

Abstract
Environmental stress is increasing worldwide, yet we lack a clear picture of how stress disrupts the stability of microbial communities and the ecosystem services they provide. Here, we present the first evidence that naturally-occurring microbiomes display network properties characteristic of unstable communities when under persistent stress. By assessing changes in diversity and structure of soil microbiomes along 40 replicate stress gradients (elevation/water availability gradients) in the Florida scrub ecosystem, we show that: (1) prokaryotic and fungal diversity decline in high stress, and (2) two network properties of stable microbial communities—modularity and negative:positive cohesion—have a clear negative relationship with environmental stress, explaining 51–78% of their variation. Interestingly, pathogenic taxa/functional guilds decreased in relative abundance along the stress gradient, while oligotrophs and mutualists increased, suggesting that the shift in negative:positive cohesion could result from decreasing negative:positive biotic interactions consistent with the predictions of the Stress Gradient Hypothesis. Given the crucial role microbiomes play in ecosystem functions, our results suggest that, by limiting the compartmentalization of microbial associations and creating communities dominated by positive associations, increasing stress in the Anthropocene could destabilize microbiomes and undermine their ecosystem services.
Subject terms: Microbial ecology, Microbial ecology
Introduction
Microbial communities play critical roles in ecosystem functioning (e.g., nitrogen and carbon cycling) and in the persistence and health of many plant and animal species important to restoration, management, and agronomy [1–3]. However, as human activity in the Anthropocene continues to disrupt natural environments and microbial processes through increased environmental stress (e.g., higher temperatures [4]), there remains an urgent need to understand the factors that undergird microbiome stability in order to mitigate potential changes to ecosystem services. Previous work has shown that stress affects microbiome composition [5–8], microbe–microbe interactions [9, 10], and microbe–host interactions [11, 12]. However, the link between environmental stress and the stability of these microbial systems is less clear.
One promising avenue for understanding how stress affects microbial community stability is network analysis. When applied to ecology, networks are mathematical representations of communities in which nodes represent individual taxa and edges represent observed correlations in abundances among taxa from which interactions may be inferred [13, 14]. Network properties (e.g., modularity, sparsity, etc.) have been used to successfully predict the stability of networks of macroorganisms such as plant-pollinator networks [15] and food webs [16], and have recently been applied to microbiomes [6, 17]. In particular, communities with certain network characteristics—greater modularity, reduced positive associations among taxa, and greater negative associations among taxa—are more stable, meaning these communities: (1) have more limited shifts in composition in response to environmental perturbations and/or (2) are more likely to return to their equilibrium composition after a perturbation [17–19].
The first of these properties, modularity, quantifies how strongly taxa are compartmentalized into groups of interacting/co-occurring taxa (i.e., a module). Modularity can reflect biological processes such as shared ecological functions among taxa in a module [20–22], spatial compartmentalization [23], or similar habitat preferences [24, 25], and can impact community stability. For instance, high modularity in food webs stabilizes communities by restricting the impact of losing a taxon to its own module, thereby preventing the effects of that taxon’s extinction from propagating to affect the rest of the network [16].
The second property that can predict stability in co-occurrence networks is the ratio of negative to positive associations between taxa. Positive relationships represent high niche overlap and/or positive interactions between taxa, while negative relationships indicate divergent niches and/or negative interactions. The relative fraction of negative:positive associations can impact community stability [17, 18, 26, 27]. For example, positive interactions can destabilize microbial communities by creating positive feedback loops between taxa supporting each others’ fitness; thus, when one member in the positive feedback loop decreases in abundance it will negatively impact the fitness of other taxa reliant on the feedback loop’s services [18].
Here, we combine network theory and high throughput sequencing across 270 soil microbial communities from 40 replicate stress gradients to provide the first investigation of how the stability of microbial networks changes with stress. To do this, we characterized soil microbial communities from the Florida scrub, an ecosystem whose topography generates a series of naturally replicated stress gradients of decreasing nutrient and water availability [11, 28] within just a few square kilometers of area and a few meters of elevation gain [29]. We then assessed prokaryotic and fungal community properties and network structure across these replicate stress gradients to: (1) relate environmental stress and microbial diversity and (2) relate environmental stress and network stability using modularity and network cohesion (i.e., the strength and number of positive and negative associations). To gain insight into whether changes in network properties could be attributed to changes in species interactions, we (3) examined how the abundance of facilitative, stress tolerant, and pathogenic taxa change along the stress gradient.
Methods
Study system and field soil surveys
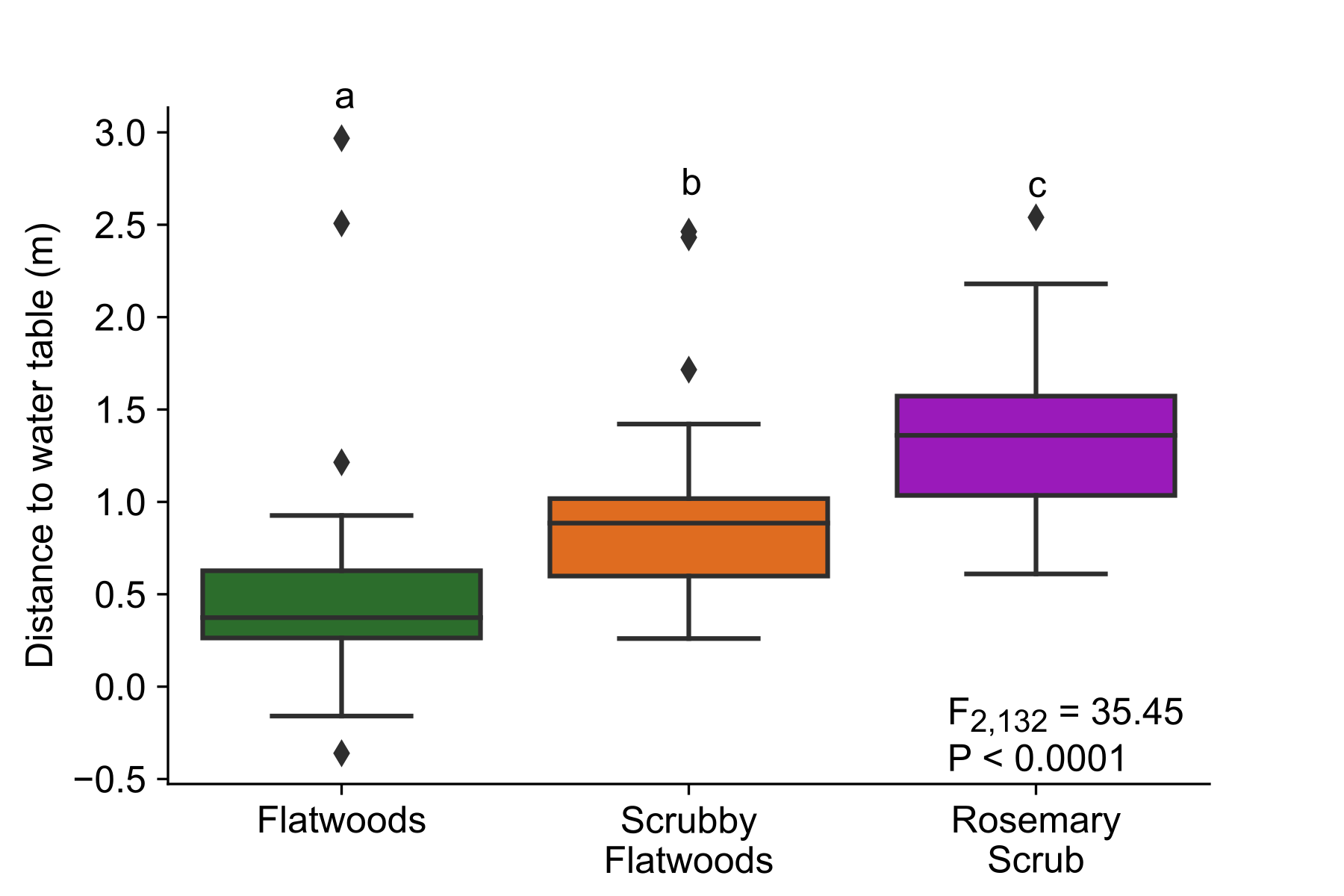
We conducted this study at Archbold Biological Station (27°11′ N, 81°21′ W) in the imperiled and endemic Florida Scrub (~15% remaining). This ecosystem, one of the oldest in Florida, is located along the Lake Wales Ridge in central Florida (Fig. 1). We focused on soil microbiomes from three scrub habitats (flatwoods, scrubby flatwoods, and rosemary scrub) that occur at increasing elevation relative to the water table (Supplementary Fig. 1; F2,132 = 35.45, P < 0.0001), and experience corresponding decreases in soil moisture [30] resulting in increasing stress [29, 31, 32]. The stress gradient associated with elevation is a strong determinant of plant community assemblages in this system [29], and recent work has shown that facilitative microbial effects on plant population dynamics increase in higher stress, higher elevation sites in the Florida Scrub [11]. Flatwoods occupy the lowest elevation sites and have the highest vegetation density followed by scrubby flatwoods (intermediate elevation and vegetation density) and rosemary scrub (highest elevation and lowest vegetation density). Rosemary scrub occurs as isolated patches across the landscape surrounded by scrubby flatwoods and flatwoods, providing naturally replicated stress gradients to test the relationship between stress and network structure.
Fig. 1. Schematic of sampling sites and stress gradients.

