

Summary
This protocol is a comprehensive guide to phage display-based selection of virus neutralizing VH antibody domains. It details three optimized parts including (1) construction of a large-sized (theoretically > 1011) naïve human antibody heavy chain domain library, (2) SARS-CoV-2 antigen expression and stable cell line construction, and (3) library panning for selection of SARS-CoV-2-specific antibody domains. Using this protocol, we identified a high-affinity neutralizing human VH antibody domain, VH ab8, which exhibits high prophylactic and therapeutic efficacy.
For complete details on the use and execution of this protocol, please refer to Li et al. (2020)
Subject areas: Cell culture, High Throughput Screening, Immunology, Microbiology, Molecular Biology, Antibody, Protein expression and purification
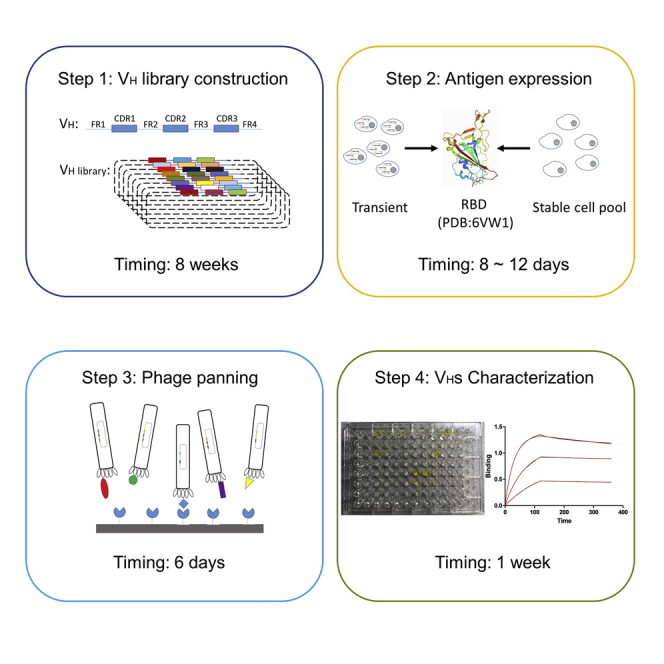
Graphical abstract
Highlights
-
•
Large-sized (> 1011) naïve human antibody heavy chain domain library construction
-
•
Quick antigen expression and stable cell pool selection with Expi293TM cells
-
•
Quick phage-display library panning (6 days)
-
•
High-affinity aggregation-resistant human antibody domain binder selection
This protocol is a comprehensive guide to phage display-based selection of virus neutralizing VH antibody domains. It details three optimized parts including (1) construction of a large-sized (theoretically > 1011) naïve human antibody heavy chain domain library, (2) SARS-CoV-2 antigen expression and stable cell line construction, and (3) library panning for selection of SARS-CoV-2-specific antibody domains. Using this protocol, we identified a high-affinity neutralizing human VH antibody domain, VH ab8, which exhibits high prophylactic and therapeutic efficacy.
Before you begin
Total RNA isolation and CDNA synthesis
Timing: 3–5 days
-
1.
Collect peripheral blood mononuclear cells (PBMCs) from 12 healthy donors’ blood samples before SARS-CoV-2 pandemic using Ficoll-Paque PLUS gradient (Sigma, Cat#GE17-1440-02) according to the manufacturer’s protocol (https://www.sigmaaldrich.com/technical-documents/protocols/biology/isolation-of-mononuclear-cells/recommended-standard-method.html). 1.5 × 109 PBMCs were collected for RNA isolation.
-
2.
Dissolving the cell pellet immediately with 60 mL RLT buffer (2.5 × 107 Cells/mL RLT buffer with 1% volume of 2-mercaptoethanol) and isolation of total RNA following the protocol of RNeasy Mini Kit.
Pause point: Total RNA in DEPC water can be kept at −80°C for short-term storage.
-
3.
Preparation of cDNA using both random hexamer and Oligo dT as primers. Total RNA was prepared, and first strand cDNA was synthesized by Superscript™ III first-strand Synthesis System (Thermo, Cat#18080044) according to the manufacturer’s protocol ( https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FsuperscriptIII_man.pdf&title=U3VwZXJTY3JpcHQgSUlJIFJldmVyc2UgVHJhbnNjcmlwdGFzZQ== ).
Synthetic primers and genes
-
4.
PCR primers used for gene amplification and VH assemble are listed in Table 1. The primers were synthesized by IDT with high quality (Purified by HPLC).
-
5.
Codon Optimize the RBD-AviTag gene (GenBank: QHD43416.1, Figure 1) by “Codon Optimization Tool” in IDT (https://www.idtdna.com/pages/tools/codon-optimization-tool?returnurl=%2FCodonOpt ). Optimized RBD-AviTag DNA was synthesized by IDT gBlock service. The gBlock DNA was diluted with ddH2O into 10 ng/μL for PCR.
Table 1.
Sequences of the primers for VH assemble and RBD-AviTag cloning
| Name | Sequence | Usage |
|---|---|---|
| ALL-F | GTTTCGCTACCGTGGCCCAGGCGGCCGAGGT GCAGCTGGTGGA |
FR1 and CDR1 |
| FR1R | ACAGGAGAGTCTCAG | |
| H1F-157 | CTGAGACTCTCCTGTAAGGSTTCT | |
| H1F-2 | CTGAGACTCTCCTGTACCKTCTCT | |
| H1F-3 | CTGAGACTCTCCTGTGYAGCCTCT | |
| H1F-4 | CTGAGACTCTCCTGTRCTGTCTCT | |
| H1F-6 | CTGAGACTCTCCTGTGCCATCTCC | |
| H1R-13 | CCTGGAGCCTGGCGGACCCAGTKCAT | |
| H1R-2 | CCTGGAGCCTGGCGGACCCAGCYCAC | |
| H1R-3 | CCTGGAGCCTGGCGGACCCAGCTCAT | |
| H1R-4 | CCTGGAGCCTGGCGGACCCAGCYCCA | |
| H1R-5 | CCTGGAGCCTGGCGGACCCAGCYGAT | |
| H1R-6 | CCTGGAGCCTGGCGGACCCAGTTCCA | |
| H1R-7 | CCTGGAGCCTGGCGGACCCAATTCAT | |
| H1R-ALL | CCTGGAGCCTGGCGGACCCA | |
| H2F167 | TGGGTCCGCCAGGCTCCAGGACAASGSCTTGAGTGG | CDR2 |
| H2F2 | TGGGTCCGCCAGGCTCCAGGGAAGGCCCTGGAGTGG | |
| H2F1345 | TGGGTCCGCCAGGCTCCAGGGAAGGGNCTRGAGTGG | |
| H2R1 | ATTGTCTCTGGAGATGGTGACCCTKYCCTGRAACTY | |
| H2R36 | ATTGTCTCTGGAGATGGTGAATCGGCCCTTCACNGA | |
| H2R24 | ATTGTCTCTGGAGATGGTGACTMGACTCTTGAGGGA | |
| H2R5 | ATTGTCTCTGGAGATGGTGACSTGGCCTTGGAAGGA | |
| H2R7 | ATTGTCTCTGGAGATGGTAAACCGTCCTGTGAAGCC | |
| FR3F | ACCATCTCCAGAGACAATTCC | FR3 |
| FR3R | GTCCTCGGCTCTCAGGCTG | |
| H3F3p | AGCCTGAGAGCCGAGGACACRGCYTTRTATTACTGT | CDR3 |
| H3F1p257p | AGCCTGAGAGCCGAGGACACAGCCAYRTATTACTGT | |
| H3Fother | AGCCTGAGAGCCGAGGACACRGCYGTRTATTACTGT | |
| H3R | GTGGCCGGCCTGGCCACTTGAGGAGACGGTGACC | |
| ALL-R | GTCGCCGTGGTGGTGGTGGTGGTGGCCGGCCTGGCCACTTG | |
| EcoRV-RBD-P1 | TCAGTGATATCCCAAATATAACCAATCTCTGCCCATTCG | RBD-AviTag-His |
| EcoRI-His-AviTag-P2 | TCAGTGAATTCCTATTAGTGATGGTGGTGGTGATG GCTACCCTCGTGCCACTCG |
Figure 1.

