

Abstract
Infections are a major complication of obesity, but the mechanisms responsible for impaired defense against microbes are not well understood. Here, we found that adipocyte progenitors were lost from the dermis during diet-induced obesity (DIO) in humans and mice. The loss of adipogenic fibroblasts from mice resulted in less antimicrobial peptide production and greatly increased susceptibility to Staphylococcus aureus infection. The decrease in adipocyte progenitors in DIO mice was explained by expression of transforming growth factor–β (TGFβ) by mature adipocytes that then inhibited adipocyte progenitors and the production of cathelicidin in vitro. Administration of a TGFβ receptor inhibitor or a peroxisome proliferator–activated receptor–γ agonist reversed this inhibition in both cultured adipocyte progenitors and in mice and subsequently restored the capacity of obese mice to defend against S. aureus skin infection. Together, these results explain how obesity promotes dysfunction of the antimicrobial function of reactive dermal adipogenesis and identifies potential therapeutic targets to manage skin infection associated with obesity.
INTRODUCTION
The excessive expansion of white adipose tissue (WAT) that is associated with obesity affects more than 20% of the global population (1–3) and promotes several human diseases (1, 4). It has been suggested that obesity can promote cardiovascular disease, cancer, and delayed wound healing by driving systemic inflammation (1, 4–7). However, although these observations provide a rational explanation for disorders related to increased inflammation and obesity, it remains unclear why the incidence of infection, an adverse event typically associated with immunosuppression, is increased in the obese population. The increasing prevalence of systemic medical complications associated with obesity requires better understanding of the mechanisms by which the expansion of adipose tissue promotes disease.
A key mechanism to combat infection is by the production of antimicrobial peptides and proteins (AMPs) (8, 9). These gene-encoded antibiotics are expressed by many organs and cell types in humans and in other animal models (8). AMPs are found in high concentrations within classical innate immune cells such as neutrophils and mast cells, as well as in epithelial cells such as keratinocytes and colonic epithelia (9, 10). Recently, the activation of dermal fibroblasts (dFBs) to differentiate into adipocytes was observed to be an important source of AMPs (11 12). Skin infection induces a subset of dFBs to differentiate into adipocytes, thus expanding the local dermal WAT (dWAT). This event of reactive adipogenesis results in the transient production of large amounts of AMPs and was found to be essential for optimal defense against deep skin infection by Staphylococcus aureus (11–13). Given its location as the deepest layer of skin, dWAT is the final defensive barrier to systemic spread of virulent pathogens.
In addition to defense against infection, dWAT also has several other important nonmetabolic functions (14–16). Changes in the dermal adipocyte and adipocyte progenitor (AP) populations have been described during hair cycling (17, 18), wound repair (15, 19–21), skin fibrosis (22), and thermogenesis (23). A better understanding of how obesity alters dermal fat function could provide insight into the increased incidence of infection in the obese population and other obesity-related skin complications such as defective wound healing and hair loss.
The reported increase in dermal adipocytes associated with obesity appears to be counterproductive to the beneficial activity of dWAT in fighting infections. To address this apparent inconsistency, we sought to determine whether obesity has a negative effect on AMP production during the differentiation of fibroblast precursors into adipocytes, and whether this may explain decreased innate immune defense against infection that is seen in diet-induced obesity (DIO). Using several independent methods, including single-cell RNA sequencing (RNA-seq) (scRNA-seq) analyses and dual-color adipocyte lineage tracing, we sought to obtain in-depth knowledge of how DIO affects dermal adipogenesis. Furthermore, as DIO promotes expansion of mature adipocytes, a coculture system that combined mature adipocytes and APs was established to determine the underlying mechanisms by which DIO may inhibit the antimicrobial function of the dermal progenitors.
