events downstream of GPVI activation. Under vehicle-treated conditions, CRP-XL stimulation upregulated the phosphorylation of several markers of GPVI activation, including Syk Y525, BTK Y551, DAPP1 Y139, PLCγ2 Y1217, Akt T308, and Akt S473, as well as generalized PKC and Akt substrates (Fig. 4). Preincubation of platelets with PP2 (a well-established inhibitor of signaling events downstream of GPVI activation) inhibited the CRP-XL-evoked phosphorylation of all substrates examined. Pretreatment of platelets with the PKC inhibitor Ro 31-8220 prevented the phosphorylation of PKC substrates in response to CRP-XL, with minimal effects on the upstream phosphorylation of Syk, BTK, and PLCγ2 as well as divergent Akt signaling pathways.
Figure 4.

Effects of Syk and BTK inhibitors on platelet activation signaling proteins. Replicate samples (n = 3) of purified, washed human platelets (1 × 109/mL) were incubated with the selected Syk and BTK inhibitors or with (0.1% DMSO) in solution and stimulated with CRP-XL. After collection into Laemmli sample buffer, platelet lysates were separated by SDS-PAGE and transferred to nitrocellulose. Western blots were conducted using antibodies for phosphorylated Syk Y525, BTK Y551, DAPP1 Y139, PLCγ2 Y1217, Akt T308, Akt S473, Akt substrates, and PKC substrates; α-tubulin serves as a loading control. Positions of molecular weight (kDa) markers relative to phosphorylated PKC and Akt substrates are indicated. Results representative of n = 4 experiments are shown. AVL, AVL-292 (spebrutinib); Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; CG, CG-806; Fen, fenebrutinib; ONO, ONO-4059 (tirabrutinib); PLCγ2, phospholipase C γ2; R406, fostamatinib; Syk, spleen tyrosine kinase; veh., vehicle; 935, BMS-935177; 986, BMS-986195; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220; CRP-XL, cross-linked collagen-related peptide; Aca, acalabrutinib.
Previous studies have examined the effects of several Syk and BTK inhibitors including R406 and ibrutinib on platelet GPVI signaling (31–33); however, comparative analyses of these and other related agents have not yet been carried out. Additionally, a number of “next generation” Syk and BTK inhibitors (i.e., TAK-659, BMS-986195) have not yet been studied for effects on platelet signaling. As seen in Fig. 4, preincubation of platelets with Syk inhibitors (entospletinib, Bay 61-3606, R406, TAK-659) eliminated CRP-XL-evoked auto-phosphorylation of Syk Y525 as well as the downstream phosphorylation of BTK Y551 and PLCγ2 Y1217. Pretreatment of platelets with all the Syk inhibitors also prevented the phosphorylation Akt and associated Akt substrates in platelets in response to GPVI activation (Fig. 4). All Syk inhibitors tested likewise prevented the phosphorylation of DAPP1 Y139 as well as PKC substrates following GPVI activation. However, the extent of inhibition suggested some inhibitor-specific variation; for example, the degree of inhibition of DAPP1 Y139 phosphorylation was reduced for entospletinib and Bay 61-3606 relative to TAK-659 and R406, despite complete inhibition of upstream activating phosphorylation of Syk Y525 and BTK Y551 by these Syk inhibitors (Fig. 4).
As seen in Fig. 4, pretreatment of platelets with eight different BTK inhibitors also prevented BTK-mediated phosphorylation of PLCγ2 Y1217 without altering upstream Syk autophosphorylation of Syk Y525 in response to CRP-XL stimulation. However, BTK inhibitors had varying effects on the phosphorylation of PKC substrates in response to CRP-XL stimulation, where AVL-292, fenebrutinib, BMS-935177, BMS-986195, and CG-806 had more potent inhibitory effects compared with ibrutinib, acalabrutinib, and ONO-4059. Overall, the effects of all BTK inhibitors on DAPP1 Y139 phosphorylation were less than those of Syk inhibitors; however, CG-806, which completely inhibited CRP-evoked DAPP1 phosphorylation, did not without fully inhibit PLCγ2 and Akt substrate phosphorylation. Together, these results demonstrate that all 12 Syk and BTK inhibitors examined in this study have inhibitory effects on GPVI-mediated activation of Syk and BTK, as expected; however, some downstream phosphorylation events may be altered in an inhibitor-specific manner.
Effects of Syk and BTK Inhibitors on Platelet Integrin Activation and Spreading on Fibrinogen
Complementary to signaling events downstream of GPVI activation, fibrinogen binding to the platelet integrin αIIbβ3 invokes a parallel “outside-in” ITAM-mediated signaling cascade also involving Syk and BTK as well as PKC, PI3K/Akt, and other related pathways to mediate cytoskeletal assembly and platelet aggregation (34). We assessed these platelet integrin activation responses in vitro by determining the ability of purified platelets to spread on surfaces of immobilized fibrinogen in the presence of Syk and BTK inhibitors. Washed, purified platelets (2 × 107/mL) were treated with selected Syk and BTK inhibitors or vehicle (0.1% DMSO) in solution, before incubation on fibrinogen-coated coverslips (37°C, 45 min). Platelets were then visualized by DIC microscopy to assess the extent of spreading on fibrinogen, determined by the average surface area per adherent platelet. As seen in Fig. 5A, platelets readily adhered to fibrinogen-coated coverslips and spread to a mean surface area of 33.3 ± 0.6 µm2 per platelet (Fig. 5B). Preincubation of platelets with PP2, Ro 31-8220, or TKIs targeting Syk and BTK significantly decreased the mean surface area of platelets adherent to fibrinogen (Fig. 5B). However, inhibitor-specific variations were observed for the distribution frequency of platelet surface areas for each inhibitor (gray curves in Fig. 5C, relative to control, white curve). For instance, although the Syk inhibitors Bay 61-3606 (22.9 ± 0.5 µm2 in surface area) and R406 (24.1 ± 0.4 µm2) inhibited platelet spreading to a similar extent, Syk inhibitor TAK-659 (20 ± 1 µm2) and entospletinib (20.3 ± 0.5 µm2) further reduced platelet spreading (Fig. 5B), shifting the distribution of platelet surface areas toward lower values relative to controls (Fig. 5C). Similarly, although BTK inhibitor ONO-4059 (25.8 ± 0.6 µm2), BMS-935177 (24.0 ± 1.8 µm2), and fenebrutinib (24.2 ± 1.0 µm2) inhibited platelet spreading to a similar extent, BTK inhibitor ibrutinib (21.1 ± 0.5 µm2), AVL-292 (20.3 ± 0.4 µm2), and BMS-986195 (20.4 ± 0.8 µm2) reduced platelet spreading to an even greater extent. Despite limiting the spreading of some platelets, the BTK inhibitors CG-806 and fenebrutinib had less pronounced effects on mean platelet surface areas (31.0 ± 0.4 µm2; Fig. 5B), while broadening the distribution of measured platelet surface areas (Fig. 5C). Together, these results demonstrate that all Syk and BTK inhibitors examined in this study have inhibitory effects on platelet spreading on fibrinogen.
