

Abstract
Background
Copy number variations (CNV) are a significant source of variation in the genome and are therefore essential to the understanding of genetic characterization. The aim of this study was to develop a fine-scaled copy number variation map for African goats. We used sequence data from multiple breeds and from multiple African countries.
Results
A total of 253,553 CNV (244,876 deletions and 8677 duplications) were identified, corresponding to an overall average of 1393 CNV per animal. The mean CNV length was 3.3 kb, with a median of 1.3 kb. There was substantial differentiation between the populations for some CNV, suggestive of the effect of population-specific selective pressures. A total of 6231 global CNV regions (CNVR) were found across all animals, representing 59.2 Mb (2.4%) of the goat genome. About 1.6% of the CNVR were present in all 34 breeds and 28.7% were present in all 5 geographical areas across Africa, where animals had been sampled. The CNVR had genes that were highly enriched in important biological functions, molecular functions, and cellular components including retrograde endocannabinoid signaling, glutamatergic synapse and circadian entrainment.
Conclusions
This study presents the first fine CNV map of African goat based on WGS data and adds to the growing body of knowledge on the genetic characterization of goats.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-021-07703-1.
Keywords: African goats, Copy number variations, Whole genome sequence
Background
Structural variations (SV) are an important source of genetic variation [1–4]. SV are generally considered to comprise a myriad of subclasses that consist of unbalanced copy number variants (CNV), which include deletions, duplications and insertions of genetic material, as well as balanced rearrangements, such as inversions and interchromosomal and intrachromosomal translocations [5]. Deletions and insertions are referred to as unbalanced SV because they result in changes in the length of the genome. Insertions or deletions in the genome are typically considered CNV when they are at least 50–1000 base-pairs (bp) long [6–11]. CNV are not as abundant as single nucleotide polymorphisms (SNP), but because of their larger sizes, they may have a dramatic effect on gene expression in individuals [12]. Duplication or deletion in or near a gene or the regulatory region of the gene may lead to modification of the function of the gene.
CNV cover about 4.5–9.8% of the human genome [13] and are associated with many Mendelian disorders [12]. Girirajan et al. [14] found that CNV significantly determine the severity and prognosis of many genetic disorders. Approximately 14% of diseases in children with intellectual disability are caused by CNV [15]. On the other hand, some CNV have been found to be associated with adaptive fitness of individuals, such as adaptation to starch diets associated in the gene encoding α-amylase [13].
Traditionally, microarray-based comparative genomic hybridization (array CGH) or SNP genotyping arrays are used to detect CNV. Several studies have been carried out using these methods to detect and map CNV in the goat genome, including studies by Fontanesi et al. [16] in four goat breeds; Nandolo et al. [17] in 13 East African goat breeds; and Liu et al. [18] in the global goat population.
Detecting CNV using array CGH and SNP genotyping arrays suffers from shortcomings that include hybridization noise, limited coverage of the genome, low resolution, and difficulty in detecting novel and rare mutations [19–21]. The development of whole-genome sequencing (WGS) technologies has made it possible for more rigorous and accurate detection of CNV.
According to Mills et al. [22], WGS-based CNV detection methods fall into four major approaches: methods based on paired-end (PE) mapping, split reads (SR), read depth (RD) and de novo assembly of a genome (AS). The PE and SR methods are useful for detection of small-scale CNV [23], and several algorithms are loosely based on them, including BreakDancer [24], Pindel [25], and Delly [26]. RD approaches are very useful for detection of larger CNV. Algorithms using this approach include CNV-Seq [27], CNVnator [28] and the event-wise testing approach (EWT) developed by Yoon et al. [29]. The methods can also be combined. For example, LUMPY [30] is able to combine two or more of the previous approaches to refine SV detection. Assembly-based approaches are computationally intensive and are therefore not generally used with WGS data [23, 31]. Most of these SV-detection algorithms have been extensively reviewed [1, 31–34].
LUMPY implements a breakpoint prediction framework, where a breakpoint is defined as a pair of genomic regions that are adjacent in a sample, but not in the reference genome. The location of the breakpoint is determined using a probability function that considers different sources of evidence supporting the existence of a breakpoint, including information from discordant read pairs and split reads. A discordant read pair occurs when sequence from two ends of an insert are inconsistent when compared to the reference genome. These inconsistencies result from differences between mapping distance or the orientation between the pairs of sequences [35, 36]. Split reads are sequences that map to the reference genome on one end only, and, as explained by Ye and Hall [33], such reads can indicate the location of a breakpoint with a high degree of certainty. There are similar algorithms that rely heavily on the use of breakpoints to determine genome rearrangements at single-nucleotide resolution, including Delly [26] and Pindel [25].
Like LUMPY, Manta [37] incorporates use of PE and SR methods. However, Manta also uses AS analysis. Manta overcomes the computational expense of AS methods by splitting the work into many smaller workflows which can be carried out in parallel. Manta scans the genome for SV and then scores, genotypes and filters the SV based on diploid germline and somatic biological models [37]. Manta can detect all structural variant types that are identifiable in the absence of copy number analysis and large-scale de-novo assembly, which is why this approach is also a good candidate for joint analysis of small sets of diploid individuals, tumor samples, and similar analyses. Both LUMPY and Manta are good at identifying SV break points with high resolution.
Many studies have been carried out to detect CNV using WGS data in various domesticated species: cattle [38], cats [39], chickens [40], dogs [41], etc. So far, there is no report of goat CNV discoveries using WGS data. The goal of this study was to identify CNV in the goat genome through the intersection of LUMPY and Manta outputs as a part of the characterization of African goats in conjunction with the ADAPTmap project [42]. Goats are a very important farm animal genetic resource for the livelihoods of African smallholders, and a deeper understanding of the goat genome is necessary to facilitate the improvement of goats in the region. This study aimed to generate a fine-scale CNV map for the goat genome.
Results
Number and distribution of CNV
The number of CNV detected depended on the filter levels (low, medium, or stringent) and the cut-off point for CNV length (3 Mb or 10 Mb) as given in Supplementary Figure 11 (Additional file 2). Using precise SV only with moderate filters (PE + SR ≥ 5), LUMPY detected 8563 duplications and 230,497 deletions while Manta detected 24,088 duplications and 320,374 deletions. A combined data set with 244,876 deletions and 8677 duplications (totaling 253,553, translating into an average of 1393 CNV per animal) was derived from the intersection of the LUMPY and Manta sets after removal of variants shorter than 50 bp or longer than 3 Mb. The combined data set had more observations than the LUMPY data set (which had fewer raw CNV) because for some individuals, many short CNV from Manta intersected with few long CNV from LUMPY.
The CNV were distributed across the 29 autosomes as shown in Fig. 1. A vast majority of the CNV (96.6%) were losses. This is not unexpected, because all CNV detection methods suffer from an inherent deficiency in detecting insertions. In the case of CNV detection using WGS data, this limitation is even more pronounced with PE methods, because they detect insertions when the mapped reads are at a distance shorter than the fragment length, so they are not able to detect insertions larger than the insert size of the reference library [43]. This has also been supported by the observation that recall percentage is lower than 2 and 5% for medium (1–100 kb) and large (100 kb-1 Mb) duplications, respectively, for most of the SV-calling algorithms currently in use, including Manta and LUMPY used in this study [44].
Fig. 1.
