

Abstract
Purpose
To evaluate the pharmacokinetics (PK) of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in mice after IV and oral administration and to develop mechanistic PK models to assess the potential enterohepatic circulation (EHC) and target-mediated drug disposition (TMDD) of AZD3965.
Methods
Female BALB/c mice were administered AZD3965 by IV injection (10, 50 and 100 mg/kg) or oral gavage (100 mg/kg). Plasma samples were analyzed using LC/MS/MS, and PK parameters determined by compartmental and non-compartmental analyses.
Results
AZD3965 exhibited a large volume of distribution and rapid oral absorption, with a high oral bioavailability. Prominent reentry peaks were observed after both oral and IV administration, suggesting potential EHC of AZD3965 or of a potential glucuronide conjugate. The dose-dependent studies indicated greater than proportional increases in exposure, an increase in the terminal half-life, and decrease in clearance and volume of distribution with increasing IV doses, indicating nonlinear pharmacokinetics and potential TMDD of AZD3965. Mechanistic compartmental models were developed to characterize the complex pharmacokinetics of AZD3965.
Conclusions
The current study represents the first comprehensive report of the pharmacokinetics of AZD3965 in mice, indicating the potential contribution of EHC and TMDD in the disposition of AZD3965.
Keywords: AZD3965, target-mediated drug disposition, enterohepatic cycling, monocarboxylate transporters, pharmacokinetics
INTRODUCTION
The transport of lactic acid across the plasma membrane is mediated by four proton-linked monocarboxylate transporters, MCT1–4, which are part of the solute carrier SLC16A gene family (1–4). Soon after the discovery that MCTs are involved in lactic acid influx and efflux from cells in the early 1980s, it was recognized that MCTs could serve a potential druggable target in cancer (5,6). With the large production of lactic acid from the glycolytic phenotype of some cancer cells, MCTs facilitate the transport of lactate and proton out of the cells, which has been recognized as one of the mechanisms contributing to cancer cell survival (7,8). Blocking the transport of lactate out of cells can result in the accumulation of lactate and protons inside cells, thereby inducing acidosis and apoptosis. Multiple other downstream effects of blocking lactate efflux have been proposed in recent years, which include restoring cancer immunosurvillance, since lactic acid is considered as the main immunosuppressive metabolite in the tumor microenvironment (9–11). Early studies with nonspecific inhibitors of MCTs such as α-cyano-4-hydroxy-cinnamic acid (CHC) and silencing RNA have provided proof of concept that inhibiting MCTs result in reduction in tumor growth (12,13).
More recently, a potent and selective class of MCT1 inhibitors was discovered by AstraZeneca (14). One of which is AZD3965 (5-[(4S)-4-hydroxy-4-methyl-1,2-oxazolidine-2-carbonyl]-1-isopropyl-3-methyl-6-{[3-methyl-5-(trifluoromethyl)-2H-pyrazol-4-yl]methyl}thieno[2,3-d]pyrimidine-2,4-dione, Fig. 1) an orally bioavailable inhibitor of MCT1, that is currently being investigated in a Phase I clinical trial in the UK for advanced solid tumors and lymphomas (NCT01791595). AZD3965 is part of a series of pyrrole pyrimidine derivatives initially developed as novel immunosuppressive agents to inhibit T lymphocyte proliferation (15). It was later discovered that these compounds are involved in inhibiting MCT1-mediated lactic acid efflux during T lymphocyte proliferation (14–16). AZD3965 is a potent inhibitor of MCT1 with a Ki value of 1.6 nM (17), with some activity against MCT2, but to a lesser extent. AZD3965 has been demonstrated to decrease cell and tumor growth in human small cell lung cancer, Raji lymphoma, diffuse large B cell lymphoma, Burkitt’s lymphoma and breast cancer xenograft models overexpressing MCT1 (17–23).
Fig. 1.
Chemical structure of AZD3965.
To our knowledge, the pharmacokinetics of AZD3965 has not been previously reported in the literature. A clinical report of patients with advanced solid tumors administered AZD3965 at total daily doses of 5–30 mg given orally once and twice daily indicated that plasma concentrations were in the preclinical efficacy range, but detailed data were not provided (24). The objectives of our current study were: (1) to investigate the pharmacokinetics and oral bioavailability of AZD3965 in mice; (2) to develop pharmacokinetic models to mechanistically characterize and assess the plasma concentration-time profiles of AZD3965; and (3) to simulate the physicochemical and ADME properties of AZD3965. The research described here will support the design and implementation of further studies to assess the therapeutic potential of AZD3965 in preclinical cancer xenograft models, and may allow for translation to humans.
MATERIALS AND METHODS
Chemicals and Reagents
AZD3965 (>98% purity) was obtained from MedKoo Biosciences (Chapel Hill, NC) and AR-C155858 (the internal standard; ≥ 98% purity) was purchased from ChemScene (Monmouth Junction, NJ). All other reagents used in the studies were purchased from Sigma-Aldrich (St. Louis, MO).
Animals
Female, BALB/c mice (Envigo, Indianapolis, IN) weighting 18–20 g were used for the experiments. Animals were housed in standard vinyl cages. Water and food provided ad libitum. Animals were maintained under the standard 12-h light/dark cycle at 22–24°C. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University at Buffalo.
Pharmacokinetic Study
Animals were acclimated to their environment for 2 weeks prior to the study. AZD3965 was administered at doses of 10, 50 and 100 mg/kg intravenously (IV) by tail vein injection. One additional group of animals was given 100 mg/kg of AZD3965 orally (po) by oral gavage. AZD3965 stock solutions (1, 5 and 10 mg/mL) were prepared in 20% w/v cyclodextrin in normal saline. All the animals had free access to food and water before and after administration of AZD3965. Blood samples of approximately 0.7–0.8 mL were collected from each mouse (n = 3–4 mice per time point) by terminal cardiac puncture while the animals were anesthetized with isoflurane. Lithium heparin was used as the anticoagulant and blood samples were collected at 10, 20, and 30 min, and 1, 2, 3, 6, 9, 12, and 24 h after oral administration and at 15, and 30 min, and 1, 2, 3, 6, 9, 12, and 24 h after IV administration. Plasma samples were stored at −80°C until analysis.
Plasma Sample Preparation and LC/MS/MS Analysis
AZD3965 plasma concentrations were measured using a recently developed and validated liquid chromatography coupled to tandem mass spectrometry (LC/MS/MS) assay (25). Briefly, samples were prepared by adding 5 μL of internal standard (I.S.) solution containing the AZD3965 analogue, AR-C155858 (15 and 200 ng/mL for low and high standard curve, respectively) to 35 μL of sample. To ensure the concentrations quantified were below the upper limit of the high standard curve, part of the plasma samples were diluted 10-fold with blank plasma prior to the sample preparation. Plasma standards and quality controls were prepared by adding 5 μL of I.S. (15 and 200 ng/mL for low and high standard curve, respectively) and 5 μL of stock solution containing AZD3965 to 30 μL of blank plasma. Plasma proteins were precipitated by the addition of 600 μL of 0.1% formic acid in acetonitrile. Samples were vortexed, followed by centrifugation at 10,000×g for 20 min at 4°C. Then 540 μL of supernatant was collected and evaporated under a stream of nitrogen gas, followed by reconstitution in 200 and 1000 μL of acetonitrile/water (40/60, v/v) for low and high standard curve, respectively.
All the analyses were performed using Shimadzu Prominence HPLC with binary pump and autosampler (Shimadzu Scientific, Marlborough, MA) connected to a Sciex API 3000 triple quadruple tandem mass spectrometer with utilizing Atmospheric Pressure Chemical Ionization source (APCI) (Sciex, Foster City, CA). The data was analyzed using Analyst version 1.4.2 (Sciex, Foster City, CA).
Chromatographic separation was conducted by injecting 4 or 15 μL of the sample on to an Xterra MS C18 column (250 × 2.1 mm i.d., 5-μm particle size; Waters, Milford, MA) for the low or high calibration curve, respectively. Mobile phase A consisted of acetonitrile/water (5/95, v/v) with 0.1% acetic acid and mobile phase B was acetonitrile/water (95/5, v/v) with 0.1% acetic acid. The flow rate was 250 μL/min with a gradient elution profile and a total run time of 15 min. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode with APCI for specific detection of AZD3965 and the I.S. by measuring the characteristic ion transition. Q1/Q3 m/z ratio for the precursor/product ion of AZD3965 and AR-C155858 was 516.4/413.2 and 462.3/373.2, respectively. The retention time of AZD3965 and AR-C155858 (I.S.) were 5.77 and 4.91 min, respectively. The chromatograph conditions, mass spectrometer parameters, linear calibration ranges, accuracy/precision, dilution integrity and recovery are detailed previously (25). The standard curves were linear over the concentration range of 0.15–500 and 100–12,000 ng/mL with r2 > 0.999. The lower and upper limit of quantification of AZD3965 was 0.15 and 12,000 ng/mL, respectively.
