

Abstract
Compared with acyanotic congenital heart disease (CHD), cyanotic CHD has an increased risk of lifelong mortality and morbidity. These adverse outcomes may be attributed to delayed cardiomyocyte maturation, since the transition from a hypoxic fetal milieu to oxygen-rich postnatal environment is disrupted. We established a rodent model to replicate hypoxic myocardial conditions spanning perinatal development, and tested the hypothesis that chronic hypoxia impairs cardiac development. Pregnant mice were housed in hypoxia beginning at embryonic day 16. Pups stayed in hypoxia until postnatal day (P)8 when cardiac development is nearly complete. Global gene expression was quantified at P8 and at P30, after recovering in normoxia. Phenotypic testing included electrocardiogram, echocardiogram, and ex vivo electrophysiology study. Hypoxic P8 animals were 47% smaller than controls with preserved heart size. Gene expression was grossly altered by hypoxia at P8 (1,427 genes affected), but normalized after recovery (P30). Electrocardiograms revealed bradycardia and slowed conduction velocity in hypoxic animals at P8, with noticeable resolution after recovery (P30). Notable differences that persisted after recovery (P30) included a 65% prolongation in ventricular effective refractory period, sinus node dysfunction, 23% reduction in ejection fraction, and 16% reduction in fractional shortening in animals exposed to hypoxia. We investigated the impact of chronic hypoxia on the developing heart. Perinatal hypoxia was associated with changes in gene expression and cardiac function. Persistent changes to the electrophysiological substrate and contractile function warrant further investigation and may contribute to adverse outcomes observed in the cyanotic CHD population.
NEW & NOTEWORTHY We utilized a new mouse model of chronic perinatal hypoxia to simulate the hypoxic myocardial conditions present in cyanotic congenital heart disease. Hypoxia caused numerous abnormalities in cardiomyocyte gene expression, the electrophysiologic substrate of the heart, and contractile function. Taken together, alterations observed in the neonatal period suggest delayed cardiac development immediately following hypoxia.
Keywords: cardiac development, cardiac electrophysiology, congenital heart disease, perinatal hypoxia
INTRODUCTION
Outcomes for patients with cyanotic congenital heart disease (CHD) remain guarded despite countless advances in clinical care strategies over the last decades. CHD affects 1% of live births (1), with one quarter of CHD representing cyanotic conditions (2). Infants with cyanotic CHD are at an eightfold increased risk of death (2) compared with acyanotic CHD, and up to 39% of all patients with CHD develop heart failure during childhood (3). With clinical advances, the CHD population is increasingly surviving to adulthood, leading to an increased burden of CHD-associated heart failure (4). Despite the high incidence of morbidity and mortality in the cyanotic CHD population, the underlying risk factors are not fully understood (3).
Normal embryology of the heart includes streaming of the most highly saturated blood to the ascending aorta, which directly supplies the head vessels and coronary arteries without mixing with desaturated blood from the ductus arteriosus (5, 6). For a fetus with complex CHD—such as hypoplastic left heart syndrome or d-looped transposition of the great arteries—this streaming pattern is disrupted and the blood in the ascending aorta is desaturated compared with the normal fetal circulation (7–10). This relative desaturation of ascending aortic blood leads to chronic hypoxic conditions in the developing brain and heart starting prenatally and persisting after birth until definitive repair. At the time of cardiac surgery, the cyanotic myocardium has depleted endogenous antioxidants (11), higher tissue lactate levels (12), more troponin I release (13), higher levels of oxidative stress (14), and less available adenosine triphosphate (ATP) (15) compared with acyanotic myocardium. Moreover, cyanotic infants exhibit more myocardial injury during bypass surgery and worse postoperative outcomes (13, 15). Little is known about the mechanisms that contribute to the cyanotic myocardium’s vulnerability to metabolic derangements during surgery.
The transition from the hypoxic fetal milieu to the oxygen-rich postnatal environment is thought to stimulate postnatal maturation (16). Accordingly, a limited number of studies suggest that hypoxia delays cardiac maturation. In mice, postnatal hypoxia prolongs the neonatal period of cardiomyocyte proliferative ability (17); and in chickens, prenatal hypoxia results in immature calcium handling (18). Moreover, cardiomyocytes sampled from human patients with hypoplastic left heart syndrome show some persistence in fetal gene programming (19). However, the direct effects of chronic perinatal hypoxia on the developmental processes of the cardiomyocyte remain largely unknown.
To the best of our knowledge, this is the first study to examine the combined effects of pre- and postnatal hypoxia on the developing heart. The current study aimed to establish a rodent model of chronic perinatal hypoxia, as would be seen in cyanotic CHD, to investigate the developmental status of the cardiomyocyte under these conditions. We hypothesized that exposure to chronic hypoxia, beginning prenatally and continuing through the neonatal period, would perturb cardiomyocyte gene expression, contractile function, and the electrophysiologic substrate of the heart. The Cardiac Safety Research Consortium has implored the research community to perform more studies of developmental cardiac physiology to better understand the substrate on which therapies may work in the pediatric population (20), and this study intended to contribute to that call for knowledge.
METHODS
Disclosure Statement and Ethical Approval
Data that support the findings of this study are available from the corresponding author upon reasonable request. Animal experiments were approved by the Institutional Animal Care and Use Committee at Children’s National Research Institute, in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Animal Model
Wild-type pregnant CD1 mice [6- to 8-wk-old Crl:CD1(ICR), Charles River Laboratories] were kept in a hypoxic chamber (BioSpherix, Redfield, NY) starting on embryonic day (E)16, a time that coincides with myocardial reliance on coronary flow for oxygen delivery and the beginning of a period of rapid growth of the ventricular myocardium, similar to the second trimester in human fetuses (21, 22) (Fig. 1). Blood in the ascending aorta is desaturated in complex CHD (7), leading to desaturated coronary arterial flow and hypoxic myocardial conditions once the myocardium is dependent on the coronary arteries for oxygenation. During the experiment, the oxygen concentration was maintained, monitored, and recorded continuously with sensors placed inside the chamber to achieve a level of 11% ± 0.5% (Pro:Ox Model 360, BioSpherix, Redfield, NY). Nitrogen gas was used to displace oxygen. This oxygen concentration corresponds to rodent pulse oximetry readings of 65%–73% (23), and was chosen to approximate ascending aortic saturations of 48% in the human fetus with cyanotic CHD (7) and target oxygen saturations of 70%–85% in the human neonate with cyanotic CHD. Dams gave birth in the hypoxic chamber (n = 11) and pups remained in hypoxia until postnatal day (P)8, when cardiomyocyte maturation is nearly complete (24, 25). Strain and age-matched normoxic dams (n = 10) were kept in normoxia and gave birth under normoxic conditions. After P8, hypoxic animals were moved to normoxic conditions and allowed to recover until further testing at P30, thus simulating normalized oxygen saturations in human infants who have undergone surgical repair of cyanotic CHD. At the end of the study, animals were anesthetized with 4% isoflurane, the heart was excised, and euthanasia ensued via exsanguination. For each experiment, normoxic and hypoxic animals (n = 185) were chosen randomly. Approximate equal distribution of males (53.6%) and females (46.3%) were used for subsequent experiments, and multiple litters were used for each end point.
Figure 1.

Chronic perinatal hypoxia mouse model. Pregnant CD1 mice were placed in hypoxia starting on embryonic day (E)16, corresponding with rapid growth of the ventricular myocardium and reliance on coronary arteries, similar to a 4 months gestation human fetus. Hypoxic pups were born and reared in hypoxia until postnatal day (P)8 when the majority of cardiomyocyte development is complete. Removal from the hypoxic chamber represents surgical repair in human neonates with cyanotic congenital heart disease (CHD). Mice recovered in normoxia until further testing at P30, representing a child with repaired cyanotic CHD and corresponding normalization in oxygen saturations. , fractional inspired oxygen concentration. Image created with Biorender.com and used with permission.
