

Key Points
Question
What are the clinicopathologic features of oculopharyngodistal myopathy (OPDM) with CGG repeat expansions in LRP12 (hereafter referred to as OPDM_LRP12)?
Findings
In this case series, 65 Japanese patients with OPDM_LRP12 had 85 to 289 CGG repeats in LRP12; most of these patients showed adult-onset myopathy with ptosis, dysphagia and dysarthria, predominant gastrocnemius and soleus muscle involvement, and rimmed vacuoles. Five patients developed respiratory insufficiency that required mechanical ventilation; 7 patients showed cardiac abnormalities.
Meaning
This study suggests that OPDM_LRP12 is the most frequent OPDM subtype in Japan and is clinically characterized by predominant gastrocnemius and soleus muscle involvement in addition to oculopharyngeal weakness.
Abstract
Importance
Repeat expansion of CGG in LRP12 has been identified as the causative variation of oculopharyngodistal myopathy (OPDM). However, to our knowledge, the clinicopathologic features of OPDM with CGG repeat expansion in LRP12 (hereafter referred to as OPDM_LRP12) remain unknown.
Objective
To identify and characterize the clinicopathologic features of patients with OPDM_LRP12.
Design, Setting, and Participants
This case series included 208 patients with a clinical or clinicopathologic diagnosis of oculopharyngeal muscular dystrophy (OPDM) from January 1, 1978, to December 31, 2020. Patients with GCN repeat expansions in PABPN1 were excluded from the study. Repeat expansions of CGG in LRP12 were screened by repeat primed polymerase chain reaction and/or Southern blot.
Main Outcomes and Measures
Clinical information, muscle imaging data obtained by either computed tomography or magnetic resonance imaging, and muscle pathologic characteristics.
Results
Sixty-five Japanese patients with OPDM (40 men [62%]; mean [SD] age at onset, 41.0 [10.1] years) from 59 families with CGG repeat expansions in LRP12 were identified. This represents the most common OPDM subtype among all patients in Japan with genetically diagnosed OPDM. The expansions ranged from 85 to 289 repeats. A negative correlation was observed between the repeat size and the age at onset (r2 = 0.188, P = .001). The most common initial symptoms were ptosis and muscle weakness, present in 24 patients (37%). Limb muscle weakness was predominantly distal in 53 of 64 patients (83%), but 2 of 64 patients (3%) had predominantly proximal muscle weakness. Ptosis was observed in 62 of 64 patients (97%), and dysphagia or dysarthria was observed in 63 of 64 patients (98%). A total of 21 of 64 patients (33%) had asymmetric muscle weakness. Aspiration pneumonia was seen in 11 of 64 patients (17%), and 5 of 64 patients (8%) required mechanical ventilation. Seven of 64 patients (11%) developed cardiac abnormalities, and 5 of 64 patients (8%) developed neurologic abnormalities. Asymmetric muscle involvement was detected on computed tomography scans in 6 of 27 patients (22%) and on magnetic resonance imaging scans in 4 of 15 patients (27%), with the soleus and the medial head of the gastrocnemius being the worst affected. All 42 muscle biopsy samples showed rimmed vacuoles. Intranuclear tubulofilamentous inclusions were observed in only 1 of 5 patients.
Conclusions and Relevance
This study suggests that OPDM_LRP12 is the most frequent OPDM subtype in Japan and is characterized by oculopharyngeal weakness, distal myopathy that especially affects the soleus and gastrocnemius muscles, and rimmed vacuoles in muscle biopsy.
This case series identifies and characterizes the clinicopathologic features of patients with oculopharyngodistal myopathy with CGG repeat expansion in LRP12.
Introduction
Oculopharyngodistal myopathy (OPDM) is a rare, clinicopathologically defined, hereditary muscle disease that was first described in 4 Japanese families by Satoyoshi and Kinoshita in 1977.1 Since then, nearly 200 patients of different ethnicities with OPDM have been reported.2,3,4,5,6,7,8,9,10,11,12 Patients with OPDM typically exhibit late-onset, slowly progressive ptosis, ophthalmoplegia, dysphagia, dysarthria, and facial muscle weakness, in addition to predominantly distal muscle involvement, and show rimmed vacuoles on muscle pathologic findings.1,7 In 2019, the expansion of CGG repeats in the noncoding region of LRP12 (OMIM 618299) was reported as a cause of OPDM.13 Subsequently, CGG repeat expansions in the 5′ untranslated region of GIPC1 (OMIM 605072) and NOTCH2NLC (OMIM 618025) were also reported as causative for OPDM.14,15,16,17 In this article, OPDM associated with the CGG repeat expansions in LRP12, GIPC1, and NOTCH2NLC will be referred to as OPDM_LRP12, OPDM_GIPC1, and OPDM_NOTCH2NLC, respectively. To our knowledge, clinicopathologic features of OPDM_LRP12 have not been well described. Therefore, we aimed to identify patients with OPDM_LRP12 and characterize their clinical and histopathologic features.
Methods
Inclusion Criteria
The National Center of Neurology and Psychiatry functions as a referral center in Japan for patients with muscular diseases. Among the patients whose samples were sent to the National Center of Neurology and Psychiatry from January 1, 1978, to December 31, 2020, for diagnostic purposes, we searched for patients who were suspected to have OPDM or oculopharyngeal muscular dystrophy (OPMD) based on oculopharyngeal weakness and/or rimmed vacuoles detected in muscle biopsy samples. We excluded patients with GCN repeat expansions in PABPN1 (OMIM 602279). A total of 208 Japanese patients from 198 unrelated families were screened for CGG repeat expansion in LRP12. Among these, muscle biopsies were available from 112 families, and rimmed vacuoles were observed in 65 patients. The remaining 86 families received a clinical diagnosis of OPDM or OPMD (Figure 1).14,15 The National Center of Neurology and Psychiatry ethical committee approved this study; all participants provided written informed consent.
Figure 1. Screening of the Patients.