Non-compartmental Pharmacokinetic Analysis
The naïve-pooled AZD3965 plasma data were analyzed by non-compartmental (NCA) and compartmental analysis using Phoenix WinNonlin version 7.0 (Pharsight, Mountain View, CA) and ADAPT V (BMSR, University of South California, Los Angeles CA), respectively. For NCA, the area under the plasma concentration-time curve (AUC) was determined using the trapezoidal method, with AUC values extrapolated to infinity. The terminal slope, λz represents the slope of the terminal regression line. Vss represents the volume of distribution estimated at steady state. The maximal concentration after oral or IV administration (Cmax) was estimated by NCA and confirmed by visual inspection. The oral bioavailability (F) of AZD3965 after IV and oral administration from the 100 mg/kg dosing group was calculated by the following equation:
| (1) |
where, AUC0−∞ is the AUC from time zero to infinity.
Pharmacokinetic Modeling for AZD3965
Based on the initial PK assessment and NCA, mechanistic models were constructed to describe the PK of AZD3965 after IV and oral administration (Fig. 2), incorporating potential enterohepatic cycling (EHC) and nonlinearity behavior with either Michaelis-Menten (M-M) elimination or target-mediated drug disposition (TMDD). Model A (Fig. 2a) describes the EHC by first-order release rate from the bile to the absorption site with M-M elimination while Model B (Fig. 2b) simplifies the EHC process with zero-order release rate from the bile to the systemic circulation. Model C (Fig. 2c) accounts for potential TMDD, describing the high-affinity, slowly reversible and saturable binding of AZD3965 to its specific pharmacological target, MCT1 (26). The differential equations derived from the pharmacokinetic models were as follows:
Fig. 2.
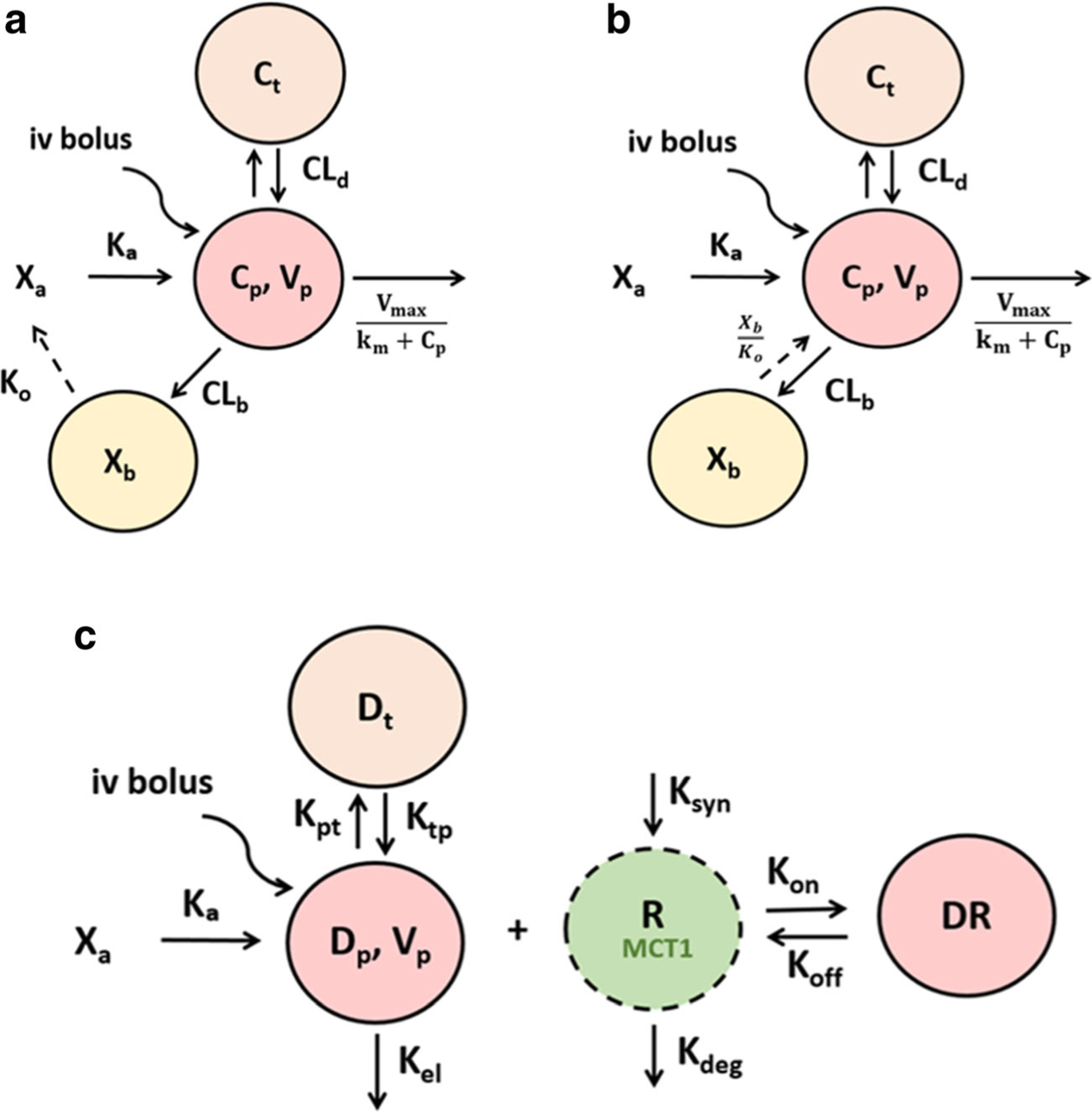
The schematic representation of the pharmacokinetic models of AZD3965 following IV and oral administration. Model A describes the EHC by first-order release rate constant from the bile to the absorption site with M-M elimination a. Model B simplifies the EHC process with zero-order release rate from the bile to the systemic circulation b. The key feature of Model C is potential target-mediated drug disposition (TMDD), where AZD3965 binds to its target, MCT1 on the cell surface (kon) to form the drug-transporter complex c. AZD3965 can dissociate from the transporter (koff). The free transporter is subject to turnover, which is characterized a zero-order production rate constant (ksyn) and first-order degradation rate constant (kdeg).
Model A (Fig. 2a). For oral administration:
| (2) |
| (3) |
| (4) |
| (5) |
For IV administration:
Where, Xa represents the drug amount at the absorption site; ka is the first-order absorption rate constant, Cp and Ct represent AZD3965 concentration in the central and peripheral compartment, respectively; Vp and Vt represent the volume of distribution of AZD3965 in the central and peripheral compartment, respectively; CLD is the distributional clearance from and to the central compartment; Vmax and km are the M-M parameters to describe AZD3965 nonlinear elimination from the central compartment; CLb is AZD3965 clearance from the central compartment to the bile compartment; Xb represents the drug amount in the bile compartment; ko is the first-order release rate constant of drug from the bile compartment back to the systemic circulation in Model A; tlag represents the lag time in the EHC process.
Model B (Fig. 2b). For oral administration:
| (6) |
| (7) |
| (8) |
| (9) |
For IV administration:
| (10) |
Where, ko is the zero-order release rate constant of the drug from the bile compartment back to the systemic circulation in Model B; tlag.iv and tlag.po represent the lag time in the EHC process. The subscripts, (iv) and (po) indicate the intravenous and oral administration routes.
Model C (Fig. 2c). For oral administration:
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
For IV administration:
| (16) |
Where, Dp and Dt are amount of drug in the central and tissue compartment, respectively; Rtotal represents transporter (i.e. MCT1) concentration; DR represents drug-transporter complex concentration; kel is the first-order elimination rate constant from the central compartment; kpt and ktp are first-order distribution rate constant from central and to tissue compartment and back, respectively; kon describes the second-order binding rate constant of the drug to the transporter; koff describes the first-order dissociation rate constant of the drug from the transporter; ksyn and kdeg are zero-order synthesis and first-order degradation rate constant of the transporter, respectively that would describe the physiological turnover of the MCT1 transporter.