Figure 5.

Effects of Syk and BTK inhibitors on platelet spreading on fibrinogen. A: replicate samples (n = 3) of washed human platelets (2 × 107/mL) were incubated with the selected Syk and BTK inhibitors or vehicle (0.1% DMSO) for 10 min and incubated on a fibrinogen coated glass cover glass at 37°C for 45 min. After fixation, adherent platelets were visualized using differential interference contrast (DIC) microscopy. Images representative of each replicate sample is shown. Scale bar = 10 µm. B: images were analyzed with Image J software to determine the per platelet surface areas (µm2) of adherent platelets per inhibitor condition. Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM. Statistical significance is indicated by one asterisk (*) for a P value < 0.05. C: frequency distribution plots numbers of platelets (y-axis) vs. measured individual platelet surface area (µm2—x axis) for each inhibitor condition tested. AVL, AVL-292 (spebrutinib); Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; CG, CG-806; Fen, fenebrutinib; ONO, ONO-4059 (tirabrutinib); R406, fostamatinib; Syk, spleen tyrosine kinase; 935, BMS-935177; 986, BMS-986195; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220; Aca, acalabrutinib.
Effects of Syk and BTK Inhibitors on PI3K Spatial Localization in Platelets
Together, experiments above suggest that TKIs targeting Syk and BTK inhibit essential platelet adhesion and secretion responses, with potential differential effects on downstream PI3K and PKC signaling events important to the kinetics of platelet activation and spreading on fibrinogen. Previous studies from our group and others have shown that SFK-driven Syk and BTK signaling regulates a (de)polymerization of platelet microtubules in manner orchestrating platelet activation (35–39). More recently, we also showed that PKC signaling events take place in physical proximity to platelet microtubules in a manner related to platelet Rho GTPase regulation and spreading on fibrinogen (18, 20, 40). Interestingly, classical studies of PI3K signaling also noted a localization of the PI3K p85 regulatory subunit at microtubules (41, 42) in a manner supporting signaling events important to cell adhesion (43). Previous studies have likewise noted a dramatic increase in PI3K p85 subunit in cytoskeletal fractions of activating platelets (44), but relationships between PI3K localization and signaling mechanisms in activating platelets have not yet been established.
To further determine the effects of Syk and BTK inhibitors on intracellular signaling events and platelet cell physiology in vitro, we examined the organization of key signaling molecules in activated platelets with fluorescence microscopy. Previous studies from our group and others have noted an association between PI3K and PKC signaling systems and microtubules in platelets and other cell types (20, 41, 42). As seen in Fig. 6, PI3K p85α regulatory subunit colocalized with microtubules (α-tubulin) in platelets adherent to fibrinogen, with a Pearson’s correlation coefficient (PCC) of 0.834 ± 0.0216 and a Mander’s overlap coefficient (MOC) of 0.76 ± 0.02. The colocalization of PI3K and α-tubulin was related to microtubule structure, as treatment of platelets with nocodazole (microtubule destabilizer) significantly decreased the association between PI3K and α-tubulin (PCC = 0.67 ± 0.02; MOC = 0.66 ± 0.04), whereas taxol (microtubule stabilizer) significantly increased their association (PCC = 0.9 ± 0.01; MOC =0.93 ± 0.01). As seen in Fig. 6, B and C, pretreatment of platelets with Syk and BTK inhibitors significantly affected the colocalization of PI3K and α-tubulin, where reversible BTK inhibitors had the strongest effects. Specifically, the PCC was decreased (in which Bay 61-3606, BMS-935177, and BMS-986195 were significant), suggesting that the correlation between tubulin and PI3K fluorescence intensity was decreased. In addition, all the Syk and BTK inhibitors tested significantly increased the MOC. This indicates a higher degree of overlap between tubulin and PI3K in platelets treated with the select inhibitors, suggesting that PI3K becomes more dispersed in relation to tubulin when treated with the select Syk and BTK inhibitors. Similarly, Syk and BTK inhibitors also had significant effects on the organization and colocalization of PKC with microtubules in platelets adherent to fibrinogen (Supplemental Fig. S2). Together, these results suggest a relationship between microtubule organization and PI3K and other signaling systems in platelets, where small molecules targeting Syk and BTK may alter the localization of platelet signaling proteins in a manner specific to the inhibitor’s pharmacology (Fig. 6 and Supplemental Fig. S2) (45).
Figure 6.