Gene Expression
Whole hearts (including both atria and ventricles) were excised, submerged in RNA later, and then subsequently frozen (−80°C). Total RNA was isolated using an RNeasy fibrous tissue kit. Verification of RNA integrity and RNA quantification were done by spectrophotometry and an RNA 6000 Nano assay (Bioanalyzer 2100, Agilent Technologies). The RNA integrity number for all samples was >6 (8.6 ± 0.19). Total RNA (250 ng) was primed for the entire length of RNA, including both poly(A) and non-poly(A) mRNA and reverse transcribed to generate sense-strand targets that were biotin-labeled using a WT Plus Reagent kit, and then hybridized to Affymetrix GeneChip Mouse Clariom S arrays for 16 h (48°C), following manufacturer’s instructions (Thermo Fisher Scientific). Hybridization cocktails were removed, and arrays were washed and stained on a Fluidics Station 450 (mouse Clariom S arrays). Arrays were scanned on the Affymetrix GCS3000 7G scanner and initial quality control data evaluated using Affymetrix Expression Console software (Thermo Fisher Scientific). Microarray data were imported and analyzed (ANOVA P < 0.05, 1.5-fold cut-off, 0.1 false discovery rate) using the Transcriptome Analysis Console (Applied Biosciences). Gene ontology (GO) enrichment analysis was performed with the GOrilla tool using a single rank-ordered gene list (26, 27). Dataset was available via the Gene Expression Omnibus (No. GSE169214).
In Vivo Electrocardiography
Noninvasive electrocardiogram (ECG) recordings were obtained using an ecgTUNNEL system (emka Technologies). ECG waveforms were recorded for 1–2 min on P8 and P30 conscious and isoflurane-sedated animals. ECG segments were quantified using ecgAuto software (emka Technologies) for heart rate, heart rate variability, atrial depolarization time (P-wave duration), atrioventricular (AV) conduction time (PR interval), ventricular depolarization time (QRS duration), and ventricular repolarization time [QT interval; uncorrected as heart rate does not significantly alter QT interval in mice (28)]. Heart rate variability was measured as a root mean square of the successive differences (RMSSD) (28, 29). Due to motion artifact, only clearly discernable ECG parameters were included in the analysis for each animal.
Ex Vivo Electrophysiology Study
P30 animals were anesthetized with 4% isoflurane, the heart was rapidly excised, and the aorta cannulated. The heart was transferred to a temperature-controlled (37°C) constant-pressure (70 mmHg) Langendorff perfusion system. Excised hearts were perfused with Krebs–Henseleit buffer bubbled with carbogen, as previously described (29, 30). A stimulation electrode was placed externally on the right atrium, and an atrial pacing protocol was used to determine Wenckebach cycle length (WBCL) and atrioventricular nodal effective refractory period (AVNERP). WBCL is the shortest pacing cycle length during atrial pacing that causes the Wenckebach phenomenon. AVNERP is the shortest extrastimulus interval during atrial pacing that fails to conduct through the atrioventricular (AV) node, as indicated by loss of ventricular capture. For ventricular pacing, a stimulation electrode was placed on the left ventricular epicardium. To determine the ventricular effective refractory period (VERP), dynamic pacing was performed with stepwise decrements in the pacing cycle length (S1–S2) until loss of capture was noted. Baseline rhythms were monitored throughout the duration of the studies for detection of dysrhythmias including ectopy, sinus node dysfunction, and AV nodal block. Sinus node dysfunction was defined as bradycardia with an irregular sinus rate.
High-Frequency Ultrasound Echocardiography
P30 animals underwent sedated transthoracic echocardiography to assess the persistent effects of chronic perinatal hypoxia on left ventricular systolic function. Anesthesia was initiated (4%) and maintained (2%–2.5%) with inhaled isoflurane. Preclinical high-frequency ultrasound systems (VisualSonics Vevo 770, 30 MHz probe and Vevo 3100, 40 MHz probe) were used to obtain M-mode images of the left ventricle at the level of the papillary muscles from a parasternal short-axis view. Fractional shortening and thickness of the interventricular septum and left ventricular posterior wall were measured on the M-mode images. Measurements from a minimum of three cardiac cycles were averaged. The Vevo 3100 was also used to obtain ECG-gated Kilohertz Visualization (EKV) images of the left ventricle from parasternal long- and short-axis views. Ejection fraction from the long-axis view was calculated offline (Vevo Lab) via a modified Simpson’s monoplane method of disks. Fractional area change was measured from the short-axis view at the level of the papillary muscles. EKV acquisitions average multiple cardiac cycles to increase temporal frame rate to 10,000 frames/s allowing for accurate analysis in animals with high-heart rates.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism. Data are presented as means ± SE. Data normality was confirmed by Shapiro–Wilk test. Datasets were compared between control and hypoxic animals using two-tailed Student’s t test (parametric data) or Mann–Whitney test (nonparametric data). Significance was defined as P < 0.05. To compare ECG parameters under awake and anesthetized conditions, two-way analysis of variance (ANOVA) was used to compare hypoxic versus normoxic animals. In the latter, significance was defined as q < 0.1 (adjusted P value after multiple comparisons testing with a false discovery rate (FDR) of 0.1).
RESULTS
Hypoxia Decreased Litter Size and Pup Weight
The number of viable pups was counted at P8, upon removal from the normoxic or hypoxic chamber. Fewer pups were present in hypoxic litters, as compared with normoxic (n ≥ 10 litters per group, 6.8 vs. 11.1 pups per litter, P < 0.01, Fig. 2A). There was no survival benefit afforded by sex between hypoxic and normoxic groups (chi-square, P = 0.97). Maternal mice who gave birth in hypoxia weighed less (32.7 ± 2 g) than controls (45.1 ± 1.7 g), assessed at 8 days after birth to coincide with removal from the hypoxia chamber (P < 0.001). Hypoxic pups (P8) also weighed less (3.3 ± 0.1 g) than normoxic control pups (6.2 ± 0.12 g, P < 0.0001, Fig. 2, B and C). Despite lower body weight, heart weight was preserved in hypoxia, resulting in a higher heart-to-body-weight ratio in hypoxic animals (13.1 ± 0.9 vs. 6.9 ± 0.3 mg/g, P < 0.0001). After P8, hypoxic animals recovered in normoxic conditions, thus simulating the return to normal oxygen saturations that occurs after surgical repair of cyanotic CHD. At P30, hypoxic animals had undergone catch-up growth such that their body weight was comparable with normoxic controls (21.9 ± 0.7 vs. 21.7 ± 0.7 g, P = 0.86, Fig. 2D). At P30, hypoxic animals maintained a slightly higher heart-to-body-weight ratio (6.4 ± 0.3 vs. 5.4 ± 0.2 mg/g, P < 0.005), albeit less pronounced than at the P8 timepoint before recovery in normoxic conditions.
Figure 2.

Number of pups and body measurements. A: hypoxia reduced the number of pups per litter, measured at postnatal day (P)8 upon removal from hypoxia. B and C: at P8, hypoxic animals were markedly smaller than control; however, heart weight was preserved, and thus heart-to-body-weight ratio was higher in hypoxic animals. D: by P30, hypoxic animals underwent catch-up growth; heart weight and heart-to-body weight ratios were higher in hypoxic animals. Biological replicates are shown and sample size indicated. Data expressed as means ± SE. *P < 0.05 hypoxic versus control via two-tailed t test. ns, not significant.