DNA sequence of RBD-AviTag
Codon optimized receptor-binding domain (RBD) of the SARS-CoV-2 spike (S) glycoprotein with AviTagTM and His-tag.
Sequences highlighted plum (RBD): Codon optimized receptor-binding domain of the SARS-CoV-2 spike (S) glycoprotein.
Sequences highlighted lavender (AviTagTM): The AviTagTM sequences (GLNDIFEAQKIEWHE) for site-specific biotinylation of purified RBD-AviTag antigen with BirA Biotin-Protein Ligase Kit.
Sequences highlighted pink (His-tag): 6×His tag sequences for RBD-AviTag purification with Ni-NTA column.
Plasmids and other materials
-
6.
Generation of an empty plasmid named pIW-Zeo (Figure 2A) that contains a CMV promotor, Intron, a secret signal peptide (SPE) for extracellular expression, multiple cloning site (MCS), internal ribosome entry site (IRES), zeocin resistant gene, woodchuck posttranscriptional regulatory elements, BGH poly A, origin of replication and ampicillin resistant gene (This plasmid was generated before starting the protocol).
-
7.
Prepare the phagemid for library construction with Qiagen plasmids Maxi-Prep kit (Qiagen Maxi-prep, Cat#12663). The phagemid we used is a modified pComb3X vector (Cat#VPT4012, Creative Biogene), in which the HA-tag was replaced by Flag-tag (Figures 2C and 2D).
-
8.
Maintain Expi293F™ cells (Thermo, Cat#A14527) with Expi293™ Expression Medium in a CO2 resistant incubator at 135 rpm, 8% CO2, 95% humidity according to the manufacturer’s protocol (https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0006283_Expi293_Cells_UG.pdf&title=VXNlciBHdWlkZTogRXhwaTI5M0YgQ2VsbHM=).
Note: Determine viability and cell clumping using the trypan blue dye exclusion method and make sure cell viability is higher than 97%. The Expi293F™ cells can maintain in Expi293™ Expression Medium for 2 months (20–30 passages) with no significant expression decrease. So, thawing a new vial of Expi293F™ cells every two months is needed.
Figure 2.

The schematic view of maps of Vectors pIW-Zeo, pIW-Zeo-RBD and pComb3X
(A) Maps of pIW-Zeo: A secret signal peptide (SPE) was inserted into the empty vector for extracellular expression.
(B) Maps of pIW-Zeo-RBD: RBD secret expression plasmid.
(C) Maps of pComb3x with an amber stop codon. TG1 is an amber codon (TAG) suppressor strain, allowing translation to read through the codon to produce a full-length VH-gene III fusion protein. HB2151 is an amber codon non-suppressor strain, Only VH gene can be translation to produce VH in this strain.
(D) Amber stop codon in pComb3x
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| TG1 Electrocompetent Cells | Lucigen | 60502-1 |
| E. cloni 5-alpha Chemically Competent Cells | Lucigen | 60602-2 |
| HB2151 bacteria | CAT Lab, University of Pittsburgh | N/A |
| M13KO7 Helper Phage | Thermo Fisher Scientific | 18311019 |
| Antibodies | ||
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) | Sigma | A8592 |
| Anti-M13 Antibody (HRP) | Sino Biological Inc. | 11973-MM05T-H |
| Chemicals, peptides, and recombinant proteins | ||
| T4 DNA Polymerase | NEB | M0203L |
| T4 DNA Ligase | NEB | M0202L |
| ACE2 Protein, Human, Recombinant (mFc Tag) | Sino Biological Inc. | 10108-H05H |
| Trypan Blue Solution, 0.4% | Thermo Fisher Scientific | 15250061 |
| UltraPure™ Agarose | Thermo Fisher Scientific | 16500500 |
| Agar for bacteriology | VWR | 97064-332 |
| Ampicillin Sodium Salt | Fisher Scientific | BP1760-25 |
| Kanamycin Sulfate | Fisher Scientific | BP906-5 |
| Dextrose (D-Glucose), Anhydrous | Fisher Scientific | D16-1 |
| SfiI | NEB | R0123L |
| EcoRV-HF | NEB | R3195S |
| EcoRI-HF | NEB | R3101S |
| Polyethylene Glycol 8000 | Fisher Scientific | BP233-1 |
| Sodium Chloride | Fisher Scientific | S271-3 |
| PEI MAX™ | Polysciences | 24765-1 |
| Zeocin™ Selection Reagent | Thermo Fisher Scientific | R25001 |
| Bovine Serum Albumin (BSA) | VWR | 97063-626 |
| Blotting-Grade Blocker (nonfat dry milk) | Bio-Rad | 1706404 |
| TWEEN® 20 | Sigma-Aldrich | P1379-500ML |
| Glycerol | Sigma-Aldrich | G5516-1L |
| Isopropanol | Fisher Scientific | AC327272500 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG) | Fisher Scientific | BP1755-10 |
| SimplyBlue™ SafeStain | Thermo fisher scientific | LC6065 |
| Polymyxin B sulfate salt | Sigma-Aldrich | P1004-50MU |
| 2-Mercaptoethanol | Thermo fisher scientific | 21985023 |
| Imidazole | Sigma-Aldrich | I2399 |
| 50× TAE Buffer (Tris-acetate-EDTA) | Fisher Scientific | FERB49 |
| Buffer RLT | QIAGEN | 79216 |
| ACK Lysing Buffer | Thermo Fisher scientific | A1049201 |
| NuPAGE™ 4–12%, Bis-Tris, 1.0 mm, Mini Protein Gel | Thermo Fisher Scientific | NP0321BOX |
| Ficoll-Paque PLUS | GE Healthcare | 17-1440-03 |
| HisPur™ Ni-NTA Resin | Thermo Fisher Scientific | 88222 |
| Expi293™ Expression Medium | Thermo Fisher Scientific | A1435101 |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190136 |
| DPBS (10×), no calcium, no magnesium | Thermo Fisher Scientific | 14200-075 |
| Nuclease-Free Water | Thermo Fisher Scientific | AM9937 |
| Teknova2-YT BROTH | Fisher Scientific | 50-843-444 |
| Teknova 2-YT AGAR | Fisher Scientific | 50-843-447 |
| Experimental models: cell line | ||
| Expi293F™ | Thermo Fisher Scientific | A14527 |
| Oligonucleotides | ||
| Primers (see Table 1) | IDT | N/A |
| Recombinant DNA | ||
| pComb3X | CAT Lab, University of Pittsburgh | N/A |
| pIW-Zeo | CAT Lab, University of Pittsburgh | N/A |
| Critical commercial assays | ||
| QIAquick Gel Extraction Kit | QIAGEN | 28706 |
| QIAprep Spin Miniprep Kit | QIAGEN | 27106 |
| HiSpeed Plasmid Maxi Kit | QIAGEN | 12663 |
| BirA Biotin-Protein Ligase Kit | Avidity | BirA-500 |
| High Fidelity PCR Master | Roche | 12140314001 |
| Phusion Flash High-Fidelity PCR Master Mix | Thermo Fisher Scientific | F548L |
| Dynabeads™ MyOne™ Streptavidin T1 | Thermo Fisher Scientific | 65602 |
| DNA Clean & Concentrator-5 | Fisher Scientific | 50-197-7310 |
| Pierce™ Protein Concentrator PES, 10K MWCO, 5–20 mL | Thermo Fisher Scientific | 88528 |
| Pierce™ Protein Concentrator PES, 10K MWCO, 2–6 mL | Thermo Fisher Scientific | 88517 |
| RNeasy Midi Kit | QIAGEN | 75144 |
| SuperScript™ III First-Strand Synthesis System | Thermo Fisher Scientific | 18080044 |
| Other | ||