RESULTS
DIO increases susceptibility of the skin to S. aureus and inhibits reactive adipogenesis
To determine how DIO influences the capacity of dermal adipocytes to defend the skin against infection, we fed mice with high-fat diet (HFD) or standard diet (SD) for 6 months and then challenged them with S. aureus by intradermal injection. After intradermal challenge with S. aureus, mice on HFD had significantly (P < 0.01) increased lesion size (Fig. 1, A and B) and significantly increased bacterial colony-forming units (CFUs) recovered from the site of infection (Fig. 1C and fig. S1A). A similar phenotype was observed in a separate study in which mice on HFD were paired with mice on a nutrient- and calorie-matched low-fat diet (LFD) as control (fig. S1, B and C). Flow cytometry analyses of cells isolated from the skin at the site of infection revealed that recruitment of CD11B+Ly6G+ neutrophils upon infection was not notably altered in HFD versus SD (Fig. 1D). However, analysis of platelet-derived growth factor receptor A–positive (PDGFRA+) dFBs after infection revealed that the activation of adipogenic dFBs identified as THY1+SCa1+ cells was prominently impaired in obese mice compared to lean controls (Fig. 1, E and F). In addition, other features of early adipogenesis including proliferation of preadipocytes (pADs) as shown by costaining for preadipocyte factor 1 (PREF1) and the proliferation marker 5-bromo-20-deoxyuridine (BrdU; fig. S1D), markers of early adipocyte differentiation phospho-CCAAT enhancer binding protein beta (pC/EBPβ) and collagen type IV (COLIV) (Fig. 1G), expression of AMP cathelicidin (Camp) and expression of pAD marker genes (Pdgfra and Sca1) (Fig. 1, H and I, and fig. S1, E and F) were lower in the dWAT layer of obese mice. Skin from mice on HFD also showed increased expression of the profibrotic marker Spp1 after infection (Fig. 1I). Together, these results show that obesity in mice results in loss of the dermal pool of pADs and inhibited capacity to initiate reactive adipogenesis and express cathelicidin AMP.