Hypoxia Altered Global Gene Expression at P8
Gene expression arrays were performed on whole heart samples isolated at P8 to assess the effects of hypoxia at the end of the neonatal period of rapid cardiomyocyte development, and again at P30 after hypoxic animals had recovered in normoxia. Principal component analysis demonstrated that experimental groups (normoxia vs. hypoxia) were well-separated by their mRNA expression profiles at P8, but this separation was negligible at P30 (Fig. 3A) (31, 32). Using a 1.5-fold expression cut-off and a 10% FDR to correct for multiple testing (33), a total of 1,427 mRNAs were differentially expressed between hypoxic and control hearts at P8 (Fig. 3B; data available via Gene Expression Omnibus). Differentially expressed genes important to cardiac functioning and development are highlighted in the Fig. 3C volcano plot. Within treatment groups, hypoxic animals exhibited a greater number of gene changes between P8 and P30 (4,593 genes; Fig. 3B) than the normoxic group (2,147 genes), suggesting that the hypoxic group underwent more developmental changes over this time period after transitioning to normoxia. Interestingly, gene expression nearly normalized in P30 animals after recovering in normoxia. Only one gene was expressed differentially between groups at P30: Wsb2 (WD repeat and SOCS box-containing 2).
Figure 3.

Global gene expression. A: principal component analysis demonstrated that experimental groups were well separated by their mRNA expression profiles at postnatal day (P)8 (n = 5 hypoxic, n = 4 control biological replicates), but this separation was negligible in P30 samples (n = 5 hypoxic, n = 4 control biological replicates). B: a total of 1,427 mRNAs were differentially expressed between hypoxic (Hx) and normoxic control (Nx) hearts at P8, with near resolution of differences by P30 [ANOVA with 1.5-fold expression cut-off, P < 0.05 and false-discovery rate (FDR) q ≤ 0.1]. C: specific genes important to cardiovascular functioning and development are highlighted in a volcano plot of all differentially expressed genes between groups at P8.
Differentially expressed genes at P8 were significantly overrepresented in >400 GO categories (319 biological processes, 42 molecular functions, 46 cellular components; data available via Gene Expression Omnibus). Categories associated with phenotypic changes observed in our experimental studies included extracellular matrix structural constituent (GO:0005201), ion channel binding (GO:0044325), glycolytic process (GO:0006096), hypoxia-inducible factor-1 alpha signaling pathway (GO:0097411), mitotic cell cycle process (GO:1903047), and cell maturation (GO:0048469) (Fig. 4).
Figure 4.

Gene ontologies. A: highlighted gene ontologies (GOs) important to cardiac development and functioning that were different between groups at postnatal day (P)8 (see Fig. 3). GOs based on differential gene expression between hypoxic (Hx) and normoxic control (Nx) hearts at P8 (ANOVA, P < 0.05, false-discovery rate q ≤ 0.1). B: heat maps of the main cluster of differentially expressed genes, as well as selected gene ontologies demonstrate differential expression at P8 between groups. Each gene is median-centered, with data displayed as fold-change. n = 5 hypoxic, n = 4 control biological replicates.
Hypoxia Altered Transmembrane Ion Channel Expression at P8
Multiple transmembrane ion channels were differentially expressed in P8 hypoxic animals, including potassium (K+), sodium (Na+), and calcium (Ca2+) channels involved in the cardiac action potential and maintenance of a stable resting membrane (Fig. 5A). In cardiac myocytes, phase 0 of the cardiac action potential is characterized by rapid depolarization via Na+ influx (INa); voltage-gated Na+ channel gene expression was upregulated with hypoxia (Scn3b: fold change +3.4, P = 0.0002, q = 0.017; Scn1b: fold change +1.3, P = 0.018, q = 0.15). Phase 1 is characterized by a fast transient outward K+ current (Ito), which was downregulated in hypoxia (Kcnd2: fold change −8.02, P = 0.0003, q = 0.019; Kcnip2: fold change −3.17, P = 0.0012, q = 0.042; Kcnd3: fold change −1.46, P = 0.0079, q = 0.10). Notably, decreased Ito current is also observed in patients with atrial fibrillation (34) and heart failure (35), and can impair electromechanical coupling and prolong action potential duration (36). Phase 1 also includes slow Na+ efflux via the Na+/Ca2+ exchanger (NCX1), which was upregulated in hypoxia (Slc8a1: fold change +2.78, P = 0.0001, q = 0.012). The plateau phase (phase 2) is primarily responsible for action potential duration. Membrane potential is held stable by balancing Ca2+ influx and K+ efflux, and both were affected by hypoxia. Ca2+ influx continues via NCX1 (upregulated in hypoxia, see Fig. 5), and L-type voltage-gated Ca2+ channels open to increase Ca2+ influx (ICaL). A regulatory subunit of ICaL channels was downregulated in hypoxia (Cacna2d1: fold change −1.96, P = 0.0014, q = 0.045). Calmodulin expression was increased (Calm3: fold change +1.67, P = 0.0009, q = 0.036) which modulates both action potential duration and excitation-contraction coupling by modifying ICaL. Slow K+ efflux (IKs) occurs via voltage-gated K+ channels, which were upregulated in hypoxia (Kcnq1: fold change +2.54, P = 0.0057, q = 0.089). Final rapid repolarization (phase 3) occurs mainly by K+ efflux (IKr). Both genes associated with IKr trended toward downregulation (Kcne2: fold change −1.47, P = 0.22, q = 0.51; Kcnh2: fold change −1.24, P = 0.28, q = 0.57). Finally, phase 4 represents the resting membrane potential, which is maintained via constant K+ efflux (IK1, IAch, IATP) through inwardly rectifying K+ channels; these genes were largely unaffected by hypoxia.
Figure 5.

Genes affecting ion channels and the contractile apparatus. A: at postnatal day (P)8, hypoxia affected genes involved with most phases of the cardiac action potential. B: hypoxia affected multiple genes important to the contractile apparatus, including both the sarcomere and calcium handling machinery, at P8. Results based on differential gene expression. Significance denoted by *P ≤ 0.05 and FDR q ≤ 0.1, or †P ≤ 0.05 and FDR q > 0.1. n = 5 hypoxic, n = 4 control biological replicates. Image created with Biorender.com.
In cardiac pacemaker cells, the T-type Ca2+ channel generates Ca2+ influx (ICaT) to initiate an action potential (gene = Cacna1g). Cacna1g had a 3.13-fold downregulation, which did not meet significance (P = 0.052, q = 0.26). Additional studies are needed to examine regional differences in T-type Ca2+ channel expression, which can result in bradycardia and sinus node dysfunction if localized to pacemaker cells (i.e., sinoatrial and atrioventricular nodes). Finally, gap junctions connect neighboring cardiomyocytes and allow for rapid spread of an action potential from one cell to the next. Gap junction expression was downregulated in hypoxia (Gja1: fold change −2.57, P = 0.0022, q = 0.056; Gja6: fold change −1.74, P = 0.0067, q = 0.096), which can lead to slower electrical conduction.
Hypoxia Altered Expression of Genes Important to the Contractile Apparatus at P8
Multiple genes important to the contractile apparatus were differentially expressed in P8 hypoxic animals, including those involved in both calcium handling and sarcomere structure (Fig. 5B). Within the cardiac sarcomere, isoform switching occurs during perinatal development for many key structural proteins. In hypoxic P8 animals, there was persistence of immature isoforms of Troponin-I (Tnni1: fold change +13.13, P = 9.71 E−05, q = 0.011), alpha-actin (Acta1: fold change +5.57, P = 0.012, q = 0.12), gamma-actin (Actg1: fold-change +3.05, P = 0.010, q = 0.12), and myosin heavy chain (Myh7: fold change +1.48, P = 0.083, q = 0.32); although only Troponin-I met the predetermined threshold for significance. The isoform switch for myosin light chain was unaffected (Myl7→Myl2), however, expression of two myosin light chain regulators were altered (Myl3: fold change −1.79, P = 0.0045, q = 0.08; Myl9: fold change +5.48, P = 0.0025, q = 0.06).