| Gene Pulser/MicroPulser Electroporation Cuvettes, 0.1 cm gap | Bio-Rad | 1652089 |
| T100™ Thermal Cycler | Bio-Rad | 1861096EDU |
| 125 mL Fisherbrand™ Shaker Flasks | Fisher Scientific | PBV12-5 |
| 250 mL Fisherbrand™ Shaker Flasks | Fisher Scientific | PBV250 |
| Pierce™ Disposable Columns, 5 mL | Thermo Fisher Scientific | 29922 |
| 6 Tube Magnetic Stand | Thermo Fisher Scientific | AM10055 |
| Corning® 96-well Half Area Clear Flat Bottom Polystyrene High Bind Microplate | Fisher Scientific | 07-200-37 |
| Amicon® Ultra-4 Centrifugal Filters | Sigma-Aldrich | UFC8030 |
| Amicon® Ultra-15 Centrifugal Filters | Sigma-Aldrich | UFC9030 |
Materials and equipment
Primers combinations for PCR amplification of CDRs
| CDRs | Human Ig VH family | Primers combinations |
|---|---|---|
| CDR1 | IGVH1 | H1F-157 and H1R-13 |
| IGVH2 | H1F-2 and H1R-2 | |
| IGVH3 | H1F-3 and H1R-13; H1F-3 and H1R-3 | |
| IGVH4 | H1F-4 and H1R-4 | |
| IGVH5 | H1F-157 and H1R-5 | |
| IGVH6 | H1F-6 and H1R-6 | |
| IGVH7 | H1F-157 and H1R-7 | |
| CDR2 | IGVH1 | H2F167 and H2R1; H2F1345 and H2R1 |
| IGVH2 | H2F2 and H2R24 | |
| IGVH3 | H2F1345 and H2R36 | |
| IGVH4 | H2F1345 and H2R24 | |
| IGVH5 | H2F1345 and H2R5 | |
| IGVH6 | H2F167 and H2R36 | |
| IGVH7 | H2F167 and H2R7 | |
| CDR3 | IGVH1 | H3F1p257p and H3R; H3Fother and H3R |
| IGVH2 | H3F1p257p and H3R | |
| IGVH3 | H3Fother and H3R | |
| IGVH4 | H3Fother and H3R | |
| IGVH5 | H3F1p257p and H3R | |
| IGVH6 | H3Fother and H3R | |
| IGVH7 | H3F1p257p and H3R; H3Fother and H3R |
Binding buffer:
| Reagent | Final concentration | Amount |
|---|---|---|
| MilliQ water | n/a | 900 mL |
| DPBS (10 ×) | 1 × | 100 mL |
| Imidazole | 5 mM | 0.34 g |
| NaCl | 300 mM | 17.53 g |
| Total | n/a | 1L |
Note: Adjust the pH to 8.0, filter with 0.45 μm bottle top filter and store at 4°C. This buffer is guaranteed for one year when stored properly.
Wash buffer:
| Reagent | Final concentration | Amount |
|---|---|---|
| MilliQ water | n/a | 900 mL |
| DPBS (10×) | 1 X | 100 mL |
| Imidazole | 20 mM | 1.36 g |
| NaCl | 300 mM | 17.53 g |
| Total | n/a | 1L |
Note: Adjust the pH to 8.0, filter with 0.45 μm bottle top filter and Store at 4°C. This buffer is guaranteed for one year when stored properly.
Elution buffer:
| Reagent | Final concentration | Amount |
|---|---|---|
| MilliQ water | n/a | 900 mL |
| DPBS (10×) | 1 X | 100 mL |
| Imidazole | 250 mM | 17.02 g |
| NaCl | 300 mM | 17.53 g |
| Total | n/a | 1L |
Note: Adjust the pH to 8.0, filter with 0.45 μm bottle top filter and store at 4°C. This buffer is guaranteed for one year when stored properly.
PEG/NaCl solution
| Reagent | Final concentration | Amount |
|---|---|---|
| MilliQ water | n/a | ≈ 900 mL |
| Polyethylene Glycol 8000 | 25 mM | 200 g |
| NaCl | 2.5 M | 150 g |
| Total | n/a | 1L |
Note: Autoclave and mix after cooling down. Keep it at RT (20°C–25°C). This buffer is guaranteed for 6 months when stored properly.
Critical Reagents: TG1 Electrocompetent Cells (Lucigen, Cat#60502-1). DNA Clean & Concentrator-5 (Fisher Scientific, Cat#50-197-7310). These two reagents are critical for library construction.
Alternatives: Throughout this protocol, we refer to several specific kit for many standard biology techniques. Investigators may substitute other commercially available kit as needed.
Step-by-step method details
VH library construction
High diversity and large size are the characteristics of a good library and the basic requirements for successful library panning leading to selection of high affinity binders. Due to the limited diversity of PBMCs B cell antibody gene, it is unlikely to generate a large size VH library by direct PCR amplification of VH region from the cDNA of PBMCs. Therefore, overlap-PCR was used to combine different antibody heavy chain complementarity-determining regions (CDRs) to increase the size and diversity of VH genes. In this protocol, a well-defined stable scaffold IGVH3-23 (Figure 3A) was chosen as basic scaffold for framework Region (FR): FR1, FR2, FR3 and FR4. The primer combinations used to amplify CDRs and overlap CDRs-FRs are listed in Table 1 and Figure 4.
-
1.
Dissolve the primers with ddH2O and adjust the concentration to 10 μM.
-
2.Assemble the VH with PCR (Figure 3A).
-
a.PCR amplify FR1, CDR1, CDR2, FR3 and CDR3 separately with High Fidelity PCR Master kit. Different primer combinations as shown in Table 1 (Figure 4) using the following cycling conditions:
-
i.50 μL PCR system:
Component Volume/Weight Final concentration PCR master mix 1 25 μL 1 × 10 μM forward primer 1 μL 0.2 μM 10 μM reverse primer 1 μL 0.2 μM cDNA template 100 ng 2 ng/μL ddH2O to 50 μL n/a Thermocycling conditions for PCR:Steps Temperature Time Initial denaturation 94°C 4 min 25 cycles 94°C 45 s 55°C 45 s 72°C 1 min Final extension 72°C 5 min Hold 4°C -
ii.Run all the PCR samples with 2% agarose gel to verify the size and purify the right size PCR products (Figure 3B) from the agarose gel with QIAquick Gel Extraction Kit.
 CRITICAL: The template cDNA is a mixture; the primers may have non-specific binding with the cDNA to PCR out some non-specific DNA bands. In the experiment, only the target bands will be purified for further overlapping PCR process to assembly full-length VH.
CRITICAL: The template cDNA is a mixture; the primers may have non-specific binding with the cDNA to PCR out some non-specific DNA bands. In the experiment, only the target bands will be purified for further overlapping PCR process to assembly full-length VH.
-
i.
-
b.Over-lapping PCR to assemble full-length VH with FR1, CDR1, CDR2, FR3 and CDR3 DNA purified from step 2a as template, ALL-F and ALL-R as primes using the following cycling conditions:
-
i.50 μL PCR system:
Component Volume/Weight Final concentration PCR master mix 1 25 μL 1 × 10 μM ALL-F primer 1 μL 0.2 μM 10 μM ALL-R primer 1 μL 0.2 μM FR1, CDR1, CDR2, FR3 and CDR3 DNA template mixture 100 ng 2 ng/μL ddH2O to 50 μL n/a Thermocycling conditions for PCR:Steps Temperature Time Initial denaturation 94°C 4 min 28 cycles 94°C 45 s 55°C 45 s 72°C 1 min Final extension 72°C 5 min Hold 4°C -
ii.Run all the PCR samples with 2% agarose gel to verify the size and purify the right size PCR products (Figure 3B) from the agarose gel with QIAquick Gel Extraction Kit.