Effects of Syk and BTK inhibitors on platelet microtubules and PI3K organization. Replicate samples (n = 3) of washed human platelets (2 × 107/mL) were treated with the selected Syk or BTK inhibitors or vehicle (0.1% DMSO) for 10 min and incubated on fibrinogen-coated cover glass at 37°C for 45 min. Platelets are fixed and stained for p85 PI3K (green) and microtubules (red) with anti-p85 PI3K (Santa Cruz) and anti-α-tubulin (Sigma T6199) primary antibodies. A: adherent platelets were visualized using fluorescence microscopy. Images were quantified using a custom MATLAB script to determine the Pearson’s correlation coefficient (B) and the Mander’s overlap coefficient (C). Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM. Statistical significance is indicated by one asterisk (*) for a P value < 0.05 and by two asterisks (**) for a P value < 0.001. Images representative of replicate (n = 3) samples are shown. Scale bar = 10 µm. Bay, Bay 61-3606; BTK, Bruton’s tyrosine kinase; ONO, ONO-4059 (tirabrutinib); PI3K, phosphoinositide 3-kinase; R406, fostamatinib; Syk, spleen tyrosine kinase; Ento, entospletinib; Ib, ibrutinib; TAK, TAK-659; Ro, Ro 31-8220.
Effects of Syk and BTK Inhibitors on Platelet under Physiological Flow
To investigate the effects of Syk and BTK inhibitors on platelet function under additional physiological conditions, we assessed for platelet adhesion and platelet aggregation under continuous human venous blood flow conditions. To do this, whole blood was pretreated with a subset of selected TKIs including entospletinib, ibrutinib, and fenebrutinib and perfused through a glass capillary tube coated with fibrillar collagen. Following blood perfusion, resulting platelet aggregates were imaged with DIC microscopy to acquire Z-stacks. As seen in Fig. 7A, platelets from whole blood treated with vehicle alone readily adhered to collagen surfaces under physiological venous flow and formed robust platelet aggregates, with an average total surface area of 4,270 ± 112 µm2 (Fig. 7B) and an average total volume of 13,600 ± 931 µm3 (Fig. 7C) per imaged field of view. Pretreatment of whole blood with TKIs entospletinib (3,640 ± 500 µm2), ibrutinib (3,310 ± 368 µm2), and fenebrutinib (4,390 ± 903 µm2) did not significantly affect the adhesion or total surface area of platelets (Fig. 7B). In contrast, when pretreated with the selected TKIs, entospletinib (9,290 ± 1,150 µm3, 31.9% reduction), ibrutinib (6,630 ± 414 µm3, 51.4% reduction), and fenebrutinib (8,560 ± 899 µm3, 37.3% reduction), all significantly reduced the platelet aggregates’ total volume (Fig. 7C). This suggests that in a more physiological setting, the subset of Syk and BTK inhibitors tested reduced platelet aggregation but preserved platelet adhesion to collagen under human venous flow.
Figure 7.

Effects of Syk and BTK inhibitors on platelet adhesion and platelet aggregation under physiological flow conditions. Replicate samples (n = 3) of human venous whole blood were pretreated with a subset of Syk and BTK inhibitors, including entospletinib, ibrutinib, and fenebrutinib (10 µM) or vehicle (0.1% DMSO) for 10 min. Sample was then perfused through a glass capillary tube (0.2 × 2 × 200 mm) coated with fibrillar collagen (100 µg/mL) at a venous shear rate of 300 s−1 for 10 min. A: following blood perfusion, resulting platelet aggregates are imaged using Z-stack microscopy. Using a custom MATLAB script, the Z-stack images were processed to determine the platelet aggregates’ total surface area (B) and total volume (C) per imaged field of view for each treatment condition. Data are shown as mean ± SE; statistical analysis were conducted using a one-way ANOVA test and a Dunnett’s multiple comparison test on GraphPad PRISM. Statistical significance is indicated by one asterisk (*) for a P value < 0.05. BTK, Bruton’s tyrosine kinase; Fen, fenebrutinib; Syk, spleen tyrosine kinase; Veh., vehicle; Ento, entospletinib; Ib, ibrutinib.
DISCUSSION
In this study, we examined the effects of 12 different tyrosine kinase inhibitors (TKIs) targeting Syk and BTK signaling on human platelet function. We find that in vitro, four different Syk inhibitors and eight different BTK inhibitors all reduced platelet adhesion to collagen and dense granule secretion while having a more varied range of effects on alpha granule secretion, integrin activation, and PI3K/Akt and PKC signaling downstream of Syk-BTK. Under physiological flow conditions, selected subset of TKIs inhibited platelet aggregation but preserved platelet adhesion. Our results provide insight on the mechanisms of Syk and BTK inhibitors on platelets, while also illuminating aspects of platelet cell physiology around the Syk-BTK-PI3K signaling axis.
Over the past decade, small molecule TKIs targeting Syk (i.e., fostamatinib) and BTK (i.e., ibrutinib) have emerged as effective agents in the treatment of hematopoietic malignancies and autoimmune and inflammatory disorders (1, 8, 9, 46). The Syk inhibitor fostamatinib (R406) was originally identified as an inhibitor FcεRI mast cell degranulation (47). The BTK inhibitor ibrutinib (PCI-32765) was developed as an inhibitor of B cell receptor signaling to treat B cell lymphoma and autoimmune diseases. Ibrutinib further demonstrated a proof of concept that small molecules that covalently target Cys residues proximal to active sites in Tec family kinases (Cys 481) could serve as a means to covalently and irreversibly inhibit kinases with high potency and selectivity (48). BTK inhibitors such as ibrutinib (32) and acalabrutinib (49) are far more commonly associated with platelet inhibition and bleeding in comparison with fostamatinib and other Syk inhibitors (12, 46) for reasons that remain unexplored.