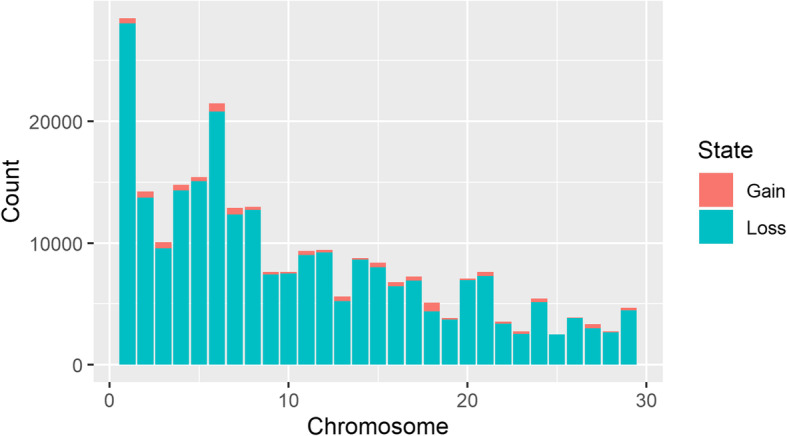
Overall numbers of CNV by chromosome and CNV state. Orange is for copy gain and blue-green is for copy loss
Overall, the mean CNV length was about 3.3 kb, with a median of 1.3 kb. The distribution of the lengths of the CNV for each population are shown in Fig. 2 by CNV length category. A summary of the descriptive statistics of the CNV for the populations are given in Table 1. Most of the CNV losses (99.92%) were less than 100 kb long while 6.3% of CNV gains were longer than 100 kb. Despite the overwhelming proportion of losses over gains, there were more CNV gains observed over 100 kb than losses. Similarly, only 1.04% of the loss CNV were longer than 10 kb, while almost one-quarter (22.99%) of all gain CNV were over 10 kb. As a result, CNV gains were longer than CNV losses and had larger range in length. Deletions and duplications averaged about 2.3 and 31.5 kb long, with median lengths of 1.3 and 1.4 kb, respectively. There were no significant differences in the distribution of CNV across the five populations as shown in the percentile and sample QQ plots in Fig. 3.
Fig. 2.

Distribution of the sizes of CNV for each population by CNV state. Orange is for copy gains while the rest of the colors for copy loss for each of the five populations (magenta for Boer; blue is for the East African; green for Madagascar; brown for Southern African and purple for West African)
Table 1.
Descriptive statistics of CNV and CNV length for each population
| Population | Number of samples | CNV | CNV length (bp) | ||||
|---|---|---|---|---|---|---|---|
| State | Number | Mean | Median | Minimum | Maximum | ||
| BOE | 9 | Loss | 9079 | 2227.1 | 1326 | 67 | 254,129 |
| Gain | 331 | 20,165.9 | 1500 | 161 | 631,262 | ||
| Overall | 9410 | 2858.1 | 1330 | 67 | 631,262 | ||
| EAF | 80 | Loss | 108,051 | 2244.7 | 1293 | 52 | 2,161,018 |
| Gain | 3544 | 30,979.2 | 1316.5 | 118 | 2,777,398 | ||
| Overall | 111,595 | 3157.2 | 1293 | 52 | 2,777,398 | ||
| MAD | 27 | Loss | 31,426 | 2475.3 | 1295 | 84 | 2,069,909 |
| Gain | 1078 | 28,384.1 | 1446 | 84 | 1,660,243 | ||
| Overall | 32,504 | 3334.6 | 1296 | 84 | 2,069,909 | ||
| SAF | 44 | Loss | 67,099 | 2368.9 | 1285 | 51 | 2,539,701 |
| Gain | 2514 | 31,000.7 | 1192 | 101 | 1,959,154 | ||
| Overall | 69,613 | 3402.9 | 1283 | 51 | 2,539,701 | ||
| WAF | 22 | Loss | 29,221 | 2491.4 | 1280 | 52 | 2,457,795 |
| Gain | 1210 | 40,255.3 | 1234 | 65 | 2,788,546 | ||
| Overall | 30,431 | 3993 | 1280 | 52 | 2,788,546 | ||
Fig. 3.
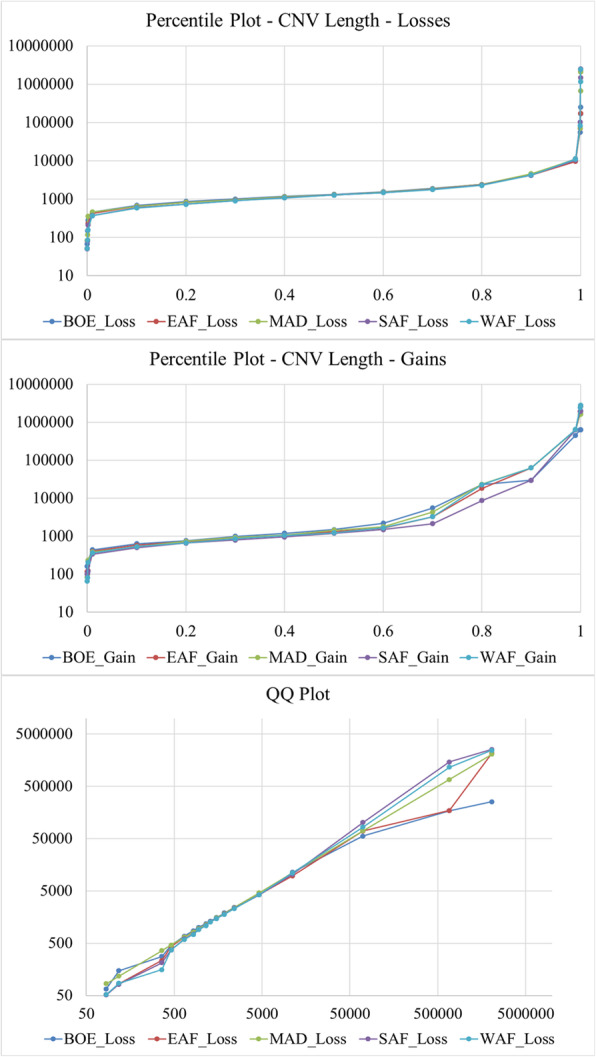
Percentile plots for CNV gains and losses and a QQ plot for CNV losses
Population CNV differentiation
Analysis of population differentiation (VST) as described by Redon et al. [11] showed that several CNV were highly differentiated between and across the populations. Some of these CNV overlapped with genes of importance in goats. Results for the pairwise population VST tests and the VST test across all the populations with their respective 99th percentile CNV VST thresholds are given in Supplementary Table 1 (Additional file 1). VST values for the pairwise tests are given in Supplementary Figures 1–10 (Additional file 2). The VST values for genes that were in CNV that were highly differentiated across all populations are shown in Fig. 4. The gene DST was in a CNV with a very high VST threshold across all the populations. DST has been associated with herpes virus and respiratory disease (BRD) in cattle [45]. Some CNV were highly differentiated both between and across populations. CNV with high differentiation between only some populations include the CNV corresponding to the genes BCO2, CCSER1 (FAM190A), COL24A1, CPNE4, CWC22, IMMP2L, KBTBD12, LAMA3, NAALADL2, RFX3, SEMA3D, SLC2A13, STPG2 (C4orf37), TAFA2 (FAM19A2), TMEM117, TMEM161B and VPS13B. The rest of the genes were in CNV that were highly differentiated across all populations.
Fig. 4.

Population CNV differentiation, estimated by VST computed across all populations, plotted for each chromosome. The dotted line represents the VST threshold value for this test (0.601)
Number and distribution of CNV regions (CNVR)
The lists of CNV regions (CNVR) by population are given in Supplementary Table 2 (Additional file 1) and their locations on the goat genome are shown in Fig. 5. Plots of the CNVR for each breed (with more than 2 animals) are given in Supplementary Figures 12 to 40 (Additional file 2). Descriptive statistics of the CNVR for each population are given in Supplementary Table 3 (Additional file 1) while a distribution of CNVR by size and populations is given in Fig. 6. Over 92% of the CNVR were copy losses. There was a wide variation in the number and sizes of the CNVR between and among the populations. The fraction of copy gains or gains and losses was highest in the group of CNVR of at least 10 kbp, with 25% copy gains and 19% for losses/gains (Fig. 6).
Fig. 5.