A total of 198 families with clinical or clinicopathologic diagnoses of oculopharyngodistal myopathy (OPDM) or oculopharyngeal muscular dystrophy (OPMD) were screened for CGG repeat expansion. Patients with GCN repeats in PABPN1 were excluded. Muscle biopsy samples were available from 112 families, and the samples from 65 families showed rimmed vacuoles. Among the samples with rimmed vacuoles, repeat expansion was identified in the samples from 54 families (42 in LRP12, 5 in GIPC1, and 7 in NOTCH2NLC). No sequence variant was identified in 47 families that did not show rimmed vacuoles in muscle biopsy samples.
aPatients with OPDM_GIPC1 and OPDM_NOTCH2NLC have been previously reported and described.14,15
Genetic Analysis
The OPDM_LRP12 in 19 patients was previously diagnosed using repeat-primed polymerase chain reaction (RP-PCR)13; the remaining patients had OPDM_LRP12 that was diagnosed by Southern blot analysis, which detects CGG repeat expansions in LRP12.13 The CGG repeats in GIPC1 and NOTCH2NLC were also evaluated by RP-PCR, fragment analysis, and/or Southern blot analysis.14,15
Clinical Information
The clinical information of patients with confirmed OPDM_LRP12 was rereviewed from the records maintained in the muscle repository. For comparison, we used the clinical data of 10 previously described patients with OPDM_GIPC1 and 7 previously described patients with OPDM_NOTCH2NLC,14,15 in addition to 1 recently identified woman with OPDM_GIPC1 (onset at age 62 years; 93 CGG repeats). We reviewed laboratory test information, including results of brain magnetic resonance imaging (MRI; n = 32) and creatine kinase level. Asymmetric muscle involvement is clinically defined by at least 1 of the following symptoms: apparent asymmetric ptosis or facial muscle weakness, 2-grade side-to-side difference on the manual muscle testing scale, and 2-fold difference in grip strength.
Imaging Data
Axial muscle computed tomography and MRI data of the lower extremities were available for 27 and 15 patients, respectively. Each side of the midthigh and distal muscles in the same patient was evaluated separately. The areas of signal hyperintensity in the MRI T1W sequences and the hypodense lesions on computed tomography scans were interpreted as areas of adipose tissue replacement. The extent of adipose tissue replacement and the distribution in muscles were evaluated by staging with a modified Mercuri scale.18,19 Asymmetric involvement was judged by comparing side-to-side modified Mercuri scale score differences. A difference of at least 2 points in at least 1 muscle was regarded as asymmetry.20
Histologic Findings
Muscle biopsy was performed for 42 patients. Although various biopsy sites were chosen by physicians for diagnostic purposes, the biceps brachii muscle was most frequently biopsied (26 of 42 [62%]) (eTable 1 in the Supplement). A battery of histochemical tests was performed for all biopsy samples. Immunohistochemistry, including that for neonatal myosin heavy chain and anti-p62/SQSTM1, was performed for 38 and 20 samples, respectively. For 4 patients with OPDM_LRP12, immunohistochemistry for SUMO-1, phospho-p62/SQSTM1 (Ser351), poly-ubiquitinated proteins, and caveolin-3 antibodies was also performed. An immunohistochemical protocol was applied as previously described.15 Glutaraldehyde-fixed, Epon-embedded samples from 5 patients were analyzed by electron microscopy.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism, version 5.03 for Windows (GraphPad Software). One-way analysis of variance with the Tukey post hoc test and the Fisher exact test was used to ascertain the differences between OPDM_LRP12, OPDM_GIPC1, and OPDM_NOTCH2NLC. A coefficient of determination was used to find the association between the expansion size of CGG repeats and the age at onset. The t test was performed to ascertain the association between the 2 variables. All P values were from 1-sided tests and results were deemed statistically significant at P < .05.
Results
Genetic Analysis and Identification of Patients With OPDM_LRP12
We identified a total of 65 patients (59 families) with OPDM_LRP12. Southern blot analysis was performed for 60 patients, while the remaining 5 patients were diagnosed only by use of RP-PCR owing to low amounts of DNA. The estimated repeat size in 60 patients ranged from 85 to 289 repeats (vs 13-45 repeats in control individuals13). Eight patients had mosaicism in CGG repeats (eTable 2 in the Supplement).
Clinical Findings
Thirty-three of 59 probands (56%) were sporadic. First-degree relatives in different generations were affected in 11 of 59 probands (19%). Fifteen of 59 probands (25%) had affected siblings or distant relatives, suggesting autosomal dominant inheritance (eTable 2 in the Supplement). Among the 52 paitents with OPDM_LRP12 who did not have mosaicism, a negative correlation was observed between the repeat size and the age at onset (r2 = 0.188; P = .001) (Figure 2A). However, the father (family 1, I-1) of patient 14 (family 1, II-3) and patient 27 (family 1, II-1), harboring mosaicism (336 and 364 repeats, respectively), did not show any symptoms (Figure 2B and C). In addition, the sibling of patient 58 (family 2, III-4) who harbored 63 CGG repeats was asymptomatic (Figure 2D and E).
Figure 2. Genetic Analysis of Patients With OPDM_LRP12.