(Left) Map of replicate stress gradient sampling sites at Archbold Biological Station, Venus, FL, USA (scale bar, 500 m). Dots indicate locations of the 40 replicate stress gradients from which soil samples were collected in the six environments (2 soil depths × 3 habitat types), and triangles represent 24 additional rosemary scrub sites that were sampled. (Right) Diagram of environmental gradient from low stress (flatwoods) to intermediate stress (scrubby flatwoods) to high stress (rosemary scrub). Images are examples of each habitat and have colored dots that represent habitat IDs used in the rest of this work (Photo Credits: D. Revillini, B. Almeida).
In July 2017, soil samples were collected from the three habitat types. We sampled from 71 of the 106 mapped rosemary scrub patches at Archbold Biological Station, as well as from the neighboring scrubby flatwoods and flatwoods for each of the 71 replicate focal locations (Fig. 1). Sampling points for each soil core were randomly selected within the target habitats using ArcGIS (Version 10.5.1). Cores were divided into two depths: crust (0–2.5 cm depth) and subterranean (9–11.5 cm depth) for a total of 426 samples (71 locations × 3 habitat types × 2 soil depths). Each sample was the aggregation of 3 cores within ~2 m of the sampling point. Samples were flash frozen in 50 mL conical tubes in the field using liquid nitrogen and stored at −80 °C.
Microbiome DNA extraction and sequencing
Total genomic DNA was extracted from 1 g soil samples using the E.Z.N.A. Soil DNA Kit (OMEGA Bio-Tek, Norcross, GA, USA) following manufacturer’s instructions with minor modifications. To increase the amount of DNA obtained from sandy soils with few microbes, homogenization was performed in 15 mL conical vials instead of microcentrifuge tubes, then the supernatant was transferred to microcentrifuge tubes for heat incubation and alkaline lysis. We subsampled available extractions, selecting 40 sextuplet (3 habitat type × 2 soil depth) sample combinations that passed our initial quality control checks using endpoint PCR and gel electrophoresis. Because of our previous work showing strong interactions between the soil microbiome and endangered rosemary scrub plants [11, 33, 34], we sequenced additional samples from the rosemary scrub (24 crust and six subterranean samples). We also sequenced six negative controls in which 1 mL of Ultrapure Water (G-Biosciences; 786-293) was used in place of soil during DNA extractions to test for potential contamination during the extraction process. In total, 276 samples were sequenced (270 soil extractions and six negative controls). Fungal and prokaryotic genomic libraries were prepared and sequenced at the University of Minnesota Genomics Center (UMGC) following the two-step dual-indexing approach in [35]. 16S_V4 and ITS1 amplicons (Earth Microbiome Project primers [36–39]) were sequenced on two Illumina MiSeq lanes (v3, 300 bp paired end) at UMGC to characterize prokaryotic and fungal communities, respectively.
Bioinformatic processing
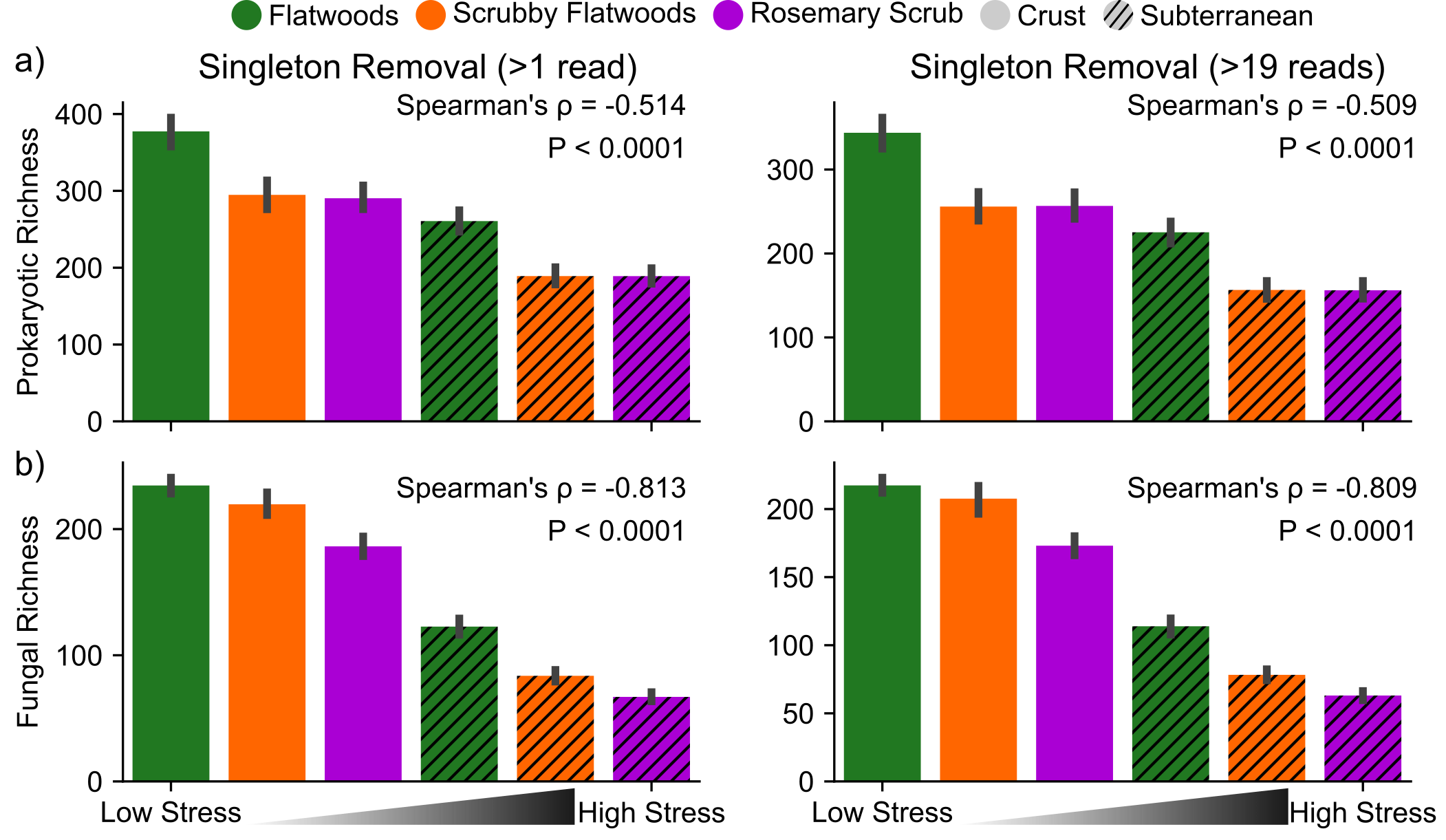
Reads were demultiplexed using bcl2fastq (Illumina). We merged reads from both lanes to counterpart fastq files, built directory structures, and created paired-end manifest files using Supplementary Files 1–4. We processed sequencing data with associated metadata (Supplementary Table 1) through QIIME2 (v2018.8; Supplementary File 5; ref. [40]) to remove sequencing adapters and chimeras, join paired-end reads, classify operational taxonomic units (OTUs), and calculate diversity metrics. OTUs, which were Exact Sequence Variants after denoising with Dada2 in QIIME2, were grouped into “species” based on 97% similarity to references in the Greengenes (v13_8; [41, 42]) and UNITE (v7_01.12.2017; [43]) databases for prokaryotic and fungal classification, respectively. Higher levels of taxonomic organization such as order and phylum were attached to species assignments. All six negative controls had <40 (20.9 ± 0.9) reads after denoising, which is consistent with a lack of contamination in our samples. Samples were rarefied to 2000 reads to calculate OTU richness, diversity (Shannon index), and evenness (Pielou’s index). Samples with <2000 reads (10 of 270 prokaryotic and 9 of 270 fungal samples) were excluded from analyses because their rarefactions curves did not reach saturation. Singletons were not removed, unless otherwise indicated, as their removal (OTUs with <2 or <20 reads) did not affect alpha diversity differences among habitats (Supplementary Fig. 2). We used combined relative abundances (i.e., taxon read count/total read counts in sample) of prokaryotes and fungi to construct co-occurrence networks [44, 45] and calculating cohesion as suggested by Herren and McMahon [17].
Microbial abundance and diversity analyses
We quantified the environmental stress experienced by microbial communities by measuring prokaryotic abundance (natural log of the number of genomic target sites for 16S_V4 primers per microliter in a sample) using a qPCR dual-indexing approach [35]. We used prokaryotic abundance as a proxy for microbial stress as it is analogous to using plant biomass to estimate stress in plant communities [46–48]. To quantify relative levels of stress between different soil depth × habitat combinations, we first performed an analysis of deviance in which a mixed model factorial ANOVA (fixed effects: habitat, depth, habitat × depth; random effect: replicate focal location) was compared to a null model containing only the random effect followed by a Tukey’s HSD test. The resulting stress gradient from least (1) to most stressful (6) environment was: flatwoods crust < scrubby flatwoods crust < rosemary crust < flatwoods subterranean < scrubby flatwoods subterranean < rosemary subterranean. We then regressed prokaryotic abundance against the stress ranking using a Spearman’s Correlation test to confirm that prokaryotic abundance decreases (i.e., stress increases) along this ranked gradient. After this confirmation, we used Spearman’s Correlations to test how richness, diversity, and evenness varied along this stress gradient.