The initial parameter estimates for Model A (Fig. 2a) and B (Fig. 2b) were obtained from NCA. Since there is no prior knowledge of drug (i.e. kon, koff) and system (i.e. ksyn, kdeg) properties of AZD3965, the initial parameter estimates for Model C (Fig. 2c) were based on the general feature of a drug that exhibits TMDD (27) which are as follows: Assuming linear PK at high doseiv, kel, kpt, ktp and Vp were obtained from fitting the data to a two-compartment model. Assuming free target, Rfree = Rtotal − DR, then at high doseiv DR approaches Rtotal and therefore, Rtotal was obtained from the high doseiv data. Assuming at a low doseiv, the initial rapid distribution phase is mainly governed by binding of the drug to its target, then, from the slope of the initial distribution phase, λ0, the kon can be approximately estimated as λ0 → − (kel + kon · Rtotal). From the NCA analysis at low doseiv, Vss was used to estimate koff as , where kD is the dissociation equilibrium constant and can be expressed as .
The pharmacokinetic models were simultaneously fitted to plasma concentration data for both the IV and oral administration using the Maximum Likelihood method. Naïve-pooled data from all replicate at each time point were analyzed since the PK data were obtained from terminal sampling and were being pooled together using the following variance model:
| (17) |
where, σ1 and σ2 are the variance parameters and Y is the model predicted output. The goodness of fit was assessed by visual inspection, diagnostic plots, precision of the parameter estimates (CV%), Akaike’s Information Criterion (AIC), and Schwarz Criterion (SC).
Physicochemical and ADME Prediction for AZD3965
For predicting the physicochemical and ADME properties of AZD3965, MedChem Designer Version 4.5.0.12 (Simulations Plus Inc. Lancaster, CA) was used which is based on the software’s built-in algorithm. AZD3965 chemical structure was loaded into MedChem Designer in the ADMET Predictor Module via the SIMILES to obtain the physicochemical and ADME properties.
RESULTS
Pharmacokinetics of AZD3965 Following Oral Administration
The time course of AZD3965 plasma concentrations following oral administration (100 mg/kg) is shown in Fig. 3a. AZD3965 concentrations could be detected in all plasma samples collected up to 24 h. AZD3965 exhibited rapid absorption with tmax occurring at 20 min. The oral bioavailability of AZD3965 calculated from AUC data was 0.827 (Table 1). From the oral profile of AZD3965, a reentry peak was visible and occurred at between 6 to 9 h (Fig. 3a), suggesting potential EHC of AZD3965.
Fig. 3.
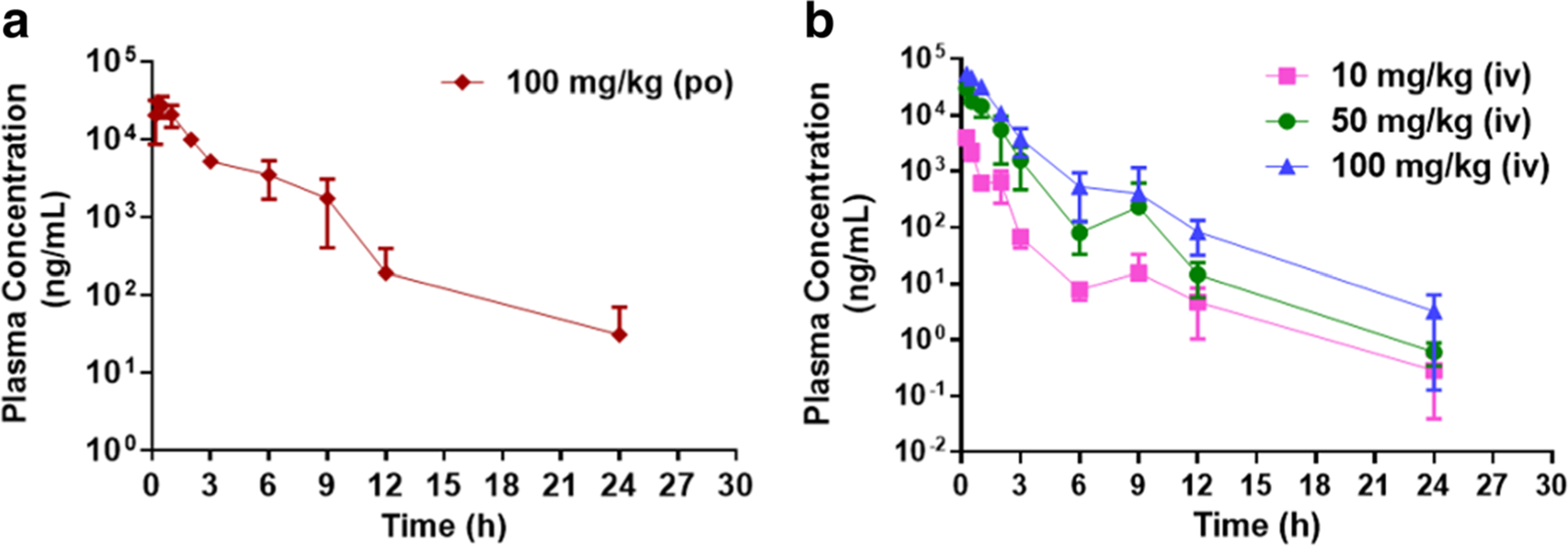
The plasma concentration profile of AZD3965 after oral a and intravenous b administration. Mice were dosed IV with 10, 50, or 100 mg/kg or orally with 100 mg/kg of AZD3965. Data are expressed as mean ± SD, n = 3–4.
Table 1.
Mean pharmacokinetic parameters for AZD3965 after intravenous and oral administration from non-compartmental analysis (NCA). The average weight of female Balb/c mice, 19 g was incorporated for the analysis
| Parameter | Intravenous |
Oral | ||
|---|---|---|---|---|
| 10 mg/kg | 50 mg/kg | 100 mg/kg | 100 mg/kg | |
| λz (h−1) | 0.238 | 0.266 (1.12) | 0.275 (1.16) | 0.156 |
| t1/2 (h) | 2.91 | 2.60 (0.89) | 2.51 (0.87) | 4.46 |
| Cmax (μg/ml) | 4.02 | 30.1 (7.49) | 54.5 (13.6) | 30.3 |
| AUC0−∞ (μg.h/ml) | 4.94 | 41.3 (8.36) | 86.4 (17.5) | 71.4 |
| CL (ml/h) | 46.4 | 23.0 (0.50) | 22 (0.47) | N/A |
| CL/F (ml/h) | 56.1 | 27.8 | 26.6 | 26.6 |
| Vss (ml) | 44.2 | 25.6 (0.58) | 28.6 (0.65) | N/A |
| Vc/F (ml) | 53.4 | 31.0 | 34.6 | 171 |
| F | N/A | N/A | N/A | 0.827 |
(): fold change from the lowest IV dose, 10 mg/kg; N/A: not applicable
Pharmacokinetics of AZD3965 Following Intravenous Administration
The time course of AZD3965 plasma concentrations following IV administration is shown in Fig. 3B. AZD3965 concentrations could be detected in all plasma samples collected after 24 h from all the IV dose groups. However, by 24 h after the low dose, the plasma concentrations (average concentration of 0.285 ng/mL) approached the lower limit of quantitation (0.15 ng/mL). AZD3965 showed at least a biexponential disposition following intravenous dosing. The dose-normalized plasma concentration-time profiles (Fig. 4a) were not superimposable, suggesting nonlinearity in the PK of AZD3965 in mice. Additionally, from the NCA, there were greater than proportional increases in exposure (i.e. AUC and Cmax) with dose. Both systemic clearance and Vss decreased with dose, appearing to reach a plateau at higher doses; although the fold changes were small (Table 1, Fig. 4b and c). The terminal slope (λz) increased and seemed to be capacity limited with increasing dose (Table 1 and Fig. 4d). These observations suggest the possibility of TMDD in the disposition of AZD3965. Similar to oral PK profile of AZD3965, the reentry peaks occurring at 6 to 9 h were present at all dose levels (Fig. 3b), suggesting potential EHC of AZD3965.
Fig. 4.