To this end, this study aims to investigate the effects of 12 clinically relevant Syk and BTK inhibitors. For this study, four Syk inhibitors (Bay 61-3606, R406, entospletinib, TAK-659), four irreversible BTK inhibitors (ibrutinib, acalabrutinib, AVL-292, tirabrutinib), and four reversible BTK inhibitors (CG-806, BMS-935177, BMS-986195, fenebrutinib) are evaluated and compared. Within each group, a range of first-generation to third-generation inhibitors are selected. For instance, ibrutinib is a first-generation irreversible BTK inhibitor with a reported IC50 value of 0.5 nM, while remaining selective with an IC50 value for Syk > 10,000 nM. Similarly, acalabrutinib is a second-generation irreversible BTK inhibitor with lesser potency, but greater specificity toward BTK. It inhibits BTK with a reported IC50 value of 3 nM, with no reported inhibition on Syk. The IC50 values for each Syk and BTK inhibitor used in this study are listed in Supplemental Table S1 detailing its specificity and potency in addition to secondary kinases that each drug inhibits.
Syk and BTK are well known components of platelet GPVI signaling. Numerous studies have demonstrated that small molecule inhibitors targeting Syk and BTK have antiplatelet effects in vivo and inhibiting platelet aggregation in vitro (4, 5, 31, 50, 51). However, although several Syk and BTK inhibitors have been examined for inhibitory effects against platelet aggregation, specific mechanistic effects of inhibitors on platelet signaling, adhesion, secretion, and other cell physiological factors have only been minimally examined or ignored for a number of compounds apart from more commonly studied agents such as ibrutinib and fostamatinib (R406). Given the key role of BTK in platelet GPVI-mediated signaling, it is unsurprising that ibrutinib use may inhibit platelet function in vivo. However, ibrutinib was early on found to be associated with bleeding in a manner perhaps beyond expected from platelet GPVI signaling alone (10), especially given the lack of bleeding in patients with BTK mutations X-linked agammaglobulinemia (XLA) as well as BTK knockout mouse models (5). Moreover, pharmacological inhibitors of Syk are not typically associated with bleeding (52), despite inhibiting BTK activation and PLCγ2 phosphorylation, including fostamatinib, which has a relatively promiscuous off-target profile. Similar unexpected off target effects such as atrial fibrillation were also noted for BTK inhibitors, but not Syk inhibitors (53). Moreover, few studies have examined the effects of inhibitors relative to one another simultaneously in parallel.
Historically, studies of Syk and BTK inhibition have helped to illuminate physiologically relevant signaling mechanisms in platelets and other blood cells through in vivo as well as in vitro studies (5, 54). In this study, we find that 12 different Syk and BTK inhibitors (Supplemental Table S1) all effectively inhibit in vitro platelet adhesion to collagen (Fig. 1), GPVI-evoked dense granule secretion (Fig. 2), and GPVI-mediated signaling (Fig. 4). All Syk and BTK inhibitors examined also similarly significantly inhibited platelet α-granule secretion and integrin activation following CRP-XL stimulation, with the exception of CG-806 (55), a next-generation reversible BTK inhibitor that does not target Tec, but does inhibit a number of therapeutically relevant kinases (FLT3, TRKA, etc.) (55). These results suggest that a range of different Syk and BTK inhibitors all similarly inhibit essential aspects of platelet function following GPVI activation. Current models of platelet signaling place Syk and BTK in key positions in signaling downstream of GPVI as well as integrins and other receptors (i.e., CLEC2, FcγRIIA) (5, 56). We found that Syk and BTK inhibitors all inhibit the initial steps of Syk and BTK signaling (Fig. 4). Although all 12 inhibitors inhibited platelet ITAM signaling downstream of GPVI and Syk→BTK activation, some inhibitor-specific variations in downstream signaling were apparent. For instance, Syk inhibitors more potently inhibited GPVI-evoked phosphorylation of PLCγ2 and DAPP1, while next-generation reversible BTK inhibitors had more pronounced effects on PKC substrate phosphorylation. Although CG-806 had the most variable effects on platelet functions, CG-806 potently inhibited signaling events downstream of BTK.
In addition to assays of GPVI-mediated platelet function in response to agonists such as CRP-XL, effects of inhibitors on platelet function are also commonly assayed by following effects of inhibitors on platelet spreading on fibrinogen. In line with other results, we found that all 12 Syk and BTK inhibitors diminished the ability of platelets to spread on fibrinogen to a similar extent. Interestingly, some differences were apparent in the numbers of platelets that were inhibited and the extent of inhibition by reversible BTK inhibitors, relative to irreversible BTK inhibitors and Syk inhibitors, suggesting different cell physiological effects of reversible BTK inhibitors on platelet responses in this context. Similar to our recent analyses of PKC signaling in platelet adherent to fibrinogen (20), we find that the regulatory p85α subunit of PI3K localizes to microtubules in activating platelets in a manner that may support orchestration of platelet activation. Interestingly, although all Syk and BTK inhibitors examined in this study perturb the colocalization of p85α PI3K and tubulin, reversible BTK have more dramatic effects. Indeed, somewhat unexpectedly, reversible inhibitors of BTK are found to be more potent inhibitors of platelet signaling and activation than ibrutinib and other inhibitors that target BTK through covalent mechanisms. These findings are in line with a recent study of the reversible BTK inhibitor fenebrutinib as more potent inhibitor of FcγRIIa-mediated platelet activation compared with ibrutinib and other inhibitors (57).