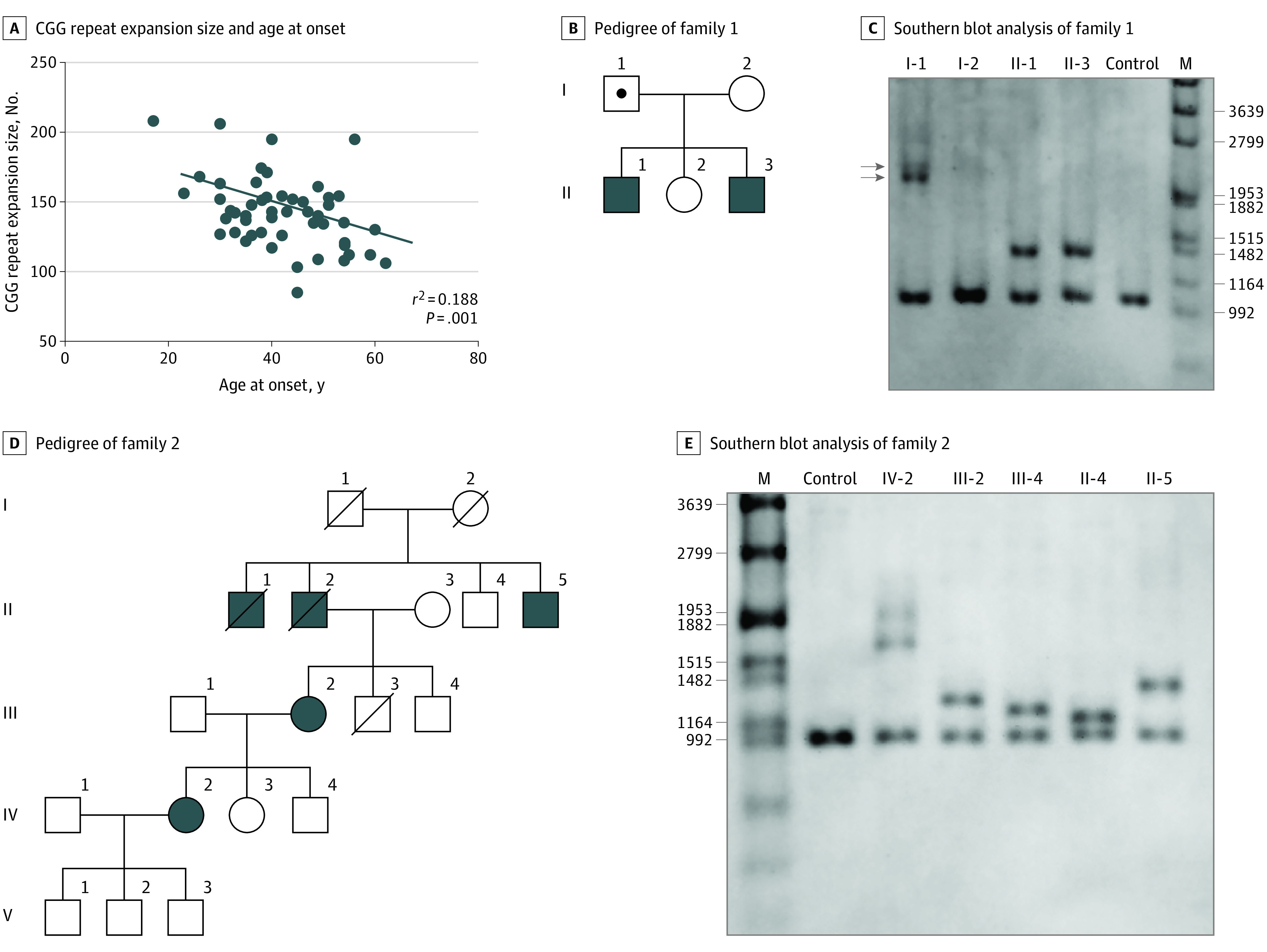
A, Regression analysis between CGG repeat expansion size and the age at onset (r2 = 0.188; P = .001; n = 52). B, Pedigree of family 1. C, Southern blot analysis of family 1; patients 14 (II-3) and 27 (II-1) showed expansions of 144 and 140 CGG repeats, respectively. Their asymptomatic father (I-1) has mosaicism (arrows) and harbors 336 and 364 CGG repeats. Their mother (I-2) does not exhibit CGG expansion. D, Pedigree of family 2. E, Southern blot analysis of family 2; patients 57 (IV-2), 58 (III-2), and 59 (II-5) revealed mosaicism (219 and 289) and expansions of 85 and 127 repeats, respectively. The other 2 family members (III-4 and II-4) without symptoms harbor expansions of 63 and 49 repeats, respectively.
Men (40 patients [62%]) were 1.6 times more frequently represented in the patient group than women (25 patients [39%]) (Table). The mean (SD) age at onset was 41.0 (10.1) years (range, 17-62 years), with first symptoms occurring in patients in their 30s or later (59 patients [91%]). No difference in the mean (SD) age at onset was observed between sexes (men: 40.9 [10.7] years; women: 41.4 [9.1] years; P = .85).
Table. Summary of Clinical Findings of 65 Patients With OPDM_LRP12 Compared With 11 Patients With OPDM_GIPC1 and 7 Patients With OPDM_NOTCH2NLC.
| Clinical finding | No./total No. (%) | P value | |||
|---|---|---|---|---|---|
| OPDM_LRP12 (n = 65) | OPDM_GIPC1 (n = 11) | OPDM_NOTCH2NLC (n = 7) | OPDM_LRP12 and OPDM_GIPC1 | OPDM_LRP12 and OPDM_NOTCH2NLC | |
| Sex | |||||
| Men | 40/65 (62) | 6/11 (55) | 4/7 (57) | .33 | >.99 |
| Women | 25/65 (39) | 5/11 (46) | 3/7 (43) | ||
| Age at onset, mean (SD) [range], y | 41.0 (10.1) [17-62] | 42.4 (13.5) [17-62] | 33.1 (23.5) [1-68] | ||
| <20 | 1/65 (2) | 1/11 (9) | 2/7 (29) | .93 | .23 |
| 20-29 | 5/65 (8) | 2/11 (18) | 2/7 (29) | ||
| 30-39 | 25/65 (39) | 1/11 (9) | 0/7 | ||
| 40-49 | 20/65 (31) | 2/11 (18) | 1/7 (14) | ||
| 50-59 | 11/65 (17) | 4/11 (36) | 1/7 (14) | ||
| ≥60 | 3/65 (5) | 1/11 (9) | 1/7 (14) | ||
| Initial symptom | |||||
| Ptosis | 24/65 (37) | 6/11 (55) | 1/7 (14) | .33 | .41 |
| Dysphagia | 5/65 (8) | 2/11 (18) | 0/7 | .27 | >.99 |
| Dysarthria or nasal voice | 7/65 (11) | 0/11 | 0/7 | .58 | >.99 |
| Limb muscle weakness | 24/65 (37) | 4/11 (36) | 3/7 (43) | >.99 | >.99 |
| Facial weakness | 7/65 (11) | 2/11 (18) | 0/7 | .61 | >.99 |
| Distribution of muscle weakness | |||||
| Distal > proximal | 53/64 (83) | 8/11 (73) | 5/7 (71) | .42 | .60 |
| Proximal > distal | 2/64 (3) | 3/11 (27) | 1/7 (14) | .02a | .27 |
| Distal = proximal | 7/64 (11) | 0/11 | 1/7 (14) | .58 | .58 |
| No weakness | 2/64 (3) | 0/11 | 0/7 | >.99 | >.99 |
| Frequency of main clinical features | |||||
| Ptosis | 62/64 (97) | 11/11 (100) | 7/7 (100) | >.99 | >.99 |
| Ophthalmoplegia | 53/64 (83) | 10/11 (91) | 7/7 (100) | .68 | .59 |
| Bulbar muscle weakness | 63/64 (98) | 10//11 (91) | 7/7 (100) | .27 | >.99 |
| Limb muscle weakness | 63/65 (97) | 11/11 (100) | 7/7 (100) | >.99 | >.99 |
| Facial weakness | 59/64 (92) | 8/11 (73) | 5/7 (71) | .09 | .14 |
| Cardiomyopathy | 2/64 (3) | 0/11 | 0/7 | >.99 | >.99 |
| Ventilator dependent | 5/64 (8) | 0/11 | 0/7 | >.99 | >.99 |
| Loss of ambulation | 14/64 (22) | 0/11 | 1/7 (14) | .11 | >.99 |
| CK level, mean (SD) [range], IU/L | 470 (331) [30-1674] | 606 (637) [11-2146] | 779 (670) [63-1886] | .58 | .16 |
Abbreviations: CK, creatine kinase; OPDM, oculopharyngodistal myopathy.