Microbiome network construction
In order to understand how the structure of microbial communities changed along the stress gradient, we constructed co-occurrence networks using SparCC (Version 2016_10_17; ref. [49]) with OTUs present in ≥10% of all samples [17] and default parameters. Significance of correlations between taxa abundances was calculated using 1000 permutations. SparCC was used in place of shuffled Pearson correlation coefficients because it accounts for sparse, compositional data. We constructed six networks (one for each habitat × depth combination) rather than one large network. Building individual co-occurrence networks enabled comparisons of network structure, like modularity, across the stress gradient, and minimized the effects of broad habitat preferences for one of the six habitat × depth combinations (e.g., microbes forming a flatwoods crust module). This also allowed for comparing how co-occurrences between taxa change along the stress gradient. For example, if pairs of taxa switch from negative to positive associations when transitioning from low to high-stress environments, this relationship would not be visible in a network in which communities from all stress levels are combined. Essentially, strong negative and positive associations at low and high-stress extremes, respectively, would cancel each other out (i.e., taxa would have weak correlations), which would be indistinguishable from weak associations between taxa that remain constant along the gradient.
To provide insight into the role of stress in microbial networks, we then quantified two network properties that have been associated with stability of ecological communities in perturbation studies: (1) the number and strength of positive correlations, and (2) how compartmentalized the network is, via cohesion and modularity analyses, respectively [6, 17, 18].
Modularity
To understand how modularity changed across the stress gradient, we first identified modules (groups of taxa whose abundances are more correlated/anti-correlated with each other than the rest of the community) using the Clauset–Newman–Moore algorithm (greedy_modularity_communities from the networkx Python package; 50). We then calculated modularity, a measure of whether connections tend to occur within or between modules, using the quality function in the networkx Python package. The Clauset–Newman–Moore algorithm identifies modules by starting with each node in its own “module” and sequentially joining pairs of these modules that increase the modularity metric (Q) the most until additional pairing no longer increases modularity [50]. Modularity (Q) is calculated using the following equation from [51]:
Where m is the number of significant pairwise correlations between taxa abundances in the network, Aij is 1 if OTUs i and j are connected and 0 if they are not, ki and kj are the number of taxa that have significant correlations with taxa i and j, respectively, and δ(ci, cj) equals 1 if i and j are in the same module and zero if they are not. The component of modularity calculates the probability that a connection exists between i and j given how well-connected i and j are to the rest of the network (i.e., Aij is the observed value and is the expected value). This probability is used to weight connections between nodes within the same module (i.e., when δ(ci, cj) is equal to 1). Large, positive modularity values (i.e., close to 1) indicate that more connections occur within, rather than between, modules compared to random chance [52]. Communities with high modularity tend to be more stable because effects from changes in abundance of one species are more strongly limited to that species’ module [16]. We calculated one value of modularity for each environment’s network for a total of six modularity values. To determine if stress can explain variation in modularity, we used a Spearman’s Correlation to test the relationship between modularity and the stress rankings of our six environments.
Cohesion
By characterizing positive and negative co-occurrences separately, cohesion provides insights into associations among taxa caused by both positive and negative species interactions and/or by both similarity and differences in the niches of microbial taxa [17]. Two cohesion values (positive and negative) are calculated for each sample (j) as the sum of the significant positive or negative correlations between taxa weighted by taxa abundances:
and
where ai is the abundance of taxon i in sample j and and are positive and negative connectedness, respectively [17]. Within a given network (i.e., one of the six habitat × depth combinations), the positive () and negative connectedness () for a given taxon i were calculated as the average of all its significant positive or negative correlations with all other microbial taxa found in the network. Negative and positive cohesion range from −1 to 0 and 0 to 1, respectively, with higher absolute values signifying more and/or stronger correlations. Communities with larger negative cohesion values tend to be more stable, as their composition is less variable over time (i.e., have lower beta diversity between sampling times; 17).
We used the two cohesion values to evaluate two metrics of the microbial networks. First, we calculated the proportion of negative to positive co-occurrences as the absolute value of negative:positive cohesion, which allowed us to determine if more stressful environments are better characterized by processes driving negative associations, which might include competition and niche divergence, than less stressful environments. We then tested the relationship between stress and negative:positive cohesion using a Spearman’s Correlation test with the stress rankings of our six environments as the explanatory variable. Second, we tested the relationship between stress and total cohesion—a metric of network complexity calculated as the sum of positive cohesion and absolute value of negative cohesion—using a Spearman’s Correlation test with the six environmental stress rankings.
Next, to determine which cohesion value (positive or negative) was more sensitive to the stress gradient, we regressed both positive cohesion and the absolute value of negative cohesion against the six ranked environments (Spearman’s Correlation test) and compared these two correlations to each other using the paired.r function (a Fisher z-transformation followed by a z-test) in the R package psych. We also calculated the slopes of the relationships of positive and negative cohesion with stress using a nonparametric Siegel regression (mblm function in the mblm R package).
Microbial composition and function analyses
To gain additional insight into how the microbial community changed across the stress gradient, we further assessed abundances of different prokaryotic and fungal taxa as well as fungal functional guilds across the stress gradient. Counts for OTUs were normalized separately for prokaryotic and fungal communities by dividing by the total number of counts within a sample. We tested if taxa abundances were affected by the stress gradient by correlating abundance with the stress rankings of our six habitats using Spearman’s Correlation tests and correcting for multiple comparisons (Benjamini–Hochberg). We also assigned fungi to functional guilds by associating “species”, as identified with 97% similarity to representative sequences in the UNITE fungal database, with guilds in the FunGuild database [53] and filtering the OTU abundance tables for guild assignments with confidence rankings “Highly Probable” and “Probable” [54, 55]. FunGuild associates taxonomic groups to functional guilds such as “wood saprotroph”, “plant pathogen”, and “ectomycorrhizal fungi”, according to an online community annotated database. Using the same procedure as for taxonomic groups, we then tested if fungal functional guild abundances were affected by the stress gradient.
Results
Quantification of the stress gradient
The qPCR data measuring prokaryotic abundance established a clear stress gradient across all six habitat/depth combinations in which stress was inversely proportional to prokaryotic abundance (Fig. 2). Subterranean soils contained lower estimated prokaryotic abundance than crust soils (Depth: F1,187 = 269.4, P < 0.0001) and prokaryotic abundance decreased across our habitat stress gradient from flatwoods to scrubby flatwoods to rosemary scrub (Habitat: F2,187 = 58.2, P < 0.0001). Expanding on a priori knowledge of the study system’s gradient of habitat types, our data supported the interpretation of relative levels of stress across this ecosystem as: flatwoods crust < scrubby flatwoods crust < rosemary crust < flatwoods subterranean < scrubby flatwoods subterranean < rosemary subterranean. This sequence of habitat and depth combinations is strongly correlated with prokaryotic abundance (Fig. 2, Spearman’s ⍴ = −0.748, P < 0.0001). Thus, hereafter, we discuss the stress gradient following this sequence of six environments (assigned values 1–6).
Fig. 2. Quantifying microbial stress.
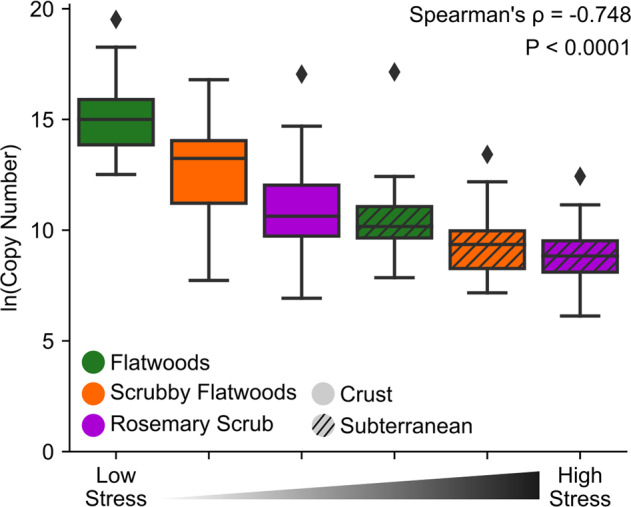
Estimation of microbial stress by prokaryotic abundance in different habitat/depth combinations. Graph depicts natural log of the number of 16S_V4 primer target sites (i.e., number of prokaryotes) in soil DNA extractions. X-axis represents the categorical stress rankings of the six environments along the stress gradient. P value is derived from a Spearman’s Correlation test. Detailed statistics are available in Supplementary Table 3.