Multiple stabilizing components of the cardiac sarcomere were also altered in P8 hypoxic animals. Titin, the cardiac myofilament responsible for passive tension, was downregulated in hypoxia (fold change −2.56, P = 0.0055, q = 0.088), which may cause an increased risk for diastolic dysfunction (37). Desmin provides strength of attachment of myofibrils to the Z-disk, and was upregulated in hypoxia (Des: fold change +2.31, P = 0.0023, q = 0.057). Desmosomes and adherens junctions provide structural integrity between cells, and both demonstrated downregulation of components in hypoxia (Pkp1: fold change −1.55, P = 0.0022, q = 0.056; Pcdh7: fold change −1.82, P = 0.0025, q = 0.061; Dsc2: fold change −2.04, P = 0.016, q = 0.14). Vimentin expression was increased (Vim: fold change +3.08, P = 0.0017, q = 0.050) indicating an increased fibroblast population, but collagen expression was decreased (extracellular matrix cluster in Fig. 4B), suggesting decreased fibroblast functioning and a less robust extracellular matrix to act as a scaffold for muscle contraction.
Calcium handling genes were also altered in hypoxic P8 animals. In mature cardiomyocytes, synchronized ryanodine receptors facilitate a rapid increase in cytosolic Ca2+ concentration which is necessary for excitation-contraction coupling. Ryanodine receptor expression was decreased in hypoxic animals (Ryr2: fold change −3.74, P = 0.0038, q = 0.073; Ryr1: fold change −1.16, P = 0.029, q = 0.20), consistent with a delay in maturation. Furthermore, extracellular Ca2+ entry may be altered in hypoxic animals due to decreased expression of L-type calcium channels (Cacna2d1, see Fig. 5) and increased expression of the Na+/Ca2+ exchanger (Slc8a1, see Fig. 5).
Hypoxia Caused Bradycardia and Slowed Conduction at P8
With the observed changes in ion channel expression on gene arrays, we collected in vivo ECG tracings at P8 (Fig. 6A) to identify alterations in the electrophysiologic substrate of the heart immediately after chronic perinatal hypoxia. During normal murine development, heart rate increases as maturation progresses. At P8, hypoxic animals were bradycardic compared with normoxic controls (375 ± 12 vs 547 ± 11 beats/min, q < 0.0001), consistent with a delay in normal postnatal maturation (Fig. 6B). We examined the effects of isoflurane sedation on ECG measurements, as cyanotic newborns with CHD are subjected to anesthesia at the time of surgical repair. Hypoxic animals remained more bradycardic than normoxic when sedated (222 ± 7 vs. 278 ± 6 beats/min, q < 0.0001, Fig. 6B). Conduction speed is expected to increase throughout development as cell-cell interactions mature. Awake measurements indicated that P8 hypoxic animals had a slightly longer P-wave duration (13.9 ± 0.5 vs. 12.4 ± 0.3 ms, q = 0.09) and PR interval (46.9 ± 2 vs. 37.3 ± 0.9 ms, q < 0.01). Heart rate variability was also decreased in hypoxic animals (RMSSD: 3.9 ± 0.5 vs. 7.1 ± 1.4, q < 0.005), a parameter that normally increases with postnatal development (29). In vivo ECG parameters were also altered under isoflurane sedation (Fig. 6B), hypoxic P8 animals exhibited longer P-wave duration (20 ± 0.9 vs. 17.4 ± 0.4, q < 0.001), PR interval (88.7 ± 3.5 vs. 57.7 ± 1.7 ms, q < 0.0001), QRS duration (22.5 ± 0.6 vs. 21.0 ± 0.6 ms, q < 0.05), and QT duration (104.3 ± 3.7 vs. 86.8 ± 3.1 ms, q < 0.005). For accuracy, QT intervals were only analyzed in sedated animals. Anesthetic agents have been reported to slow atrioventricular conduction in animal models (38, 39), and human case reports (40, 41), and our results suggest that perinatal hypoxia may exaggerate this effect.
Figure 6.

In vivo electrocardiogram measurements. A: representative electrocardiogram (ECG) traces (isoflurane-sedation shown). B: awake hypoxic postnatal day (P)8 animals had lower heart rates compared to control, and hypoxia prolonged ECG intervals, consistent with slowed conduction speed. Sedated hypoxic P8 animals exhibited slower heart rate and conduction slowing in most ECG parameters compared with control. C: at P30, most ECG measurements had normalized or improved. Hypoxic animals had slight bradycardia and longer QT intervals under isoflurane-sedation. Biological replicates are shown. Data expressed as means ± SE. *q < 0.1 hypoxic versus control, two time points assessed by two-way ANOVA with multiple comparisons testing (0.1 FDR). *P < 0.05 hypoxic versus control via two-tailed t test. Due to motion artifact, only clearly discernable ECG parameters were included in the analysis. ns, not significant.
In Vivo ECG Measurements Normalized at P30 after a Period of Recovery in Normoxia
Since there was resolution of gene expression differences at P30 after recovering in normoxia, ECG measurements were repeated at P30 to determine if electrophysiologic differences had also normalized. We observed a slight decrease in the heart rate of P30 hypoxic animals under both awake (734.7 ± 9 vs. 779 ± 8 beats/min, q < 0.005) and sedated (398.2 ± 9 vs. 441.1 ± 11 beats/min, q < 0.005) conditions, although heart rate slowing was less pronounced than at the P8 timepoint. We also observed slight lengthening of the QT interval in hypoxic P30 animals (62.4 ± 2.1 vs. 56.2 ± 1.7 ms, P < 0.05), indicating a longer repolarization time. No significant difference in P-wave duration (awake: 12.3 ± 0.6 vs. 11.9 ± 0.4 ms; sedated: 14.3 ± 0.8 vs. 13.7 ± 0.5 ms), PR duration (awake: 29.1 ± 0.5 vs. 29.7 ± 0.4 ms; sedated: 40.9 ± 1.6 vs. 44.5 ± 2.1 ms), or heart rate variability (awake RMSSD: 11.7 ± 1.6 vs. 15.9 ± 3.3; sedated RMSSD: 9.5 ± 1.3 vs. 9.1 ± 1.3) was observed between hypoxic and control animals at P30 (Fig. 6C). The significant differences in ECG parameters observed at P8 largely abated after the 22-day period of recovery in normoxia, consistent with gene expression data (Fig. 3, A and B).
Ex Vivo Electrophysiology Study Revealed Persistent Underlying Changes to the Electrophysiologic Substrate after Hypoxia
Although ECG measurements largely normalized by P30, we conducted more rigorous testing of the cardiac electrophysiologic substrate in the absence of autonomic influences. Programmed electrical stimulation protocols were implemented to pinpoint tissue refractoriness, which may not be observed using baseline ECG recordings. Electrophysiology studies performed at P30 revealed that perinatal hypoxia caused persistent prolongation of VERP (76.5 ± 6.7 vs. 46.5 ± 5.3 ms, P < 0.05), a parameter that normally decreases with rodent age (Fig. 7A). Such an increase in ventricular refractoriness can alter tissue excitability and slow repolarization, which is in agreement with QT lengthening in vivo. No difference in atrioventricular conduction was observed between groups, measured by WBCL (84.0 ± 3.7 vs. 84.5 ± 5.6 ms) and AVNERP (72.8 ± 3.1 vs. 70.5 ± 6.4 ms) (Fig. 7A). Ex vivo studies also revealed sinus node dysfunction in all four of the hypoxic hearts, as opposed to 25% of the normoxic control hearts (chi-square, P = 0.029; Fig. 7, B and C). Cyanotic CHD carries a high incidence of sinus node dysfunction (42, 43), and our results suggest that hypoxia may play a role in creating the substrate for sinus node dysfunction.
Figure 7.