Fig. 1. Dermal reactive adipogenesis upon S. aureus infection is lost in DIO mice.
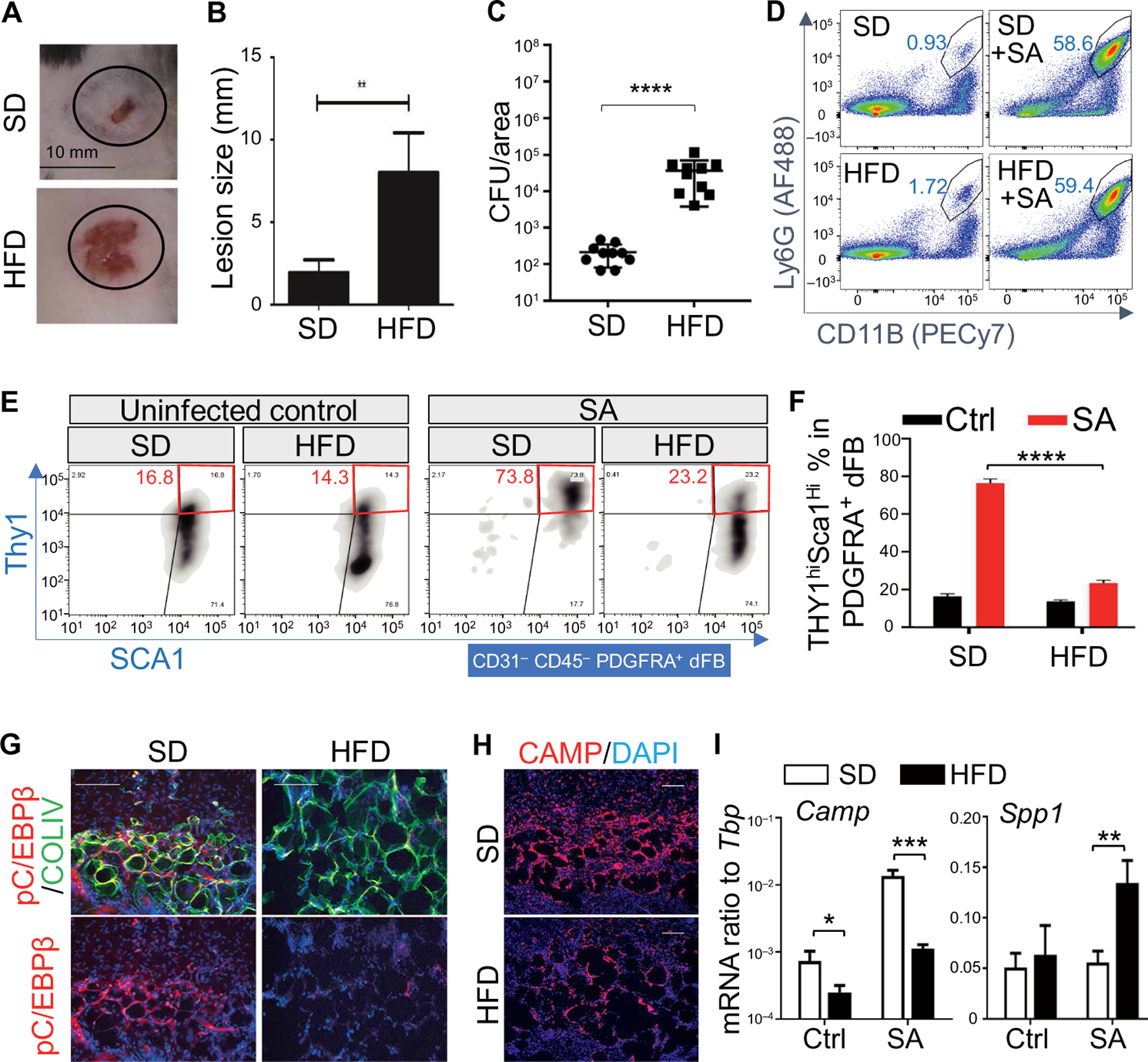
(A and B) SD or HFD mice were infected intradermally for 3 days with S. aureus (SA), and infected skin was collected for lesion size measurement (n = 10 per group). (C) Measurement of bacterial CFU from the infected edge area of the skin from SD or HFD mice (n = 10 per group). (D) FACS plots for neutrophils (gated as viable Ly6GhiCD11Bhi cells) in the skin of uninfected or S. aureus–infected mice fed with SD or HFD (representative of n = 3 per group). (E) FACS plots for Thy1 and Sca1 expression on CD31−CD45−PDGFRA+ dFBs (representative of n = 3 per group). (F) Quantification of the percentage of THY1hiSca1hi cells in PDGFRA+ dFBs (n = 3 per group). (G and H) Skin sections of S. aureus–infected SD or HFD skin were stained with pC/EBPβ (red) and COLIV (green) to measure adipocyte differentiation (G), or CAMP [red in (H)] as indicated. Nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bars, 100 µm. (I) qRT-PCR analysis of Camp and Spp1 mRNA expression in control (Ctrl) or S. aureus–infected SD or HFD skin (ratio to Tbp) (n = 3 to 5 per group). Scale bars, 100 µm (G and H). All error bars indicate mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (one-way ANOVA).
scRNA-seq reveals defective activation of dFBs in obese mice
To further define how obesity alters the dermal immune response, we performed 3′-end scRNA-seq on cells isolated from whole dorsal skin of SD and HFD mice challenged with S. aureus skin infection (Fig. 2A) (24). In line with our fluorescence-activated cell sorting (FACS) results, scRNA-seq analyses (Fig. 2B and fig. S2A) demonstrated that the recruitment of Ptprc/Cd45+ Cd11b+S100a8+neutrophils after S. aureus infection was not impaired in HFD skin, whereas Pdgfra+ dFB clusters were greatly depleted in HFD mouse skin compared to SD mouse skin after infection (Fig. 2B).
Fig. 2. scRNA-seq analysis reveals defective activation of dFB in S. aureus–infected skin in DIO mice.