Microbiome diversity and network properties across the stress gradient
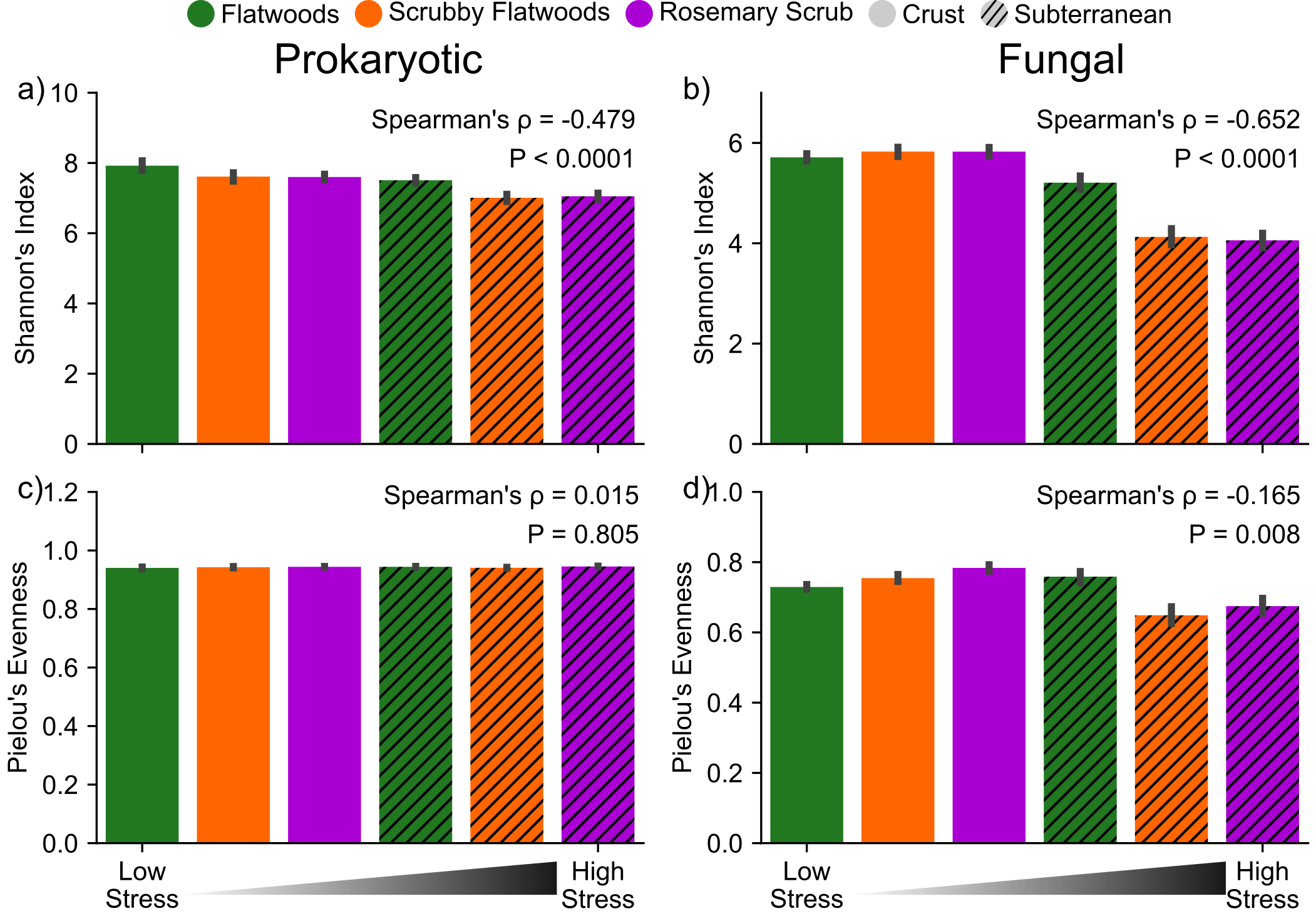
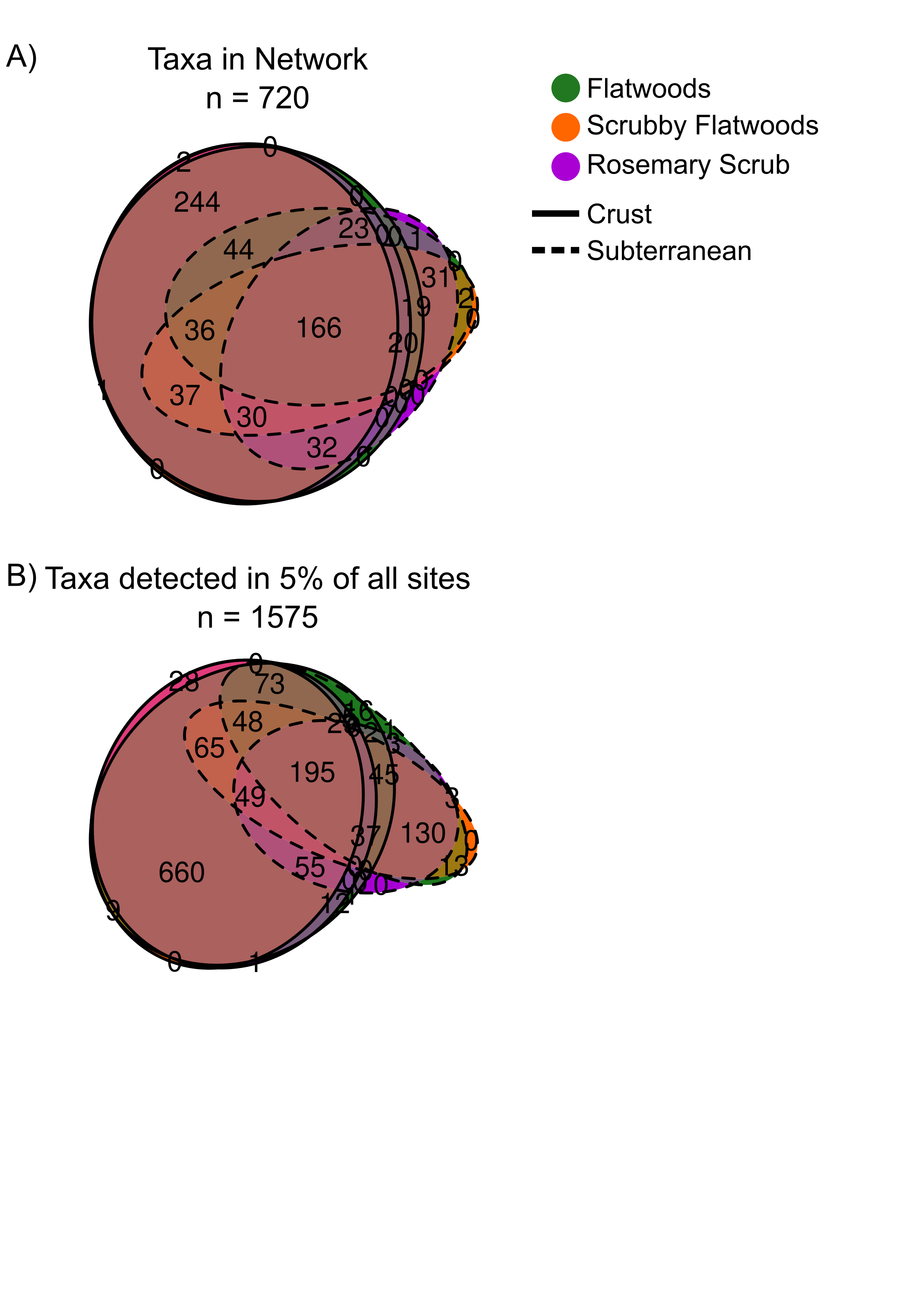
To assess how microbial community properties change along the stress gradient, we compared richness and diversity across these six environments. Overall, fungal and prokaryotic OTU richness decreased with increasing stress across the six environments (Fig. 3a, b; Prokaryotes: Spearman’s ⍴ = −0.517, P < 0.0001; Fungi: Spearman’s ⍴ = −0.807, P < 0.0001). This decreasing relationship with the stress gradient was significant, but weaker for Shannon diversity (Supplementary Fig. 3a, b; Prokaryotes: Spearman’s ⍴ = −0.479, P < 0.0001, Fungi: Spearman’s ⍴ = −0.652, P < 0.0001), because species evenness remained fairly constant across habitats (Supplementary Fig. 3c, d; Prokaryotes: Spearman’s ⍴ = 0.015, P = 0.805, Fungi: Spearman’s ⍴ = −0.165, P = 0.008). The taxa in our co-occurrence networks that are present in our high stress (low diversity) sites are largely a subset of those present in our low stress (high diversity) sites (Supplementary Fig. 4). Only 4.6% (33/720) of taxa whose abundances were used to construct the six co-occurrence networks are unique to the high-stress subterranean samples (Supplementary Fig. 4a).
Fig. 3. Characterizing microbial diversity and network properties across the stress gradient.

a, b Decrease in prokaryotic (a) and fungal (b) community richness (observed OTUs) across the stress gradient. Error bars depict standard error of the mean. c Decrease in modularity of microbiome networks across the stress gradient. A single value of modularity was calculated for each environment. d Decrease in ratio of negative:positive cohesion across the stress gradient. Box plots show inner quartiles and median negative:positive cohesion. Both negative correlations of modularity and negative:positive cohesion with stress are consistent with decreasing microbiome stability across the stress gradient. X-axis represents the categorical stress rankings of the six environments along the stress gradient. Detailed statistics are available in Supplementary Table 3.
Furthermore, stressed microbial communities had lower network stability as evidenced by our analyses of modularity and cohesion. Microbial community networks decreased in modularity along the stress gradient (Fig. 3c; Spearman’s ⍴ = −0.886, P = 0.033), indicating that microbial communities in high-stress environments are less compartmentalized than in low stress environments. We also found that the ratio of negative:positive cohesion decreased with stress (Fig. 3d; Spearman’s ⍴ = −0.712, P < 0.0001), indicating that positive associations between taxa, rather than negative associations, dominate in high-stress environments. Similarly, total cohesion (sum of positive cohesion and the absolute value of negative cohesion) decreased with increasing stress (Fig. 4a, Spearman’s ⍴ = −0.896, P < 0.0001), indicating that community networks become less complex across the stress gradient.
Fig. 4. Changes in cohesion across the stress gradient.