As BTK associates with several components within the LAT signalosome (i.e., PLCγ2, SLP-76) critical for platelet function (56, 58), we suspected that permanent, irreversible, covalent chemical modification of BTK with irreversible inhibitors such as ibrutinib may disrupt organization and alter platelet function in a manner apart from BTK kinase activities, where BTK may have roles as an adaptor protein or molecular scaffold (50). Although such mechanisms have not yet been reported for ibrutinib and BTK, mechanisms are emerging as the field of covalent inhibitors develops. For instance, the Cravatt group has recently shown that dimethyl fumarate (DMF) inhibits inflammatory signaling by covalently modifying IRAK4 to disrupt intermolecular interactions with MyD88 to inhibit cytokine production and systemic inflammation (59). In addition to interactions with the LAT signalosome (51), other potential scaffolds around BTK signaling include microtubules, which are known to serve as platforms or adaptors for related signaling processes in a number of cell types, including PI3K signaling in cell lines (41, 42) and Ca2+-mediated PKC signaling in platelets (20, 60); however, scaffolding of PI3K/Akt signaling at microtubules in platelets remains largely unexplored. Interestingly, we found that PI3K p85 localizes to microtubules in activating platelets in a manner related to PI3K/Akt signaling. Recent efforts by other groups aim to target platelet inflammatory signaling with colchicine (61, 62) as well as a cooperativity of microtubule polymerization inhibitors in treating TKI-resistant cancers (and vice versa) (63). Accordingly, a cooperativity between BTK signaling and microtubule regulation may be impacted by specific TKIs in a manner relevant to platelet function. Future studies that target BTK for degradation with protac molecules will further help to resolve structural versus enzymatic roles for BTK in GPVI-mediated platelet function (64, 65).
Given the key roles of Syk and BTK in platelet signaling events, a number of physiological studies have aimed to better understand how these kinases and drugs against them (i.e., ibrutinib) cause platelet inhibition associated with bleeding. Recent efforts suggest that these effects may be therapeutically exploited, as BTK inhibitors may serve as specific antiplatelet agents for cardiovascular disease (66) and platelet-related inflammatory conditions (57, 67). Roles for Syk inhibitors remain unexplored but may emerge as trials of other inhibitors (many explored in this study) are evaluated for safety and efficacy in inflammatory and other conditions. Interestingly, a recent study of BTK inhibitors as potential agents against heparin-induced thrombocytopenia (HIT) similarly found that fenebrutinib—a reversible BTK not associated with bleeding complications in clinical studies—is a more potent inhibitor of BTK signaling than ibrutinib (57). We similarly find that fenebrutinib is a potent inhibitor of GPVI-mediated platelet function. Previous studies have also demonstrated that a highly selective reversible BTK inhibitor RN486 more potently inhibits platelet GPVI responses in vitro, but without off target effects of ibrutinib.
In conclusion, this study demonstrates that a range of pharmacologically distinct Syk and BTK inhibitors similarly disrupt Syk-BTK signaling events in platelets and essential ITAM-mediated platelet responses. In addition to expanding knowledge of roles for Syk and BTK in platelet cellular physiology, the results from this study may help to understand how “off target” or undesired effects of TKIs on platelets and physiologically relevant cells come about. Models following from our results may also inform efforts to target platelet GPVI signaling in immunothrombosis, where Syk and BTK signaling likely have roles in pathologies ranging from atherothrombosis (66) to COVID-19 (68). As TKIs targeting Syk, BTK, and other kinases are further developed and implemented for an increasing number of inflammatory, oncogenic, and other conditions, studies such as our work herein will help to address a growing need to better understand the effects of such compounds on essential molecular machinery around Syk-BTK signaling in platelets and other physiologically relevant cell types.
GRANTS
This work was supported by the Medical Research Foundation of Oregon, a Scholar Award from the American Society of Hematology (to J. E. Aslan) and the National Institutes of Health (R01HL146549 to J. E. Aslan, R01HL101972 to O. J. T. McCarty, R01HL130274 to M. T. Hinds, and R01HL144113 to M. T. Hinds and O. J. T. McCarty). E. R. Lofurno is a 2019 Oregon State University Johnson Scholar.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.J.Z. and J.E.A. conceived and designed research; T.J.Z., E.R.L., A.R.M., H.H.S.L., J.P., and T.C.L.K. performed experiments; T.J.Z., J.E.A., E.R.L., H.H.S.L., K.G.P., M.E.F., and A.T.P.N. analyzed data; T.J.Z. and J.E.A. interpreted results of experiments; T.J.Z. and J.E.A. prepared figures; T.J.Z., J.E.A., and M.E.F. drafted manuscript; T.J.Z., J.J.S., M.T.H., O.J.T.M., J.E.A., and H.H.S.L. edited and revised manuscript; T.J.Z., J.J.S., M.T.H., O.J.T.M., J.E.A., E.R.L., A.R.M., H.H.S.L., J.P., K.G.P., M.E.F., T.C.L.K., and A.T.P.N. approved final version of manuscript.