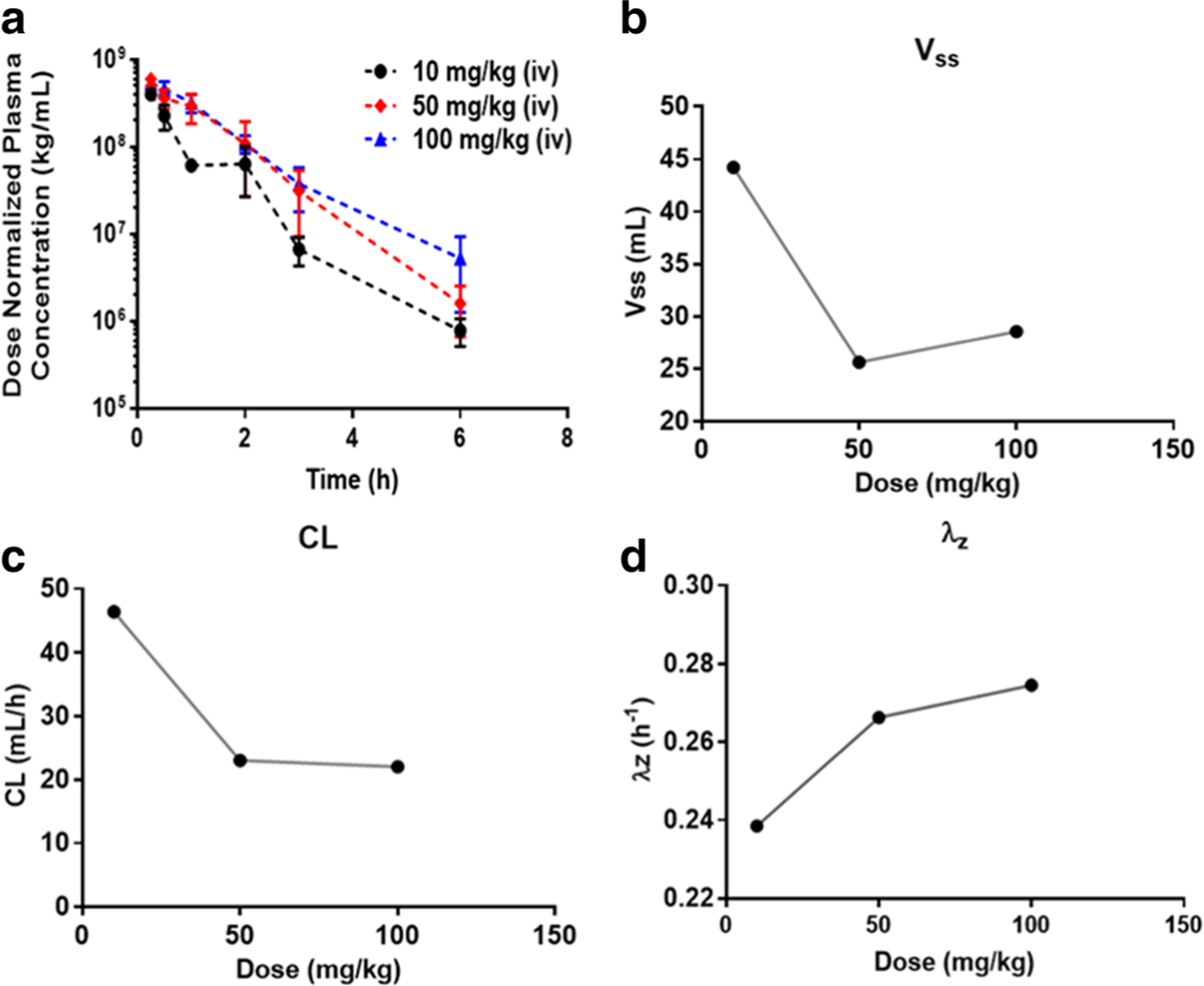
The dose-normalized plasma concentration of AZD3965 in mice after IV administration of doses of 10, 50 or 100 mg/kg a. The effect of increasing AZD3965 dose on the volume of distribution; Vss b, elimination clearance; CL c and terminal slope; λz d where the symbols represent the mean parameter estimate obtained from NCA, Table 1. Data are presented as mean ± SD, n = 3–4.
Compartmental Pharmacokinetics Modeling of AZD3965
Based on the initial PK assessment of AZD3965, different mechanistic models were constructed to simultaneously fit the plasma concentration profiles following IV and oral administration. In model development, one-, two-, and three-compartment models with linear or M-M elimination was examined. A two-compartment model with M-M elimination was chosen as the base model.
Since reentry peaks were observed at all dose levels following both IV and oral administration, we incorporated a bile compartment to the base model to describe potential EHC process. We evaluated the reabsorption of AZD3965 from the absorption site (i.e. gut) to the systemic circulation and directly to the systemic circulation by zero- or first- order reabsorption rates with a lag time or a sine function to represent the periodical changes as described by Wajima et al. (28) and Moon et al. (29). Model A describes the EHC process by the release of bile (first-order rate constant) from the gallbladder to the gut prior to the reabsorption of the drug and/or its conjugate back to the systemic circulation with a time lag (Fig. 2a). Tlag in Model A was initially estimated and fixed to 5.92 h in the final estimation, as it remains largely unchanged during the model converging process. In Model B, a simplified EHC process was proposed whereby the release of bile (zero-order rate constant) allows the drug and/or its conjugate to directly circulate back to the systemic circulation bypassing the gut (Fig. 2b). Two separate lag times were used to better describe the EHC following IV and oral doses. Both model fitting and the diagnostic plots for Model A and Model B are presented in Figs. 5 and 6, respectively. The final parameter estimates and their corresponding CV%, estimated sigma variance terms (σ), AIC and SC values are summarized in Table 2. Overall, based on the goodness- of-fit criteria, both models provide similar overall fits and reasonably capture both intravenous and oral data with Model B being able to predict the peak and trough of the reentry peaks. From the observed versus predicted diagnostic plots (Fig. 5e and 6e), both models capture the data well except for a few high concentrations where the models over predicted. The plot of standardized residuals versus model predicted concentrations in model B (Fig. 5f) shows less biased distribution than model A, indicating reasonable fittings (Fig. 6f). The final estimates for km and Vmax differ substantially between Model A and B. The estimated km value from Model A (9.59 μg/ml) is within the observed concentration range from our studies, indicating a better estimate than Model B.
Fig. 5.
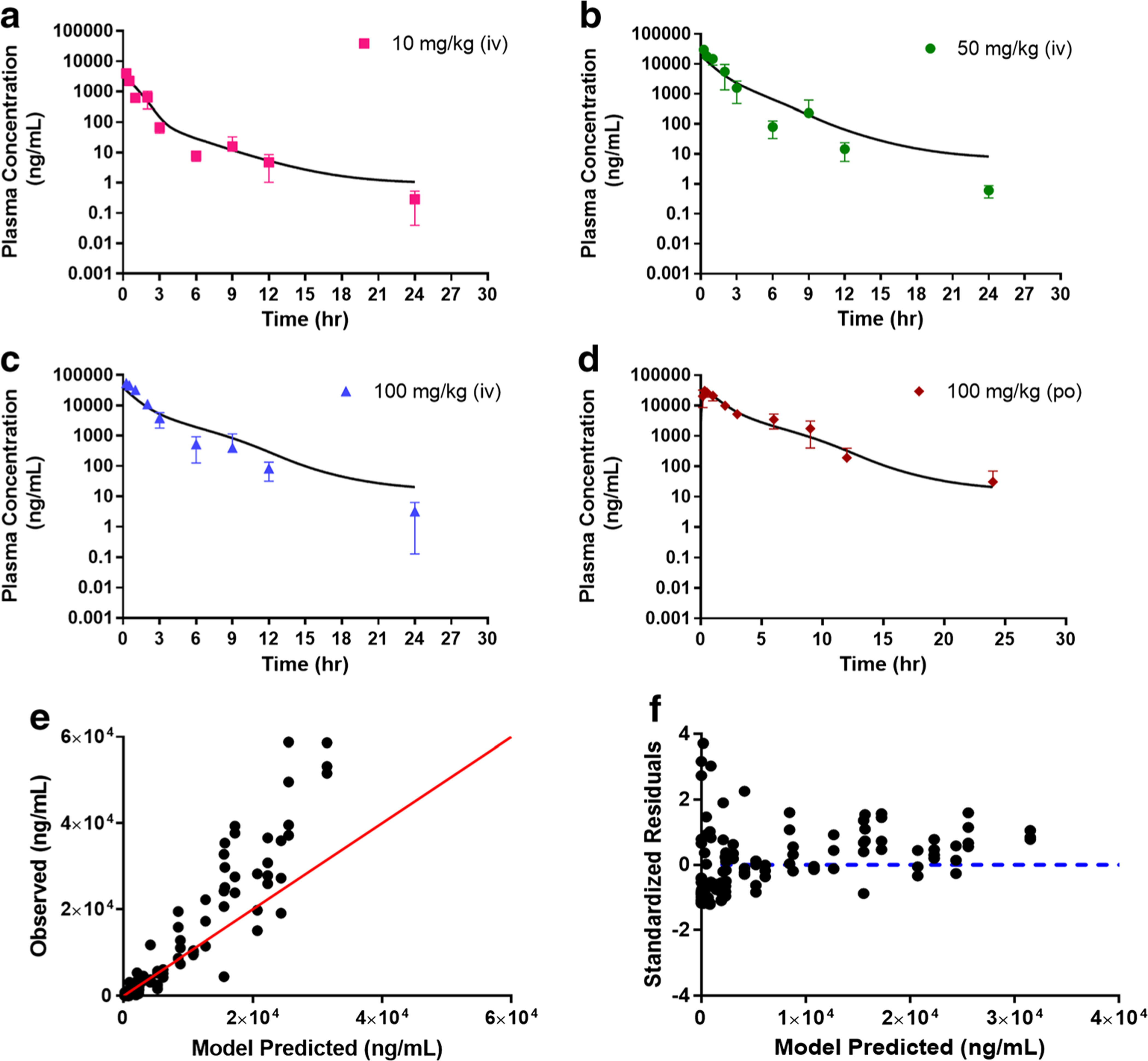
Simultaneous Model A fitting of the plasma AZD3965 concentration-time profile following intravenous (10 a, 50 b, and 100 mg/kg c) and oral (100 mg/kg d) administration. Observed versus predicted plot e and residual plot f. The proposed Model A accounts for potential EHC occurring from the release of bile to the absorption site and back to the systemic circulation with M-M elimination and tlag. Data are presented as mean ± SD, n = 3–4. Symbols depict the observed mean data and the lines represent the model fitted results.