-
i.
-
a.
Note: If the over-lapping PCR productivity is low. Assemble the fragments with two steps (troubleshooting 1).
-
3.Digest the pComb3X plasmid and VH gene repertoires with SfiI restriction enzyme in PCR tubes at 50°C for 12 h with the following conditions:
-
a.100 μL digest system:
Component Volume/Weight Final concentration CutSmart buffer 10 μL 1 × plasmid or VH DNA 2 μg 0.02 μg/μL SfiI 1 μL for pComb-3X DNA
4 μL for VH DNA0.2 units/μL (pComb-3X)
0.8 units/μL (VH DNA)ddH2O to 100 μL n/a Total 1.5 mg pComb3X and 500 μg VH gene needed.CRITICAL: The size of pComb3X plasmid is ∼9 fold larger than VH DNA. With the same amount of DNA, VH has higher molarity. So, more SfiI is needed to digest the VH DNA completely. -
b.Run all the digested DNA with 1% agarose gel to verify the size and purify the digested products (Figure 3C) from the agarose gel with QIAquick Gel Extraction Kit.
-
a.
-
4.
Ligation of purified VH fragments to pComb3X vector using a molar ratio of 3:1 VH fragments to pComb3X at 16°C for 60 h with the following conditions:
Figure 3.

VH antibody domain library construction and agarose gel results
(A) An overview of VHs library construction. The CDR1, CDR2 and CDR3 are amplified from human PBMC cDNA and grafted into a well-defined stable scaffold IGVH3-23.
(B) CDR PCR results of different immunoglobulin heavy-chain variable region gene (IGVH) subgroups (IGHV1 to IGHV7) and overlap-PCR assembled VH. Below are illustrations of the results:
The CDR1 PCR products subgroups (∼74 bp) in each row (Row 1: IGHV1. Row 2: IGHV2. Row 3: IGHV3. Row 4: IGHV4. Row 5: IGHV5. Row 6: IGHV6. Row 7: IGHV7).
The CDR2 PCR products subgroups (∼114 bp) in each row (Row 1: IGHV1. Row 2: IGHV2, 4. Row 3: IGHV3, 6. Row 4: IGHV5. Row 5: IGHV7).
The CDR3 PCR products subgroups (∼132 bp) in each row (Row 1–3: IGHV1, 2, 5, 7. Row 4: IGHV3. Row 5: other subgroups).
Overlap-PCR assembled VH results (Row 1–5: IGHV1∼7 mixture, ∼430 bp).
(C) Digest results of VH and pComb3x. The size of digested VH∼370 bp. The size of digested pComb3x∼3300 bp
Figure 4.

Schematic diagram of the assembled VH and the primers for PCR
Degenerated bases: R = A,G; Y = C,T; M = A,C; K = G,T; S =C,G; N = A,C,G,T. The restriction sites were marked in yellow, complementary sequences were highlighted by underline, overlapping sequences were marked in red.
100 μL ligation system:
| Component | 100 μL reaction |
|---|---|
| T4 DNA Ligase Buffer (10 ×) | 10 μL |
| pComb3X | 750 ng |
| VH | 250 ng |
| T4 DNA ligase | 5 μL |
| ddH2O | To 100 μL |
Total 50 mL ligation is needed for a 1011 size library construction.
-
5.
Recover the ligation DNA with DNA Clean & Concentrator-5 according to the manufacturer’s protocol (https://files.zymoresearch.com/protocols/_d4003t_d4003_d4004_d4013_d4014_dna_clean_concentrator_-5.pdf).
-
6.
Pool all the eluted DNA and determine the DNA concentration by Nanodrop Lite.
-
7.Electroporate TG1 Electrocompetent Cells with the following conditions:
-
a.Add 1 μg ligation DNA into each vial of TG1 cells (1 μg DNA in 50 μL competent bacteria per vial). Stir with tips 20 times and keep on ice 5–10 min.
-
b.Transfer the bacteria into ice pre-chilled 0.1 cm cuvette.
-
c.Electroporate using the pre-set program with setting at 1.8 kV/ 200 ohms/25 μF.
-
d.Wash the cuvette with 1 mL pre-warm 2-YT medium three times and transfer the bacteria into 50 mL pre-warm 2-YT medium.
-
a.
Note: During large scale electroporation, 10 electroporated/transformed TG1 vials are resuspended with 500 mL pre-warm 2-YT medium in a 2 L shake flask. Around 150 electroporation vials are needed for > 1011 size library construction, thus 15 × 2 L shake flasks with 7.5 L pre-warm 2-YT medium is needed to resuspend all the electroporated/transformed TG1 cells.
-
8.
Recover the bacteria at 37°C, 200 rpm for 30 min, Aliquot 1/105 bacteria (5 μL culture medium) from each bottle into a 1.5 mL centrifuge tube which contain 995 μL fresh 2-YT medium for titration. 10-fold serial dilute the bacteria and take 1/107, 1/108 and 1/109 of total bacteria (100 μL of 10-, 102- and 103-fold diluted samples) from the dilutions, plate onto 2-YT-Agar plates with 100 μg/mL ampicillin. Select the transformants by adding 100 μg/mL ampicillin and 2% glucose, shaking at 37°C, 200 rpm for 2–3 h till the OD600 reach to ∼0.6–0.8.
-
9.
Add M13KO7 helper phage into the cells with multiplicity of infection (MOI) = 10:1, incubate at 37°C, 45 min. Mix every 15 min during incubation.
Note: MOI means the ratio of phages added to bacteria. OD600 of 1 corresponds to approximately 5 × 108 TG1 cells per ml. If the OD600 ≈ 0.6, total TG1 cells equals 0.6 × 5 × 108/mL × 7500 mL ≈ 2.25 × 1012, and ≈ 2.25 × 1013 M13KO7 helper phage are needed for the infection.
-
10.
Centrifuge the bacteria at 5,000 g for 5 min at 4°C. Resuspend the bacterial pellet with fresh 2-YT contain 100 μg/mL ampicillin, 50 μg/mL kanamycin.
-
11.
Shaking at 30°C, 200 rpm overnight (12–15 h).
-
12.
Spin down the bacteria at 8,000 g for 15 min at 4°C, transfer the supernatant into new bottles, add 25% volume of PEG/NaCl solution into the supernatant and incubate on ice for 1 h.
-
13.
Centrifugation of the mixture at 11,000 g for 20 min at 4°C. Discard supernatant and resuspend the pellet with 50 mL ice cold DPBS per liter of culture.
-
14.
Centrifugation again at 10,000 g for 10 min at 4°C to eliminate the bacterial contamination.
-
15.
Transfer all the supernatant into a new bottle, add 20% glycerol and aliquot into 1 mL/vial, stock at −80°C for long-term storage. Determine the phage titer by detecting OD268 (1 OD ≈ 5 × 1012 phage)(Durr, Nothaft et al. 2010). The phage titer should above 5 × 1012/mL
-
16.Quality checks of the library:
-
a.Randomly pick > 100 colonies and scale up in 4 mL 2-YT medium with 100 μg/mL ampicillin, shaking at 200 rpm overnight (12–15 h).
-
b.Purify the plasmid with QIAprep Spin Miniprep Kit and elute with 30 μL water.
-
c.Send all the plasmid for sequencing and analyze all the sequences.
-
a.
Note: If repeat VHs are detected, the quality of the library is not good and library size might be smaller than 1011, so re-construction of the library to make the size >1011 is needed.
Note: We are using two asymmetric SfiI sites for library construction. Sequencing results showed there is no issue of orientation of VH insert in our pComb3X system.