Ex vivo electrophysiology study. A: ventricular effective refractory period (VERP) was prolonged at postnatal day (P)30 in animals exposed to hypoxia. There was no difference for Wenckebach cycle length (WBCL) or atrioventricular nodal effective refractory period (AVNERP). Data expressed as means ± SE. *P < 0.05 hypoxic (n = 4) versus control (n = 4) via two-tailed t test. B: baseline rhythm during electrophysiology studies is displayed for each animal. C: 100% of hypoxic and 25% of control animals had sinus node dysfunction during the study; significance assessed using chi-squared analysis. *P < 0.05. Biological replicates are shown. ns, not significant.
Perinatal Hypoxia Caused a Persistent Decrease in Contractile Function
Gene expression changes indicated differences in the sarcomere and calcium handling at P8. Although animals were too small to obtain measurements at P8, we measured phenotypic contractile function at P30 after recovery in normoxia to assess persistent changes. Transthoracic echocardiography at P30 demonstrated worse contractile function in animals exposed to hypoxia as compared with normoxic controls, as measured by fractional shortening (33.6% ± 1% vs. 40.3% ± 1.1%, P < 0.0001), fractional area change (53.1% ± 1.1% vs. 60.8% ± 1.3%, P < 0.0005) and ejection fraction (55.1% ± 1.2% vs. 71.6% ± 1.3%, P < 0.0001, Fig. 8). There was no difference in interventricular septum thickness or left ventricular posterior wall thickness between groups at both systole and diastole (Fig. 8C).
Figure 8.
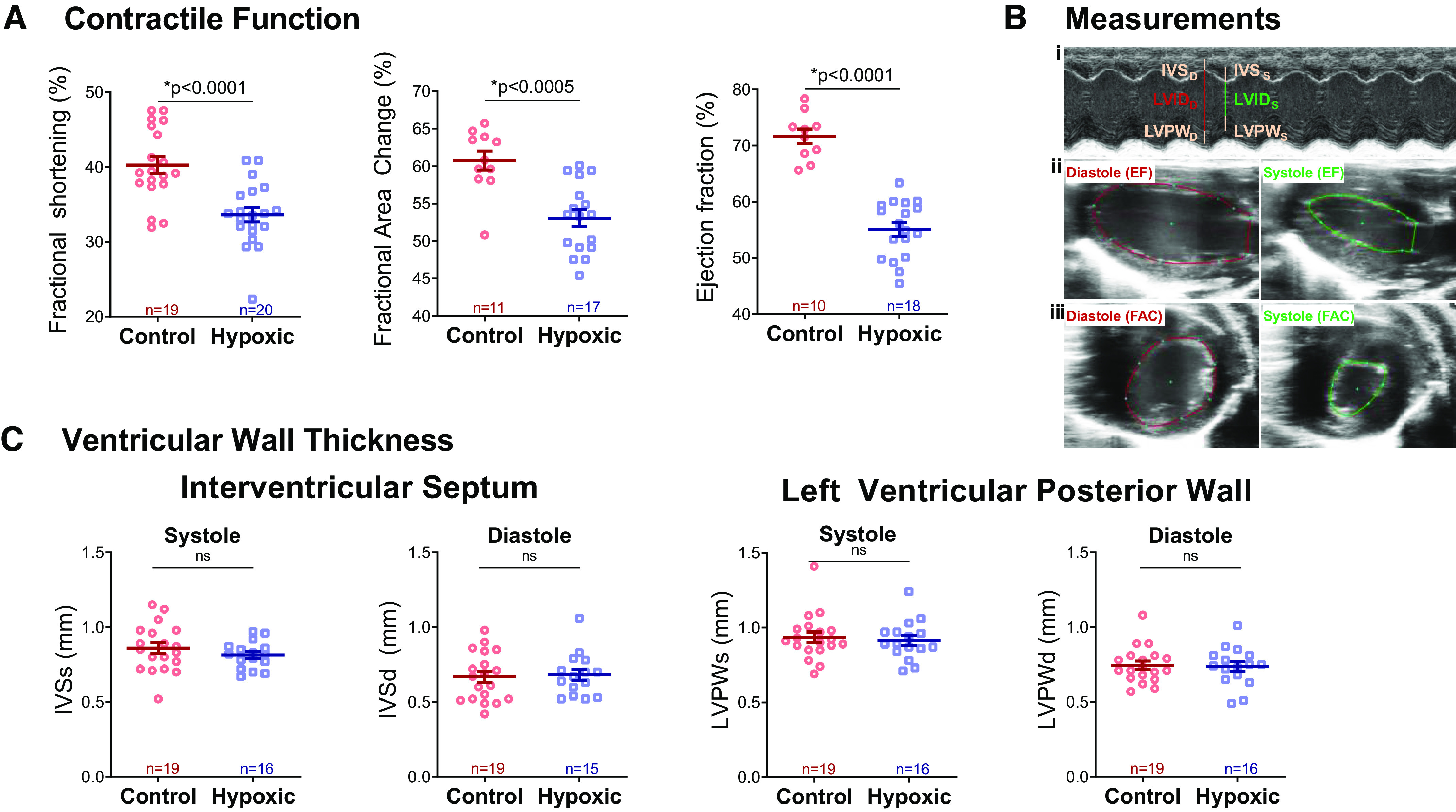
Contractile function. A: hypoxic animals had decreased contractile function at P30 compared with control, as measured by fractional shortening (FS), fractional area change (FAC), and ejection fraction (EF). B: left ventricular internal diameter (LVID), interventricular septum (IVS), and left ventricular posterior wall (LVPW) were measured in systole and diastole from short-axis M-mode images (i); long-axis traces of the endocardial border were performed in systole and diastole for calculation of EF via a modified Simpson’s monoplane method (ii); and short-axis traces of the endocardial border were performed in systole and diastole for calculation of fractional area change (iii). C: there was no difference between groups in either interventricular septal thickness or left ventricular posterior wall thickness in both systole (IVSs, LVPWs) and diastole (IVSd, LVPWd). Data expressed as means ± SE. *P < 0.05 via two-tailed t test. Biological replicates are shown. ns, not significant.
DISCUSSION
Our results support our hypothesis that cardiac maturation is perturbed in a mouse model of chronic perinatal hypoxia. Chronic perinatal hypoxia altered both the electrophysiologic substrate and the contractile apparatus. Although many of the differences detected at P8 normalized after recovering in normoxia, there were persistent alterations at P30 that may contribute to lifelong mortality and morbidity in the cyanotic CHD population.
Numerous genetic and phenotypic differences were detected at P8. Hypoxia altered global gene expression and ECG parameters, including bradycardia, slowed conduction, and decreased heart rate variability. In our animal model, P8 represents the time of surgical repair, and therefore, phenotypic differences in hypoxic animals may have implications for surgical outcomes and the immediate postsurgical course. Disturbances in ion channel expression may explain ECG disturbances at P8 and may predispose the hypoxic heart to arrhythmias. Specifically, reduced Ito current and an altered plateau phase can prolong the action potential, and reduced gap junctions can slow electrical conduction across the heart. Likewise, decreases in extracellular matrix collagen and alterations in the contractile apparatus have the potential to affect the strength of cardiac contraction. Electromechanical coupling development was especially delayed in hypoxic animals, as demonstrated by decreased L-type Ca2+ channel and ryanodine receptor expression, both of which can contribute to a blunted increase in cytosolic Ca2+ concentration and therefore weaker contraction. Increased dependence on glycolysis (Fig. 4) may reduce myocardial energy reserves, as observed in infants undergoing surgical repair (11–15). Furthermore, if myocardial growth continues by cell proliferation instead of hypertrophy, there is an increased risk of cell structure abnormalities. Notably, some differentially expressed genes in our study are associated with clinical sudden arrhythmic death syndromes (44) (long QT syndrome, Brugada syndrome, arrhythmogenic right ventricular dysplasia, catecholaminergic polymorphic ventricular tachycardia) and clinical cardiomyopathies (45) (dilated, hypertrophic, left ventricular noncompaction).