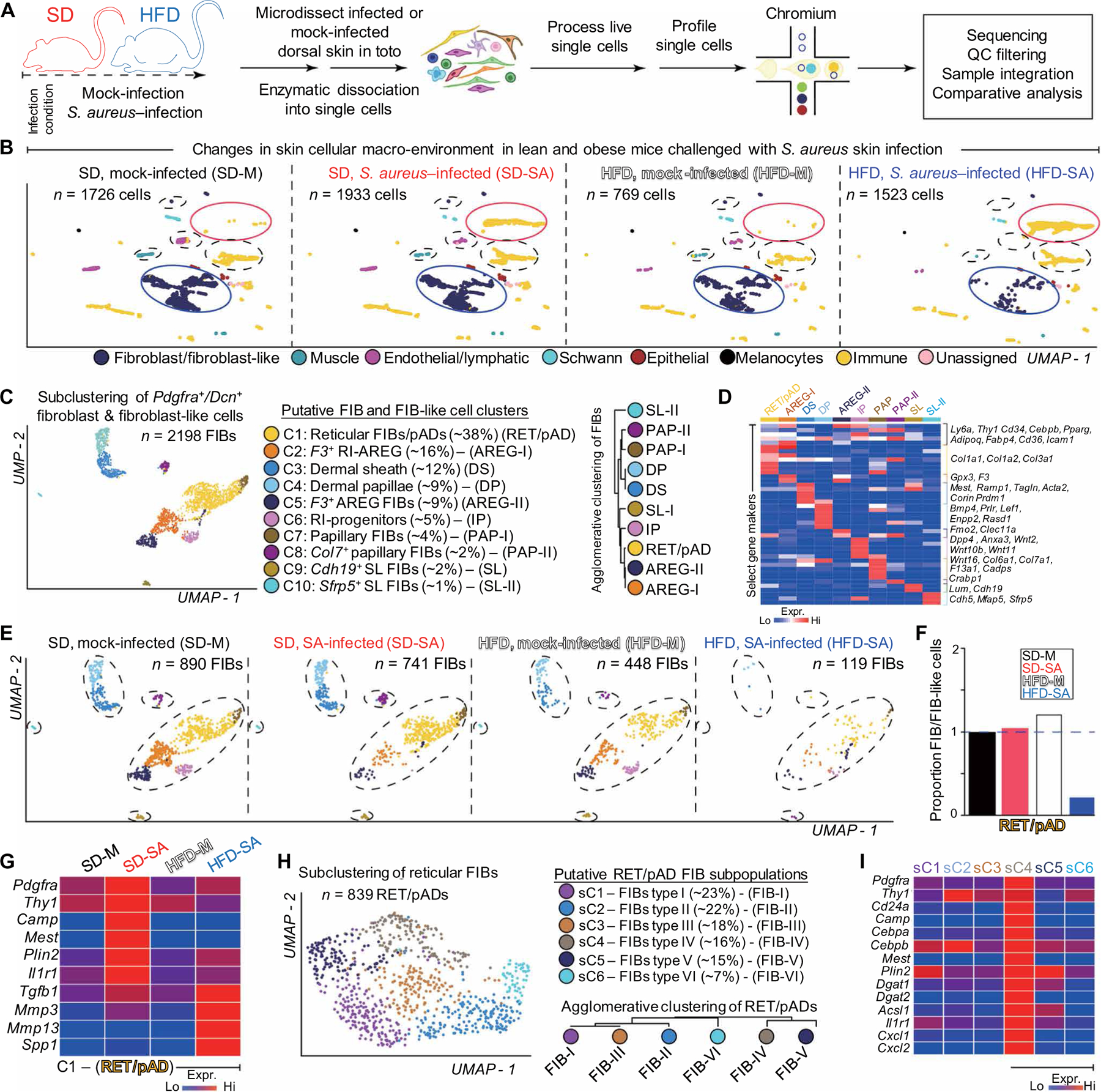
(A) Schematic of scRNA-seq experiment. Single cells were isolated from dorsal skin of SD or HFD mice challenged with or without S. aureus (SA) skin infection. Isolation, processing, and capture of single cells by droplet-based device, 3′-end scRNA-seq, and downstream cc. QC, quality control. (B) Assembly of multiple distinct datasets (SD, S. aureus–infected; HFD, mock-infected; HFD, S. aureus–infected) into a reference dataset (SD, mock-infected) visualized with uniform manifold approximation and projection (UMAP) showing changes in fibroblast/fibroblast-like and immune cell composition in DIO mice challenged with S. aureus infection. Putative cell community identity is defined on the bottom. Neutrophil cluster is shown in yellow and highlighted by a red circle; dFB clusters are shown in blue and highlighted in blue circle. (C) Subclustering of Pdgfra+/Dcn+ dFBs and fibroblast-like cells. Ten putative cell clusters were identified, and each cluster is coded with a distinct color. Putative cell cluster identity was defined by bona fide biomarker expression. Agglomerative clustering demonstrates hierarchical relationships between Pdgfra+/Dcn+ fibroblast and fibroblast-like cells. (D) Heatmap showing enriched genes for each subcluster of Pdgfra+/Dcn+ fibroblasts and fibroblast-like cells. (E) Anchored Pdgfra+/Dcn+ fibroblasts and fibroblast-like cells split by condition and visualized in UMAP space. SD, mock-infected (n = ~890 viable cells); SD, S. aureus–infected (n = ~741 viable cells); HFD, mock-infected (n = ~448 viable cells); and HFD, S. aureus–infected (n = ~119 viable cells) cell clusters are color coded. Broken circles demarcate changes in Pdgfra+/Dcn+ cells across conditions. (F) Proportion