Fig. 6.
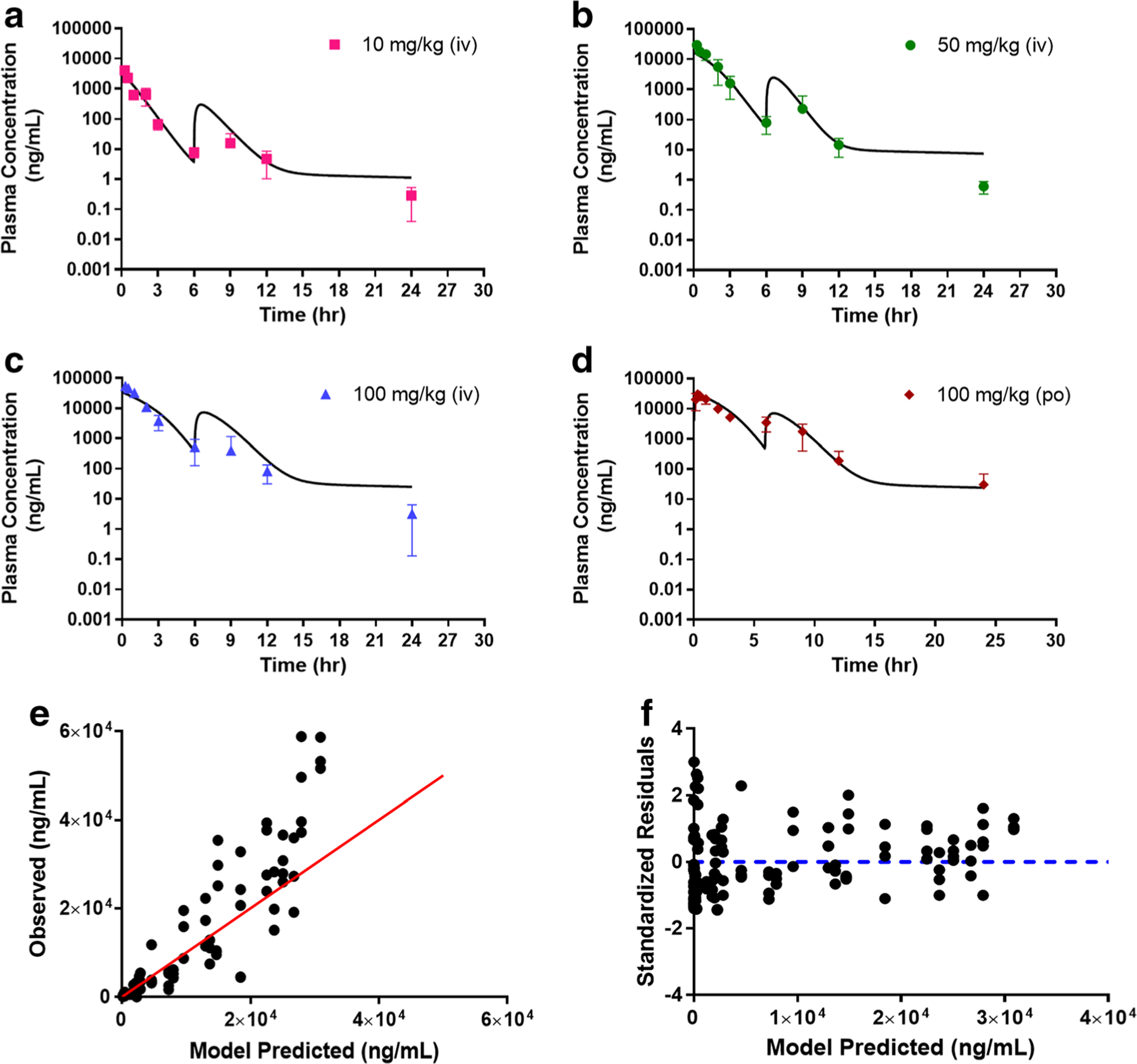
Simultaneous Model B fitting of the plasma AZD3965 concentration-time profile following intravenous (10 a, 50 b, and 100 mg/kg c) and oral (100 mg/kg d) administration. Observed versus predicted plot e and residual plot f. The proposed Model B simplifies potential EHC process through the release of bile directly back to the systemic circulation with tlag. Data are presented as mean ± SD, n = 3–4. Symbols depict the observed mean data and the lines represent the model fitted results.
Table 2.
Model A, B and C final parameter estimates for AZD3965 obtained from simultaneous model fitting of IV and oral data
| Parameter | Mean ± SD (CV%) |
||
|---|---|---|---|
| Model A | Model B | Model C | |
| Vp (ml) | 48.6 ± 5.86 (12.1) | 55.8 ± 5.20 (9.35) | 2.04 (Fixed) |
| ka (h−1) | 3.39 ± 1.11 (32.7) | 5.13 ± 1.98 (38.6) | 34.4 ± 4.80 (14.0) |
| F | 1.00 ± 0.003 (0.32) | 0.998 ± 0.003 (0.31) | 1.00 (Fixed) |
| CLd (ml/h) | 0.332 ± 0.064 (19.3) | 0.023 ± 0.003 (11.1) | N/A |
| Vmax (μg/h) | 26.4 ± 8.30 (31.7) | 411 ± 42.0 (10.3) | N/A |
| km (μg/ml) | 9.59 ± 4.63 (48.3) | 385 ± 58.0 (15.0) | N/A |
| CLb (ml/h) | 0.529 ± 0.074 (13.9) | 0.178 ± 0.019 (10.8) | N/A |
| ko (ng/h or h−1) | 0.0015 ± 0.0004 a (29.0) | 0.386 ± 0.146 b (37.9) | N/A |
| Tlag (h) | 5.92 (Fixed) | N/A | N/A |
| Tlag.iv (h) | N/A | 6.00 ± 0.0006 (0.01) | N/A |
| Tlag.po (h) | N/A | 5.90 ± 0.06 (1.05) | N/A |
| kpt (h−1) | N/A | N/A | 0.302 ± 0.074 (24.7) |
| ktp (h−1) | N/A | N/A | 0.452 ± 0.041 (9.12) |
| kel (h−1) | N/A | N/A | 1.13 ± 0.09 (7.87) |
| kon (μM−1 h−1)c | N/A | N/A | 6.21 ± 5.92 (95. 3) |
| Rtotal (μg/ml) | N/A | N/A | 54.8 ± 4.90 (8.92) |
| koff (h−1) | N/A | N/A | 23.08 ± 3.19 (13.8) |
| σ: 0.816 (9.67%) | σ: 0.686 (9.08%) | σ: 0.806 (9.37%) | |
| intercept: 0.1 (Fixed) | intercept: 0.176 (Fixed) | intercept: 0.01 (Fixed) | |
| AIC: 2179.74; SC: 2205.62 | AIC: 2120.25; SC: 2151.88 | AIC: 2149.08; SC: 2172.09 | |
: first-order rate constant;
: zero-order rate constant;
: kon was converted to μM−1 • h−1 based on the molecular weight of AZD3965; SD:standard deviation; CV%: percent coefficient of variation, N/A: not applicable
Model A: EHC model; Model B: simplified EHC model; Model C: full TMDD model
To investigate whether incorporating TMDD would further improve the model, we proposed Model C (Fig. 2c), incorporating high-affinity, slowly reversible and saturable binding of AZD3965 to the pharmacological target, MCT1 (26). In developing the TMDD model, implementing the quasi-equilibrium approximation method as described by Mager and Krzyzanski (30), did not improve the overall model fit (data not shown); therefore, a full TMDD model was proposed. As expected incorporating the potential internalization or the removal of drug-transporter complex process via endocytosis, a process that is more commonly anticipated with biologics than small molecule drugs, did not improve the overall model fit (data not shown). Thus, this component was not included in the proposed TMDD model. We also incorporated a bile compartment into Model C to account for potential EHC and it did not improve the overall model fit (data not shown).