SI conversion factor: To convert CK to microkatals per liter, multiply by 0.0167.
P < .05.
The Table summarizes the clinical features of patients with OPDM_LRP12 and their comparison with patients with other OPDM subtypes. The 2 most common initial presenting symptoms were limb muscle weakness (n = 24 [37%]) and ptosis (n = 24 [37%]). Fifty-three of 64 patients (83%) showed predominantly distal muscle weakness; however, 2 of 64 patients (3%) presented with predominantly proximal muscle weakness. Both patients who presented with predominantly proximal muscle weakness initially received a diagnosis of OPMD. Ptosis developed during the course of the disease in 62 of 64 patients (97%), bulbar muscle weakness developed in 63 of 64 patients (98%), and limb muscle weakness developed in 63 of 65 patients (97%) patients. Ophthalmoplegia was observed in 53 of 64 patients (83%). The results of physical examination revealed asymmetric muscle involvement in 21 of 64 patients (33%). Fourteen of 64 patients (22%) lost their ambulation within the age range of 40 to 81 years (mean [SD] age, 53.9 [11.0] years), and the disease duration until becoming nonambulatory ranged between 7 and 41 years (mean [SD] duration, 19.4 [9.1] years). The serum creatine kinase level was mildly elevated at a mean (SD) of 470 (331) IU/L (range, 30-1674 IU/L; to convert to microkatals per liter, multiply by 0.0167) in 48 of 62 patients (77%) (eTable 2 in the Supplement).
Fifty-nine of 64 patients (92%) showed no significant central nervous system manifestations. One patient developed dementia in their 80s; another developed idiopathic Parkinson disease in their 70s. Three patients had mild cognitive impairment between 50 and 60 years of age (eTable 2 in the Supplement). Of 32 patients who underwent brain MRI, 22 showed no apparent abnormalities, 8 showed only age-related abnormalities (including mild cortical atrophy), and 2 had a lesion corresponding to a previous ischemic stroke.
Respiratory symptoms were observed in 22 of 64 patients (34%). Five of 64 patients (8%) between 48 and 80 years of age (mean [SD] age, 61.4 [12.9] years) developed respiratory insufficiency that required noninvasive positive pressure ventilation. Three of these patients were ambulatory at the time of becoming ventilator dependent. Tracheostomy and/or laryngectomy was performed for 3 patients. Eleven of 64 patients (17%) had at least 1 episode of aspiration pneumonia (Table; eTable 2 in the Supplement).
Two patients developed cardiomyopathy of undetermined cause. Five patients experienced atrial fibrillation, and 3 of them developed chronic heart failure, second-degree atrioventricular block, and dilated cardiomyopathy, respectively. One patient (patient 19) developed acute heart failure; an echocardiogram after clinical improvement revealed no abnormal ventricular motion (Table; eTable 2 in the Supplement). The remaining 57 of 64 patients (89%) showed no significant cardiac abnormalities.
Muscle Imaging
Distal leg muscles were more seriously affected than the thigh muscles, with mean (SD) modified Mercuri scale scores of 2.34 (0.55) vs 1.59 (0.38) for computed tomography (P < .001) and 2.06 (0.60) vs 1.46 (0.49) for MRI (P < .001). Asymmetric muscle involvement was observed on computed tomography scans of 6 of 27 patients (22%) and on MRI scans of 4 of 15 patients (27%). Among the thigh muscles, the long head of the biceps femoris, the adductor magnus, and the semimembranosus were the most affected, whereas the gracilis, the rectus femoris, and the adductor longus were the least affected (Figure 3A and B).
Figure 3. Muscle Imaging Results of Patients With OPDM_LRP12.