of C1 RET/pAD, normalized to library size after quality control, across conditions. (G) Heatmap showing expression of selected markers in C1 RET/pAD fibroblasts per condition. (H) Subclustering of RET/pAD fibroblasts. Six putative cell clusters were identified and color coded. Putative cell cluster identity was defined. Agglomerative clustering demonstrates hierarchical relationships between RET/pAD fibroblast subtypes. (I) Heatmap showing expression of enriched gene markers in sC1~sC6 FIB subtypes from the RET/pAD fibroblasts under SD-SA condition. SD, standard diet; HFD, high-fat diet; M, mock; FIB, fibroblasts; RET, reticular; pAD, preadipocyte; RI, reticular interstitial; AREG, adipogenesis-regulatory cells; DS, dermal sheath; DP, dermal papillae; RI-P, RI progenitor; PAP, papillary.
We next identified 10 subclusters within the Pdgfra+;Dcn+ dFB population based on marker gene expression (Fig. 2, C and D). The top eight major subclusters constituted 97% of all dFB cells and were defined as reticular/pAD (RET/pAD) dFBs (C1, ~38%, Col1a1+;Cd36+;Icam1+; Cebpb+;Pparg+), RET interstitial (RI) adipogenesis-regulatory (AREG) cells–I (C2, ~16%, F3+;GPX3+) (25), dermal sheath (C3, ~12%, Tagln+;Mest+), dermal papillae (C4, ~10%, Corin+;Prdm1+;Bmp4+; Lef1+), RI-AREG-II (C5, ~9%, F3+; Clec11a+;Gpx3+) (25), RI progenitors (C6, ~5%, Wnt2+;Anxa3+;Dpp4+) (26), and papillary (PAP) dFB (C7 and C8, ~4%). In samples from SD mice, three RI clusters (C2, C5, and C6) became depleted after infection, whereas the RET/pAD C1 cluster was slightly enriched compared to mock control (Fig. 2, E and F). In contrast, almost all dFB clusters (fig. S2B), including the RET/pAD cluster (Fig. 2F), were depleted in HFD skin after S. aureus infection.