The simultaneous fitting for Model C is shown in Fig. 7 and the final parameter estimates are summarized in Table 2. The overall fitting of the model is reasonable as demonstrated by the observed versus model predicted diagnostic plot (Fig. 7e) and in the precision of the overall parameter estimates. There is some biased distribution in the residual plot (Fig. 7f) and the kon was estimated with some uncertainty, which is likely due to the lack of knowledge of the target binding kinetics, as well as the limited availability of low dose and oral dose data. In the model converging process, the oral bioavailability, F and Vp remained largely unaffected and thus, subsequently fixed to 1 and 2.04 ml, respectively, in our final analysis to better characterize the data and reducing the number of parameters in the model.
Fig. 7.
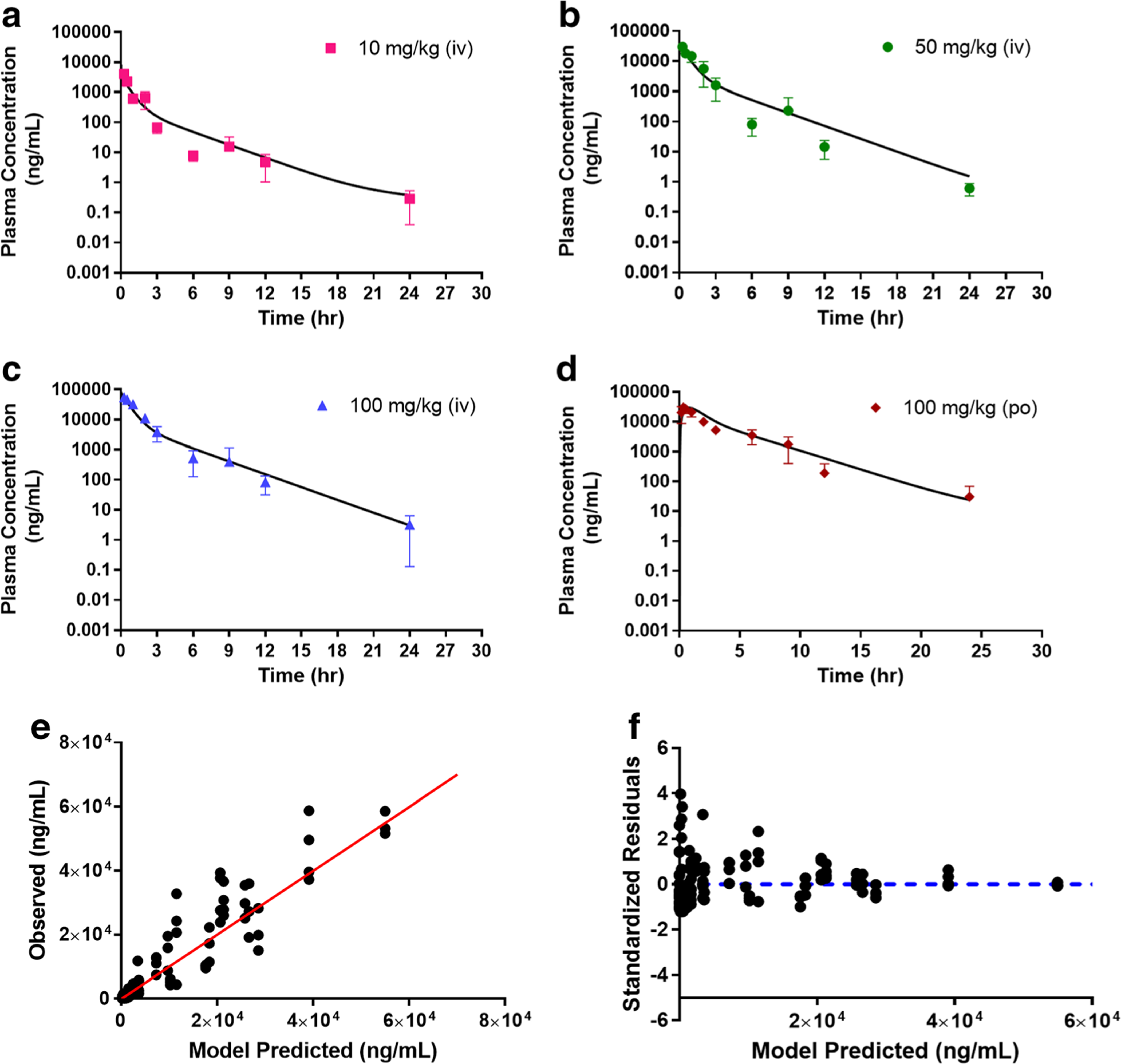
Simultaneous Model C fitting of the plasma AZD3965 concentration-time profile following intravenous (10 a, 50 b, and 100 mg/kg c) and oral (100 mg/kg d) administration. Observed versus predicted plot e and residual plot f. The proposed Model C imparts nonlinearity by potential high-affinity and saturable binding of AZD3965 to its target, MCT1 to form a drug-transporter complex (TMDD behavior). Data are presented as mean ± SD, n = 3–4. and the lines represent the model fitted results.
To further evaluate our TMDD model, we compared the Model C fitting to a two-compartment model with M-M kinetic and linear clearance (Suppl. Fig. 1a; Model D), a frequently applied model that has been shown to fit the TMDD kinetic data well or sometime preferred over a full TMDD model, as shown by Yan et al. (31). Here the M-M kinetics was used as an approximation for TMDD behavior. As shown by the fitting and the observed versus predicted plot in Suppl. Fig. 1, Model D (dashed line) poorly captured the data compared to our proposed TMDD model (solid line). Since the PK at higher doses (50 and100 mg/kg) appears to approach linearity, we also retrospectively compared our proposed TMDD model (Model C) to a two-compartment model with linear clearance (Model E, Suppl. Fig. 2a) that we had tested during our model development. Overall the Model E prediction almost closely overlapped with that of the proposed TMDD model except for the low dose (10 mg/kg) where Model E clearly over predicted (Suppl. Fig. 2). For the parameter estimates, Model D and E were estimated with fewer parameters and good CV% (Suppl. Table 1). Among Model C, D and E, the proposed TMDD model (Model C) appears to be superior based on AIC and SC values (Table 2 and Suppl. Table 1).
Taken together, based on AIC and SC values the models can be ranked as follows: Model B < Model C < Model A < Model D < Model E. While Model B provides the best fit based on AIC and SC values, Model A predicted more reasonable parameter estimates for km than Model B. Unlike the EHC models (Model A and B), the proposed TMDD model (Model C) was able to capture both the terminal IV and oral data well (Fig. 7). Overall, through model assessment and comparison, the proposed TMDD model appears to give the second best-fitting suggesting potential relevance of TMDD in the disposition of AZD3965.
Prediction of Physicochemical and ADME Properties of AZD3965
The prediction of physicochemical and ADME properties of AZD3965 by MedChem Designer are summarized in Table 3. The simulation is based on the built-in algorithm of the software and provided initial assessment into the PK properties of AZD3965. Based on the chemical structure, AD3965 predicted log P of 1.78, pKa of 9.95, and fraction unbound in plasma (fup) of 0.217 and 0.0622 in rat and human, respectively. From this analysis, AZD3965 was indicated to be a substrate for UGT2B7 with high probability (100%). The chance that that AZD3965 is also a substrate for p-glycoprotein (P-gp) was 58%, and a substrate for CYP3A4 was 98%, respectively.
Table 3.
The physicochemical and absorption, distribution, metabolism and elimination (ADME) properties of AZD3965, predicted from MedChem Designer. The prediction was based on the chemical structure of AZD3965. % represents the chance AZD3965 will be a substrate for the transporter or metabolizing enzyme
| Name | AZD3965 |
|---|---|
| pKa | 9.95 |
| Log P | 1.78 |
| Human fup (%) | 6.22 |
| Rat fup (%) | 21.7 |
| Pgp substrate | Yes (58%) |
| CYP3A4 substrate | Yes (98%) |
| UGT2B7 substrate | Yes (100%) |
DISCUSSION
AZD3965 represents the first-in-class monocarboxylate transporter 1 (MCT1) inhibitor that is under evaluation in a Phase I clinical trial in the UK for solid tumor and lymphoma treatment, based on its chemotherapeutic properties assessed in various preclinical xenograft animal models. (17–23). Limited data from this study has been reported (24) and the pharmacokinetics, physicochemical and ADME properties of AZD3965 remain largely unknown. In the present work, we performed pharmacokinetic studies of AZD3965 following IV and oral administration in mice, and developed mechanistic PK models to further characterize the potential EHC and TMDD properties of AZD3965.