In our model, P30 represents recovery after early surgical repair of cyanotic CHD. Gene expression and in vivo ECG differences observed at P8 mostly resolved by P30, suggesting that the heart was able to complete development after recovery. This is an optimistic sign that many of the observed effects from chronic perinatal hypoxia may be reversible with early repair. Although, some persistent changes in contractility and electrophysiology were observed in older animals after recovery in normoxia. In vivo ECG recordings revealed slightly slower heart rates and longer QT intervals in P30 hypoxic animals, albeit to a lesser extent than at the P8 timepoint. Sinus node dysfunction and increased ventricular tissue refractoriness were also observed in ex vivo studies using programmed electrical stimulation. Notably, ventricular tissue refractoriness can diminish cardiomyocyte excitability and also lengthen repolarization time. Hypoxic P30 animals also had worse contractile function, which is in agreement with previous animal studies that observed systolic and diastolic dysfunction following either pre- or postnatal hypoxia alone (18, 46–48). Indeed, the literature suggests that prenatal hypoxia may imprint on a fetus and cause lifelong changes to the cardiovascular system, such as increased susceptibility to systemic hypertension and metabolic syndrome and worse response to myocardial infarction (49). Unfortunately, in the present study, we were not able to perform ex vivo electrophysiology or echocardiography studies at the earlier P8 timepoint due to small animal size (hypoxic: 3.3 g). We can only speculate that cardiac dysfunction was further exacerbated at this younger age and the persistent changes we observed at P30 were a significant improvement, which would be in line with our gene expression data between the two timepoints. Clearly, additional work is needed to fully address the underlying mechanisms that are responsible for the persistent phenotype observed at P30, which could be attributed to alterations in protein expression, localization, post-translational modifications, alterations in ion channel current, and/or myofilament architecture.
To date, there has been limited investigation into the effects of hypoxia on the developing heart, and thus there is no established animal model. The main embryological progression of heart development is the same between humans and rodents (50). The early embryonic heart is thin-walled and relies on diffusion of oxygen from the chambers until the coronaries connect to the aorta (50), at which point the myocardium starts to grow and thicken (24, 50). This timeline was the rationale for starting hypoxia on embryonic day 16, when the coronary development is complete (22, 51). This timing captures the period of rapid ventricular growth that occurs in both species once the myocardium is reliant on the coronary circulation for oxygen delivery. For both humans and rodents, there are similar changes in myocyte proteins for the remainder of gestation, and both demonstrate rapid and marked development in the first postnatal week, reaching definitive adult cell function and morphology quickly after the postnatal change in loading conditions and oxygenation (24, 25).
Our study replicated a finding seen in previous investigations of perinatal hypoxia: overall decreased size of animals with preserved heart size and increased heart-to-body weight ratios (16). In hypoxic conditions, the organism is able to maintain growth of the heart via preferential perfusion over other organ systems; however, chronic hypoxia still causes cardiac abnormalities at the cellular level. In one study, postnatal hypoxia from P1 to P7 caused increased heart-to-body weight ratios in mice, and cell size quantification revealed smaller and more numerous cardiomyocytes with preserved replication capabilities that might explain this finding (17). In human neonates with intrauterine growth restriction (IUGR), heart size is not only preserved, but in some studies found to be increased compared with neonates who were appropriate weight for gestational age. In one review of IUGR infants (52), the authors note that although the heart is larger relative to body size, the cardiomyocytes are altered on a cellular level; specifically suggesting that the observed developmentally delayed and smaller cardiomyocytes may be an adaptive response to limited availability of oxygen and nutrients.
Our study aimed to be a proof of concept that chronic perinatal hypoxia disrupts the process of normal cardiac development. To our knowledge, this is the first study to include both pre- and postnatal hypoxia to model the range of cardiac development affected by hypoxia in cyanotic CHD. Furthermore, we incorporated a period of recovery to simulate surgical repair of cyanotic CHD. Limitations of our model include the inherent constraints of using small animals to model human disease, and the risk of introducing maternal stress into gestation. Pup number and size were reduced in the hypoxic litters, which may indicate maternal stress. Furthermore, the maternal mice exposed to hypoxia weighed less than controls following the hypoxic period. The effects of fetal undernutrition are documented to include alterations in cardiovascular health over the lifetime, specifically inducing metabolic syndrome, hypertension, and ventricular hypertrophy. The majority of these effects, however, are documented to occur in adulthood, sparing juveniles (53, 54). Furthermore, in humans, these effects are most pronounced when maternal malnutrition occurs in early gestation (55). By comparison, our animal model introduced hypoxia for only the last 4 days of a 20-day gestation. Regardless, animal models of maternal malnutrition for the entirety of the second half of gestation have revealed alterations in cardiac tissue metabolism (56), thus it may be prudent to interpret results from the current study with caution regarding cardiac tissue metabolism.
One way to avoid the confounding effects of maternal stress in animal models of hypoxia is to use hypoxic incubation of chicken eggs. Results of such studies have revealed prenatal hypoxia to cause delays in calcium handling development and decreased ventricular systolic and diastolic function (18). The counterargument to using birds is that they are not mammals and results may be more difficult to generalize to humans. Overall, maternal stress and decreased availability of fetal nutrients are not counter to the physiology of human severe CHD. Indeed, maternal stress has emerged as an important predictor of poor outcomes in infants with CHD (57, 58). Although the presence of maternal stress and undernutrition may confound our specific investigation of the effects of prenatal hypoxia on the heart, it may be consistent with the overall physiology of the fetus with cyanotic CHD. Intrauterine growth restriction (IUGR) can result from CHD, placental insufficiency, and maternal malnutrition, among numerous other causes. IUGR of any etiology, not solely maternal malnutrition, is linked to an increased prevalence of adult cardiovascular disease (e.g., metabolic syndrome, hypertension, atherosclerosis) (59). Thus, induction of low birthweight in our animals may indicate consistency with the human condition of cyanotic CHD.
Another potential limitation of our study is that the comparative effect of degree of hypoxia between species is unknown. We chose 11% for the degree of hypoxia, as fractional inspired oxygen concentrations () of 9% and 12% correlate with pulse oximetry readings of 65% and 73%, respectively, in rodents (23). For comparison, human fetuses with cyanotic CHD have a mean oxygen saturation of 48% in the ascending aorta (7), and neonates have target oxygen saturation ranges of 70%–85%. Despite the unknowns between species, we believe this study is a first step toward understanding the impact of hypoxia on cardiac development.
The Cardiac Safety Research Consortium has implored the research community to perform more studies of developmental cardiac physiology to better understand the substrate on which cardiac therapies may work in the pediatric population (20). Further studies regarding the effects of hypoxia on cardiac development may allow us to better target cardiac therapeutics for the cyanotic CHD population. A better understanding of the effects of chronic perinatal hypoxia on cardiac development could lead to improved surgical outcomes and overall improved cardiovascular health in the cyanotic CHD population.
GRANTS
This work was supported by the National Institutes of Health Grants R01HL139472, R01HL139712, R01HL146670, and S10OD028619, Children’s National Heart Institute, and the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH2010199 (to N. Ishibashi). This publication was also supported by the Gloria and Steven Seelig family.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R., Z.D., N.I., and N.G.P. conceived and designed research; J.R., D.G., Z.D., C.M., M.R., L.S., N.V., M.R., and L.L. performed experiments; J.R., D.G., Z.D., C.M., L.L. and N.G.P. analyzed data; J.R., D.G., Z.D., L.L., and N.G.P. interpreted results of experiments; J.R., D.G., and N.G.P. prepared figures; J.R., D.G., and N.G.P. drafted manuscript; J.R., D.G., N.I., and N.G.P. edited and revised manuscript; J.R., D.G., Z.D., C.M., M.R., L.S., N.V., M.R., L.L., N.I., and N.G.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge Dr. Susan Knoblach, Karuna Panchapakesan, and the Children’s National Research Institute Genomics and Bioinformatics Core for assistance with microarray experiments and acknowledge Tomas Prudencio and Shreya Chandran for assistance with electrocardiogram recordings and mouse husbandry. We also acknowledge Dr. Norman Lee for assistance with gene expression and heatmap analysis and Dr. Christopher Spurney and Dr. Charles Berul for helpful discussions.