Note: Beside randomly picked > 100 colonies for sequencing, we also evaluated the library quality and performance by panning with more than 5 antigens. For a high-quality library, several dozens of different VH binders binding to each antigen were enriched. Multiple binders and the diversity of the selected VH binders revealed good quality of library.
Molecular cloning, antigen expression, and protein purification
Compare with the microbial expression system, mammalian cell expression has the advantage of protein expression, more advanced peptide folding and post-translational modifications (Gray 2001, Khan 2013). In general, antigens expressed from mammalian cells which have proper folding and post-translational modifications are essential for full biological activity and successful selection of high affinity specific binders. Here we used the SARS-CoV-2 receptor-binding domain (RBD) as an example. This section includes molecular cloning, expression, and purification (Figures 1, 2, and 5).
Figure 5.

RBD-AviTag antigen expression
(A) An overview of RBD-AviTag transient expression and stable cell pool selection. There are three steps in RBD-AviTag expression: Step 1: Molecular cloning of the pIW-Zeo-RBD plasmid (4 days were needed). Step 2: Overnight transfection of the Expi293TM cell with pIW-Zeo-RBD plasmid (1 day was needed). Step 3: After transfection, cells were maintained at 37°C in incubator shaker rotating at 135 rpm with 8% CO2 and 85% humidity for 3–7 days for RBD-AviTag transient expression. Or select stable cell pool with Zeocin antibiotic (step 18e, optional).
(B) SDS-PAGE results of RBD-AviTag with or W/O DTT. Line 1 (L1) and Line 2 (L2): Both RBD-AviTag samples with or W/O DDT shown three bands. Line 3 (L3) and line 4 (L4): After deglycosylation, only one band was detected in both L3 and L4. The results show multiple bands due to glycosylation of the RBD-AviTag protein. Figure reprinted with permission from Liu, X. et al., 2020.
Molecular cloning
-
17.Clone the RBD into expression plasmid pIW-Zeo (Figure 2A):
-
a.We have previously generated an expression plasmid named pIW-Zeo
-
b.Amplify the SARS-CoV-2 RBD gene by the EcoRV-RBD-P1/EcoRI-His-AviTag-P2 primers (which contain the EcoRV and EcoRI restriction sites) with the Phusion Flash High-Fidelity PCR Master Mix according to the manufacturer’s protocol (https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0012774_Phusion_Flash_HiFi_PCR_MasterMix_100rxn_UG.pdf&title=VXNlciBHdWlkZTogUGh1c2lvbiBGbGFzaCBIaWdoLUZpZGVsaXR5IFBDUiBNYXN0ZXIgTWl4), using the following cycling conditions:
-
i.50 μL PCR system:
Component Volume/Weight Final concentration 2 × Phusion Flash PCR Master Mix 25 μL 1 × 10 μM EcoRV-RBD-P1 primer 2 μL 0.4 μM 10 μM EcoRI-His-AviTag-P2 primer 2 μL 0.4 μM gBlock RBD-AviTag 20 ng 0.4 ng/μL ddH2O to 50 μL n/a Thermocycling conditions for PCR:Steps Temperature Time Initial denaturation 98°C 20 s 34 cycles 98°C 5 s 55°C 10 s 72°C 15 s Final extension 72°C 5 min Hold 4°C -
ii.Run all the PCR product on a 1% agarose gel to verify the size and purify the PCR RBD-AviTag product from the agarose gel with QIAquick Gel Extraction Kit.
-
i.
-
c.Digest the pIW-Zeo plasmid and the PCR amplified RBD-AviTag gene in separate reactions. Incubate each mixture at 37°C for 2 h with the following conditions:50μL digest system:
Component Volume/Weight Final concentration CutSmart buffer 5 μL 1 × pIW-Zeo or RBD-AviTag 1 μg 0.02 μg/μL EcoRV-HF 2 μL 0.8 units/μL EcoRI-HF 2 μL 0.8 units/μL ddH2O to 50 μL n/a -
d.Run a 1% agarose gel, the digested PCR product and plasmid bands should be ∼700 bp and ∼5,000 bp, respectively.
-
e.Recovery the digested PCR product and plasmid DNA from the agarose gel. Elute with 10 μL water.
-
f.Ligate the digested RBD-AviTag gene into the pIW-Zeo plasmid. Leave the ligation reaction at 37°C for 15 min:10 μL ligation system:
Component 10 μL reaction T4 DNA Ligase Buffer (10 ×) 1 μL pIW-Zeo 1 μL RBD-AviTag 2 μL T4 DNA ligase 1 μL ddH2O to 10 μL -
g.Transform 2 μL ligation reaction with 20 μL of E. coli DH5α competent cells. After recovering for 45–60 min at 37°C (shaking at 200 rpm), spread cells onto a 2-YT agar plate supplemented with 100 μg/mL ampicillin. Incubate the plate in a 37°C incubator overnight (12–15 h).
-
h.Randomly pick several colonies and scale up in 4 mL 2-YT medium with 100 μg/mL ampicillin, shaking at 200 rpm overnight (12–15 h).
-
i.Purify the plasmid with QIAprep Spin Miniprep Kit and elute with 50 μL water. Verification of the plasmid by DNA sequencing and keep the right clones (pIW-Zeo-RBD, Figure 2B).
-
a.
RBD-AviTag antigen expression
-
18.Transfection and Protein Expression (30 mL expression, Figure 5A):
-
a.The day before transfection, seed the cells at a density of 2.0 × 106 viable cells/mL and incubate at 37°C in incubator shaker rotating at 135 rpm with 8% CO2 and 85% humidity.
-
b.On the day of transfection, determine number and viability of the cells using an automated cell counter. Dilute the cells to 3 × 106 viable cells/mL with Expi293™ Expression Medium.
-
c.Add 27 mL cell suspension into a 125 mL Erlenmeyer shaker flask. Return the cells to the incubator.
-
d.prepare DNA-PEI complexes as follows:
-
i.Dilute 30 μg of plasmid DNA into 1.5 mL Expi293™ Expression Medium, mix gently and incubate for 5 min at RT (20°C–25°C).
-
ii.Dilute 120 μg of PEI into 1.5 mL Expi293™ Expression Medium, mix gently and incubate for 5 min at RT (20°C–25°C).
-
iii.After 5 min incubation, mix the plasmid DNA with the PEI. Incubate at RT (20°C–25°C) for 10–20 min.
-
iv.After the DNA-PEI complex incubation is complete, add the complex into shaker flask from step 18c. Gently swirl the flask.
-
v.Return and incubate the cells in the incubator. Maintain 7 days at 37°C.Note: Some expressed proteins might have degradation, denaturation, or aggregation during several days culturing at 37°C. Short time culture can protect protein from degradation, denaturation or aggregation to get proteins of higher quality, while long time culturing has higher yield. For the RBD-AviTag expression, the yield of 3 days culture is ≈ 5mg/L, and of 7 days is higher than 10 mg/L. We also found the activity of RBD-AviTag purified at day 3 is better than that of day 7. Thus, according to our experience, 3 days of culture is sufficient for RBD-AviTag expression.
-
i.
-
e.(optional) 24 h after transfection, the transfected cells can be used for stable cell pool selection with the following steps (Figure 5A):
-
i.Take 5 mL transfected cells into 50 mL centrifugation tube. Centrifugation at 300 g, 3 min. Return the supernatant into the expression bottle.
-
ii.Resuspend the cells with 5 mL Expi293™ Expression Medium contain 250 μg/mL Zeocin. Return and incubate the cells in the incubator.
-
iii.Change medium at day 1, day 2, day 3 and day 4 with 5 mL fresh Expi293™ Expression Medium containing 250 μg/mL Zeocin.Note: Because of the transfection efficiency variation, the viability of the cells at day 4 might be different. If the viability is < 30% at day 4, reduce the Zeocin concentration to 50 μg/mL for the following three days selection.In our RBD-AviTag stable cell pool selection process, the viability is ∼64% at day 4. So, 250 μg/mL Zeocin was used for 7 days selection.