Our results demonstrated that AZD3965 exhibited good oral bioavailability, rapid absorption, and nonlinear elimination and distribution, and may be involved in EHC. Analysis revealed that the terminal slope (λz), systemic clearance and Vss change with increasing dose. Both systemic clearance and Vss decreased, whereas the terminal slope increased with dose. At higher doses of 50 and 100 mg/kg, the profiles became more superimposable, with smaller changes in clearance and volume of distribution, suggesting that the PK of AZD3965 approached linearity at higher doses. These findings are consistent with the expected behavior for a drug that exhibits target-mediated disposition. It has been previously proposed by Dr. Gerhard Levy in 1994 that “potent and specifically acting drugs are substances that are bound to with high affinity to pharmacologic target sites such as receptors and enzymes” (32) and transporters (27) are likely to exhibit pharmacologic TMDD.
Although compared to biologics that are known to exhibit TMDD behavior, TMDD with small-molecule drugs is somewhat less anticipated. However, with the growing development of new small-molecule drugs that act potently on highly specific targets, the prevalence of TMDD in small-molecule drugs is expected to increase (33–35). Therefore, given that AZD3965 represents a potent and selective inhibitor of MCT1 with high binding affinity (17), TMDD behavior of AZD3965 may be anticipated. Additionally, we have shown that AZD3965 exhibited rapid inhibition in cellular L-lactate uptake studies (maximal inhibition occurred after a 5 min-pre-incubation period) and the inhibition was prolonged up to 5 h in a murine 4T1 breast tumor cell line expressing only MCT1 (26). We also demonstrated that inhibition by AZD3965 was slowly reversible. After the removal of AZD3965 from washing the cells with ice-cold buffer, inhibition of L-lactate uptake was only completely reversed after 12 h (26). Through PK modeling of the current data, we were able to demonstrate the potential relevance of TMDD in the disposition of AZD3965 (Model C) as shown by reasonable overall data fitting, despite some uncertainty in the kon estimate, which is likely due to a lack of AZD3965 binding and low dose data. Additionally, the estimated total transporter concentration (Rtotal = 54.8 ± 4.90 μg/ml) was very close to the Cmax (54.5 μg/ml) at the highest dose, 100 mg/kg IV, where the system appears to be saturated and approaches linearity, indicating adequate fit. Of note, Vp which was initially estimated and fixed to 2.04 ml, approaches blood volume in mice which is one of the general feature seen for drugs exhibiting TMDD (34). Here a full TMDD model was needed, as opposed to the other TMDD approximation models, to better capture the data. Together these results support the hypothesis that the high affinity binding of AZD3965 to its target site, MCT1 could likely impact its disposition in vivo. Although the current TMDD modeling suggests internalization of AZD3965-MCT1 complex via endocytosis upon binding is unlikely, further experiments using antibody labeling and biotinylation approaches are needed to verify this. Additionally, pharmacokinetic studies in a wide range of doses and in MCT1 knockout animals are needed in order to fully discern the potential TMDD properties (27,33) and guide the PK exposure and efficacy for a clinically relevant dose.
The prominent reentry peaks (occurring at 6–9 h) present in the plasma concentration-time profiles after both IV and oral dosing, suggest that AZD3965 may be involved in EHC. EHC occurs when a compound and/or its conjugate undergoes biliary excretion and is reabsorbed back into the systemic circulation from the small intestine (29,36). Compounds that are susceptible to EHC often have a high (MW >450) molecular weight (36). This is the case with AZD3965 as its molecular weight of 515.5 is above the general MW threshold cut-off (375–400 and 425 in rats and humans, respectively) for a drug that undergoes biliary excretion (36). Based on the simulated physicochemical properties of AZD3965, the compound is likely a substrate for CYP3A4 and UGT2B7. Consistent with our simulation results, a previous report in a series of analogues of AZD3965 has indicated that these compounds are prone to metabolism by glucuronidation on the hydroxyl group as well as cytochrome P450 metabolism (14). Furthermore, our simulation also predicted that AZD3965 could be a substrate of P-gp (a ABC transporter present at the canalicular membrane), which may be responsible for the efflux of AZD3965 into the bile. This simulation provided an initial assessment of the metabolism and transport properties of AZD3965; future studies are necessary to verify these predictions. Taken together, these results provided evidence that AZD3965 itself could undergo EHC, or it may undergo biliary excretion as a glucuronide conjugate, with hydrolysis and reabsorption of the parent compound.
Based on the initial assessment of AZD3965 kinetic profiles, physiologically relevant EHC models were also constructed to characterize the kinetic behavior, where the nonlinearity was described by M-M elimination (Model A and B). Model B simplified the EHC process whereby the release of bile (zero-order rate constant) allows the drug and/or its conjugate to directly circulate back to the systemic circulation bypassing the gut. Based on the goodness- of-fit criteria, both models provide similar overall fits and reasonably capture data with Model A providing a more reasonable km estimate that is within the observed concentration range. The EHC modeling suggests that the presence of re-entry peaks may be contributing to the changes that occurred in the PK parameters across the dose groups. As the larger doses of AZD3965, the changes in the PK parameters (Vss, CL and terminal slope) were small; 0.47–1.16 fold change as the dose increased (Table 1). One potential reason why the fold-changes in PK parameters were small could be attributed to saturation of MCT1 over the current dose range. However, it is also possible that the periodic recycling of the parent compound back to the systemic circulation results in a shallower terminal slope and changes in the half-life.
The bioavailability (F) estimated across models agrees well with the calculated value (0.827) and is supported by the results of a previous report (17). Overall the models predicted rapid oral absorption but the ka varies substantially across models and this is most likely due to the limited oral dose data available. The high values of Vss were consistently estimated throughout our models but vary across models; nevertheless, they suggest that AZD3965 may be extensively tissue bound. This finding is also consistent with our breast tumor xenograft study, where we observed a 25.5-fold higher AZD3965 concentration in the 4T1 tumor than in the plasma (25).
Although our simulation study predicted AZD3965 undergoes conjugation by UGT2B7(glucuronidation),Model B assumed the recirculation of the parent compound back to the systemic circulation was rapid and directly from the bile compartment, due to our lack of metabolite data. This approach has been previously used by Wajima et al. (28), as well as used to described the PK of morphine and its glucuronide conjugate that undergoes EHC (37). Our model development was also limited due to the evaluation of only one oral dose and the lack of bile and metabolite data. The potential contribution of EHC on the PK behavior of AZD3965 needs to be further evaluated in animals with bile duct cannulation, as the increase in exposure could have an impact if there is significant EHC occurring at the clinically relevant dose and the extent of the reabsorption from the gut.
CONCLUSIONS
In conclusion, the current study comprehensively evaluated the pharmacokinetics of AZD3965 in mice following both intravenous and oral administration and to computationally assess the physicochemical and ADME properties of AZD3965. The pharmacokinetics analysis of AZD3965 indicated rapid oral absorption, nearly complete oral bioavailability, nonlinear clearance and distribution, and suggested the presence of EHC. The PK of AZD3965 following IV and oral administration was characterized using different PK models to describe the nonlinearity and the reentry peaks observed in the pharmacokinetic profiles. Additionally, our present study also revealed that the terminal slope (λz), systemic clearance and Vss values were similar at higher doses, but differed from the low dose parameters, which suggest that the nonlinear kinetic behavior of AZD3965 may reflect the influence of TMDD, consistent with the high affinity and slowly reversible binding of AZD3965 to MCT1. Further PK studies are necessary to better understand the potential TMDD properties of AZD3965 and involvement of AZD3965 in EHC, which will enable incorporation of more mechanistic modeling to guide the design of preclinical cancer xenograft studies. Finally, the relevance of such information may also allow for potential translation to humans.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Donald E. Mager for his insightful comments and helpful suggestions on the manuscript. This work was funded by the National Institute of Health National Institute on Drug Abuse [grant DA023223]. X.G. was funded in part by an Allen Barnett Fellowship.