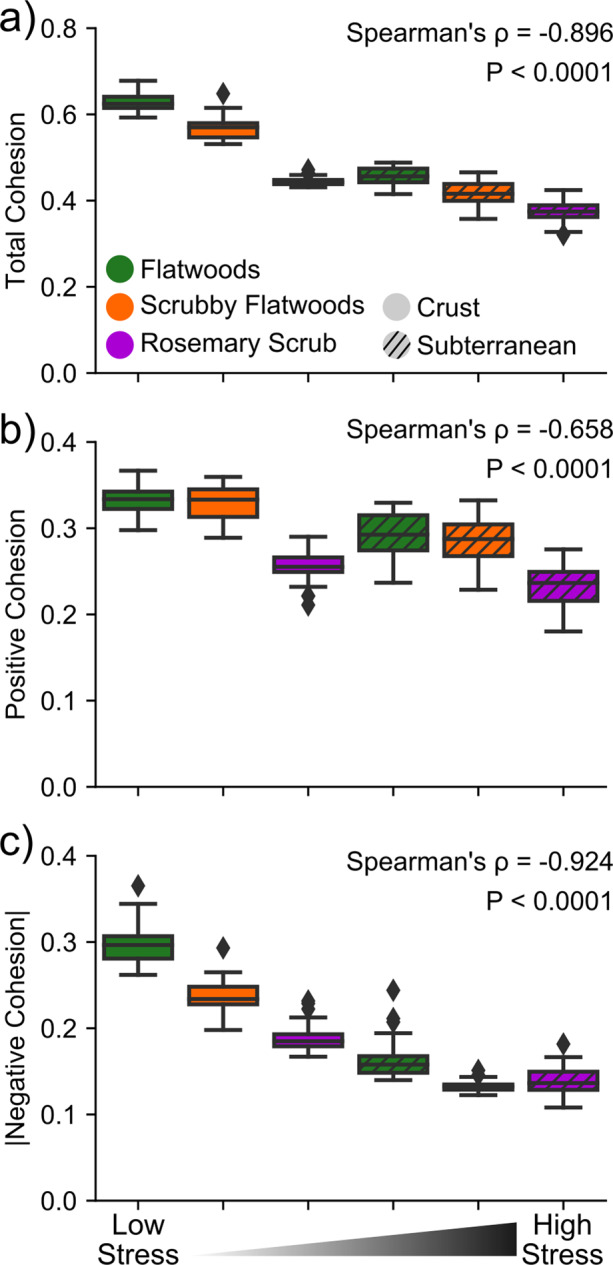
a Decrease in total cohesion (sum of positive cohesion and the absolute value of negative cohesion) across the stress gradient. b Decrease in positive cohesion across the stress gradient. c Decrease in the absolute value of negative cohesion across the stress gradient. Negative cohesion decreases more strongly across the stress gradient than does positive cohesion (P < 0.0001), driving the decrease in negative:positive cohesion seen in Fig. 3d. Box plots show inner quartiles and median values. X-axis represents the categorical stress rankings of the six environments along the stress gradient. P values are derived from Spearman’s Correlation tests. Detailed statistics are available in Supplementary Table 3.
This reduced complexity of microbial networks (i.e., decreased total cohesion) was attributable to both positive (Fig. 4b, Spearman’s ⍴ = −0.658, P < 0.0001) and negative (Fig. 4c, Spearman’s ⍴ = −0.924, P < 0.0001) cohesion decreasing in magnitude across the stress gradient; however, negative co-occurrences were a much stronger driver of decreasing total cohesion than positive co-occurrences (Z = 9.25, P < 0.0001). In fact, the decrease in negative cohesion (Siegel slope = −0.0303; 95% CI (−0.0296, −0.0320)) was twofold higher than that of positive cohesion (Siegel slope = −0.0141; 95% CI (−0.0138, −0.0158)), thereby driving the decrease in negative:positive cohesion. This amounted to negative cohesion declining in magnitude by 53% between the lowest stress environment (flatwoods crust) and the highest stress environment (rosemary subterranean).
Characterization of taxonomic and functional groups across the stress gradient
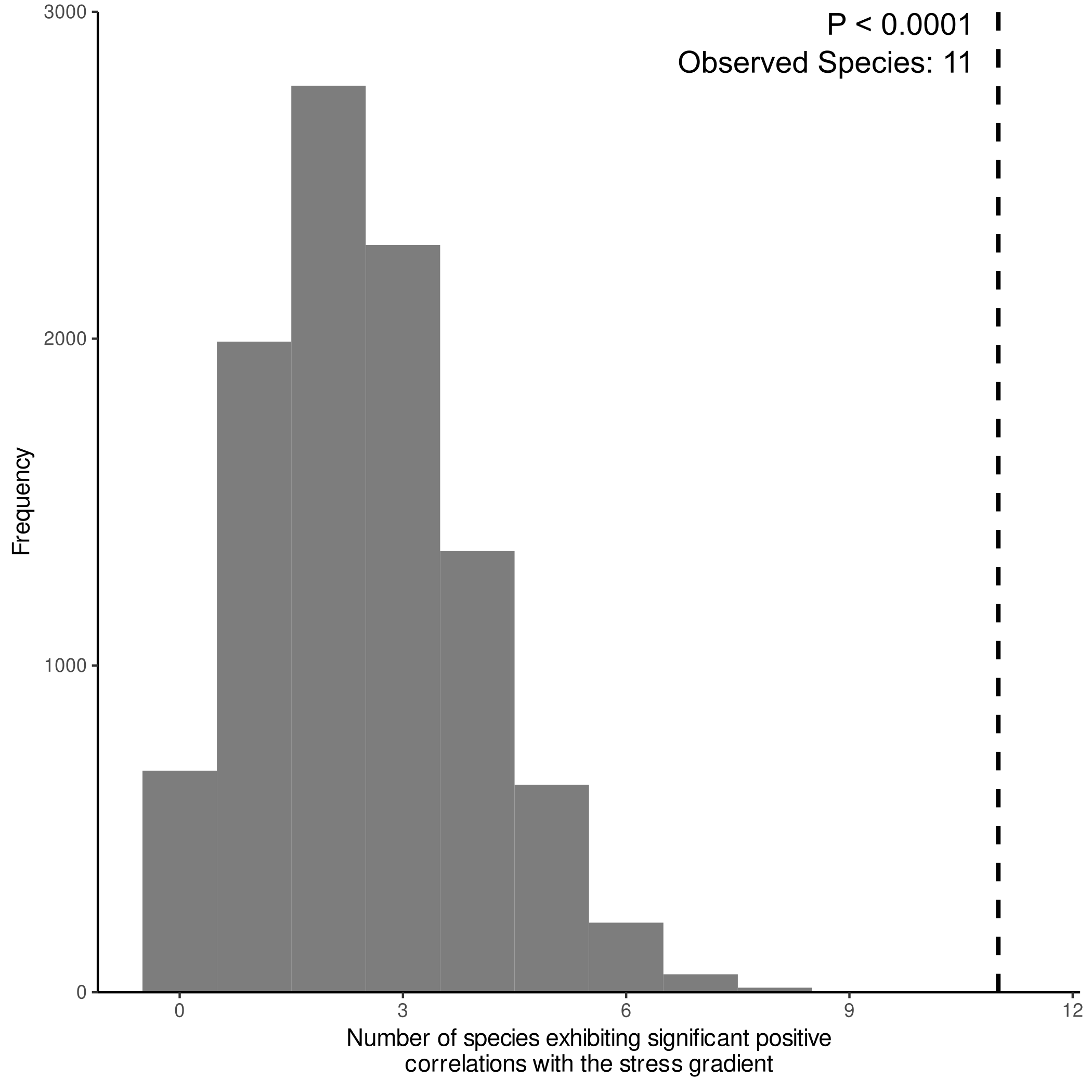
We then quantified changes in prokaryotic taxa, fungal taxa, and fungal functional guilds across the stress gradient to understand which groups are impacted by the stress gradient. In general, we found that several oligotrophic (i.e., stress tolerant) taxa and plant mutualists increased in relative abundance along the stress gradient (Supplementary File 7). Oligotrophs positively associated with the stress gradient included the bacterial phylum Acidobacteria (Spearman’s ⍴ = 0.686, PFDR < 0.0001) and the bacterial order Xanthomonadales (Fig. 5a, Spearman’s ⍴ = 0.695, PFDR < 0.0001; most of our OTUs belonged to the oligotrophic family Sinobacteraceae and not the plant pathogen genera Xanthomonas or Xylella). Plant–mutualist groups positively associated with stress included the nitrogen-fixing Rhizobiales (Fig. 5a, Spearman’s ⍴ = 0.650, PFDR < 0.0001), the ectomycorrhizal fungal orders Russulales (Fig. 5b, Spearman’s ⍴ = 0.385, PFDR < 0.0001) and Boletales (Fig. 5b, Spearman’s ⍴ = 0.453, PFDR < 0.0001), and the arbuscular mycorrhizal fungal order Glomerales (Spearman’s ⍴ = 0.162, PFDR = 0.016). Conversely, plant pathogens tended to decrease with stress. The fungal order Capnodiales, of which our OTUs were predominantly from the pathogenic family Mycosphaerellacea, had the strongest negative correlation with the stress gradient (Fig. 5b, Spearman’s ⍴ = −0.700, PFDR < 0.0001). Abundance of Capnodiales decreased from making up a substantial portion of communities in the least stressful environment (7.7% ± 0.2%, mean ± s.e.m.) to 0.2% (±0.009% s.e.m.) in the most stressful habitats. Consistent with the taxonomic assessment above, we found that the plant pathogenic functional guild assigned using FunGuild decreased in abundance across the stress gradient (Spearman’s ⍴ = −0.701, PFDR < 0.0001), while ectomycorrhizae were the fungal guild most positively correlated with the stress gradient (Spearman’s ⍴ = 0.495, PFDR < 0.0001). In fact, the percentage of fungal species classified as members of mutualistic guilds that are positively associated with the stress gradient was ~4.5 times greater than among randomly drawn subsets of fungal species (P < 0.0001, see Supplementary Fig. 5 for details).