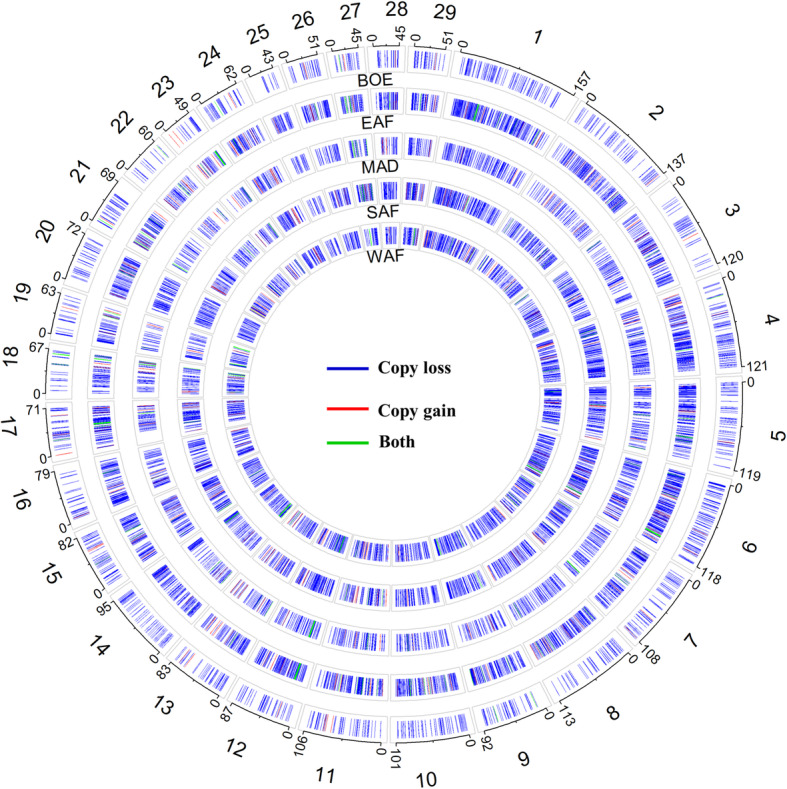
Location of the CNVR for the 29 autosomes by population. The outermost numbers are the autosomes, and the other numbers are the start and end positions of each autosome
Fig. 6.

Distribution of size of CNVR (in kbp) for each population. Orange is for copy gains and red is for CNVR with both copy gains and losses. The rest of the colours for copy loss for each of the five populations (magenta for Boer; blue is for the East African; green for Madagascar; brown for Southern African and purple for West African)
Number and distribution of global CNVR
Global CNVR for different levels of SV filter parameters are given in Supplementary Figures 41 to 64 (Additional file 2). Only the PE and SR filter levels and the CNV length cut-off point affected CNVR coverage. Inclusion of imprecise SV led to an increase in the proportion of called duplications, but the additional duplications were much longer than the upper cut-off point for CNV length. A total of 6231 global CNVR were found across all animals. A list of the global CNVR is given in Supplementary Table 4 (Additional file 1) and a summary is given in Table 2. There were 5742 CNVR with copy losses, 280 with copy gains and 209 with both copy losses and gains in different individuals. The locations of the global CNVR are given in Fig. 7. CNVR with both gains and losses were much longer (mean 185.8 kb) and constituted a significant proportion of the total CNVR coverage (65.6%). Sixteen of these were longer than 1 Mb (on chromosomes 1, 2, 6, 7, 12, 14 (two regions), 17, 19, 21, 23 (two regions), 27 and 29).
Table 2.
CNVR summary statistics for each CNV state based on CNV occurring in at least 2 individuals
| Copy state | Number of CNVR | Length (bp) | CNVR coverage (bp) | |||
|---|---|---|---|---|---|---|
| Mean | Median | Minimum | Maximum | |||
| Loss | 5742 | 3041.3 | 1140.5 | 52 | 1,177,087 | 17,463,236 |
| Gain | 280 | 10,377.9 | 1008.0 | 302 | 236,347 | 2,905,806 |
| Both | 209 | 185,755.2 | 1731.0 | 616 | 2,956,746 | 38,822,839 |
| Overall | 6231 | 9499.6 | 1157.0 | 52 | 2,956,746 | 59,191,881 |
Fig. 7.

Location of the global CNVR across the 29 autosomes. Blue is for loss; red is for gain and green is for both loss and gain
Overall, the CNVR covered about 59.2 Mb of the goat genome. Previous work on genome-wide CNV discovery in goats using SNP data done by Liu et al. [18] showed that CNVR cover approximately 262 Mb of the goat genome. Of the 978 CNVR reported in that study, 540 CNVR intersected with 819 CNVR identified in our study. The amount of the overlap between the CNVR in the two studies was 217.1 Mb, covering 38.6 Mb (65.1%) in this study, and 194.2 Mb (74.1%) in the other study.
Common and rare CNVR
Most of the CNVR (> 95.9%) were found in at least 2 breeds. Out of the 6231 CNVR, 98 (1.6%) were present in all the 34 breeds and 1790 (28.7%) were present in all the populations (Fig. 8a and b). The most frequent CNVR observed was on chromosome 6 from 115,822,332 bp to 115,825,687 bp with a frequency of 96.2%. There were 259 CNVR private to 30 breeds, and 1018 private to all 5 populations, distributed as shown in Fig. 8c and Fig. 8d. BOE (Tanzania and Zimbabwe), KEF (Ethiopia) and MLY (Tanzania) breeds had the highest numbers of private CNVR (20, 21 and 31, respectively).
Fig. 8.
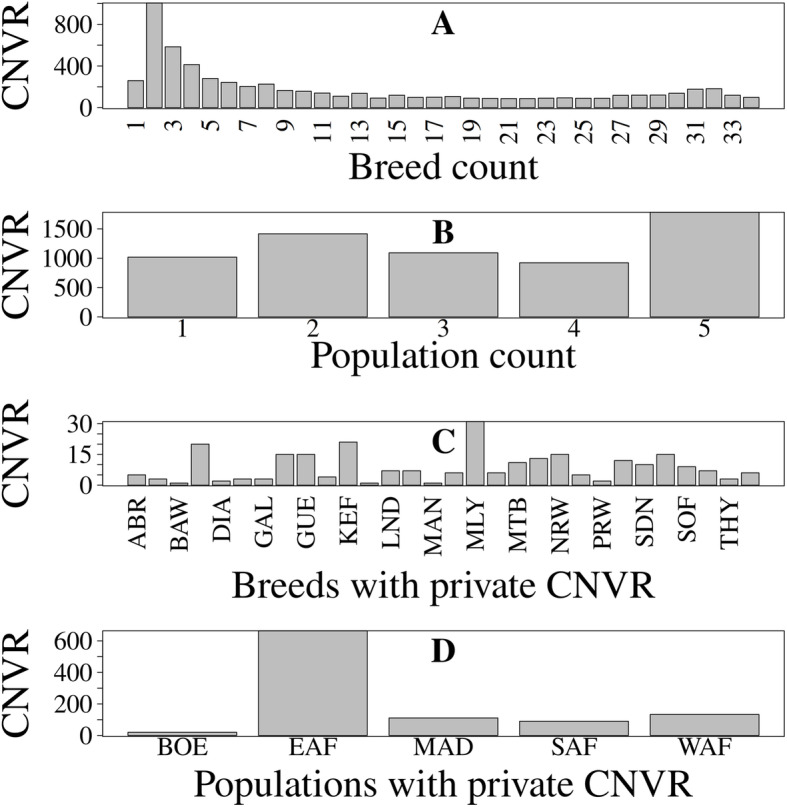
Distribution of the CNVR. a, b Number of CNVR found in different numbers of breeds and populations, respectively. c, d Distribution of CNVR found in only a single breed and only a single population only, respectively. In C, only 30 breeds had private CNVR
Functional annotation and gene enrichment analysis
Functional annotation was carried out for genes in global and private CNVR. Up to 2980 genes overlapped with the 6321 CNVR identified in this study. Up to 755 of these genes formed 24 clusters, with enrichment scores ranging from 0.0 to 1.89. Higher enrichment scores imply higher overrepresentation of the genes in the gene set for the gene enrichment term [46]. The top 3 clusters with the highest enrichment scores are given in Table 3 while the full list is given in Supplementary Table 5 (Additional file 1). The most significant GO terms identified in the analysis included retrograde endocannabinoid signaling; glutamatergic synapse; circadian entrainment; dopaminergic synapse; gastric acid secretion; long-term potentiation; salivary secretion; and calcium signaling pathway.