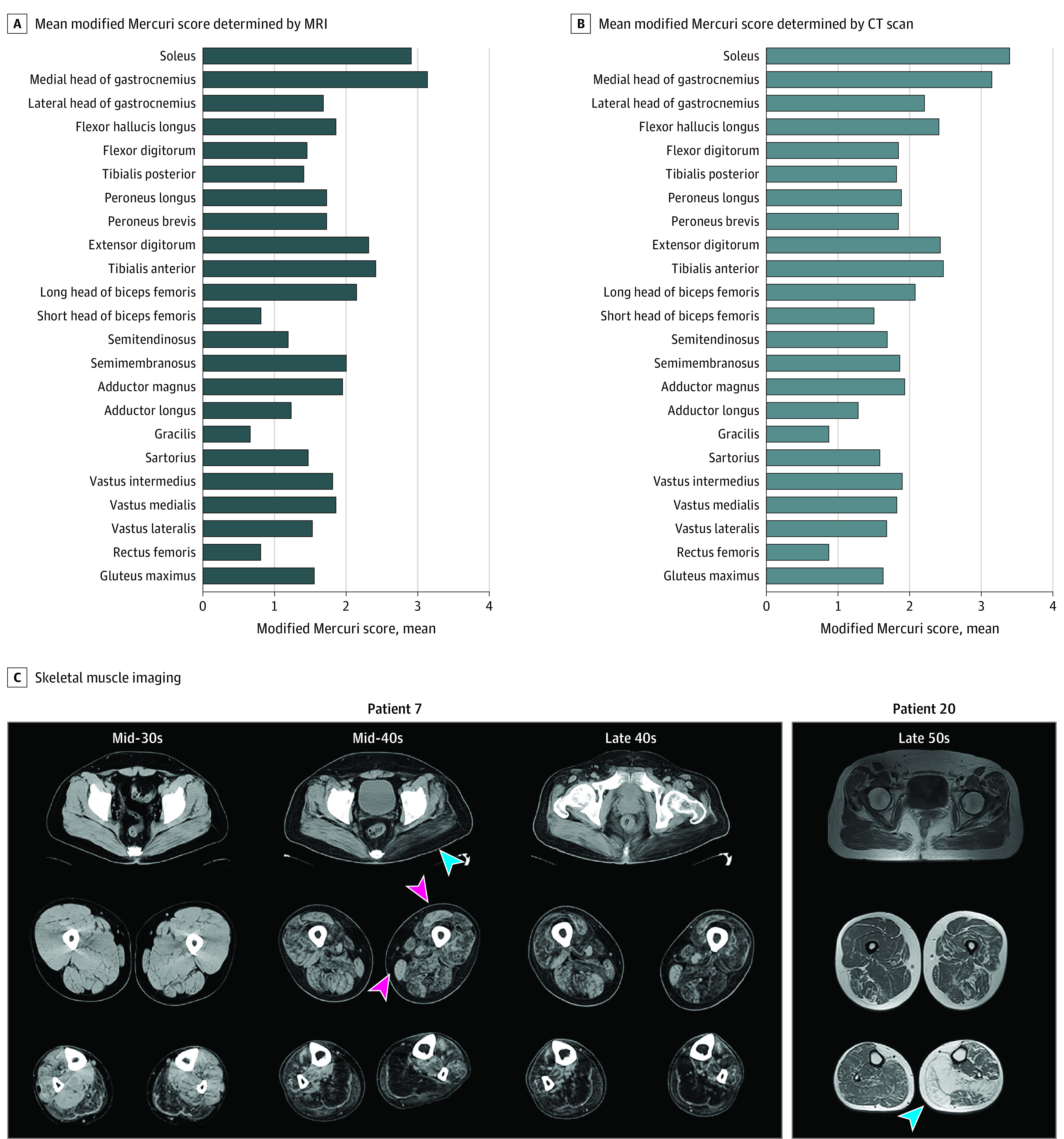
A, Mean modified Mercuri scale score determined by T1-weighted magnetic resonance imaging (MRI) of 15 patients with OPDM_LRP12. B, Mean modified Mercuri scale score determined by computed tomography (CT) for 27 patients with OPDM_LRP12. C, Skeletal muscle imaging for patients with OPDM with LRP12 variations. Muscle imaging scans for patients 7 and 20 are shown. The distal muscles were affected earlier and more severely than the proximal muscles. Asymmetrical involvement was observed in the gluteus muscle and the lower legs (blue arrowheads). In the lower legs, the earliest and most severe involvement was observed in the soleus and gastrocnemius muscles. In the upper legs, the adductor magnus, semimembranosus, and short head of the biceps femoris were more affected, whereas the rectus femoris and gracilis muscles were usually preserved in patients with OPDM_LRP12 (pink arrowheads).
Among the distal leg muscles, the soleus and the medial head of the gastrocnemius were most affected, whereas the tibialis posterior, the flexor digitorum, and the peroneus brevis were the least affected (Figure 3A and B). The chronological imaging data from 1 patient (patient 7) showed that the rectus femoris and gracilis muscles were relatively well preserved, even at the advanced stage (Figure 3C).
Pathologic Findings
Chronic myopathic changes with rimmed vacuoles and small angular fibers were observed in all 42 patients. A few necrotic fibers were observed, but only in 2 of 42 patients (5%). Variable degrees of endomysial fibrosis and fat infiltration were observed, especially during the later stages of the disease. Fibers with internal nuclei were also observed in all 42 patients, and type 2C fibers were observed in 39 of 42 patients (93%). Immunohistochemistry revealed a dotlike deposition of p62, most of which corresponded to rimmed vacuoles, in all 20 patients tested (Figure 4A-C; eTable 3 in the Supplement). Neonatal myosin heavy chain–positive fibers were observed in all 38 patients. Intramyonuclear inclusions were stained by anti-SUMO1, anti–phospho-p62/SQSTM1, and anti–poly-ubiquitinated protein antibodies in all 4 patients (Figure 4D-O).
Figure 4. Histopathologic and Electron Microscopy Findings in Patients With OPDM_LRP12 Patients.

A, B, and C, Biopsies from the left biceps brachii of patient 21, who has had the disease for 8 years, are shown. Scale bar: 20 μm. A, Moderate to marked fiber size variation and fibers with internal nuclei are seen on hematoxylin-eosin stain. B, Fibers with rimmed vacuole and moderate fibrous tissue infiltration are seen on modified Gomori trichrome stain. C, The dotlike deposition of p62 can be observed in muscle fibers. Staining with anti–SUMO-1 antibody (D), anti–caveolin-3 antibody (E, I, M), 4′,6-diamidino-2-phenylindole (F, J, N), anti-p62 (H), and anti–poly-ubiquitinated protein antibody (L). G, K, and O show merged immunohistochemistry. D to O, Scale bar: 10 μm. P and Q, Electron microscopy images of the biopsied left biceps brachii muscle from patient 10, in their late 60s. Scale bars are 1 μm for P and 200 nm for Q: Intranuclear tubulofilamentous inclusions (mean [SD] diameter, 17.3 [1.4] nm) are shown. R and S, Electron microscopy images of the biopsied left deltoid from patient 14, in their late 30s. Scale bars are 1 μm for R and 200 nm for S: Cytoplasmic tubulofilamentous inclusions (diameter, 10 nm) are shown.
All samples analyzed by electron microscopy showed nuclear abnormalities, including hyperchromicity and an irregular, fragmented appearance, albeit only in less than 1% of the nuclei. Tubulofilamentous intranuclear inclusions were observed in only 1 patient (patient 10), and the mean (SD) diameter of the inclusions was 17.3 (1.4) nm. Only patient 14 had cytoplasmic filaments of approximately 10-nm diameter, instead of intranuclear inclusions (Figure 4P-S).