REFERENCES
- 1.Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr 153: 807–813, 2008. doi: 10.1016/j.jpeds.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oster ME, Lee KA, Honein MA, Riehle-Colarusso T, Shin M, Correa A. Temporal trends in survival among infants with critical congenital heart defects HHS public access. Pediatrics 131: e1502–e1508, 2013. doi: 10.1542/peds.2012-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinton RB, Ware SM. Heart failure in pediatric patients with congenital heart disease. Circ Res 120: 978–994, 2017. doi: 10.1161/CIRCRESAHA.116.308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinardo JA. Heart failure associated with adult congenital heart disease. Semin Cardiothorac Vasc Anesth 17: 44–54, 2013. doi: 10.1177/1089253212469841. [DOI] [PubMed] [Google Scholar]
- 5.Rudolph AM. Congenital Diseases of the Heart: Clinical-Physiological Considerations (3rd ed.). Oxford, UK: Wiley-Blackwell, 2009. doi: 10.1002/9781444311822. [DOI] [Google Scholar]
- 6.Yagel S, Silverman NH, Gembruch U (Editors). Fetal Cardiology: Embryology, Genetics, Physiology, Echocardiographic Evaluation, Diagnosis, and Perinatal Management of Cardiac Diseases (3rd ed.). Boca Raton: CRC Press, 2019. [Google Scholar]
- 7.Sun L, Macgowan CK, Sled JG, Yoo S-J, Manlhiot C, Porayette P, Grosse-Wortmann L, Jaeggi E, McCrindle BW, Kingdom J, Hickey E, Miller S, Seed M. Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation 131: 1313–1323, 2015. doi: 10.1161/CIRCULATIONAHA.114.013051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudolph AM. Congenital cardiovascular malformations and the fetal circulation. Arch Dis Child Fetal Neonatal Ed 95: 132–136, 2010. doi: 10.1136/adc.2007.128777. [DOI] [PubMed] [Google Scholar]
- 9.Lauridsen MH, Uldbjerg N, Henriksen TB, Petersen OB, Stausbøl-Grøn B, Matthiesen NB, Peters DA, Ringgaard S, Hjortdal VE. Cerebral oxygenation measurements by magnetic resonance imaging in fetuses with and without heart defects. Circ Cardiovasc Imaging 10: e006459, 2017. doi: 10.1161/CIRCIMAGING.117.006459. [DOI] [PubMed] [Google Scholar]
- 10.Sun L, Marini D, Saini B, Schrauben E, MacGowan CK, Seed M. Understanding fetal hemodynamics using cardiovascular magnetic resonance imaging. Fetal Diagn Ther 47: 354–362, 2020. doi: 10.1159/000505091. [DOI] [PubMed] [Google Scholar]
- 11.Teoh KH, Mickle DA, Weisel RD, Li RK, Tumiati LC, Coles JG, Williams WG. Effect of oxygen tension and cardiovascular operations on the myocardial antioxidant enzyme activities in patients with tetralogy of Fallot and aorta-coronary bypass. J Thorac Cardiovasc Surg 104: 159–164, 1992. [PubMed] [Google Scholar]
- 12.Modi P, Suleiman M-S, Reeves BC, Pawade A, Parry AJ, Angelini GD, Caputo M. Basal metabolic state of hearts of patients with congenital heart disease: the effects of cyanosis, age, and pathology. Ann Thorac Surg 78: 1710–1716, 2004. doi: 10.1016/j.athoracsur.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Imura H, Caputo M, Parry A, Frcs A, Pawade Frcs GD, Angelini Frcs M-S, Suleiman P. Age-dependent and hypoxia-related differences in myocardial protection during pediatric open heart surgery improved surgical outcome after fetal diagnosis of hypoplastic left heart syndrome. Circulation 103: 1551–1556, 2001. doi: 10.1161/01.cir.103.11.1551. [DOI] [PubMed] [Google Scholar]
- 14.Ercan S, Cakmak A, Kösecik M, Erel O. The oxidative state of children with cyanotic and acyanotic congenital heart disease. Anadolu Kardiyol Derg 9: 486–490, 2009. [PubMed] [Google Scholar]
- 15.Najm HK, Wallen WJ, Belanger MP, Williams WG, Coles JG, Van Arsdell GS, Black MD, Boutin C, Wittnich C. Does the degree of cyanos1is affect myocardial adenosine triphosphate levels and function in children undergoing surgical procedures for congenital heart disease? J Thorac Cardiovasc Surg 119: 515–524, 2000. doi: 10.1016/S0022-5223(00)70131-0. [DOI] [PubMed] [Google Scholar]
- 16.Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med 10: 653–666, 2010. doi: 10.2174/156652410792630643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157: 565–579, 2014. [Erratum in Cell 157: 1243, 2014]. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonker SS, Giraud GD, Espinoza HM, Davis EN, Crossley DA. Effects of chronic hypoxia on cardiac function measured by pressure-volume catheter in fetal chickens. Am J Physiol Regul Integr Comp Physiol 308: R680–R689, 2015. doi: 10.1152/ajpregu.00484.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohlmeyer TJ, Helmke S, Ge S, Lynch J, Brodsky G, Sederberg JH, Robertson AD, Minobe W, Bristow MR, Perryman MB. Hypoplastic left heart syndrome myocytes are differentiated but possess a unique phenotype. Cardiovasc Pathol 12: 23–31, 2003. doi: 10.1016/S1054-8807(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 20.Bates KE, Vetter VL, Li JS, Cummins S, Aguel F, Almond C, Dubin AM, Elia J, Finkle J, Hausner EA, Joseph F, Karkowsky AM, Killeen M, Lemacks J, Mathis L, McMahon AW, Pinnow E, Rodriguez I, Stockbridge NL, Stockwell M, Tassinari M, Krucoff MW. Pediatric cardiovascular safety: challenges in drug and device development and clinical application. Am Heart J 164: 481–492, 2012. doi: 10.1016/j.ahj.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Olivey HE, Compton LA, Barnett JV. Coronary vessel development: the epicardium delivers. Trends Cardiovasc Med 14: 247–251, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Theveniau-Ruissy M, Perez-Pomares J-M, Parisot P, Baldini A, Miquerol L, Kelly RG. Coronary stem development in wild-type and Tbx1 null mouse hearts. Dev Dyn 245: 445–459, 2016. doi: 10.1002/dvdy.24380. [DOI] [PubMed] [Google Scholar]
- 23.Morgan BJ, Adrian R, Bates ML, Dopp JM, Dempsey JA. Quantifying hypoxia-induced chemoreceptor sensitivity in the awake rodent. J Appl Physiol (1985) 117: 816–824, 2014. doi: 10.1152/japplphysiol.00484.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porter GA, Hom JR, Hoffman DL, Quintanilla RA, Bentley KDM, Sheu SS. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog Pediatr Cardiol 31: 75–81, 2011. doi: 10.1016/j.ppedcard.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scuderi GJ, Butcher J. Naturally engineered maturation of cardiomyocytes. Front Cell Dev Biol 5: 50, 2017. doi: 10.3389/fcell.2017.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48, 2009. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3: e39, 2007. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.TaskForce of the European Society of Electrophysiology . Heart rate variability: standards of measurement, physiological interpretation, and clinical use. Circulation 93: 1043–1065, 1996. doi: 10.1161/01.CIR.93.5.1043. [DOI] [PubMed] [Google Scholar]