Table 3.
Functional annotation clusters of the genes found in the global CNVR based on analysis in DAVID
| Cluster (Enrichment score, database) | Enrichment term | Gene count | p-value |
|---|---|---|---|
| 1 (1.89, KEGG_PATHWAY) | chx04020: Calcium signaling pathway | 31 | 0.001 |
| chx04970: Salivary secretion | 16 | 0.011 | |
| chx04022: cGMP-PKG signaling pathway | 25 | 0.016 | |
| chx04270: Vascular smooth muscle contraction | 19 | 0.037 | |
| chx04261: Adrenergic signaling in cardiomyocytes | 21 | 0.046 | |
| 2 (1.62, KEGG_PATHWAY) | chx04723: Retrograde endocannabinoid signaling | 24 | 0.000 |
| chx04724: Glutamatergic synapse | 24 | 0.000 | |
| chx04713: Circadian entrainment | 21 | 0.001 | |
| chx04728: Dopaminergic synapse | 25 | 0.002 | |
| chx04971: Gastric acid secretion | 16 | 0.004 | |
| chx04720: Long-term potentiation | 14 | 0.009 | |
| chx04970: Salivary secretion | 16 | 0.011 | |
| chx04925: Aldosterone synthesis and secretion | 15 | 0.014 | |
| chx04730: Long-term depression | 13 | 0.014 | |
| chx04540: Gap junction | 16 | 0.019 | |
| chx04750: Inflammatory mediator regulation of TRP channels | 18 | 0.020 | |
| chx04921: Oxytocin signaling pathway | 23 | 0.028 | |
| chx04922: Glucagon signaling pathway | 17 | 0.028 | |
| chx04972: Pancreatic secretion | 16 | 0.033 | |
| chx04270: Vascular smooth muscle contraction | 19 | 0.037 | |
| chx04725: Cholinergic synapse | 18 | 0.043 | |
| chx04911: Insulin secretion | 14 | 0.053 | |
| chx04726: Serotonergic synapse | 17 | 0.072 | |
| chx04915: Estrogen signaling pathway | 15 | 0.091 | |
| chx04961: Endocrine and other factor-regulated calcium reabsorption | 8 | 0.094 | |
| chx04912: GnRH signaling pathway | 13 | 0.131 | |
| chx04918: Thyroid hormone synthesis | 11 | 0.137 | |
| chx04924: Renin secretion | 10 | 0.161 | |
| chx04611: Platelet activation | 16 | 0.273 | |
| chx04916: Melanogenesis | 11 | 0.458 | |
| chx04310: Wnt signaling pathway | 14 | 0.599 | |
| 3 (1.14, KEGG_PATHWAY) | chx05204: Chemical carcinogenesis | 12 | 0.033 |
| chx00980: Metabolism of xenobiotics by cytochrome P450 | 10 | 0.075 | |
| chx00982: Drug metabolism - cytochrome P450 | 10 | 0.075 | |
| chx00830: Retinol metabolism | 9 | 0.152 |
CNVR private to populations and breeds overlapped with 172 and 620 genes, respectively. The GO terms associated with these genes based on functional analysis are listed in Supplementary Table 6 (Additional file 1). The genes that overlapped with the CNVR private to breeds were not significantly enriched in biological processes, molecular functions and cellular components, while the ones that overlapped with the CNVR private to populations were significantly enriched (P ≤ 0.05) with such terms as aldosterone synthesis and secretion; glucagon signaling pathway; insulin secretion; glutamatergic synapse; thyroid hormone synthesis; gastric acid secretion and phosphatidylinositol signaling system. The most common CNVR (chr6:115,822,332-115,825,687) includes the gene TMEM129 (transmembrane protein 129) that has been reported to be responsible for ubiquitination and proteasome-mediated degradation of misformed or unassembled proteins in the cytosol [47–49], and belongs to a network responsible for cellular assembly and organization, cellular function and maintenance, and cell cycle [50].
Discussion
This study identified CNV and CNVR in the goat genome using WGS data. Use of WGS for CNV detection is highly encouraged, because it overcomes many of the shortcomings of the other CNV detection methods such as the ones using array CGH and SNP data [19–21]. Genome-wide studies to discover CNV have already been done in other domesticated species, such as in Sus scrofa [51], Bos taurus [38, 52] and Felis catus [39]. Here we provide a first glimpse of the goat genome CNV map at a dense genome coverage, using animals from 34 diverse breeds from the African continent. This addition is an important contribution, as goats are an important source of income and high-quality animal protein for small holder farmers in Africa.
We used two software suites (LUMPY [30] and Manta [37]) for detecting SV to increase our confidence in the SV calls. Both software packages use split read and read-pair methods. They complement each other in that LUMPY makes use of read depth methods, while Manta draws heavily on genome assembly methods. Taking the intersection of SV calls from the two methods gives us confidence that the number of false positives in the SV calls was kept to a minimum, although this means that some true SV were possibly filtered out.
This study has shown that there are wide variations in the number and sizes of CNV in the goat genome between chromosomes, individuals and breeds. However, considering the small and variable numbers of samples within breeds, breed comparisons are not particularly meaningful. The results suggest that there are negligible differences in the sizes of CNV between populations. Some of the CNV displayed large differences between populations, suggestive of population-specific selective pressures.
A large proportion of the global CNVR identified in this study (65.1%) are within the CNVR reported by Liu et al. [18]. The remaining 34.9% may comprise false positive CNVR and CNVR that were missed by the PennCNV algorithm used in the other study, considering the limitation of CNV detection using SNP data, which include limited coverage for genome, low resolution, and difficulty in detecting novel and rare mutations. The CNVR coverage of 2.4% (59.2 Mb of about 2466 Mb of autosomal genome) found in this study is lower than the 4.8–9.5% SV coverage in the human genome [13], comparable to 55.6 Mb (2.0%) reported for cattle [38], later revised to 87.5 Mb (3.1%) [53].
VST analysis showed that several CNV were highly differentiated among and across the populations. The genes in the highly differentiated CNV included BCO2 (Madagascar vs West African population differentiation), CCSER1 (FAM190A) (Boer vs East African), FAM155A (across all populations), GNRHR (Boer vs Madagascar; Boer vs West African), IMMP2L (East vs Southern African), LAMA3 (East African vs Madagascar), NAALADL2 (East vs Southern African), TAFA2 (FAM19A2) (East vs Southern African) and TOMM70 (across all the populations). Våge and Boman [54] reported that BCO2 is associated with the accumulation of carotenoids in the adipose tissue of sheep, leading to the yellow fat syndrome. The quality of semen (including total sperm motility, average path velocity and beat cross frequency) in Holstein-Friesian bulls has been associated with CCSER1 (FAM190A) as well as FAM155A [55]. GNRHR has been associated with number of days to first service after calving in dairy cattle [56] while IMMP2L is associated with cow conception rate [57]. The partial deletion of LAMA3 is responsible for epidermolysis bullosa in horses [58]; NAALADL2 is believed to be responsible for immune homeostasis [59], and TAFA2 (FAM19A2) is believed to be responsible for the regulation of feed intake and metabolic activities in mice [60]. Yamano et al. [61] reported that TOMM70 is responsible for integral mitochondrion proteins and for metabolism.