S. aureus infection induced the expression of a panel of proadipogenic genes, including AMP Camp, pAD markers Pdgfra and Thy1, adipocyte markers Mest and Plin2, and innate immune receptor Il1r1 in the C1 RET/pAD dFB subcluster in SD mice but not in HFD mice (Fig. 2G). Instead, HFD RET/pAD dFBs gained higher expression of profibrotic genes including Tgfb1, Mmp3, Mmp13, and Spp1 (Fig. 2G) after infection. To further characterize Camp-producing RET/pAD cells, the C1 RET/pAD subcluster was reclustered into six additional subclusters (sC1 to sC6) (Fig. 2H). Camp expression was detected only in subcluster sC4 (Fig. 2I), which was characterized by an adipogenic gene signature (fig. S2, C to E) and was enriched with other genes associated with reactive adipogenesis and innate immune function such as Pdgfra, Thy1, Cd24a, Cebpa, Il1r1, Mest, Plin2, Dgat1/2, Il1r1, Cxcl1, and Cxcl2 (Fig. 2I). These results support the conclusion that HFD results in a loss of the subpopulation of dFB that have the capacity to become mature adipocytes and express AMP upon S. aureus challenge.
Lineage tracing of dermal adipocytes during HFD feeding
We next sought to characterize how HFD alters the adipogenic function of dFBs. Histologic assessment of the back skin of mice fed with HFD for 6 months showed thinning of the dermis, expansion of dermal adipocytes, and loss of dermal cells compared to back skin of SD control mice (Fig. 3, A and B, and fig. S3, A and B), suggesting that HFD promotes hyperplastic growth of adipocytes from dFBs followed by hypertrophic expansion of these adipocytes. To characterize this further, we crossed adipocyte-specific, tamoxifen (TAM)–inducible Adiponectin-Cre estrogen receptor driver (Adipoq-CreER) mice with a dual-fluorescent reporter mouse (mTmG). Upon TAM induction, existing adipocytes become labeled with green fluorescence (membrane-targeted green fluorescent protein, mG), whereas new adipocytes formed after TAM withdrawal become red fluorescent [membrane-targeted Tomato (mT)] (Fig. 3C). This permitted us to distinguish between adipocyte hyperplasia and hypertrophy. When existing adipocytes were pulse labeled with TAM during the first week of HFD, >95% of adipocytes at 6 months of HFD were new (red; Fig. 3D). In contrast, only a small portion (<20%) of dWAT adipocytes were new at 6 months of HFD when TAM was given at 2 months of HFD, and almost all adipocytes were green when TAM was given after 4 months of HFD. These results indicate that HFD triggers an initial hyperplastic growth of adipocytes from their dermal progenitors, which may lead to depletion of dermal AD progenitor pool. However, after 2 to 4 months of HFD, adipocyte hypertrophy accounts for the majority of the expansion seen in dWAT, leading to domination of mature adipocytes in the dermis.
Fig. 3. Hypertrophic expansion of adipocytes promotes loss of their antimicrobial potential.