- 29.Swift LM, Burke M, Guerrelli D, Reilly M, Ramadan M, McCullough D, Prudencio T, Mulvany C, Chaluvadi A, Jaimes R, Posnack NG. Age-dependent changes in electrophysiology and calcium handling: implications for pediatric cardiac research. Am J Physiol Circ Physiol 318: H354–H365, 2020. doi: 10.1152/ajpheart.00521.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaimes R, McCullough D, Siegel B. Plasticizer interaction with the heart: chemicals used in plastic medical devices can interfere with cardiac electrophysiology. Circ Arrhythmia Electrophysiol 12: e007294, 2019. doi: 10.1161/CIRCEP.119.007294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moshtagh N. Plot an ellipse in “center form,” 2021. https://www.mathworks.com/matlabcentral/fileexchange/13844-plot-an-ellipse-in-center-form, MATLAB Central File Exchange. Retrieved March 15, 2021. [Google Scholar]
- 32.Li A. Approximate Lowner Ellipsoid, 2021. https://www.mathworks.com/matlabcentral/fileexchange/21930-approximate-lowner-ellipsoid, MATLAB Central File Exchange. Retrieved March 15, 2021. [Google Scholar]
- 33.Posnack NGG, Lee NHH, Brown R, Sarvazyan N. Gene expression profiling of DEHP-treated cardiomyocytes reveals potential causes of phthalate arrhythmogenicity. Toxicology 279: 54–64, 2011. doi: 10.1016/j.tox.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandt MC, Priebe L, Böhle T, Südkamp M, Beuckelmann DJ. The ultrarapid and the transient outward K+ current in human atrial fibrillation. Their possible role in postoperative atrial fibrillation. J Mol Cell Cardiol 32: 1885–1896, 2000. doi: 10.1006/jmcc.2000.1221. [DOI] [PubMed] [Google Scholar]
- 35.Beuckelmann DJ, Näbauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73: 379–385, 1993. doi: 10.1161/01.RES.73.2.379. [DOI] [PubMed] [Google Scholar]
- 36.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol 48: 12–25, 2010. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewinter MM, Granzier H. Cardiac titin—a multifunctional giant. Circulation 121: 2137–2145, 2010. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato K, WaKai J, ozaWa K, SeKiguchi M, Katahira K. Different sensitivity to the suppressive effects of isoflurane anesthesia on cardiorespiratory function in SHR/Izm, WKY/Izm, and Crl:CD (SD) rats. Exp Anim 65: 393–402, 2016. doi: 10.1538/expanim.16-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raatikainen PMJ, Morey TE, Druzgala P, Milner P, Gonzalez MD, Dennis DM. Effects of volatile anesthetics on atrial and AV nodal electrophysiological properties in guinea pig isolated perfused heart. Anesthesiology 89: 434–442, 1998. doi: 10.1097/00000542-199808000-00020. [DOI] [PubMed] [Google Scholar]
- 40.Mamiya K, Aono J, Manabe M. Complete atrioventricular block during anesthesia. Can J Anaesth 46: 265–267, 1999. doi: 10.1007/BF03012607. [DOI] [PubMed] [Google Scholar]
- 41.Kamatani T, Akizuki A, Kondo S, Shirota T. Second-degree atrioventricular block occurring after tooth extraction. Anesth Prog 63: 156–159, 2016. doi: 10.2344/15-00042.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson SK, Patel AR, Chang PM. Bradyarrhythmias in congenital heart disease. Card Electrophysion Clin 9: 177–187, 2017. doi: 10.1016/j.ccep.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Triedman JK. Arrhythmias in adults with congenital heart disease. Heart 87: 383–389, 2002. doi: 10.1136/heart.87.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fulgent Genetics. Comprehensive Arrhythmia NGS Panel test information website. Accessed March 15, 2021. https://www.fulgentgenetics.com/comprehensive-arrhythmia. [Google Scholar]
- 45.Fulgent Genetics. Comprehensive Cardiomyopathy NGS Panel test information website. Accessed March 15, 2021. https://www.fulgentgenetics.com/comprehensive-cardiomyopathy. [Google Scholar]
- 46.Thompson LP, Chen L, Polster BM, Pinkas G, Song H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am J Physiol Regul Integr Comp Physiol 315: R1232–R1241, 2018. doi: 10.1152/ajpregu.00224.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindgren I, Altimiras J. Prenatal hypoxia programs changes in-adrenergic signaling and postnatal cardiac contractile dysfunction. Am J Physiol Regul Integr Comp Physiol 305: 1093–1101, 2013. doi: 10.1152/ajpregu.00320.2013. [DOI] [PubMed] [Google Scholar]
- 48.Corno AF, Giuseppina Milano F, Samaja M, Tozzi P, von Segesser LK. Chronic hypoxia: a model for cyanotic congenital heart defects Corno. Surgery for congenital heart disease, J Thorac Cardiovasc Surg 124: 105, 2002. doi: 10.1067/mtc.2002.121302. [DOI] [PubMed] [Google Scholar]
- 49.Giussani DA, Davidge ST. Developmental programming of cardiovascular disease by prenatal hypoxia. J Dev Orig Health Dis 4: 328–337, 2013. doi: 10.1017/S204017441300010X. [DOI] [PubMed] [Google Scholar]
- 50.Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics 15: 165–176, 2003. doi: 10.1152/physiolgenomics.00033.2003. [DOI] [PubMed] [Google Scholar]
- 51.Savolainen SM, Foley JF, Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol 37: 395–414, 2009. doi: 10.1177/0192623309335060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen E, Wong FY, Horne RSC, Yiallourou SR. Intrauterine growth restriction: Impact on cardiovascular development and function throughout infancy. Pediatr Res 79: 821–830, 2016. doi: 10.1038/pr.2016.24. [DOI] [PubMed] [Google Scholar]
- 53.Remacle C, Bieswal F, Bol V, Reusens B. Developmental programming of adult obesity and cardiovascular disease in rodents by maternal nutrition imbalance. Am J Clin Nutr 94: 1846S–1852S, 2011. doi: 10.3945/ajcn.110.001651. [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez-Rodríguez P, López de Pablo AL, García-Prieto CF, Somoza B, Quintana-Villamandos B, Gómez de Diego JJ, Gutierrez-Arzapalo PY, Ramiro- Cortijo D, Carmen-Gonzalez M, Arribas SM. Long term effects of fetal undernutrition on rat heart. Role of hypertension and oxidative stress. PLoS One 12: e0171544, 2017. doi: 10.1371/journal.pone.0171544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, van Montfrans GA, Michels RP, Bleker OP. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–45. Heart 84: 595–598, 2000. doi: 10.1136/heart.84.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beauchamp B, Thrush AB, Quizi J, Antoun G, MacIntosh N, Al-Dirbashi OY, Patti ME, Harper ME. Undernutrition during pregnancy in mice leads to dysfunctional cardiac muscle respiration in adult offspring. Biosci Rep 35: 1–10, 2015. doi: 10.1042/BSR20150007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steurer MA, Peyvandi S, Baer RJ, Oltman SP, Chambers CD, Norton ME, Ryckman KK, Moon-Grady AJ, Keller RL, Shiboski SC, Jelliffe-Pawlowski LL. Impaired fetal environment and gestational age: what is driving mortality in neonates with critical congenital heart disease? J Am Heart Assoc 8: e013194, 2019. doi: 10.1161/JAHA.119.013194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaynor JW, Parry S, Moldenhauer JS, Simmons RA, Rychik J, Ittenbach RF, Russell WW, Zullo E, Ward JL, Nicolson SC, Spray TL, Johnson MP. The impact of the maternal-foetal environment on outcomes of surgery for congenital heart disease in neonates. Cardiothorac Surg 54: 348–353, 2018. doi: 10.1093/ejcts/ezy015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma D, Shastri S, Sharma P. Intrauterine growth restriction: antenatal and postnatal aspects. Clin Med Insights Pediatr 10: CMPed.S40070, 2016. doi: 10.4137/CMPed.S40070. [DOI] [PMC free article] [PubMed] [Google Scholar]