Functional annotation and clustering analysis revealed that the CNVR identified in the study have genes that are significantly enriched with many biological processes, molecular functions and cellular components, some of the most significant of which are retrograde endocannabinoid signaling, circadian entrainment and long-term potentiation. The retrograde endocannabinoid signaling system is a complex and diverse regulator of synaptic function [62], and is responsible for many diseases in the nervous system and peripheral organs. In the human genome, this system is widely considered as a potential target for treating conditions such as alcoholism [63]. A CNVR in the cannabinoid receptor 2 (CNR2) region has been reported in the human genome, but its effect has not been fully characterized [64]. Zajkowska et al. [65] suggested that there is need to explore genetic variation in the system from the perspective of copy number of variations.
Circadian entrainment is an important aspect of animal behavior and adaptation, especially considering the wide range of environmental conditions the animals are exposed to. An example of goat adaptation to the environment is their ability to rapidly change the size of their foreguts in response to changes in the environment [66]. Goats tend to be active during some parts of the day only [67], and this varies with season [67], suggesting a considerable amount of circadian entrainment. The increased importance of the biological process “response to stimulus” (GO:0050896) in the highly differentiated CNV may also support the hypothesis of the importance of circadian entrainment in goats.
Conclusions
This study presents the first fine CNV map of the African goats based on WGS data. This information will prove invaluable for further improvement of goats, especially on African continent, as more phenotype data becomes available, through CNV or CNVR association analyses and other approaches.
Methods
Sample description
The data used in this study was generated from 182 goats representing 34 breeds from 9 Sub-Saharan African countries (Ethiopia, Kenya, Madagascar, Malawi, Mali, Mozambique, Tanzania, Uganda, and Zimbabwe), and these countries were grouped into four populations based on geographic locations and a fifth population of Boer goats obtained in Tanzania and Zimbabwe. The Boer goat is a special breed widely used in Africa and much of the world [68]. The samples were previously genotyped using the Illumina Goat SNP50 BeadChip [69] as described by Bertolini et al. [70], Cardoso et al. [71] and Colli et al. [72], and some of them were also used for detection of CNV using 50 K SNP chip data, as reported by Liu et al. [18]. A list of the breeds, populations and samples sizes used in the analysis is given in Table 4.
Table 4.
List of the breeds used in the analysis
| Population | Population Code | Breed name | Breed Code | Country | Number of samples |
|---|---|---|---|---|---|
| Boer | BOE | Boer | BOE | Tanzania | 2 |
| Boer | BOE | Boer | BOE | Zimbabwe | 7 |
| East African | EAF | Abergelle | ABR | Ethiopia | 6 |
| East African | EAF | Galla | GAL | Kenya | 7 |
| East African | EAF | Gogo | GOG | Tanzania | 7 |
| East African | EAF | Gumez | GUM | Ethiopia | 4 |
| East African | EAF | Keffa | KEF | Ethiopia | 7 |
| East African | EAF | Landin | LND | Mozambique | 5 |
| East African | EAF | Maasai | MAA | Tanzania | 7 |
| East African | EAF | Manica | MAN | Mozambique | 3 |
| East African | EAF | Malya | MLY | Tanzania | 7 |
| East African | EAF | Norwegian | NRW | Tanzania | 3 |
| East African | EAF | Pare White | PRW | Tanzania | 6 |
| East African | EAF | Saanen | SAA | Tanzania | 4 |
| East African | EAF | Small East African | SEA | Kenya | 7 |
| East African | EAF | Small East African | SEA | Mozambique | 6 |
| East African | EAF | Sonjo | SNJ | Tanzania | 2 |
| East African | EAF | Woyito Guji | WYG | Ethiopia | 7 |
| Madagascar | MAD | Androy | AND | Madagascar | 4 |
| Madagascar | MAD | Diana | DIA | Madagascar | 3 |
| Madagascar | MAD | Menabe | MEN | Madagascar | 7 |
| Madagascar | MAD | Sofia | SOF | Madagascar | 6 |
| Madagascar | MAD | SudOuest | SOU | Madagascar | 7 |
| Southern African | SAF | Balaka-Ulongwe | BAW | Malawi | 2 |
| Southern African | SAF | Dedza | DZD | Malawi | 4 |
| Southern African | SAF | Lilongwe | LGW | Malawi | 3 |
| Southern African | SAF | Mashona | MSH | Zimbabwe | 7 |
| Southern African | SAF | Matebele | MTB | Zimbabwe | 7 |
| Southern African | SAF | Nsanje | NSJ | Malawi | 6 |
| Southern African | SAF | Thyolo | THY | Malawi | 7 |
| West African | WAF | Guerra | GUE | Mali | 6 |
| West African | WAF | Maure | MAU | Mali | 1 |
| West African | WAF | Naine | NAI | Mali | 5 |
| West African | WAF | Peulh | PEU | Mali | 1 |
| West African | WAF | Soudanaise | SDN | Mali | 7 |
| West African | WAF | Targui | TAR | Mali | 2 |
Sample processing was done by Edinburgh Genomics using the Edinburgh Clinical Genomics method. This approach uses Illumina SeqLab products and services, including, Illumina TruSeq library preparation, Illumina cBot2 cluster generation, Illumina HiSeqX sequencing, Hamilton Microlab STAR integrative automation, and Genologics Clarity LIMS X Edition as outlined in Supplementary Table 7 (Additional file 1). Quality control information for the samples is given in Supplementary Table 8 (Additional file 1).
Sequence alignment
Sequence alignment was done using the Burrow-Wheeler Alignment (BWA) tool version 0.7.13-r1126 with the maximal exact matches (MEM) “mem” option [73]. The reads were aligned to the ARS1 Capra hircus (goat) reference assembly (https: //www.ncbi.nlm.nih.gov/assembly/GCF_001704415.1/) [74]. The aligned reads were processed into binary sequence alignment map (BAM) format using SAMTools version 1.8 [75].
Detection of SV
SV were detected using LUMPY version 0.2.13–85-gc1bcea1 and Manta version 1.5.1, which are two of the most used algorithms for detecting SV. In LUMPY, the “lumpyexpress” script was used. This script runs automated breakpoint detection for standard analyses. It uses SAMBLASTER [76] to extract split and discordant reads from BWA-MEM-aligned Binary Sequence Alignment Map (BAM) files. Default options were used, including minimum non-overlap and minimum sample weight set to 20 and 4, respectively. In Manta, the “configManta.py” script was used to process each sample, with default options including minimum variant candidate size (8); minimum candidate spanning count (3); minimum scored variant size (50); minimum diploid variant score (10); minimum diploid variant score pass point (20); minimum somatic score (10); and minimum somatic score pass point (30). The “runWorkflow.py” scripts were run in parallel to extract the SV for each sample.
Post-processing of SV
SV from LUMPY were genotyped with svtyper version 0.6.1 [77], which uses a Bayesian maximum likelihood algorithm to determine the most likely genotype of each base-pair. Variant call format (VCF) files from the two software packages were converted to browser extensible data (BED) format for downstream analysis using svtools version 0.5.0 [78]. Various levels of SV post-processing parameters were used to come up with the CNV calls from the SV calls. The parameters included: 1) precision of SV calls (whether imprecise SV were included in computation of the CNV calls); 2) point of application of the lower SV length cut-off point (before or after merging Manta and LUMPY SV); 3) stringency of the SV call filters (low, medium, and high stringency); and 4) upper SV length cut-off (3 or 10 Mb). Stringency of SV call filters was in terms of the number of PE and SR required as evidence supporting an SV. Consensus SV were obtained by identifying the intersection of the SV from LUMPY and Manta using BEDTools version 2.26.0 [79] with default settings.
Derivation of copy number variations
CNV were defined as SV duplications and deletions longer than 50 bp [80]. SV longer that 3 Mb were also filtered out, because putative CNV in the goat genome are usually much shorter than this length. Visualization of the SV was done using R [81] package circlize version 0.4.7 [82].