-
iv.Change medium at day 7 and resuspend the cells with 20 mL Expi293™ Expression Medium with 50 μg/mL Zeocin. Add the suspension cell into a 125 mL Erlenmeyer shaker flask, Return and incubate the cells in the incubator maintain 3–6 days (Stable cell pool is ready for expression).
-
i.
-
a.
RBD-AviTag-His purification with Ni-NTA column
-
19.Protein purification
-
a.Prepare the Ni-NTA gravity column:
-
i.Transfer 1 mL His Pur™ Ni-NTA Resin (10 mg protein/ 1 mL beads) into a gravity column.
-
ii.Allow the resin to settle, then let the excess buffer drain through the column by gravity flow.
-
iii.Wash the resin with 20 mL Milli Q water.
-
iv.Wash the resin with 20 mL binding buffer.
-
i.
-
b.Cell culture media preparation:
-
i.Transfer the expression medium at step 18 into a 50 mL conical tubes, spin down at 300 g for 5 min at 4°C.
-
ii.Pour supernatant into new conical tubes. Spin down at 12,000 g for 10 min at 4°C.
-
i.
-
c.Protein loading and elution:
-
i.Load all the media into the column at step 19a.
-
ii.Once the medium has completely entered the column, wash the column with 40 mL wash buffer.
-
iii.Elution the RBD-AviTag with 4 mL elution buffer.
-
iv.Change buffer with DPBS with 10 kD ultra-filter. Follow instructions in the manufacturer’s protocol (https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0015695_2162596_PierceProteinConcentrator_2_6mLPI.pdf&title=VXNlciBHdWlkZTogUGllcmNlIFByb3RlaW4gQ29uY2VudHJhdG9yLCBQRVMgLSAzSywgMTBLLCAzMEsgYW5kIDEwMEsgTVdDTzsgMi02bUw= ).
-
v.Measure the protein concentration on NanoDrop Lite. Run SDS-PAGE gel to characterize the purified RBD-AviTag (Figure 5B. Adapted from (Liu, Drelich et al. 2020)).
-
i.
-
a.
Note: The productivity of RBD-AviTag is ∼10 mg/L. 1 μL purified RBD-AviTag sample is needed for concentration detection with NanoDrop Lite.
Phage-displayed library panning
After obtaining the RBD-AviTag antigen, its quality was checked by human angiotensin-converting enzyme 2 (ACE2) binding ELISA. Then it is ready for bio-panning. This section describes streptavidin-magnetic beads-based library panning strategy.
-
20.
Biotin-label the RBD-AviTag (RBD-Bio) antigen with BirA Biotin-Protein Ligase Kit according to the manufacture’s protocol (https://www.avidity.com/showpdf.asp?N=B9B6C28E-1976-4CFE-89DE-E37D1D8DB2CA ).
-
21.Library panning with streptavidin magnetic Beads:
-
a.Thaw a phage library aliquot (1 mL/aliquot, ≈ 5 × 1012 phage/mL). Add 250 μL (VPEG/NaCl:Vphage = 1:4) PEG/NaCl solution into the phage, incubate on ice for 20 min.
-
b.Centrifugation at 12,000 g for 10 min at 4°C. Discard supernatant and resuspend the pellet with 200 μL DPBS.
-
c.Take 2 × 1012 library phage diluted into 1 mL 3% BSA-DPBS. Add 10 μg RBD-Bio antigen, rotation 1.5 h at RT (20°C–25°C).CRITICAL: The amount of antigen used for panning are different in each round while the incubation time did not change. In our protocol we used 10 μg for 1st round, 2nd round 5 μg, 3rd round 1 μg, 4th round 0.2 μg. (Use high concentration antigen at 1st round can help to enrich more binders. Lower concentration of antigens in the following panning process will help to enrich high affinity VH domains).
-
d.At the same time, pick single colony from TG1 bacterial plate (LB-agar plate) or directly take 1 μL TG1 from commercial stock and scale up in 20 mL 2-YT medium at 37°C, 200 rpm until OD600 ≈ 0.5.
-
e.Take 25 μL streptavidin beads into a 1.5 mL centrifugation tube, wash the beads with 1 mL DPBS twice with magnets stand.Note: Blocking the beads with 3% BSA-DPBS for 1 hour at RT (20°C–25°C) before use is good for panning. Because we have an extra step of depletion with streptavidin beads before panning (start from 2nd round), Streptavidin beads blocking is optional.
-
f.Transfer the phage at step 21c into washed streptavidin beads, rotate the tubes at 10 RPM/min for 1 h at RT (20°C–25°C).
-
g.Wash the beads with 1 mL 0.05% PBST (0.05% Tween-20 in DPBS) for 5 times, then wash with DPBS twice.Note: Wash the beads with 0.05% PBST as follows: 1st round 5 times, 2nd round 8 times, 3rd round 15 times and 4th wash 20 times. (Increasing the washing numbers throughout the phage display selection can help to decrease low affinity binders. So that high affinity antibodies can be enriched efficiently).
-
h.Resuspend the beads with 5 mL TG1 cells (OD600 ≈ 0.5) in 15 mL culture tube, incubate the mixture at 37°C, 45 min. Mix every 15 min during incubation.CRITICAL: The phage on the beads can infect TG1 cells effectively, so the elution step is not required.
-
i.Take 1/103 infected TG1 cells (5 μL culture medium) into a 1.5 mL centrifuge tube which contain 995 μL fresh 2-YT medium. 10-fold serial dilute the bacteria with 2-YT medium and take 1/104, 1/105 and 1/106 of total bacteria plate onto 2-YT-Agar plates with 100 μg/mL ampicillin. The rest add 100 μg/mL ampicillin and 2% glucose, shaking at 37°C, 200 rpm for 2 h.
-
j.Add 10 μL M13KO7 helper phage (Cat#18311019, Thermo) into the cells (10 μL/5 mL, phage titer: 1011/mL), incubate at 37°C, 45 min. Mix every 15 min during incubation.
-
k.Centrifuge at 4,000 g for 10 min at RT (20°C–25°C). Discard supernatant and resuspend the pellet with 50 mL 2-YT contain 100 μg/mL ampicillin, 50 μg/mL kanamycin. shaking at 30°C, 200 rpm 12–15 h.
-
a.
-
22.Phage purification and start of the next round:
-
a.Check the colonies the next day.
-
b.Transfer the cell culture into a 50 mL centrifuge tube, Spin down the bacteria at 8,000 g for 10 min at 4°C and collect the supernatant with a new 50 mL centrifugation tube.
-
c.Add 25% volume of PEG/NaCl solution into the supernatant, mix and incubate on ice for 1 h.
-
d.Centrifuge at 11,000 g for 20 min. Discard supernatant and resuspend the pellet with 2 mL cold DPBS.
-
e.Separate the phage into two 1.5 mL tubes and centrifugation at 15,000 g for 1 min at 4°C to eliminate the bacteria.Note: This bacterial eliminate step is critical before storage, it will eliminate most of the bacteria in the phage. The rest bacteria will be eliminated by multiple rounds of wash during the next round of phage panning.
-
f.Transfer the phage to two new tubes and determined the titer by detecting OD268 (1 OD ≈ 5 × 1012 phage).CRITICAL: If the phage titer is lower than 1011/mL, do not start next round of panning. Check the quality of TG1 cell and M13KO7 helper phage to confirm both are good for experiment. Repeat the first round of panning and culture 15 h before purifying the phage.
-
g.Add 20% glycerol into one tube and stock in −80°C. Take 1012 phage from the other tube for next round of panning.
-
a.
Note: If the phage purified from previous round is lower than 1012, use > 1011 phage for panning. The phage input can be less in the later rounds of panning due to the decreased diversity.