ABBREVIATIONS
- AIC
Akaike’s Information Criterion
- AUC
Area under the plasma concentration-time curve
- Cmax
Maximum plasma concentration
- EHC
Enterohepatic cycling
- F
Oral bioavailability
- Ki
Inhibitory constant
- LC/MS/MS
Liquid chromatography coupled to tandem mass spectroscopy
- MCT1
Monocarboxylate transporter 1
- M-M
Michaelis-Menten
- NCA
Non-compartmental analysis
- PK
Pharmacokinetics
- SC
Schwarz Criterion
- TMDD
Target-mediated drug disposition
- Vss
Volume of distribution at steady state
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
Conflict of Interest The authors declare no conflict of interests. This research was performed during the time X.G. was in the Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo. X.G. is currently an employee of AbbVie Inc.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s11095–019-2735-z) contains supplementary material, which is available to authorized users.
REFERENCES
- 1.Morris ME, Felmlee MA. Overview of the proton-coupled MCT (SLC16A) family of transporters: characterization, function and role in the transport of the drug of abuse gamma-hydroxybutyric acid. AAPS J. 2008;10(2):311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Archiv Eur J Physiol. 2004;447(5):619–28. [DOI] [PubMed] [Google Scholar]
- 3.Halestrap AP. The monocarboxylate transporter family–structure and functional characterization. IUBMB Life. 2012;64(1):1–9. [DOI] [PubMed] [Google Scholar]
- 4.Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life. 2012;64(2):109–19. [DOI] [PubMed] [Google Scholar]
- 5.Belt JA, Thomas JA, Buchsbaum RN, Racker E. Inhibition of lactate transport and glycolysis in Ehrlich ascites tumor cells by bioflavonoids. Biochemistry. 1979;18(16):3506–11. [DOI] [PubMed] [Google Scholar]
- 6.Johnson JH, Belt JA, Dubinsky WP, Zimniak A, Racker E. Inhibition of lactate transport in Ehrlich ascites tumor cells and human erythrocytes by a synthetic anhydride of lactic acid. Biochemistry. 1980;19(16):3836–40. [DOI] [PubMed] [Google Scholar]
- 7.Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. Role of monocarboxylate transporters in human cancers: state of the art. J Bioenerg Biomembr. 2012;44(1):127–39. [DOI] [PubMed] [Google Scholar]
- 8.Baltazar F, Pinheiro C, Morais-Santos F, Azevedo-Silva J, Queiros O, Preto A, et al. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol Histopathol. 2014;29(12):1511–24. [DOI] [PubMed] [Google Scholar]
- 9.Kouidhi S, Ben Ayed F, Benammar EA. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol. 2018;9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18(10):1319–30. [DOI] [PubMed] [Google Scholar]
- 11.Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71(22):6921–5. [DOI] [PubMed] [Google Scholar]
- 12.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colen CB, Seraji-Bozorgzad N, Marples B, Galloway MP, Sloan AE, Mathupala SP. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: an in vitro study. Neurosurgery. 2006;59(6):1313–23. discussion 23–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guile SD, Bantick JR, Cheshire DR, Cooper ME, Davis AM, Donald DK, Evans R, Eyssade C, Ferguson DD, Hill S, Hutchinson R, Ingall AH, Kingston LP, Martin I, Martin BP, Mohammed RT, Murray C, Perry MW, Reynolds RH, Thorne PV, Wilkinson DJ, Withnall J. Potent blockers of the monocarboxylate transporter MCT1: novel immunomodulatory compounds. Bioorg Med Chem Lett. 2006;16(8):2260–5. [DOI] [PubMed] [Google Scholar]
- 15.Pahlman C, Qi Z, Murray CM, Ferguson D, Bundick RV, Donald DK, et al. Immunosuppressive properties of a series of novel inhibitors of the monocarboxylate transporter MCT-1. Trans Int Off J Eur Soc Organ Trans. 2013;26(1):22–9. [DOI] [PubMed] [Google Scholar]
- 16.Bueno V, Binet I, Steger U, Bundick R, Ferguson D, Murray C, et al. The specific monocarboxylate transporter (MCT1) inhibitor, AR-C117977, a novel immunosuppressant, prolongs allograft survival in the mouse. Transplantation. 2007;84(9):1204–7. [DOI] [PubMed] [Google Scholar]
- 17.Curtis NJ, Mooney L, Hopcroft L, Michopoulos F, Whalley N, Zhong H, Murray C, Logie A, Revill M, Byth KF, Benjamin AD, Firth MA, Green S, Smith PD, Critchlow SE. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget. 2017;8(41):69219–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, Smith PD, Critchlow SE, Stratford IJ. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther. 2014;13(12):2805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, Sasaguri Y, Nishizawa S, Kohno K. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011;102(5):1007–13. [DOI] [PubMed] [Google Scholar]
- 20.Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20(4):926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Yang C, Doherty JR, Roush WR, Cleveland JL, Bannister TD. Synthesis and structure-activity relationships of pteridine dione and trione monocarboxylate transporter 1 inhibitors. J Med Chem. 2014;57(17):7317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble RA, Bell N, Blair H, Sikka A, Thomas H, Phillips N, Nakjang S, Miwa S, Crossland R, Rand V, Televantou D, Long A, Keun HC, Bacon CM, Bomken S, Critchlow SE, Wedge SR. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. 2017;102(7):1247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, Alavi M, Bagryanova L, Krotee PAL, Gardner BK, Behbahan IS, Horvath S, Chia D, Mellinghoff IK, Hurvitz SA, Dubinett SM, Critchlow SE, Kurdistani SK, Goodglick L, Braas D, Graeber TG, Christofk HR. MCT1 modulates Cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep. 2016;14(7):1590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarah ER. Halford PJ, Wedge S, Hirschberg S, Katugampola S, Veal G, Payne G, Bacon C, Potter S, Griffin M, Chenard-Poirier, Petrides G, Holder G, Keun HC, Banerji U, Plummer ER. A first-in-human first-in-class (FIC) trial of the monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with advanced solid tumours. American Society of Clinical Oncology Annual Meeting; Monday, June 5, 2017, 2017. [Google Scholar]
- 25.Guan X, Ruszaj D, Morris ME. Development and validation of a liquid chromatography tandem mass spectrometry assay for AZD3965 in mouse plasma and tumor tissue: application to pharmacokinetic and breast tumor xenograft studies. J Pharm Biomed Anal. 2018;155:270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan X, Rodriguez-Cruz V, Morris ME. Cellular uptake of MCT1 inhibitors AR-C155858 and AZD3965 and their effects on MCT-mediated transport of L-lactate in murine 4T1 breast tumor Cancer cells. AAPS J. 2019;21(2):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32. [DOI] [PubMed] [Google Scholar]
- 28.Wajima T, Yano Y, Oguma T. A pharmacokinetic model for analysis of drug disposition profiles undergoing enterohepatic circulation. J Pharm Pharmacol. 2002;54(7):929–34. [DOI] [PubMed] [Google Scholar]
- 29.Moon YJ, Sagawa K, Frederick K, Zhang S, Morris ME. Pharmacokinetics and bioavailability of the isoflavone biochanin a in rats. AAPS J. 2006;8(3):E433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mager DE, Krzyzanski W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res. 2005;22(10):1589–96. [DOI] [PubMed] [Google Scholar]
- 31.Yan X, Mager DE, Krzyzanski W. Selection between Michaelis-Menten and target-mediated drug disposition pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2010;37(1):25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy G Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248–52. [DOI] [PubMed] [Google Scholar]
- 33.An G Small-molecule compounds exhibiting target-mediated drug disposition (TMDD): a minireview. J Clin Pharmacol. 2017;57(2):137–50. [DOI] [PubMed] [Google Scholar]
- 34.An G, Liu W, Dutta S. Small-molecule compounds exhibiting target-mediated drug disposition - a case example of ABT-384. J Clin Pharmacol. 2015;55(10):1079–85. [DOI] [PubMed] [Google Scholar]
- 35.An G, Liu W, Katz DA, Marek GJ, Awni W, Dutta S. Population pharmacokinetics of the 11beta-hydroxysteroid dehydrogenase type 1 inhibitor ABT-384 in healthy volunteers following single and multiple dose regimens. Biopharm Drug Dispos. 2014;35(7):417–29. [DOI] [PubMed] [Google Scholar]
- 36.Yang X, Gandhi YA, Duignan DB, Morris ME. Prediction of biliary excretion in rats and humans using molecular weight and quantitative structure-pharmacokinetic relationships. AAPS J. 2009;11(3):511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahlstrom BE, Paalzow LK. Pharmacokinetic interpretation of the enterohepatic recirculation and first-pass elimination of morphine in the rat. J Pharmacokinet Biopharm. 1978;6(6):505–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.