Population CNV differentiation
A measure of population differentiation (VST) as described by Redon et al. [11] was computed based on normalized read count values for each CNV, similar to the method used in PECNV as described by Liu et al. [83], which was in turn based on clustering algorithms described by Cridland et al. [84] and transposable element detection algorithms described by Rogers et al. [85]. Read count values were corrected for size of the consensus CNV, batch effect, variable GC content and genomic mappability as described by Liu et al. [83]. Regional and batch effect correction was done by computing reads per kb per million mapped reads (RPKM) as described by Mortazavi et al. [86], where , where RC is the read count of a region, S is the size of the region and TRC is the total number of mapped reads in the library. GC content and mappability correction was done on the RPKM using the formula used by Yoon et al. [29], where adjusted read count is given by where mGC is the median GC content of all regions with the same read count and m is the median GC of all regions. This approach is similar to the read depth approaches used in CNVnator [28] and in CNVcaller [87]. The normalized read count values were treated as proxies of log R ratio (LRR) values normally obtained from array analysis. As defined by Redon et al. [11], VST was computed as , where VT is the variance in LRR among all unrelated individuals and VS is the average variance in LRR within each population. CNV VST testing was done pairwise (for each combination of two populations) and (separately) across all the 5 populations. CNV with VST values above the 99th percentile of all VST values for each comparison were treated as being highly differentiated. We searched for these highly differentiated CNV in the Golden Helix Genome Browse® software (version 3.0.0) (https://www.goldenhelix.com/) using the ARS1 caprine genome reference assembly to identify the genes in the CNV.
Determination of CNV regions
CNV regions (CNVR) were obtained by merging CNV that overlapped by at least 1 bp within populations (population CNVR) and across all the individuals (global CNVR) using the “merge” function in BEDTools version 2.26.0 [79].
CNVR functional annotation and gene enrichment analysis
A list of genes for the goat genome was downloaded from the NCBI website (https://www.ncbi.nlm.nih.gov/gene). The Database for Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources (version 6.8) [88–90] was used to identify if genes in the CNVR have significant biological, cellular or molecular function. Functional analysis was done using default parameters, with significance of enriched terms determined at P ≤ 0.05. Further information about various genes was obtained from the GeneCards (www.genecards.org) database.
Supplementary Information
Additional file 1 Supplementary Table 1 Genes in CNV with VST values above the 99th percentile for each comparison. Supplementary Table 2 List of CNVR by population. Supplementary Table 3 Summary of CNVR by population. Supplementary Table 4 List of global CNVR with frequency above 1%. Supplementary Table 5 Functional annotation and clustering analysis for global CNVR. Supplementary Table 6 Functional analysis for CNVR private to breeds and to populations. Supplementary Table 7 Sample processing details. Supplementary Table 8 Quality control details for the samples used in the study.
Additional file 2 Supplementary Figures 1–10 CNV differentiation between the following populations; respectively: Boer and East African; Boer and Madagascar; Boer and Southern African; Boer and West African; East African and Madagascar; East African and Southern African; East African and West African; Madagascar and Southern African; Madagascar and West African and Southern African and Western African. Supplementary Figures 11–39 CNVR for the following goat breeds, respectively: Abergelle (Ethiopia), Androy (Madagascar), Balaka-Ulongwe (Malawi), Boer (Tanzania and Zimbabwe), Dedza (Malawi), Diana (Madagascar), Galla (Kenya), Gogo (Tanzania), Guera (Mali), Gumez (Ethiopia), Keffa (Ethiopia), Landin (Mozambique), Maasai (Tanzania), Malya (Tanzania), Manica (Mozambique), Mashona (Zimbabwe), Matebele (Zimbabwe), Menabe (Madagascar), Naine (Mali), Norwegian (Tanzania), Nsanje (Malawi), Pare White (Tanzania), Saanen (Tanzania), Small East African (Kenya and Mozambique), Sofia (Madagascar), Soudanaise (Mali), SudOuest (Madagascar), Thyolo (Malawi) and Woyito Guji (Ethiopia). Supplementary Figures 40–64 Global CNVR with variable SV calling parameters.
Acknowledgements
The VarGoats Consortium - http://www.goatgenome.org/vargoats.html
Whole genome sequencing libraries were prepared and sequenced by Edinburgh Genomics and funded via Biotechnology and Biological Sciences Research Council research grant (BBS/OS/GC/000012F) ‘Reference genome and population sequencing of African goats’ awarded to The Roslin Institute.
Abbreviations
- ABR
Abergelle
- AND
Androy
- BAM
Binary Sequence Alignment Map
- BAW
Balaka-Ulongwe
- BED
Browser Extensible Data
- BOE
Boer
- BWA
Burrow-Wheeler Alignment
- CGH
Comparative genomic hybridization
- CNV
Copy number variation
- CNVR
Copy number variation region
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- DIA
Diana
- DZD
Dedza
- EAF
East African
- EWT
Event-wise testing
- GAL
Galla
- GO
Gene Ontology
- GOG
Gogo
- GSEA
Gene set enrichment analysis
- GUE
Guerra
- GUM
Gumez
- KEF
Keffa
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LGW
Lilongwe
- LND
Landin
- MAA
Maasai
- MAD
Madagascar
- MAN
Manica
- MAU
Maure
- MEM
Maximal exact matches
- MEN
Menabe
- MLY
Malya
- MSH
Mashona
- MTB
Matebele
- NAI
Naine
- NRW
Norwegian
- NSJ
Nsanje
- PE
Paired-end mapping
- PEU
Peulh
- PR
Paired-end reads
- PRW
Pare White
- RD
Read depth
- RPKM
Reads per kb per million mapped reads
- SAA
Saanen
- SAF
Southern African
- SDN
Soudanaise
- SEA
Small East African
- SNJ
Sonjo
- SNP
Single nucleotide polymorphisms
- SOF
Sofia
- SOU
SudOuest
- SR
Split reads
- SV
Structural variation
- TAR
Targui
- THY
Thyolo
- VCF
Variant call format
- WAF
West African
- WGS
Whole genome sequencing
- WYG
Woyito Guji
Authors’ contributions
CPVD, BDR, GM and JS conceived the idea of this work. ELK and the VarGoats Consortium carried out the sequencing of the samples and pre-processing of the sequence data. WN, BDR, CPVT, GM and JS carried out the analysis and drafted the manuscript. WN, GM, MW, LJB, TNG, HAM, HNN, ELK, MJWG, ML, GEL, CPVT, BDR and JS reviewed the manuscript. The authors read and approved the final manuscript.
Funding
This research was done with funding from the Feed the Future project: Improving Livestock Productivity through Enhanced Breeding Programs (Agricultural Research Service/USDA Project No.: 8042–31000-001-02S). The data were collected by the African Goat Improvement Network (AGIN) (https://www.ars.usda.gov/office-of-international-research-programs/ftf-livestock-improvement/).
This research used resources provided by the SCINet project of the USDA Agricultural Research Service, ARS project number 0500–00093–001-00-D.
Availability of data and materials
The dataset supporting the conclusions of this article is available at the National Center for Biotechnology Information Sequence Read Archive as detailed in Additional file 1 (Supplementary Table 7).