-
23.Complete round 2, round 3 and round 4 (optional) phage panning.CRITICAL: Before starting next round of panning, take 1011 to 1012 previous round purified phage, dilute the phage into 1 mL 5% Milk-DPBS (second and fourth rounds panning blocking the phage with 5% Milk-DPBS, third round panning blocking the phage with 3% BSA-DPBS) and incubate with 50 μL streptavidin-magnetic beads at 10 rpm for 1 hour at RT (20°C–25°C). Clean the beads with magnets. The beads depleted phage is used for next round of panning.Note: Depending on the enrichment, in some cases round 4 is necessary to enrich high-affinity binders and narrow down the whole hits.Optional: polyclonal phage ELISA to detect the enrichment of binders:
-
a.Coat the ELISA plates with RBD-AviTag (5 μg/mL in DPBS, 50 μL/well) overnight (12–15 h) at 4°C.
-
b.Wash plates 3 times with 0.05% PBST, block with 100 μL 5% Milk-DPBS 2 h.
-
c.Wash plates 3 times with 0.05% PBST.
-
d.Add 100 μL 5% Milk-DPBS diluted phage (∼1011 phage from each round), shake 1 h with 200 rpm/min at RT (20°C–25°C).
-
e.Wash plates with 0.05% PBST, 4 times.
-
f.Add 100 μL Anti-M13 Antibody (HRP) (Sino Biological Inc, Cat#11973-MM05T-H, 1:2000 diluted in 5% Milk-DPBS), shake 45 min with 200 rpm/min at RT (20°C–25°C).
-
g.Wash plates with 0.05% PBST, 5 times.
-
h.Add 50 μL TMB into each well and reaction 5–10 min.
-
i.Stop the reaction with 50 μL 2 M H2SO4, read the optical density (OD) with a microplate reader at 450 nm.
-
a.
-
24.
Monoclonal ELISA Screening
After three rounds of panning, colonies will be picked for expression of heavy chain antibody domains which will be further screened by supernatant ELISA (Figure 6A. (Sun, Chen et al. 2020)).-
a.Add 180 μL 2-YT medium with 100 μg/mL ampicillin to each well of 96-well plate.
-
b.Pick single colony into each well, Incubate the 96-well plates at 37°C, 200 rpm until OD600 ≈ 0.5.Note: Take 200 μL OD600 = 0.2, 0.4, 0.6, 0.8 TG1 cells into a 96 well plates, detect the OD600 with Synergy HTX Multi-Mode Reader to get a standard curve. Compare the absorbance with the standard curve. Once most wells reach to OD600 ≈ 0.5, add IPTG to induce VH expression.
-
c.Add 20 μL IPTG stock (10 mM) into each well (final IPTG concentration = 1 mM) to induce VH expression. Incubate the plates at 30°C, 200 rpm 12–15 h.
-
d.Coat the ELISA plates with RBD-AviTag (2 μg/mL in DPBS, 50 μL/well) overnight (12–15 h) at 4°C.
-
e.Wash plates 3 times with 0.05% PBST, and block with 100 μL 3% BSA-DPBS 2 h.
-
f.Wash plates 3 times with 0.05% PBST.
-
g.Add 50 μL 6% BSA-DPBS into each well of the RBD-AviTag coated plates. Spin the bacteria expression plates (step 24c) at 4,000 g for 5 min at 4°C, transfer 50 μL supernatant into each well, shake 2 h with 200 rpm/min at RT (20°C–25°C).Note: Keep the bacterial pellet for scale up and plasmid purification.
-
h.Wash plates with 0.05% PBST, 4 times.
-
i.Add 100 μL Monoclonal ANTI-FLAG® M2-Peroxidase (1:2500 diluted in 3% BSA-DPBS), shake 45 min with 200 rpm/min at RT (20°C–25°C).
-
j.Wash plates with 0.05% PBST, 5 times.
-
k.Add 50 μL TMB into each well and reaction 5–10 min.
-
l.Stop the reaction with 50 μL 2 M H2SO4, read the optical density (OD) with a microplate reader at 450 nm.
-
m.Take 10 μL bacterial from the positive wells, scale up with 2 mL 2-YT medium with 100 μg/mL ampicillin and incubate at 37°C, 200 rpm overnight (12–15 h). Purify the plasmid of selected positive clones and send for sequencing. Keep clones with unique sequences for VHs preparation and characterization.
-
a.
-
25.VH antibody domain expression and purification
-
a.Transform HB2151 competent cells with selected plasmid DNA by heat-shock 1 min at 42°C. Recovering 45–60 min at 37°C, 200 rpm, plate the transformed cells onto 2-YT-Agar plates with 100 μg/mL ampicillin and 1% glucose. Incubate at 37°C overnight (12–15 h).CRITICAL: The pComb3X has an amber stop codon (TAG) between flag tag and gene III (Figure 2D). TG1 is an amber codon (TAG) suppressor strain, allowing translation to read through the codon and to produce a full-length VH-gene III fusion protein. While the HB2151 is an amber codon non-suppressor strain, Only VH gene can be translation to produce VH in this strain. So, HB2151 is chosen for VH expression without need to re-clone the VH gene into another expression plasmid.
-
b.Pick single colony from the fresh transformed plate into 50 mL centrifugation tube which has 20 mL 2-YT medium containing 100 μg/mL of ampicillin, Incubate at 37°C, 200 rpm until OD600 ≈ 0.5.
- Add 20 μL IPTG stock (1 M) into each tube (final IPTG concentration = 1 mM). Incubate at 30°C, 200 rpm 12–15 h.
-
c.Centrifuge the bacteria at 5,000 g for 5 min at 4°C. Resuspend the bacterial pellet in 10 mL of DPBS, add 0.1 million units polymyxin B (5 MU polymyxin B for 1 L culture) and incubate at 37°C, 200 rpm for 30 min.Note: Polymyxin B works by interacting with lipopolysaccharide (LPS), alters membrane permeability of gram-negative bacteria, ultimately leading to cell lysis. It is a simple and robust way to lysis outer membrane of gram-negative bacteria for periplasmic protein purification.
-
d.Centrifugation at 16,000 g for 30 min at 4°C. Transfer the supernatant into a new tube.
-
e.Purify the VHs with HisPur™ Ni-NTA Resin.
-
f.ELISA to confirm the binding of selected VH antibody domains and detect their EC50 (Figure 6B).
-
g.Dynamic light scattering (DLS) and size exclusion chromatography (SEC) analysis to detect the aggregates of VHs candidates. Non-aggregates and low aggregates VHs are chosen for further study.
-
h.ELISA to detect the VH antibody domains competition with ACE2 to RBD binding.
-
i.The VH antibody domains competition with ACE2 are chosen for SPR to determine affinity and virus neutralization.
-
a.
Figure 6.

ELISA results of selected clones
(A) Plate based monoclonal ELISA Screening results. Clear wells mean no VHs or very low concentration (undetectable with ELISA) VHs binding with the coated plate. Blue color wells shown high concentration VHs binding with the RBD-AviTag coated plate. The colonies which get blue color are selected and expanded for sequencing.
(B) ELISA results of different purified VH binders. The plate was coated with RBD-AviTag. 3-fold serial diluted VH binders (start concentration=1 μM) were added into each well to detect the binding of VHs to RBD-AviTag. The results shown all the VHs have good binding with RBD-AviTag.
Expected outcomes
For VH antibody domain library construction, each electroporation will result in > 109 colonies and 150 electroporation will make > 1011 size VH library. Transient expression productivity of RBD-AviTag with Expi293 system should yield > 10 mg/L and the RBD-AviTag expression stable cell pool will be generated within 10 days. High affinity RBD VH binders which compete with human ACE2 for binding to RBD will be selected after three rounds of panning. We have got 16 unique VH binders with this protocol and the equilibrium dissociation constant of these binders is from 300 nM to 4 nM.