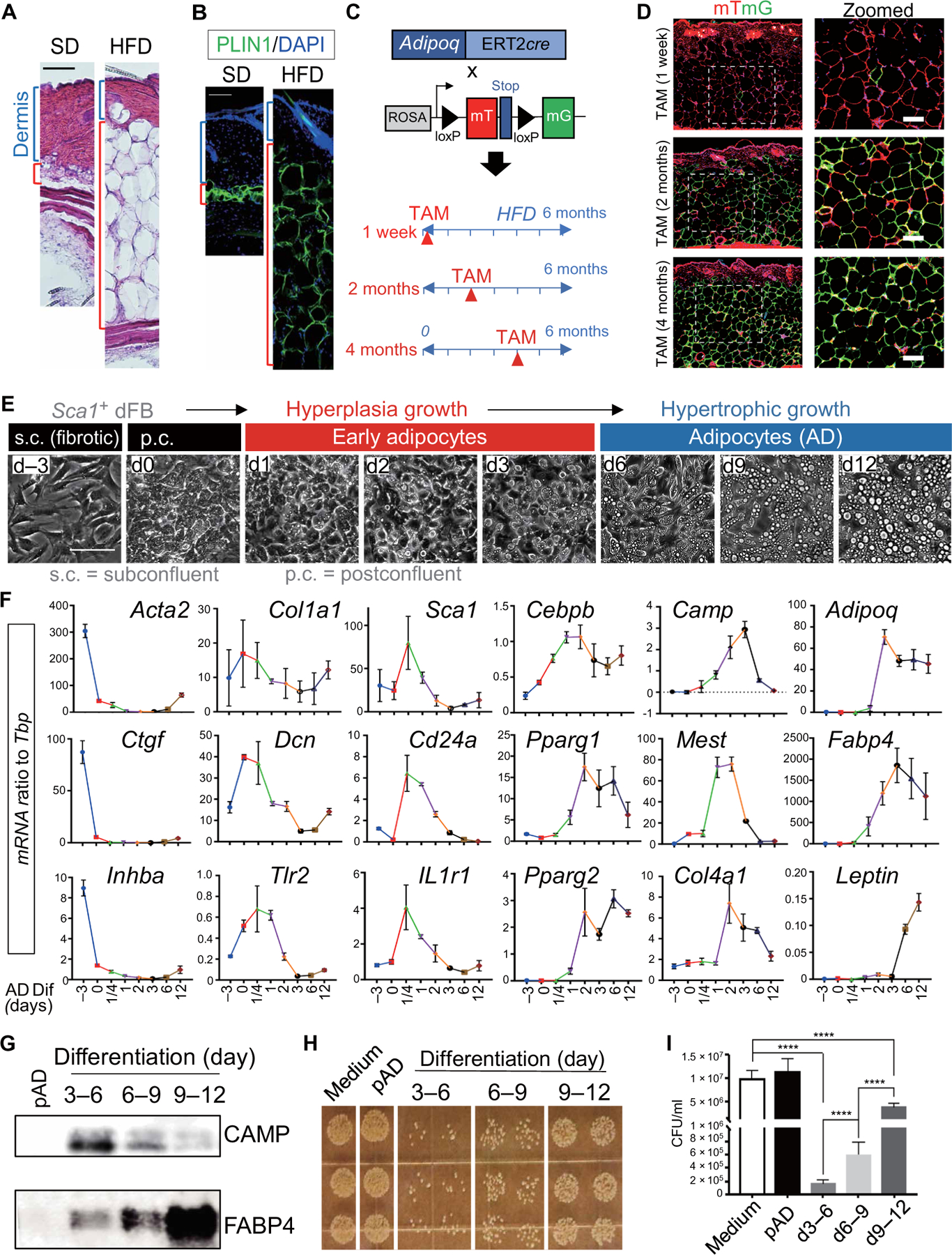
(A and B) Representative images for hematoxylin and eosin (A) or perilipin staining [green; (B)] of skin sections from mice at SD or HFD (n = 3 to 6 per group). Blue bracket indicates the dermis, and red bracket indicates the dermal fat layer. Scale bars, 100 µm. (C) Experimental scheme for analysis of adipocyte hyperplasia or hypertrophy during HFD feeding using the Adipoq-CreER;mTmG dual-fluorescence mouse model. (D) Representative images (n = 3 per group) of skin sections from Adipoq-CreER;mTmG mice at 6 months after HFD feeding after adipocytes were pulse labeled by TAM for 1 week at indicated time during HFD feeding. Scale bars, 100 µm. (E) Magnetic cell sorter–purified Sca1+ primary neonatal dermal FB culture subjected to adipocyte differentiation. Phase contrast images showing cell morphology changes and lipid droplet formation through the differentiation time course. s.c., subconfluent; p.c., postconfluent. (F) qRT-PCR analyses showing the mRNA expression kinetics of listed fibrosis markers (Acta2, Ctgf, and Inhba), extracellular matrix (Col1a1 and Dcn), innate immune receptors (Tlr2 and IL1r1), pAD (Sca1 and CD24a), and AD (Cebpb, Pparg1, Pparg2, Camp, Mest, Col4a1, Adipoq, Fabp4, and Leptin) as indicated. n = 3 per group. (G to I) Conditioned medium collected from undifferentiated pAD or differentiating adipocytes at indicated days (d) were subjected to Western blotting for CAMP and FABP4 antibodies (G), and an in vitro antimicrobial assay against S. aureus (H). (I) Quantification of bacterial CFU shown in (H) (n = 3 per group). All error bars indicate mean ± SEM. ****P < 0.0001 (one-way ANOVA).