REFERENCES
- 1.Mócsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 10: 387–402, 2010. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohamed AJ, Yu L, Bäckesjö C-M, Vargas L, Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF, Edvard Smith CI. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 228: 58–73, 2009. doi: 10.1111/j.1600-065x.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 3.Packham MA. Role of platelets in thrombosis and hemostasis. Can J Physiol Pharmacol 72: 278–284, 1994. doi: 10.1139/y94-043. [DOI] [PubMed] [Google Scholar]
- 4.Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest 129: 12–23, 2019. doi: 10.1172/JCI122955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol 8: 1137–1140, 1998. doi: 10.1016/S0960-9822(98)70471-3. [DOI] [PubMed] [Google Scholar]
- 6.Manne BK, Badolia R, Dangelmaier C, Eble JA, Ellmeier W, Kahn M, Kunapuli SP. Distinct pathways regulate Syk protein activation downstream of immune tyrosine activation motif (ITAM) and hemITAM receptors in platelets. J Biol Chem 290: 11557–11568, 2015. doi: 10.1074/jbc.M114.629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci 35: 414–422, 2014. doi: 10.1016/j.tips.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer 14: 219–232, 2014. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 9.Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, Thareja S. The development of Bruton's tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem 151: 315–326, 2018. doi: 10.1016/j.ejmech.2018.03.062. [DOI] [PubMed] [Google Scholar]
- 10.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369: 32–42, 2013. [Erratum in N Engl J Med 370: 786, 2014]. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med 353: 172–187, 2005. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 12.Shatzel JJ, Olson SR, Tao DL, McCarty OJT, Danilov AV, DeLoughery TG. Ibrutinib‐associated bleeding: pathogenesis, management and risk reduction strategies. J Thromb Haemost 15: 835–847, 2017. doi: 10.1111/jth.13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369: 507–516, 2013. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAdoo SP, Tam FWK. Fostamatinib disodium. Drugs Future 36: 273, 2011. doi: 10.1358/dof.2011.036.04.1588554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babur O, Melrose AR, Cunliffe JM, Klimek J, Pang J, Sepp A-LI, Zilberman-Rudenko J, Tassi Yunga S, Zheng T, Parra-Izquierdo I, Minnier J, McCarty OJT, Demir E, Reddy AP, Wilmarth PA, David LL, Aslan JE. Phosphoproteomic quantitation and causal analysis reveal pathways in GPVI/ITAM-mediated platelet activation programs. Blood 136: 2346–2358, 2020. doi: 10.1182/blood.2020005496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rigg RA, Aslan JE, Healy LD, Wallisch M, Thierheimer MLD, Loren CP, Pang J, Hinds MT, Gruber A, McCarty OJT. Oral administration of Bruton's tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol 310: C373–C380, 2016. doi: 10.1152/ajpcell.00325.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigg RA, Healy LD, Chu TT, Ngo ATP, Mitrugno A, Zilberman-Rudenko J, Aslan JE, Hinds MT, Vecchiarelli LD, Morgan TK, Gruber A, Temple KJ, Lindsley CW, Duvernay MT, Hamm HE, McCarty OJT. Protease-activated receptor 4 activity promotes platelet granule release and platelet-leukocyte interactions. Platelets 30: 126–135, 2019. doi: 10.1080/09537104.2017.1406076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aslan JE. Platelet Rho GTPase regulation in physiology and disease. Platelets 30: 17–22, 2019. doi: 10.1080/09537104.2018.1475632. [DOI] [PubMed] [Google Scholar]
- 19.Aslan JE, Itakura A, Gertz JM, McCarty OJ. Platelet shape change and spreading. Methods Mol Biol 788: 91–100, 2012. doi: 10.1007/978-1-61779-307-3_7. [DOI] [PubMed] [Google Scholar]
- 20.Ngo ATP, Thierheimer MLD, Babur Ö, Rocheleau AD, Huang T, Pang J, Rigg RA, Mitrugno A, Theodorescu D, Burchard J, Nan X, Demir E, McCarty OJT, Aslan JE. Assessment of roles for the Rho-specific guanine nucleotide dissociation inhibitor Ly-GDI in platelet function: a spatial systems approach. Am J Physiol Cell Physiol 312: C527–C536, 2017. doi: 10.1152/ajpcell.00274.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232, 2006. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 22.Manders EM, Stap J, Brakenhoff GJ, Van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci 103: 857–862, 1992. [DOI] [PubMed] [Google Scholar]
- 23.Peters CG, Michelson AD, Flaumenhaft R. Granule exocytosis is required for platelet spreading: differential sorting of α-granules expressing VAMP-7. Blood 120: 199–206, 2012. doi: 10.1182/blood-2011-10-389247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itakura A, Aslan JE, Kusanto BT, Phillips KG, Porter JE, Newton PK, Nan X, Insall RH, Chernoff J, McCarty OJT. p21-Activated kinase (PAK) regulates cytoskeletal reorganization and directional migration in human neutrophils. PLoS One 8: e73063, 2013. doi: 10.1371/journal.pone.0073063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aslan JE, Itakura A, Haley KM, Tormoen GW, Loren CP, Baker SM, Pang J, Chernoff J, McCarty OJT. P21 activated kinase signaling coordinates glycoprotein receptor VI–mediated platelet aggregation, lamellipodia formation, and aggregate stability under shear. Arterioscler Thromb Vasc Biol 33: 1544–1551, 2013. doi: 10.1161/ATVBAHA.112.301165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJT. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood 118: 3129–3136, 2011. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallisch M, Lorentz CU, Lakshmanan HHS, Johnson J, Carris MR, Puy C, Gailani D, Hinds MT, McCarty OJT, Gruber A, Tucker EI. Antibody inhibition of contact factor XII reduces platelet deposition in a model of extracorporeal membrane oxygenator perfusion in nonhuman primates. Res Pract Thromb Haemost 4: 205–216, 2020. doi: 10.1002/rth2.