Fig. 5. Microbial orders most positively and negatively impacted by the stress gradient.

a, b Top ten prokaryotic (a) and fungal (b) orders whose abundances are most positively or negatively correlated with the stress gradient (all correlations with P < 0.0001 after FDR correction). Abundances of taxa from kingdom to species identification are included in Supplementary File 6.
Discussion
In this study, we found support for environmental stress destabilizing microbial community networks: microbes in higher stress habitats were less diverse, less modular, and dominated by positive co-occurrences (i.e., having lower negative:positive cohesion) compared to communities in low stress habitats. We also found that negative cohesion (i.e., strength/number of negative associations) decreased more strongly along the gradient than positive cohesion (i.e., strength/number of positive associations). To determine if the increased representation of positive correlations in high stress could be due, at least in part, to more positive interactions as opposed to greater niche overlap, we compared changes in abundance of mutualists between low and high-stress sites, finding that mutualistic taxa have strong positive relationships with the stress gradient while pathogenic fungi have strong negative relationships. Given that microbial communities often experience more species turnover (i.e., are less stable) when negative correlations are less frequent [17, 18, 56] and when network modularity is reduced by removal of taxa important to modular structure [19, 57], our results indicate that stressed microbial communities have lower network stability. This outcome is concerning, as it suggests that increased anthropogenic stress could select for microbial network topologies that are more prone to rapid fluctuations and regime shifts. Below we discuss ecological mechanisms through which environmental stress can destabilize microbial networks: (1) how changes in taxonomic and functional group composition can lead to increasing representation of positive correlations in high-stress sites, and (2) how decreasing modularity and negative:positive cohesion (i.e., fewer and/or weaker negative correlations relative to positive correlations) in stressed environments can combine to destabilize microbial communities.
Loss of antagonistic interactions and increased environmental filtering as drivers of decreasing negative:positive cohesion in stressful environments
Within microbiome networks, correlations between taxa can be driven by two non-mutually exclusive mechanisms: species interactions and environmental filtering [13, 58]. In the case of species interactions, positive correlations can be caused by facilitation/mutualism among taxa and negative correlations can result from competition [44, 59]. In the case of environmental filtering, positive correlations among taxa reflect ecological or functional similarity [60], while taxa with divergent niche requirements are negatively correlated. While both ecological mechanisms could explain the dominance of positive cohesion in high-stress habitats, our data support a role for species interactions. We observe decreases in relative abundance of pathogens and increases in relative abundance of microbial mutualists along our stress gradient, which agrees with previous complementary in vitro work demonstrating increased facilitation among microbes in stressful environments [9, 10]. We discuss both alternatives below.
The Stress Gradient Hypothesis [61], primarily developed to describe plant-plant interactions, provides general predictions about how species interactions change with increasing stress. It posits that as stress intensifies, the frequency of competitive interactions decreases and that of facilitative interactions increases, which is consistent with the decreasing negative:positive cohesion along our stress gradient. This outcome can occur through two mechanisms: (1) “competitive” taxa that engage in many antagonistic interspecific interactions being replaced by slow-growing, stress-tolerant species (e.g., oligotrophic microbes) as stress increases [62, 63], and (2) species that provide direct benefits to other community members increasing as stress increases [64]. Our data support the former. For example, we found that the oligotrophic phylum Acidobacteria [65] showed the strongest positive correlation with the stress gradient.
It is also likely that facilitative taxa increased in relative abundance along the stress gradient while antagonistic taxa decreased. In our study, plant–microbial mutualists are some of the taxa most positively correlated with the stress gradient across both fungi and prokaryotes. For example, ectomycorrhizal fungi were the fungal guild most positively correlated with the stress gradient, increasing from only 21% (±3.3% s.e.m.) in the least stressful environment to 64% (±4.7% s.e.m.) in the highest stress environment. We found a similar relationship between stress and plant–bacterial mutualists, such as the order Rhizobiales and family Sinobacteraceae [66]. We also saw concomitant decreases in species that engage in antagonistic interactions. For instance, the plant fungal pathogen guild showed one of the strongest negative relationships with environmental stress (Spearman’s ⍴ = −0.701). These results demonstrate that relative abundances of facilitative microbes increase and antagonistic microbes decrease with stress for at least one major group in the community, plants. Whether this extends to how microbes interact with each other is less clear because there is far less information about how different microbial taxa affect one another. However, experimental exposure of microbial communities to stress has demonstrated that microbes switch from competition to facilitative interactions when placed in toxic media [9] and engage in metabolic cross-feeding when a common nutrient is depleted [10]. As a result, it is likely that the increase in facilitative potential seen between microbes and plants in our system [33, 34] also extends to interactions between microbes themselves.
The alternative ecological mechanism, environmental filtering, may also drive the decrease in negative:positive cohesion in high-stress conditions. For example, the dominance of ectomycorrhizal fungi in high-stress environments may reflect that in xeric conditions very few niches are available for fungi, and that plant roots constitute a major proportion of available plant habitat in these circumstances. However, given that our data support both ecological mechanisms underlying the Stress Gradient Hypothesis (i.e., greater representation of stress-tolerant taxa and increasing facilitation), a decrease in the ratio of negative:positive biotic interactions is likely to be at least part of the microbial response to stress, even if it is not entirely independent of abiotic factors selecting for higher niche overlap.
Low modularity and positive associations as processes destabilizing stressed communities
While increasing dominance of positive correlations or reduction in negative associations in microbiomes can destabilize communities alone [17, 18, 56], the lower modularity in high-stress sites may exacerbate these consequences. For example, Agler et al. [19] found that removal of taxa important for structuring community subnetworks (i.e., modules) led to greater variation in microbiome composition than when those taxa are present (i.e., when modular structure is likely maintained). Modular organization of microbial communities can reflect: (1) groups of taxa that interact with each other more than with other groups, or (2) groups of taxa that share a niche distinct from other groups. In either scenario, a modular organization insulates these groupings from disturbances by ensuring that fluctuations in abundances of sensitive taxa within one module are unlikely to spread to taxa in other modules due to limited linkages between them [16, 67].
A major argument for the destabilizing effects of positive co-occurrence/interactions in microbial communities centers around the role of positive feedback loops in which microbes that support each others’ fitness outcompete other microbes [18]. As a result, the community as a whole becomes dependent on maintaining those feedback loops, and perturbations experienced by any of these loops’ members can rapidly propagate through the entire system [18]. In contrast, negative co-occurrences/interactions can lead to negative feedback loops that dampen not only perturbations that their own members experience, but also those experienced by linked positive feedback loops [68]. Low modularity in high-stress environments indicates that cross-module associations among taxa are likely to be more prevalent than in low stress environments; thus, environmental perturbations impacting taxa in one module would be more likely to propagate to other modules because there are relatively more associations (i.e., interactions or niche similarities) between taxa in different modules at high-stress sites (Fig. 6). For example, in our system, plant–microbial mutualists such as ectomycorrhizal fungi, Rhizobiales, and Sinobacteraceae could support each other by facilitating their shared plant partners, and the accompanying low modularity would mean that any perturbations experienced by this dominant positive feedback loop would be easily propagated to the rest of the microbial community. The increased connectivity between modules could be due to increased interactions with the plant–mutualist module (if linkages in the microbial network represent species interactions), or reliance on the same environmental factors (if linkages in the microbial network represent high niche overlap). For example, in our system, the importance of plant roots as a microbial niche may increase with increasing stress. As a result, environmental effects on their shared plant root niche (e.g., replacement of compatible plants with incompatible plants, destruction of the plant community, etc.) will impact more taxa under high-stress than low stress, because more microbes share plant roots as a niche requirement.
Fig. 6. Role for environmental stress in destabilization of microbial networks.
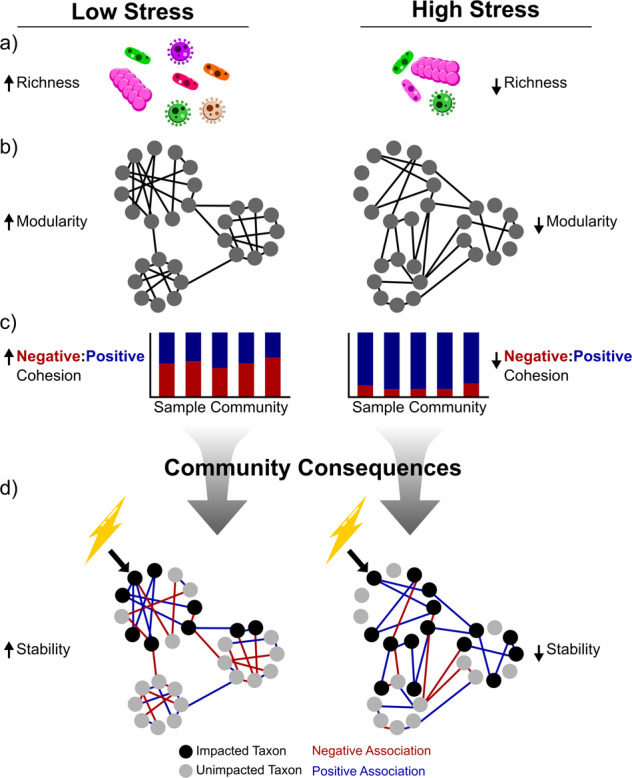
Communities in high-stress environments (right) compared to communities in low stress environments (left) have less stable network properties. Communities under high stress are less species rich (a), less modular (b), and are less dominated by negative associations (lower negative:positive cohesion, c). The consequences for microbiomes (d) are that the reduction or loss of a taxon in response to an environmental disturbance (lightning bolt) can more easily propagate to the rest of the community (impacted taxa in black, unimpacted in gray). Positive associations (blue) are important pathways through which the effects on one taxon can cascade to impact other taxa in the community because: (1) the loss of a facilitator reduces the facilitated taxon’s fitness and/or (2) positively associated taxa are likely to be impacted by the same environmental factors (i.e., their niches strongly overlap).