Discussion
We have so far identified 65, 11, and 7 Japanese patients with OPDM_LRP12, OPDM_GIPC1, and OPDM_NOTCH2NLC, respectively (Table).14,15 Therefore, we believe that CGG expansion in LRP12 may be the most common cause of OPDM in Japan, accounting for 78% of patients with genetically confirmed OPDM. Although patients with OPDM have been reported in various countries, OPDM_LRP12 has only been reported from Japan, except for 2 patients from China where OPDM_GIPC1 seems to be the most common subtype.2,3,4,5,6,7,8,9,10,11,12,14,15,16,17 Ishiura et al13 reported that patients with OPDM_LRP12 share a specific haplotype, suggesting a founder effect, which may explain the high frequency of OPDM_LRP12 in Japan.
Approximately 56% of the probands in our cohort were sporadic. However, we believe that the inheritance pattern of OPDM_LRP12 is autosomal dominant because 11 probands had affected relatives in consecutive generations. Furthermore, the father of 1 proband (family 1 I-1) harbored longer CGG repeats than those in the proband but remained asymptomatic. In contrast, genetic anticipation was observed in family 2, in which a daughter had a longer expansion than the mother (patients III-2 and IV-2). The presence of genetic anticipation or incomplete penetrance due to epigenetic factors may explain why many patients have been described as sporadic.21
Among the 65 patients with OPDM_LRP12, men were 1.6 times more frequently represented than women. A similar male predominance was not observed in patients with OPDM_GIPC1 (6 of 11 [55%]) or in patients with OPDM_NOTCH2NLC (4 of 7 [57%]), suggesting that male predominance may be a distinguishing clinical feature of OPDM_LRP12.14,15 Previously, 17β-estradiol was shown to regulate the expression of LRP12 during osteoblast differentiation.22 One plausible hypothesis is that differences in sex hormone levels may affect the penetrance in OPDM_LRP12.
Some patients with OPDM_LRP12 initially presented with proximal muscle weakness, which may be indistinguishable from OPMD. In contrast, some patients with OPMD presented with predominantly distal muscle weakness.23 These results demonstrate the phenotypic overlap between OPDM and OPMD, indicating the necessity of genetic analysis for differentiating patients with OPDM from patients with OPMD.
Respiratory muscle involvement is a complication in several hereditary muscle diseases.24 In the early stage of the disease, none of the patients with OPDM_LRP12 exhibited respiratory symptoms. However, respiratory insufficiency was observed during later stages of the disease. The onset of respiratory compromise may occur before the ambulatory loss that may reflect the notable respiratory muscle involvement in OPDM_LRP12. The prominent weakness of bulbar muscles in patients with OPDM_LRP12 may be associated with aspiration and upper airway obstruction, which further compromises the pulmonary function of these patients. Therefore, attention needs to be given to the pulmonary function of patients with OPDM_LRP12, especially those with advanced stages of the disease.
Asymmetric muscle involvement was diagnosed clinically by physical examination in 33% of patients with OPDM_LRP12 and in 22% to 27% of the patients by imaging. Similar findings were observed in a study from China examining patients with OPDM_GIPC1 (21%) and patients with OPDM_NOTCH2NLC (33%), suggesting that the OPDM subtypes may induce myodegeneration by a similar mechanism.5,15
For patients with OPDM_LRP12, skeletal muscle imaging revealed more prominent involvement of the distal muscles. The soleus and the medial heads of the gastrocnemius muscles were the earliest and most severely affected. This pattern of muscle involvement was similar to that observed in the Chinese cohort of patients with OPDM_GIPC1 or OPDM_NOTCH2NLC, which implies that this pattern of muscle involvement may be common to all or most OPDM subtypes rather than being specific to OPDM_LRP12.5,14,15,16
Cardiac muscles express high levels of LRP12.25 Although 57 of 64 patients (89%) had no cardiac abnormalities, the other patients developed atrial fibrillation, cardiomyopathy, or heart failure, suggesting that cardiac muscles are also affected in a small proportion of patients with OPDM_LRP12. However, any conclusions cannot be drawn at the moment because our study has certain limitations. For example, cardiac function was not fully studied in all patients, which warrants further studies with long-term follow-up.
All patients with OPDM_NOTCH2NLC exhibited central nervous system abnormalities, such as leukodystrophy, hearing loss, and cerebellar symptoms.15 However, most patients (>90%) with OPDM_LRP12 showed no central nervous system abnormalities. The frequency of patients with OPDM_LRP12 and mild cognitive impairment (5%) did not exceed the frequency in the healthy population.26,27 Only 1 patient developed dementia from an unspecified cause at 81 years of age. Ma et al28 reported an association between CGG repeat expansion in NOTCH2NLC and typical idiopathic Parkinson disease. Nonetheless, no definite correlation could be concluded in OPDM_LRP12 because only 1 patient experienced Parkinson disease. In addition, brain MRI scans for 32 patients with OPDM_LRP12 revealed no significant abnormalities except for age-related changes or a previous stroke lesion. Thus, compared with OPDM_NOTCH2NLC, OPDM_LRP12 is more likely to be a pure myopathy.
So far, all patients with genetically confirmed OPDM_LRP12, OPDM_GIPC1, or OPDM_NOTCH2NLC have shown rimmed vacuoles in muscle fibers except for 1 patient from China.2,14,16,29 In our cohort, 83% of patients with OPDM with the presence of rimmed vacuoles had variations in either LRP12, GIPC1, or NOTCH2NLC (Figure 1).14,15 In contrast, all 47 families who were clinically suspected of having OPDM but who lacked rimmed vacuoles in muscle biopsy samples did not have CGG repeat expansions in any of these 3 genes, suggesting that the existence of rimmed vacuoles is a distinctive feature of OPDM compared with its mimics.