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker-Groberg SM, Phillips KG, McCarty OJT. Quantification of volume, mass, and density of thrombus formation using brightfield and differential interference contrast microscopy. J Biomed Opt 18: 16014, 2013. doi: 10.1117/1.JBO.18.1.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker SM, Phillips KG, McCarty OJT. Development of a label-free imaging technique for the quantification of thrombus formation. Cell Mol Bioeng 5: 488–492, 2012. doi: 10.1007/s12195-012-0249-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sittampalam GS, Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP, Baell J, Bejcek B, Caaveiro JM, Chung TD (Editors). Assay Guidance Manual [Internet]. Bethesda, MD: Eli Lilly & Company and the National Centre for Advancing Translational Sciences, 2004. [PubMed] [Google Scholar]
- 31.Denzinger V, Busygina K, Jamasbi J, Pekrul I, Spannagl M, Weber C, Lorenz R, Siess W. Optimizing platelet GPVI inhibition versus haemostatic impairment by the Btk inhibitors ibrutinib, acalabrutinib, ONO/GS-4059, BGB-3111 and evobrutinib. Thromb Haemost 119: 397–406, 2019. doi: 10.1055/s-0039-1677744. [DOI] [PubMed] [Google Scholar]
- 32.Levade M, David E, Garcia C, Laurent P-A, Cadot S, Michallet A-S, Bordet J-C, Tam C, Sie P, Ysebaert L, Payrastre B. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood 124: 3991–3995, 2014. doi: 10.1182/blood-2014-06-583294. [DOI] [PubMed] [Google Scholar]
- 33.Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost 7: 1192–1199, 2009. doi: 10.1111/j.1538-7836.2009.03451.x. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 30: 2341–2349, 2010. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aslan JE, Phillips KG, Healy LD, Itakura A, Pang J, McCarty OJT. Histone deacetylase 6-mediated deacetylation of α-tubulin coordinates cytoskeletal and signaling events during platelet activation. Am J Physiol Cell Physiol 305: C1230–C1239, 2013. doi: 10.1152/ajpcell.00053.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouaziz A, Amor NB, Woodard GE, Zibidi H, Lopez JJ, Bartegi A, Salido GM, Rosado JA. Tyrosine phosphorylation/dephosphorylation balance is involved in thrombin-evoked microtubular reorganisation in human platelets. Thromb Haemost 98: 375–384, 2007. doi: 10.1160/TH07-01-0061. [DOI] [PubMed] [Google Scholar]
- 37.Cimmino G, Tarallo R, Conte S, Morello A, Pellegrino G, Loffredo FS, Cali G, De Luca N, Golino P, Trimarco B, Cirillo P. Colchicine reduces platelet aggregation by modulating cytoskeleton rearrangement via inhibition of cofilin and LIM domain kinase 1. Vascul Pharmacol 111: 62–70, 2018. doi: 10.1016/j.vph.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Redondo PC, Harper AGS, Sage SO, Rosado JA. Dual role of tubulin-cytoskeleton in store-operated calcium entry in human platelets. Cell Signal 19: 2147–2154, 2007. doi: 10.1016/j.cellsig.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Sadoul K. New explanations for old observations: marginal band coiling during platelet activation. J Thromb Haemost 13: 333–346, 2015. doi: 10.1111/jth.12819. [DOI] [PubMed] [Google Scholar]
- 40.Aslan JE, McCarty OJT. Rho GTPases in platelet function. J Thromb Haemost 11: 35–46, 2013. [Erratum in J Thromb Haemost 11: 1209, 2013]. doi: 10.1111/jth.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapeller R, Chakrabarti R, Cantley L, Fay F, Corvera S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3' kinase complexes: potential interactions with the microtubule cytoskeleton. Mol Cell Biol 13: 6052–6063, 1993. doi: 10.1128/mcb.13.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kapeller R, Toker A, Cantley LC, Carpenter CL. Phosphoinositide 3-kinase binds constitutively to α/β-tubulin and binds to γ-tubulin in response to insulin. J Biol Chem 270: 25985–25991, 1995. doi: 10.1074/jbc.270.43.25985. [DOI] [PubMed] [Google Scholar]
- 43.Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol 6: 1279–1289, 1996. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Fry MJ, Waterfield MD, Jaken S, Liao L, Fox JE, Rittenhouse SE. Activated phosphoinositide 3-kinase associates with membrane skeleton in thrombin-exposed platelets. J Biol Chem 267: 4686–4692, 1992. [PubMed] [Google Scholar]
- 45.Aslan JE. Platelet shape change. In: Platelets in Thrombotic and Non-Thrombotic Disorders, edited by Gresele P, López JA, Kleiman NS, and Page CP.. New York: Springer, 2017. doi: 10.1007/978-3-319-47462-5_24. [DOI] [Google Scholar]
- 46.Tan S-L, Liao C, Lucas MC, Stevenson C, DeMartino JA. Targeting the SYK–BTK axis for the treatment of immunological and hematological disorders: recent progress and therapeutic perspectives. Pharmacol Ther 138: 294–309, 2013. doi: 10.1016/j.pharmthera.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Matsubara S, Koya T, Takeda K, Joetham A, Miyahara N, Pine P, Masuda ES, Swasey CH, Gelfand EW. Syk activation in dendritic cells is essential for airway hyperresponsiveness and inflammation. Am J Respir Cell Mol Biol 34: 426–433, 2006. doi: 10.1165/rcmb.2005-0298OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA 107: 13075–13080, 2010. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghia P, Pluta A, Wach M, Lysak D, Kozak T, Simkovic M, Kaplan P, Kraychok I, Illes A, de la Serna J, Dolan S, Campbell P, Musuraca G, Jacob A, Avery E, Lee JH, Liang W, Patel P, Quah C, Jurczak W. ASCEND: phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol 38: 2849–2861, 2020. doi: 10.1200/JCO.19.03355. [DOI] [PubMed] [Google Scholar]