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Escaramís G, Docampo E, Rabionet R. A decade of structural variants: description, history and methods to detect structural variation. Brief Funct Genomics. 2015;14(5):305–314. doi: 10.1093/bfgp/elv014. [DOI] [PubMed] [Google Scholar]
- 2.Matsuzaki H, Wang PH, Hu J, Rava R, Fu GK. High resolution discovery and confirmation of copy number variants in 90 Yoruba Nigerians. Genome Biol. 2009;10:1–18. doi: 10.1186/gb-2009-10-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan S, Meyer A. Evolution of genomic structural variation and genomic architecture in the adaptive radiations of African cichlid fishes. Front Genet. 2014;5(JUN):163. doi: 10.3389/fgene.2014.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39(S7):S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho SS, Urban AE, Mills RE. Structural variation in the sequencing era. Nat Rev Genet. 2019;21(3):171–189. doi: 10.1038/s41576-019-0180-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25(12):528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 8.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 9.Feuk L, Marshall CR, Wintle RF, Scherer SW. Structural variants: changing the landscape of chromosomes and design of disease studies. Hum Mol Genet. 2006;15(suppl_1):R57–R66. doi: 10.1093/hmg/ddl057. [DOI] [PubMed] [Google Scholar]
- 10.Huang CRL, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, Wheelan SJ, Ji H, Boeke JD, Burns KH. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141(7):1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8(8):639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 13.Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
- 14.Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, Nezarati MM, Asamoah A, Jackson KE, Gowans GC, Martin JA, Carmany EP, Stockton DW, Schnur RE, Penney LS, Martin DM, Raskin S, Leppig K, Thiese H, Smith R, Aberg E, Niyazov DM, Escobar LF, el-Khechen D, Johnson KD, Lebel RR, Siefkas K, Ball S, Shur N, McGuire M, Brasington CK, Spence JE, Martin LS, Clericuzio C, Ballif BC, Shaffer LG, Eichler EE. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontanesi L, Martelli PL, Beretti F, Riggio V, Dall’Olio S, Colombo M, et al. An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC Genomics. 2010;11(1):639. doi: 10.1186/1471-2164-11-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandolo W, Lamuno D, Banda L, Gondwe T, Mulindwa H, Nakimbugwe H, et al. Proceedings of the World Congress on Genetics Applied to Livestock Production. World Congress on Genetics Applied to Livestock Production. 2018. Distribution of copy number variants in the genomes of east African goat breeds; p. 470. [Google Scholar]
- 18.Liu M, Zhou Y, Rosen BD, Van Tassell CP, Stella A, Tosser-Klopp G, et al. Diversity of copy number variation in the worldwide goat population. Heredity. 2019;122(5):636–646. doi: 10.1038/s41437-018-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, Law S, Myambo K, Palmer J, Ylstra B, Yue JP, Gray JW, Jain AN, Pinkel D, Albertson DG. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet. 2001;29(3):263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 20.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 21.Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(Suppl 11):S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Chen G, Lima L, Fang H, Jimenez L, Li M, et al. HadoopCNV- A dynamic programming imputation algorithm to detect copy number variants from sequencing data. bioRxiv. 2017:124339. 10.1101/124339.
- 24.Fan X, Abbott TE, Larson D, Chen K. Current protocols in bioinformatics. Hoboken: Wiley; 2014. BreakDancer: identification of genomic structural variation from paired-end read mapping; pp. 15.6.1–15.6.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10(1):80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19(9):1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Layer RM, Chiang C, Quinlan AR, Hall IM, Quinlan AR. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15(6):R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirooznia M, Goes F, Zandi PP. Whole-genome CNV analysis: Advances in computational approaches. Front Genet. 2015;6(MAR):138. doi: 10.3389/fgene.2015.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tattini L, D’Aurizio R, Magi A. Detection of genomic structural variants from next-generation sequencing data. Front Bioeng Biotechnol. 2015;3:92. doi: 10.3389/fbioe.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye K, Hall G, G H KY. Structural Variation Detection from Next Generation Sequencing. J Gener Seq Appl. 2015. 10.4172/2469-9853.S1-007.
- 34.Guan P, Sung WK. Structural variation detection using next-generation sequencing data: a comparative technical review. Methods. 2016;102:36–49. doi: 10.1016/j.ymeth.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Conroy JM, Morrison CD, Odunsi AO, Qin M, Wei L, Trump DL, Johnson CS, Liu S, Wang J. Structural variation discovery in the cancer genome using next generation sequencing: computational solutions and perspectives. Oncotarget. 2015;6(8):5477–5489. doi: 10.18632/oncotarget.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthur JG, Chen X, Zhou B, Urban AE, Wong WH. Detection of complex structural variation from paired-end sequencing data. bioRxiv. 2018:200170. 10.1101/200170.
- 37.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 38.Bickhart DM, Liu GE, Eichler EE, Matukumalli LK, Song J, Van Tassell CP, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22(4):778–790. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genova F, Longeri M, Lyons LAA, Bagnato A, Gandolfi B, Aberdein D, et al. First genome-wide CNV mapping in FELIS CATUS using next generation sequencing data. BMC Genomics. 2018;19:1–2. doi: 10.1186/s12864-018-5297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohrabi SS, Mohammadabadi M, Wu D-D, Esmailizadeh A. Detection of breed-specific copy number variations in domestic chicken genome. Genome. 2018;61(1):7–14. doi: 10.1139/gen-2017-0016. [DOI] [PubMed] [Google Scholar]
- 41.Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10(1):e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stella A, Nicolazzi EL, Van Tassell CP, Rothschild MF, Colli L, Rosen BD, et al. AdaptMap: exploring goat diversity and adaptation. Genet Sel Evol. 2018;50(1):61. doi: 10.1186/s12711-018-0427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teo SM, Pawitan Y, Ku CS, Chia KS, Salim A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics. 2012;28(21):2711–2718. doi: 10.1093/bioinformatics/bts535. [DOI] [PubMed] [Google Scholar]
- 44.Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20(1):117. doi: 10.1186/s13059-019-1720-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neupane M, Hoff JL, Taylor JF, Seabury CM, Womack JE, Bovine T, et al. Refinement of the DST locus associated with bovine respiratory disease complex in Holstein calves. J Anim Sci. 2016;94(suppl_5):361. doi: 10.2527/jam2016-0753. [DOI] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van De Weijer ML, Bassik MC, Luteijn RD, Voorburg CM, Lohuis MAM, Kremmer E, et al. A high-coverage shrna screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat Commun. 2014;5(1):3832. doi: 10.1038/ncomms4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van De Weijer ML, Van Muijlwijk GH, Visser LJ, Costa AI, Wiertz EJHJ, Lebbink RJ. The E3 ubiquitin ligase TMEM129 is a tri-spanning transmembrane protein. Viruses. 2016;8:309. doi: 10.3390/v8110309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Boomen DJH, Timms RT, Grice GL, Stagg HR, Skodt K, Dougan G, Nathan JA, Lehner PJ. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc Natl Acad Sci. 2014;111(31):11425–11430. doi: 10.1073/pnas.1409099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu F, Shen XY, Fan L, Yu ZC. Genome-wide analysis of genetic variations assisted by Ingenuity Pathway Analysis to comprehensively investigate potential genetic targets associated with the progression of hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2014;18:2102. [PubMed] [Google Scholar]