Using immunohistochemistry and electron microscopy, we found p62-stained intramyonuclear inclusions in patients with OPDM_LRP12; these were also reported in patients with OPDM_GIPC1 or OPDM_NOTCH2NLC.14,15 Despite being observed in only 1 patient, the intranuclear tubulofilamentous inclusion (mean [SD] diameter, 17.3 [1.4] nm) was larger than the 8.5 nm and 12.6 nm reported for OPMD and OPDM_NOTCH2NLC, respectively.15,30 Furthermore, filaments in OPDM_NOTCH2NLC show an electron light halo around the nucleus, whereas filaments in OPDM_LRP12 do not, suggesting that the inclusion components may differ among OPDM_LRP12, OPMD, and OPDM_NOTCH2NLC.15
The fact that CGG repeat expansions have been found in all 3 OPDM subtypes indicates that the subtypes share a common pathomechanism, albeit the details are unknown. One possible hypothesis may be an RNA-dependent gain-of-function mechanism, which has been described in disorders with repeat expansions in the 5′ untranslated region, such as fragile X–associated tremor/ataxia syndrome and fragile X syndrome.31,32 The expanded CGG repeat may trigger the formation of RNA foci, which sequester RNA-binding proteins and are cytotoxic.33 Alternatively, repeat-associated non-ATG (RAN) translation, which produces the toxic RAN protein, is also pathogenic in fragile X–associated tremor/ataxia syndrome and fragile X syndrome.34 In these conditions, RAN proteins produced from CGG repeats, such as polyglycine, co-localize with the ubiquitinated inclusions.34 Because similar intranuclear inclusions are seen in OPDM, it may be reasonable to hypothesize that RAN proteins are pathogenic in OPDM as well.
Moreover, the father of patient 14 and patient 27 in the present study remained asymptomatic despite having the longest CGG repeat length among the family members. A possible explanation is that a longer expansion beyond a certain length may cause hypermethylation in the promoter region of LRP12, thereby silencing the gene transcription, which would otherwise result in the production of toxic mRNA.35
Triplet repeat disorders commonly show a correlation between disease onset and the repeat length.36,37,38,39,40 In our cohort, a negative correlation between the repeats and the age at onset was observed by regression analysis (r2 = 0.188; P = .001). A similar correlation between repeat length and age at onset was also reported in patients with OPDM_GIPC1 but was not observed in those with OPDM_NOTCH2NLC.14,15,16 This observation suggests that OPDM_LRP12 may have a pathogenic mechanism more similar to that of OPDM_GIPC1 than to that of OPDM_NOTCH2NLC.
Limitations
This study has some limitations. First, detailed clinical information, including cardiac function and long-term follow-up data, was not available from all patients, which may have influenced the results of this study. Second, this is a single-center study and thus needs confirmation by other groups. Third, the pathomechanism of the disease was not investigated.
Conclusions
This study suggests that OPDM_LRP12 is the most frequent OPDM subtype in Japan. It is clinicopathologically characterized by predominant gastrocnemius and soleus muscle involvement, in addition to oculopharyngeal weakness and rimmed vacuoles.
eTable 1. List of Biopsied Muscles in 42 OPDM_LRP12 Patients
eTable 2. Detailed Clinical Findings of 65 OPDM_LRP12 Patients From 59 Families
eTable 3. Summary of Histopathological Findings in 42 OPDM_LRP12 Patients
References
- 1.Satoyoshi E, Kinoshita M. Oculopharyngodistal myopathy. Arch Neurol. 1977;34(2):89-92. doi: 10.1001/archneur.1977.00500140043007 [DOI] [PubMed] [Google Scholar]
- 2.Durmus H, Laval SH, Deymeer F, et al. Oculopharyngodistal myopathy is a distinct entity: clinical and genetic features of 47 patients. Neurology. 2011;76(3):227-235. doi: 10.1212/WNL.0b013e318207b043 [DOI] [PubMed] [Google Scholar]
- 3.Minami N, Ikezoe K, Kuroda H, Nakabayashi H, Satoyoshi E, Nonaka I. Oculopharyngodistal myopathy is genetically heterogeneous and most cases are distinct from oculopharyngeal muscular dystrophy. Neuromuscul Disord. 2001;11(8):699-702. doi: 10.1016/S0960-8966(01)00227-9 [DOI] [PubMed] [Google Scholar]
- 4.Lu H, Luan X, Yuan Y, Dong M, Sun W, Yan C. The clinical and myopathological features of oculopharyngodistal myopathy in a Chinese family. Neuropathology. 2008;28(6):599-603. doi: 10.1111/j.1440-1789.2008.00924.x [DOI] [PubMed] [Google Scholar]
- 5.Zhao J, Liu J, Xiao J, et al. Clinical and muscle imaging findings in 14 mainland Chinese patients with oculopharyngodistal myopathy. PLoS One. 2015;10(6):e0128629. doi: 10.1371/journal.pone.0128629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uyama E, Uchino M, Chateau D, Tomé FM. Autosomal recessive oculopharyngodistal myopathy in light of distal myopathy with rimmed vacuoles and oculopharyngeal muscular dystrophy. Neuromuscul Disord. 1998;8(2):119-125. doi: 10.1016/S0960-8966(98)00002-9 [DOI] [PubMed] [Google Scholar]
- 7.Tomé FM, Chateau D, Helbling-Leclerc A, Fardeau M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord. 1997;7(suppl 1):S63-S69. doi: 10.1016/S0960-8966(97)00085-0 [DOI] [PubMed] [Google Scholar]
- 8.van der Sluijs BM, ter Laak HJ, Scheffer H, van der Maarel SM, van Engelen BG. Autosomal recessive oculopharyngodistal myopathy: a distinct phenotypical, histological, and genetic entity. J Neurol Neurosurg Psychiatry. 2004;75(10):1499-1501. doi: 10.1136/jnnp.2003.025072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SY, Lim JK, Park YE, Lee CH, Kim HS, Kim DS. Oculopharyngodistal myopathy in three unrelated Korean patients: early distal limb weakness is a characteristic finding in oculopharyngodistal myopathy. Neurol Asia. 2011;16(4):315-320. [Google Scholar]
- 10.Mignarri A, Carluccio MA, Malandrini A, et al. The first Italian patient with oculopharyngodistal myopathy: case report and considerations on differential diagnosis. Neuromuscul Disord. 2012;22(8):759-762. doi: 10.1016/j.nmd.2012.03.010 [DOI] [PubMed] [Google Scholar]
- 11.Jaspar HH, Bastiaensen LA, ter Laak HJ, Joosten EM, Horstink MW, Stadhouders AM. Oculopharyngodistal myopathy with early onset and neurogenic features. Clin Neurol Neurosurg. 1977;80(4):272-282. doi: 10.1016/S0303-8467(78)80018-3 [DOI] [PubMed] [Google Scholar]