Conclusions and implications
Our study provides the first evidence that naturally-occurring microbiomes display network properties characteristic of unstable communities when under persistent stress. Because microbes can explain variation in traits important to plant distributions (e.g., growth rates, seedling establishment, secondary compound synthesis, etc. [69–74]) and undergird a plethora of ecosystem services (e.g., nutrient cycling; [3, 75, 76]), understanding how the stability of microbiomes is impacted by stress has important implications for modeling species’ distributions [77], maintaining ecosystem services [3, 77], and improving agriculture [1]. However, the development of general and testable principles for how these important, microscopic communities respond to even common environmental stress gradients is still in its early stages [3]. By characterizing microbiome network structure along a highly replicated, natural stress gradient, we demonstrate that, in high-stress environments, microbial taxa co-occur more frequently and the modularity of their communities breaks down.
As anthropogenic disturbances increasingly stress ecosystems, microbial communities may become destabilized. Our study system is one example of a naturally-occurring stress gradient that can serve as a model for environmental stressors that are predicted to increase in the Anthropocene (e.g., lower water availability; 4). While we find dramatic relationships between environmental stress and stable network properties (78% and 51% of variation in modularity and negative:positive cohesion was explained by our stress gradient, respectively), we encourage testing these relationships in a wider variety of ecosystems, including soil communities of other terrestrial biomes and microbiomes from aquatic habitats. We hope this study will provide a roadmap for characterizing microbiome network structures in a diverse range of study systems to generate broad insight into whether a negative relationship between stress and microbiome stability is a unifying principle across microbial communities. We also advocate for future studies elucidating how the consequences of unstable microbial communities cascade upwards to impact socially important goals such as restoration efforts and agriculture.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank A. Wilson, J. Dallman, and S. Wuchty for feedback on the paper, the UMGC for sequencing support, V. Sclater for GIS assistance, and L. Otaño Velazco for field and laboratory assistance. Research was funded by the Mycological Society of America’s Forest Fungal Ecology Research Award to ASD, University of Miami research funds to MEA and CAS, and the National Science Foundation (DEB-1922521). DH was supported by the University of Miami Maytag Fellowship and Julia Morton Research Fellowship.
Data availability
Demultiplexed sequence data are available at NCBI (BioProject: PRJNA559142). Details of programming languages, module versions, and virtual environments used for analyses outside of QIIME2 are listed in Supplementary Table 2. Extended ANOVA tables and detailed statistics for analyses are available in Supplementary Table 3. OTU tables, taxonomic assignments, taxonomic abundances, FunGuild fungal functional group abundances, network edge lists, modularity, and cohesion values necessary for analyses are provided in Supplementary File 6.
Code availability
The files necessary to prcoess the demultiplexed sequence data through QIIME2 are provided in Supplementary Files 1–5.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Damian J. Hernandez, Aaron S. David
These authors jointly supervised this work: Christopher A. Searcy, Michelle E. Afkhami
Supplementary information
The online version of this article (10.1038/s41396-020-00882-x) contains supplementary material, which is available to authorized users.
References
- 1.Toju H, Peay KG, Yamamichi M, Narisawa K, Hiruma K, Naito K, et al. Core microbiomes for sustainable agroecosystems. Nat Plants. 2018;4:247–57.. doi: 10.1038/s41477-018-0139-4. [DOI] [PubMed] [Google Scholar]
- 2.de Zelicourt A, Al-Yousif M, Hirt H. Rhizosphere microbes as essential partners for plant stress tolerance. Mol Plant. 2013;6:242–5. doi: 10.1093/mp/sst028. [DOI] [PubMed] [Google Scholar]
- 3.Naylor D, Sadler N, Bhattacharjee A, Graham EB, Anderton CR, McClure R, et al. Soil microbiomes under climate change and implications for carbon cycling. Annu Rev Environ Resour. 2020;45:29–59. [Google Scholar]
- 4.IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change In: Core Writing Team, Pachauri RK, Meyer LA, editors. Geneva, Switzerland: IPCC; 151 pp.
- 5.Nguyen LT, Broughton K, Osanai Y, Anderson IC, Bange MP, Tissue DT, et al. Effects of elevated temperature and elevated CO2 on soil nitrification and ammonia-oxidizing microbial communities in field-grown crop. Sci Total Environ. 2019;675:81–9. doi: 10.1016/j.scitotenv.2019.04.181. [DOI] [PubMed] [Google Scholar]
- 6.de Vries FT, Griffiths RI, Bailey M, Craig H, Girlanda M, Gweon HS, et al. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun. 2018;9:3033. doi: 10.1038/s41467-018-05516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaneveld JR, McMinds R, Thurber RV. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat Microbiol. 2017;2:1–8. doi: 10.1038/nmicrobiol.2017.121. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed HI, Herrera M, Liew YJ, Aranda M. Long-term temperature stress in the coral model Aiptasia supports the “Anna Karenina Principle” for bacterial microbiomes. Front Microbiol. 2019;10:975. doi: 10.3389/fmicb.2019.00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piccardi P, Vessman B, Mitri S. Toxicity drives facilitation between 4 bacterial species. Proc Natl Acad Sci USA. 2019;116:15979–84.. doi: 10.1073/pnas.1906172116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldford JE, Lu N, Bajić D, Estrela S, Tikhonov M, Sanchez-Gorostiaga A, et al. Emergent simplicity in microbial community assembly. Science. 2018;361:469–74.. doi: 10.1126/science.aat1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.David AS, Quintana-Ascencio PF, Menges ES, Thapa-Magar KB, Afkhami ME, Searcy CA. Soil microbiomes underlie population persistence of an endangered plant species. Am Nat. 2019;194:488–94.. doi: 10.1086/704684. [DOI] [PubMed] [Google Scholar]
- 12.Worchel ER, Giauque HE, Kivlin SN. Fungal symbionts alter plant drought response. Microb Ecol. 2013;65:671–8. doi: 10.1007/s00248-012-0151-6. [DOI] [PubMed] [Google Scholar]
- 13.Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51.. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Layeghifard M, Hwang DM, Guttman DS. Disentangling Interactions in the microbiome: a network perspective. Trends Microbiol. 2017;25:217–28.. doi: 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olesen JM, Bascompte J, Dupont YL, Jordano P. The modularity of pollination networks. Proc Natl Acad Sci. 2007;104:19891–6. doi: 10.1073/pnas.0706375104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stouffer DB, Bascompte J. Compartmentalization increases food-web persistence. Proc Natl Acad Sci USA. 2011;108:3648–52. doi: 10.1073/pnas.1014353108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herren CM, McMahon KD. Cohesion: a method for quantifying the connectivity of microbial communities. ISME J. 2017;11:2426–38.. doi: 10.1038/ismej.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: networks, competition, and stability. Science. 2015;350:663–6. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- 19.Agler MT, Ruhe J, Kroll S, Morhenn C, Kim S-T, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:e1002352. doi: 10.1371/journal.pbio.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391. doi: 10.1038/ncomms10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guidi L, Chaffron S, Bittner L, Eveillard D, Larhlimi A, Roux S, et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature. 2016;532:465–70.. doi: 10.1038/nature16942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cram JA, Xia LC, Needham DM, Sachdeva R, Sun F, Fuhrman JA. Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes. ISME J. 2015;9:2573–86.. doi: 10.1038/ismej.2015.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang B, Zhang J, Liu Y, Shi P, Wei G. Co-occurrence patterns of soybean rhizosphere microbiome at a continental scale. Soil Biol Biochem. 2018;118:178–86.. [Google Scholar]
- 26.Suweis S, Grilli J, Maritan A. Disentangling the effect of hybrid interactions and of the constant effort hypothesis on ecological community stability. Oikos. 2014;123:525–32.. [Google Scholar]
- 27.Mougi A, Kondoh M. Diversity of interaction types and ecological community stability. Science. 2012;337:349–51.. doi: 10.1126/science.1220529. [DOI] [PubMed] [Google Scholar]
- 28.Weekley CW, Tucker J, Valligny S, Menges ES. Germination ecology of Liatris ohlingerae (SF Blake) BL Rob.(Asteraceae), an endangered herb endemic to Florida scrub. Castanea. 2008;73:235–50.. [Google Scholar]
- 29.Abrahamson WG, Johnson AF, Layne JN, Peroni PA. Vegetation of the Archbold Biological Station, Florida: an example of the Southern Lake Wales Ridge. Fla Sci. 1984;47:209–50. [Google Scholar]
- 30.Menges ES, Gallo NP. Water relations of scrub oaks on the Lake Wales Ridge, Florida. Fla Sci. 1991;54:69–79. [Google Scholar]
- 31.Menges ES, Hawkes CV. Interactive effects of fire and microhabitat on plants of Florida scrub. Ecol Appl. 1998;8:935–46.. [Google Scholar]
- 32.Quintana‐Ascencio PF, Koontz SM, Smith SA, Sclater VL, David AS, Menges ES. Predicting landscape‐level distribution and abundance: Integrating demography, fire, elevation and landscape habitat configuration. J Ecol. 2018;106:2395–408.. [Google Scholar]
- 33.David AS, Thapa-Magar KB, Afkhami ME. Microbial mitigation-exacerbation continuum: a novel framework for microbiome effects on hosts in the face of stress. Ecology. 2018;99:517–23.. doi: 10.1002/ecy.2153. [DOI] [PubMed] [Google Scholar]
- 34.David AS, Thapa‐Magar KB, Menges ES, Searcy ES, Afkhami ME. Do plant–microbe interactions support the Stress Gradient Hypothesis? Ecology. 2020;101:e03081. doi: 10.1002/ecy.3081. [DOI] [PubMed] [Google Scholar]
- 35.Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 2016;34:942–9. doi: 10.1038/nbt.3601. [DOI] [PubMed] [Google Scholar]
- 36.Apprill A, McNally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol. 2015;75:129–37.. [Google Scholar]
- 37.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14.. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 38.Smith DP, Peay KG. Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PloS ONE. 2014;9:e90234. doi: 10.1371/journal.pone.0090234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White TJ, Bruns T, Lee SJ, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. 1990;18:315–22.. [Google Scholar]
- 40.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilsson RH, Larsson K-H, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47:D259–64. doi: 10.1093/nar/gky1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durán P, Thiergart T, Garrido-Oter R, Agler M, Kemen E, Schulze-Lefert P, et al. Microbial interkingdom interactions in roots promote arabidopsis survival. Cell. 2018;175:973–83.e14. doi: 10.1016/j.cell.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Araya JP, González M, Cardinale M, Schnell S, Stoll A. Microbiome dynamics associated with the Atacama Flowering Desert. Front Microbiol. 2019;10:3160. doi: 10.3389/fmicb.2019.03160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grime JP, Campbell BD. Growth rate, habitat productivity, and plant strategy as predictors of stress response. In: Mooney HA, Winner WE, Pell EJ, editors. Response of plants to multiple stresses. NY, USA: Academic Press, Inc: 1991, pp. 143–59.