- 51.Wang K, Hadley D, Liu R, Bucan M, Li M, Glessner J, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, Moore SS. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12(1):559. doi: 10.1186/1471-2164-12-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bickhart DM, Xu L, Hutchison JL, Cole JB, Null DJ, Schroeder SG, Song J, Garcia JF, Sonstegard TS, van Tassell CP, Schnabel RD, Taylor JF, Lewin HA, Liu GE. Diversity and population-genetic properties of copy number variations and multicopy genes in cattle. DNA Res. 2016;23(3):253–262. doi: 10.1093/dnares/dsw013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Våge DI, Boman IA. A nonsense mutation in the beta-carotene oxygenase 2 (BCO2) gene is tightly associated with accumulation of carotenoids in adipose tissue in sheep (Ovis aries) BMC Genet. 2010;11(1):10. doi: 10.1186/1471-2156-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borowska A, Szwaczkowski T, Kamiński S, Hering DM, Kordan W, Lecewicz M. Identification of genome regions determining semen quality in Holstein-Friesian bulls using information theory. Anim Reprod Sci. 2018;192:206–215. doi: 10.1016/j.anireprosci.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 56.Lirón JP, Prando AJ, Fernández ME, Ripoli MV, Rogberg-Muñoz A, Goszczynski DE, et al. Association between GNRHR, LHR and IGF1 polymorphisms and timing of puberty in male Angus cattle. BMC Genet. 2012;13(1):26. doi: 10.1186/1471-2156-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang J, Ma L, Prakapenka D, VanRaden PM, Cole JB, Da Y. A large-scale genome-wide association study in U.S. Holstein cattle. Front Genet. 2019;10(MAY):412. doi: 10.3389/fgene.2019.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuen WY, Pasmooij AMG, Stellingsma C, Jonkman MF. Enamel defects in carriers of a novel LAMA3 mutation underlying epidermolysis bullosa. Acta Derm Venereol. 2012;92(6):695–696. doi: 10.2340/00015555-1341. [DOI] [PubMed] [Google Scholar]
- 59.Onouchi Y. Genetics of Kawasaki disease: - what we know and don’t know. Circ J. 2012;76(7):1581–1586. doi: 10.1253/circj.CJ-12-0568. [DOI] [PubMed] [Google Scholar]
- 60.Okada J, Yamada E, Saito T, Ozawa A, Nakajima Y, Pessin JE, Okada S, Yamada M. Analysis of FAM19A2/TAFA-2 function. Physiol Behav. 2019;208:112581. doi: 10.1016/j.physbeh.2019.112581. [DOI] [PubMed] [Google Scholar]
- 61.Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016;17(3):300–316. doi: 10.15252/embr.201541486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76(1):70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Basavarajappa BS. The endocannabinoid signaling system: a potential target for next-generation therapeutics for alcoholism. Mini Rev Med Chem. 2007;7(8):769–779. doi: 10.2174/138955707781387920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ishiguro H, Leonard CM, Sgro S, Onaivi ES. Cannabinoid receptor gene variations in neuropsychiatric disorders. In: Murillo-Rodríguez E, editor. Endocannabinoids: molecular, pharmacological, behavioral and clinical features. 1. Baltimore: Bentham Science Publishers; 2013. pp. 3–24. [Google Scholar]
- 65.Zajkowska ZE, Englund A, Zunszain PA. Towards a personalized treatment in depression: endocannabinoids, inflammation and stress response. Pharmacogenomics. 2014;15(5):687–698. doi: 10.2217/pgs.14.40. [DOI] [PubMed] [Google Scholar]
- 66.Silanikove N. The physiological basis of adaptation in goats to harsh environments. Small Rumin Res. 2000;35(3):181–193. doi: 10.1016/S0921-4488(99)00096-6. [DOI] [Google Scholar]
- 67.Piccione G, Giannetto C, Casella S, Caola G. Seasonal change of daily motor activity rhythms in Capra hircus. Can J Anim Sci. 2008;88(3):351–355. doi: 10.4141/CJAS07125. [DOI] [Google Scholar]
- 68.Malan SW. The improved Boer goat. Small Rumin Res. 2000;36(2):165–170. doi: 10.1016/S0921-4488(99)00160-1. [DOI] [PubMed] [Google Scholar]
- 69.Tosser-Klopp G, Bardou P, Bouchez O, Cabau C, Crooijmans R, Dong Y, Donnadieu-Tonon C, Eggen A, Heuven HCM, Jamli S, Jiken AJ, Klopp C, Lawley CT, McEwan J, Martin P, Moreno CR, Mulsant P, Nabihoudine I, Pailhoux E, Palhière I, Rupp R, Sarry J, Sayre BL, Tircazes A, Jun Wang, Wang W, Zhang W, and the International Goat Genome Consortium Design and characterization of a 52K SNP chip for goats. PLoS One. 2014;9(1):e86227. doi: 10.1371/journal.pone.0086227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bertolini F, Cardoso TF, Marras G, Nicolazzi EL, Rothschild MF, Amills M. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet Sel Evol. 2018;50(1):59. doi: 10.1186/s12711-018-0424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cardoso TF, Amills M, Bertolini F, Rothschild M, Marras G, Boink G, et al. Patterns of homozygosity in insular and continental goat breeds. Genet Sel Evol. 2018;50:1–11. doi: 10.1186/s12711-018-0374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Colli L, Milanesi M, Talenti A, Bertolini F, Chen M, Crisà A, et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet Sel Evol. 2018;50(1):58. doi: 10.1186/s12711-018-0422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bickhart DM, Rosen BD, Koren S, Sayre BL, Hastie AR, Chan S, Lee J, Lam ET, Liachko I, Sullivan ST, Burton JN, Huson HJ, Nystrom JC, Kelley CM, Hutchison JL, Zhou Y, Sun J, Crisà A, Ponce de León FA, Schwartz JC, Hammond JA, Waldbieser GC, Schroeder SG, Liu GE, Dunham MJ, Shendure J, Sonstegard TS, Phillippy AM, van Tassell CP, Smith TPL. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet. 2017;49(4):643–650. doi: 10.1038/ng.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30(17):2503–2505. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, Hall IM. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12(10):966–968. doi: 10.1038/nmeth.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Larson DE, Abel HJ, Chiang C, Badve A, Das I, Eldred JM, et al. Svtools: Population-Scale Analysis of Structural Variation. Bioinformatics. 2019;35:4782. doi: 10.1093/bioinformatics/btz492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu GE, Bickhart DM. Copy number variation in the cattle genome. Funct Integr Genomics. 2012;12(4):609–624. doi: 10.1007/s10142-012-0289-9. [DOI] [PubMed] [Google Scholar]
- 81.R Development Core team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for statistical Computing; 2016. [Google Scholar]
- 82.Gu Z, Gu L, Eils R, Schlesner M, Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 83.Liu X, Li A, Xi J, Feng H, Wang M. Detection of copy number variants and loss of heterozygosity from impure tumor samples using whole exome sequencing data. Oncol Lett. 2018;16(4):4713–4720. doi: 10.3892/ol.2018.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cridland JM, Macdonald SJ, Long AD, Thornton KR. Abundance and distribution of transposable elements in two drosophila QTL mapping resources. Mol Biol Evol. 2013;30(10):2311–2327. doi: 10.1093/molbev/mst129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, Thornton KR. Landscape of standing variation for tandem duplications in drosophila yakuba and drosophila simulans. Mol Biol Evol. 2014;31(7):1750–1766. doi: 10.1093/molbev/msu124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 87.Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, Jiang Y. CNVcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. GigaScience. 2017;6(12):1–12. doi: 10.1093/gigascience/gix115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dennis G, Sherman B, Hosack D, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 89.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Supplementary Table 1 Genes in CNV with VST values above the 99th percentile for each comparison. Supplementary Table 2 List of CNVR by population. Supplementary Table 3 Summary of CNVR by population. Supplementary Table 4 List of global CNVR with frequency above 1%. Supplementary Table 5 Functional annotation and clustering analysis for global CNVR. Supplementary Table 6 Functional analysis for CNVR private to breeds and to populations. Supplementary Table 7 Sample processing details. Supplementary Table 8 Quality control details for the samples used in the study.
Additional file 2 Supplementary Figures 1–10 CNV differentiation between the following populations; respectively: Boer and East African; Boer and Madagascar; Boer and Southern African; Boer and West African; East African and Madagascar; East African and Southern African; East African and West African; Madagascar and Southern African; Madagascar and West African and Southern African and Western African. Supplementary Figures 11–39 CNVR for the following goat breeds, respectively: Abergelle (Ethiopia), Androy (Madagascar), Balaka-Ulongwe (Malawi), Boer (Tanzania and Zimbabwe), Dedza (Malawi), Diana (Madagascar), Galla (Kenya), Gogo (Tanzania), Guera (Mali), Gumez (Ethiopia), Keffa (Ethiopia), Landin (Mozambique), Maasai (Tanzania), Malya (Tanzania), Manica (Mozambique), Mashona (Zimbabwe), Matebele (Zimbabwe), Menabe (Madagascar), Naine (Mali), Norwegian (Tanzania), Nsanje (Malawi), Pare White (Tanzania), Saanen (Tanzania), Small East African (Kenya and Mozambique), Sofia (Madagascar), Soudanaise (Mali), SudOuest (Madagascar), Thyolo (Malawi) and Woyito Guji (Ethiopia). Supplementary Figures 40–64 Global CNVR with variable SV calling parameters.
Data Availability Statement
The dataset supporting the conclusions of this article is available at the National Center for Biotechnology Information Sequence Read Archive as detailed in Additional file 1 (Supplementary Table 7).