- 12.Amato AA, Jackson CE, Ridings LW, Barohn RJ. Childhood-onset oculopharyngodistal myopathy with chronic intestinal pseudo-obstruction. Muscle Nerve. 1995;18(8):842-847. doi: 10.1002/mus.880180807 [DOI] [PubMed] [Google Scholar]
- 13.Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. 2019;51(8):1222-1232. doi: 10.1038/s41588-019-0458-z [DOI] [PubMed] [Google Scholar]
- 14.Deng J, Yu J, Li P, et al. Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am J Hum Genet. 2020;106(6):793-804. doi: 10.1016/j.ajhg.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogasawara M, Iida A, Kumutpongpanich T, et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol Commun. 2020;8(1):204. doi: 10.1186/s40478-020-01084-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xi J, Wang X, Yue D, et al. 5′ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain. 2021;144(2):601-614. doi: 10.1093/brain/awaa426 [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Deng J, Guo X, et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain. 2021;awab077. doi: 10.1093/brain/awab077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007;25(2):433-440. doi: 10.1002/jmri.20804 [DOI] [PubMed] [Google Scholar]
- 19.Engelke K, Museyko O, Wang L, Laredo JD. Quantitative analysis of skeletal muscle by computed tomography imaging—state of the art. J Orthop Translat. 2018;15:91-103. doi: 10.1016/j.jot.2018.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasca G, Monforte M, Ottaviani P, et al. Magnetic resonance imaging in a large cohort of facioscapulohumeral muscular dystrophy patients: pattern refinement and implications for clinical trials. Ann Neurol. 2016;79(5):854-864. doi: 10.1002/ana.24640 [DOI] [PubMed] [Google Scholar]
- 21.Longshore J, Tarleton J. Dynamic mutations in human genes: a review of trinucleotide repeat diseases. J Genet. 1996;75(2):193-217. doi: 10.1007/BF02931762 [DOI] [Google Scholar]
- 22.Gui Y, Duan Z, Qiu X, et al. Multifarious effects of 17-β-estradiol on apolipoprotein E receptors gene expression during osteoblast differentiation in vitro. Biosci Trends. 2016;10(1):54-66. doi: 10.5582/bst.2016.01006 [DOI] [PubMed] [Google Scholar]
- 23.Nakashima D, Nakajima H, Ishida S, Sugino M, Kimura F, Hanafusa T. Preferential distal muscle involvement in case of oculopharyngeal muscular dystrophy with (GCG) 13 expansion. Article in Japanese. Rinsho Shinkeigaku. 2003;43(9):560-563. [PubMed] [Google Scholar]
- 24.Shahrizaila N, Kinnear WJ, Wills AJ. Respiratory involvement in inherited primary muscle conditions. J Neurol Neurosurg Psychiatry. 2006;77(10):1108-1115. doi: 10.1136/jnnp.2005.078881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battle MA, Maher VM, McCormick JJ. ST7 is a novel low-density lipoprotein receptor-related protein (LRP) with a cytoplasmic tail that interacts with proteins related to signal transduction pathways. Biochemistry. 2003;42(24):7270-7282. doi: 10.1021/bi034081y [DOI] [PubMed] [Google Scholar]
- 26.Ward A, Arrighi HM, Michels S, Cedarbaum JM. Mild cognitive impairment: disparity of incidence and prevalence estimates. Alzheimers Dement. 2012;8(1):14-21. doi: 10.1016/j.jalz.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 27.Das SK, Bose P, Biswas A, et al. An epidemiologic study of mild cognitive impairment in Kolkata, India. Neurology. 2007;68(23):2019-2026. doi: 10.1212/01.wnl.0000264424.76759.e6 [DOI] [PubMed] [Google Scholar]
- 28.Ma D, Tan YJ, Ng ASL, et al. Association of NOTCH2NLC repeat expansions with Parkinson disease. JAMA Neurol. 2020;77:1559-1563. doi: 10.1001/jamaneurol.2020.3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito R, Shimizu H, Miura T, et al. Oculopharyngodistal myopathy with coexisting histology of systemic neuronal intranuclear inclusion disease: clinicopathologic features of an autopsied patient harboring CGG repeat expansions in LRP12. Acta Neuropathol Commun. 2020;8(1):75. doi: 10.1186/s40478-020-00945-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomé FM, Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980;49(1):85-87. doi: 10.1007/BF00692226 [DOI] [PubMed] [Google Scholar]
- 31.Oostra BA, Willemsen R. A fragile balance: FMR1 expression levels. Hum Mol Genet. 2003;12(Spec No 2):R249-R257. doi: 10.1093/hmg/ddg298 [DOI] [PubMed] [Google Scholar]
- 32.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74(5):805-816. doi: 10.1086/386296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin P, Zarnescu DC, Zhang F, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39(5):739-747. doi: 10.1016/S0896-6273(03)00533-6 [DOI] [PubMed] [Google Scholar]
- 34.Cleary JD, Pattamatta A, Ranum LPW. Repeat-associated non-ATG (RAN) translation. J Biol Chem. 2018;293(42):16127-16141. doi: 10.1074/jbc.R118.003237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oostra BA, Willemsen R. FMR1: a gene with three faces. Biochim Biophys Acta. 2009;1790(6):467-477. doi: 10.1016/j.bbagen.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richard P, Trollet C, Stojkovic T, et al. ; Neurologists of French Neuromuscular Reference Centers CORNEMUS and FILNEMUS . Correlation between PABPN1 genotype and disease severity in oculopharyngeal muscular dystrophy. Neurology. 2017;88(4):359-365. doi: 10.1212/WNL.0000000000003554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799-808. doi: 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- 38.Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. 1993;4(4):398-403. doi: 10.1038/ng0893-398 [DOI] [PubMed] [Google Scholar]
- 39.Budworth H, McMurray CT. A brief history of triplet repeat diseases. Methods Mol Biol. 2013;1010:3-17. doi: 10.1007/978-1-62703-411-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulson H. Repeat expansion diseases. Handb Clin Neurol. 2018;147:105-123. doi: 10.1016/B978-0-444-63233-3.00009-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. List of Biopsied Muscles in 42 OPDM_LRP12 Patients
eTable 2. Detailed Clinical Findings of 65 OPDM_LRP12 Patients From 59 Families
eTable 3. Summary of Histopathological Findings in 42 OPDM_LRP12 Patients