Limitations
Compared with single human B cell isolation, phage display is based on bacterial- expression system. In general, it has limitations on protein expression, folding and post-translational modification. Our VH antibody domain library is generated from healthy human donors with the CDRs naturally grafted from human PBMC cDNA. It may lower the possibility of non-specific binding to human cells compare with synthetic library. However, non-specific binding was found in some of the selected VH antibody domains.
Compare with scFv, Fab and VHH libraries, due to large size of our VH library, we have selected out many high affinity VH binders (nM range affinity). There are no significant affinity limitations compare with other libraries. However, the VH antibody domains are much easier to aggregation and aggregations are detected in most of the selected antibody domains. So, characterization of the selected domains one by one to figure out the best functional candidates for further therapeutic development is needed.
Troubleshooting
Problem 1
The productivity of full-length VH assemble by over-lapping PCR is low (step 2).
Potential solution
Assemble the full-length VH with two steps:
-
1.
Over-lapping PCR to assemble FR1, CDR1 and CDR2 with ALL-F/H2R1, H2R24, H2R36, H2R57 primers to get FR1-CDR1-CDR2-FR3. Assemble FR3 and CDR3 with FR3F/All-R primers to get FR3-CDR3-FR4 using the following cycling conditions:
50 μL PCR system:
| Component | Volume/Weight | Final concentration |
|---|---|---|
| PCR master mix 1 | 25 μL | 1 × |
| 10 μM forward primer | 1 μL | 0.2 μM |
| 10 μM reverse primer | 1 μL | 0.2 μM |
| FR1, CDR1, CDR2 or FR3 and CDR3 DNA template mixture | 100 ng | 2 ng/μL |
| ddH2O | to 50 μL | n/a |
Thermocycling conditions for PCR:
| Steps | Temperature | Time |
|---|---|---|
| Initial denaturation | 94°C | 4 min |
| 25 cycles | 94°C | 45 s |
| 55°C | 45 s | |
| 72°C | 1 min | |
| Final extension | 72°C | 5 min |
| Hold | 4°C |
Run all the PCR samples with 2% agarose gel to verify the size and purify the right size PCR products from the agarose gel with QIAquick Gel Extraction Kit.
-
2.
Over-lapping PCR to assemble full-length VH with FR1-CDR1-CDR2-FR3 and FR3-CDR3-FR4 DNA as template, ALL-F and ALL-R as primes using the following cycling conditions:
50 μL PCR system:
| Component | Volume/Weight | Final concentration |
|---|---|---|
| PCR master mix 1 | 25 μL | 1 × |
| 10 μM forward primer | 1 μL | 0.2 μM |
| 10 μM reverse primer | 1 μL | 0.2 μM |
| FR1-CDR1-CDR2-FR3 and FR3-CDR3 DNA template mixture | 100 ng | 2 ng/μL |
| ddH2O | to 50 μL | n/a |
Thermocycling conditions for PCR:
| Steps | Temperature | Time |
|---|---|---|
| Initial denaturation | 94°C | 4 min |
| 28 cycles | 94°C | 45 s |
| 55°C | 45 s | |
| 72°C | 1 min | |
| Final extension | 72°C | 5 min |
| Hold | 4°C |
Run all the PCR samples with 2% agarose gel to verify the size and purify the right size PCR products from the agarose gel with QIAquick Gel Extraction Kit.
Problem 2
Low efficiency of electroporation, hard to generate large size phage library (step 7).
Potential solution
The most common reason of low efficiency is the poor quality of digested VH antibody domains (step 3) or DNA degradation by over- digestion and ligation (step 4). Long extra protection bases (such as 15 bp) in primers can be added in front of the SfiI restriction site. This will help improve the digestion of SfiI to get high quality digested VH.
Run agarose gel to check the ligation DNA quality. If degradation is detected, shorten the ligation time to 48 h.
Problem 3
Low affinity of the selected VH antibody domains (step 25f).
Potential solution
Use lower concentration of antigens for panning and increase the washing times to enrich high affinity domains.
Problem 4
Selected VH domain candidates aggregate (step 25g).
Potential solution
Human variable domains rapidly aggregate when heated to 80°C–85°C (this condition well above their melting temperatures). The aggregated phage can be eliminated by centrifugation while the non-aggregation phage remains in the supernatant. Use the heat-treated phage supernatant for panning can help to get non-aggregating VH domains (Dudgeon, Rouet et al. 2012).
Before panning (start from second round), heat the phage at 80°C for 10min, then keep on ice for 10 min and centrifugation for 10 min at 15,000 g (white pellet can be found at the bottom of the centrifuge tube after centrifugation). Collect the supernatant into a new tube to eliminate the aggregated VHs for panning.
Problem 5
During Expi293F™ cell maintain, cell clumping was detected, and cell viability is lower than 97% (In “before you begin, step 8”).
Potential solution
Thaw a new Expi293F™ should always be the first choice. If clumping still detected, check the shake speed of the CO2 Resistant incubator, and subculture the cells every two days with fresh medium.
Check the cell viability with trypan blue. If viability is lower than 97%, do not let the cell density over 5 × 106/mL and do not maintain the cells more than 5 days without subculture.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contacts, Chuan Chen (CHC316@pitt.edu), Wei Li (liwei171@pitt.edu), and Dimiter S. Dimitrov (mit666666@pitt.edu).
Materials availability
For cell lines and plasmid please contact Dimiter S. Dimitrov (mit666666@pitt.edu). All other materials are available commercially.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
We would like to thank the members of our group Dontcho Jelev, Megan Shi, Cynthia Adams, Du-San Baek, Ye-Jin Kim, and Xiaojie Chu for their helpful discussions. We also thank our previous colleagues Drs. Weizao Chen, Tianlei Ying, and Zhongyu Zhu for their helpful discussion. This work was supported by the University of Pittsburgh Medical Center.
Author contributions
C.C. drafted the manuscript, and the manuscript was edited by W.L., Z.S., X.L., and D.S.D.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Chuan Chen, Email: CHC316@pitt.edu.
Wei Li, Email: liwei171@pitt.edu.
Dimiter S. Dimitrov, Email: mit666666@pitt.edu.
References
- Dudgeon K., Rouet R., Kokmeijer I., Schofield P., Stolp J., Langley D., Stock D., Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. U S A. 2012;109:10879–10884. doi: 10.1073/pnas.1202866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr C., Nothaft H., Lizak C., Glockshuber R., Aebi M. The Escherichia coli glycophage display system. Glycobiology. 2010;20:1366–1372. doi: 10.1093/glycob/cwq102. [DOI] [PubMed] [Google Scholar]
- Gray D. Overview of protein expression by mammalian cells. Curr. Protoc. Protein Sci. 2001;Chapter 5 doi: 10.1002/0471140864.ps0509s10. Unit5 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K.H. Gene expression in Mammalian cells and its applications. Adv. Pharm. Bull. 2013;3:257–263. doi: 10.5681/apb.2013.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Schafer A., Kulkarni S.S., Liu X., Martinez D.R., Chen C., Sun Z., Leist S.R., Drelich A., Zhang L. High Potency of a Bivalent Human VH Domain in SARS-CoV-2 Animal Models. Cell. 2020;183:429–441.e16. doi: 10.1016/j.cell.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Drelich A., Li W., Chen C., Sun Z., Shi M., Adams C., Mellors J.W., Tseng C.T., Dimitrov D.S. Enhanced elicitation of potent neutralizing antibodies by the SARS-CoV-2 spike receptor binding domain Fc fusion protein in mice. Vaccine. 2020;38:7205–7212. doi: 10.1016/j.vaccine.2020.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Chen C., Li W., Martinez D.R., Drelich A., Baek D.S., Liu X., Mellors J.W., Tseng C.T., Baric R.S., Dimitrov D.S. Potent neutralization of SARS-CoV-2 by human antibody heavy-chain variable domains isolated from a large library with a new stable scaffold. MAbs. 2020;12:1778435. doi: 10.1080/19420862.2020.1778435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.