- 47.Grime JP. Competitive exclusion in herbaceous vegetation. Nature. 1973;242:344–7. [Google Scholar]
- 48.Grime JP. Evidence for the existence of three primary strategies in plants and its relevance to ecological and evolutionary theory. Am Nat. 1977;111:1169–94.. [Google Scholar]
- 49.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clauset A, Newman ME, Moore C. Finding community structure in very large networks. Phys Rev E. 2004;70:066111. doi: 10.1103/PhysRevE.70.066111. [DOI] [PubMed] [Google Scholar]
- 51.Newman M. Networks: an introduction. Oxford: OUP; 2010. p. 784.
- 52.Newman ME, Girvan M. Finding and evaluating community structure in networks. Phys Rev E. 2004;69:026113. doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016;20:241–8. [Google Scholar]
- 54.del Pilar Martínez-Diz M, Andrés-Sodupe M, Bujanda R, Díaz-Losada E, Eichmeier A, Gramaje D. Soil-plant compartments affect fungal microbiome diversity and composition in grapevine. Fungal Ecol. 2019;41:234–44.. [Google Scholar]
- 55.Nuske SJ, Anslan S, Tedersoo L, Bonner MT, Congdon BC, Abell SE. The endangered northern bettong, Bettongia tropica, performs a unique and potentially irreplaceable dispersal function for ectomycorrhizal truffle fungi. Mol Ecol. 2018;27:4960–71.. doi: 10.1111/mec.14916. [DOI] [PubMed] [Google Scholar]
- 56.Danczak RE, Johnston MD, Kenah C, Slattery M, Wilkins MJ. Microbial community cohesion mediates community turnover in unperturbed aquifers. Msystems. 2018;3:e00066–18.. doi: 10.1128/mSystems.00066-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stein RR, Bucci V, Toussaint NC, Buffie CG, Rätsch G, Pamer EG, et al. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput Biol. 2013;9:e1003388. doi: 10.1371/journal.pcbi.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freilich MA, Wieters E, Broitman BR, Marquet PA, Navarrete SA. Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology. 2018;99:690–9. doi: 10.1002/ecy.2142. [DOI] [PubMed] [Google Scholar]
- 59.Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci USA. 2015;112:6449–54.. doi: 10.1073/pnas.1421834112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaffron S, Rehrauer H, Pernthaler J, von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20:947–59.. doi: 10.1101/gr.104521.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bertness MD, Callaway R. Positive interactions in communities. Trends Ecol Evolution. 1994;9:191–3. doi: 10.1016/0169-5347(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 62.Garcia MO, Templer PH, Sorensen PO, Sanders-DeMott R, Groffman PM, Bhatnagar JM. Soil microbes trade-off biogeochemical cycling for stress tolerance traits in response to year-round climate change. Front Microbiol. 2020;11:616. doi: 10.3389/fmicb.2020.00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Männistö M, Ganzert L, Tiirola M, Häggblom MM, Stark S. Do shifts in life strategies explain microbial community responses to increasing nitrogen in tundra soil? Soil Biol Biochem. 2016;96:216–28.. [Google Scholar]
- 64.Michalet R, Brooker RW, Cavieres LA, Kikvidze Z, Lortie CJ, Pugnaire FI, et al. Do biotic interactions shape both sides of the humped-back model of species richness in plant communities? Ecol Lett. 2006;9:767–73.. doi: 10.1111/j.1461-0248.2006.00935.x. [DOI] [PubMed] [Google Scholar]
- 65.Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–64.. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Ludewig U. Nitrogen‐dependent bacterial community shifts in root, rhizome and rhizosphere of nutrient‐efficient Miscanthus x giganteus from long‐term field trials. GCB Bioenergy. 2019;11:1334–47.. [Google Scholar]
- 67.Teng J, McCann KS. Dynamics of compartmented and reticulate food webs in relation to energetic flows. Am Nat. 2004;164:85–100. doi: 10.1086/421723. [DOI] [PubMed] [Google Scholar]
- 68.Fontaine C, Guimarães PR, Jr, Kéfi S, Loeuille N, Memmott J, van der Putten WH, et al. The ecological and evolutionary implications of merging different types of networks. Ecol Lett. 2011;14:1170–81.. doi: 10.1111/j.1461-0248.2011.01688.x. [DOI] [PubMed] [Google Scholar]
- 69.Nuñez MA, Horton TR, Simberloff D. Lack of belowground mutualisms hinders Pinaceae invasions. Ecology. 2009;90:2352–9. doi: 10.1890/08-2139.1. [DOI] [PubMed] [Google Scholar]
- 70.Grotkopp E, Rejmánek M, Rost TL. Toward a causal explanation of plant invasiveness: seedling growth and life-history strategies of 29 pine (Pinus) species. Am Nat. 2002;159:396–419. doi: 10.1086/338995. [DOI] [PubMed] [Google Scholar]
- 71.Thiet RK, Boerner REJ. Spatial patterns of ectomycorrhizal fungal inoculum in arbuscular mycorrhizal barrens communities: implications for controlling invasion by Pinus virginiana. Mycorrhiza. 2007;17:507–17.. doi: 10.1007/s00572-007-0123-8. [DOI] [PubMed] [Google Scholar]
- 72.Callaway RM, Thelen GC, Rodriguez A, Holben WE. Soil biota and exotic plant invasion. Nature. 2004;427:731–3. doi: 10.1038/nature02322. [DOI] [PubMed] [Google Scholar]
- 73.Vogelsang KM, Bever JD. Mycorrhizal densities decline in association with nonnative plants and contribute to plant invasion. Ecology. 2009;90:399–407. doi: 10.1890/07-2144.1. [DOI] [PubMed] [Google Scholar]
- 74.Pringle A, Bever JD, Gardes M, Parrent JL, Rillig MC, Klironomos JN. Mycorrhizal symbioses and plant invasions. Annu Rev Ecol Evolution Syst. 2009;40:699–715. [Google Scholar]
- 75.Hestrin R, Hammer EC, Mueller CW, Lehmann J. Synergies between mycorrhizal fungi and soil microbial communities increase plant nitrogen acquisition. Commun Biol. 2019;2:233. doi: 10.1038/s42003-019-0481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dai Z, Liu G, Chen H, Chen C, Wang J, Ai S, et al. Long-term nutrient inputs shift soil microbial functional profiles of phosphorus cycling in diverse agroecosystems. ISME J. 2020;14:757–70.. doi: 10.1038/s41396-019-0567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Classen AT, Sundqvist MK, Henning JA, Newman GS, Moore JA, Cregger MA, et al. Direct and indirect effects of climate change on soil microbial and soil microbial‐plant interactions: what lies ahead? Ecosphere. 2015;6:1–21.. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Demultiplexed sequence data are available at NCBI (BioProject: PRJNA559142). Details of programming languages, module versions, and virtual environments used for analyses outside of QIIME2 are listed in Supplementary Table 2. Extended ANOVA tables and detailed statistics for analyses are available in Supplementary Table 3. OTU tables, taxonomic assignments, taxonomic abundances, FunGuild fungal functional group abundances, network edge lists, modularity, and cohesion values necessary for analyses are provided in Supplementary File 6.
The files necessary to prcoess the demultiplexed sequence data through QIIME2 are provided in Supplementary Files 1–5.