- 50.Nicolson PLR, Hughes CE, Watson S, Nock SH, Hardy AT, Watson CN, Montague SJ, Clifford H, Huissoon AP, Malcor J-D, Thomas MR, Pollitt AY, Tomlinson MG, Pratt G, Watson SP. Inhibition of Btk by Btk-specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica 103: 2097–2108, 2018. doi: 10.3324/haematol.2018.193391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin αIIbβ3 signaling in platelets. J Thromb Haemost 3: 1752–1762, 2005. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 52.Connell NT, Berliner N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood 133: 2027–2030, 2019. doi: 10.1182/blood-2018-11-852491. [DOI] [PubMed] [Google Scholar]
- 53.Thorp BC, Badoux X. Atrial fibrillation as a complication of ibrutinib therapy: clinical features and challenges of management. Leuk Lymphoma 59: 311–320, 2018. doi: 10.1080/10428194.2017.1339874. [DOI] [PubMed] [Google Scholar]
- 54.Poole A, Gibbins J, Turner M, Van Vugt MJ, Van de Winkel JG, Saito T, Tybulewicz VL, Watson SP. The Fc receptor γ‐chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J 16: 2333–2341, 1997. doi: 10.1093/emboj/16.9.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim E, Zhang H, Sivina M, Vaca A, Thompson PA, Jain N, Ferrajoli A, Estrov ZE, Keating MJ, Wierda WG, Rice WG, Andreeff M, Burger JA. CG-806, a first-in-class Pan-FLT3/Pan-BTK inhibitor, exhibits broad signaling inhibition in chronic lymphocytic leukemia cells. Blood 134: 3051, 2019. doi: 10.1182/blood-2019-124473. [DOI] [Google Scholar]
- 56.Pasquet JM, Gross B, Quek L, Asazuma N, Zhang W, Sommers CL, Schweighoffer E, Tybulewicz V, Judd B, Lee JR, Koretzky G, Love PE, Samelson LE, Watson SP. LAT is required for tyrosine phosphorylation of phospholipase Cγ2 and platelet activation by the collagen receptor GPVI. Mol Cell Biol 19: 8326–8334, 1999. doi: 10.1128/mcb.19.12.8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldmann L, Duan R, Kragh T, Wittmann G, Weber C, Lorenz R, von Hundelshausen P, Spannagl M, Siess W. Oral Bruton tyrosine kinase inhibitors block activation of the platelet Fc receptor CD32a (FcgammaRIIA): a new option in HIT? Blood Adv 3: 4021–4033, 2019. [Erratum in Blood Adv 4: 112, 2020]. doi: 10.1182/bloodadvances.2019000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gross BS, Lee JR, Clements JL, Turner M, Tybulewicz VL, Findell PR, Koretzky GA, Watson SP. Tyrosine phosphorylation of SLP-76 is downstream of Syk following stimulation of the collagen receptor in platelets. J Biol Chem 274: 5963–5971, 1999. doi: 10.1074/jbc.274.9.5963. [DOI] [PubMed] [Google Scholar]
- 59.Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, Studer S, de la Torre JC, Casanova J-L, Cravatt BF, Teijaro JR. Dimethyl fumarate disrupts human innate immune signaling by targeting the IRAK4-MyD88 complex. J Immunol 202: 2737–2746, 2019. doi: 10.4049/jimmunol.1801627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walford T, Musa FI, Harper AG. Nicergoline inhibits human platelet Ca2+ signalling through triggering a microtubule-dependent reorganization of the platelet ultrastructure. Br J Pharmacol 173: 234–247, 2016. doi: 10.1111/bph.13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shah B, Allen N, Harchandani B, Pillinger M, Katz S, Sedlis SP, Echagarruga C, Samuels SK, Morina P, Singh P, Karotkin L, Berger JS. Erratum to: effect of colchicine on platelet-platelet and platelet-leukocyte interactions: a pilot study in healthy subjects. Inflammation 39: 501, 2016. doi: 10.1007/s10753-015-0266-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tardif J-C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, Lopez-Sendon J, Ostadal P, Koenig W, Angoulvant D, Gregoire JC, Lavoie M-A, Dube M-P, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin M-C, Roubille F. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 381: 2497–2505, 2019. doi: 10.1056/NEJMoa1912388. [DOI] [PubMed] [Google Scholar]
- 63.Pandey MK, Gowda K, Sung S-S, Abraham T, Budak-Alpdogan T, Talamo G, Dovat S, Amin S. A novel dual inhibitor of microtubule and Bruton’s tyrosine kinase inhibits survival of multiple myeloma and osteoclastogenesis. Exp Hematol 53: 31–42, 2017. doi: 10.1016/j.exphem.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 64.Gabizon R, Shraga A, Gehrtz P, Livnah E, Shorer Y, Gurwicz N, Avram L, Unger T, Aharoni H, Albeck S, Brandis A, Shulman Z, Katz B-Z, Herishanu Y, London N. Efficient targeted degradation via reversible and irreversible covalent PROTACs. J Am Chem Soc 142: 11734–11742, 2020. doi: 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, Zhu H, Farley KA, Ding W, Schiemer J, Feng X, Chang JS, Uccello DP, Young JA, Garcia-Irrizary CN, Czabaniuk L, Schuff B, Oliver R, Montgomery J, Hayward MM, Coe J, Chen J, Niosi M, Luthra S, Shah JC, El-Kattan A, Qui X, West GM, Noe MC, Shanmugasundaram V, Gilbert AM, Brown MF, Calabrese MF. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci USA 115: E7285–E7292, 2018. doi: 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Busygina K, Jamasbi J, Seiler T, Deckmyn H, Weber C, Brandl R, Lorenz R, Siess W. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood 131: 2605–2616, 2018. doi: 10.1182/blood-2017-09-808808. [DOI] [PubMed] [Google Scholar]
- 67.Aslan JE. Platelet proteomes, pathways, and phenotypes as informants of vascular wellness and disease. Arterioscler Thromb Vasc Biol 41: 999–1011, 2021. doi: 10.1161/ATVBAHA.120.314647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parra-Izquierdo I, Aslan JE. Perspectives on platelet heterogeneity and host immune response in COVID-19. Semin Thromb Hemost 46: 826–830, 2020. doi: 10.1055/s-0040-1715093. [DOI] [PMC free article] [PubMed] [Google Scholar]