Mature adipocytes lose their antimicrobial potential
We next evaluated how adipocytes change their capacity to express AMPs during their maturation in vitro. Primary mouse AP cells (purified on the basis of Sca1+ from neonatal mouse dFBs) were cultured from subconfluent (s.c.; day −3) to postconfluent (p.c.; day 0) conditions and then differentiated into early (days 2 to 5) and fully mature adipocytes (days 6 to 12) as seen by the appearance of large intracellular lipid droplets (Fig. 3E). The s.c. cells exhibited a spindle cell morphology and expressed high amounts of Acta2, Ctgf, and Inhba (Fig. 3, E and F). The expression of these profibrotic genes was down-regulated when cells became p.c. (designated as day 0). p.c. cells exhibited some typical pAD characteristics, including the presence of small lipid droplets, rounded cell morphology, and elevated expression of Dcn, Cebpb, Mest, Tlr2, and Col4a1 compared to s.c. cells (Fig. 3, E and F). Upon initiation of adipocyte differentiation by the addition of dexamethasone, 3-isobutyl-1-methylxanthine (IBMX), indomethacin, and insulin, the expression of Col1a1 and Dcn decreased and the expression of Sca1, Tlr2, Il1r1, and Cd24a transiently increased during the first-day postdifferentiation. This was then followed by induction of early adipocyte genes including Camp and Mest until day 3 (Fig. 3F). The expression of antimicrobial gene Camp decreased with the later increase in mature adipocyte markers, including Adipoq, Fabp4, and Leptin (Fig. 3F). In line with this transcriptional response, cathelicidin protein (CAMP) was secreted in the early phase of differentiation, whereas later, more mature adipocytes lost cathelicidin expression and secreted more fatty acid binding protein 4 (FABP4), an adipokine produced by mature adipocytes (Fig. 3G). Antimicrobial function correlated with cathelicidin expression as culture supernatant from early adipocytes inhibited the growth of S. aureus, whereas supernatant from mature adipocytes did not (Fig. 3, H and I). These data demonstrate that innate antimicrobial defense is optimal in early adipocytes but as adipocytes further mature, they then undergo a loss of this host defense function. These in vitro observations were consistent with the loss of antimicrobial defense function observed in obese mice whose dermis is dominated by mature adipocytes at the expense of pADs.
Mature adipocytes inhibit the adipogenic potential of dFBs
To determine whether the accumulation of mature adipocytes might also feedback to inhibit the capacity of the pAD pool of dFBs to acquire antimicrobial function, we tested the effect of mature adipocytes on the function of the immature dFB/AP cells (Fig. 4A). Three days of coculture of dFB/AP cells with mature adipocytes led dFB/AP cells to develop a notable spindle cell morphology characterized by cell stratification (Fig. 4B, left panel). Furthermore, dFB/AP cells exposed to mature adipocytes lost their adipogenic potential (Fig. 4B, right two panels), production of cathelicidin (Fig. 4C), and capacity to inhibit the growth of S. aureus (Fig. 4D and fig. S4A). These observations suggest that signaling from mature adipocytes to dFBs inhibits the capacity of adipocyte precursors to produce antimicrobial activity.
Fig. 4. Adipocytes inhibit the adipogenic function of dFBs.
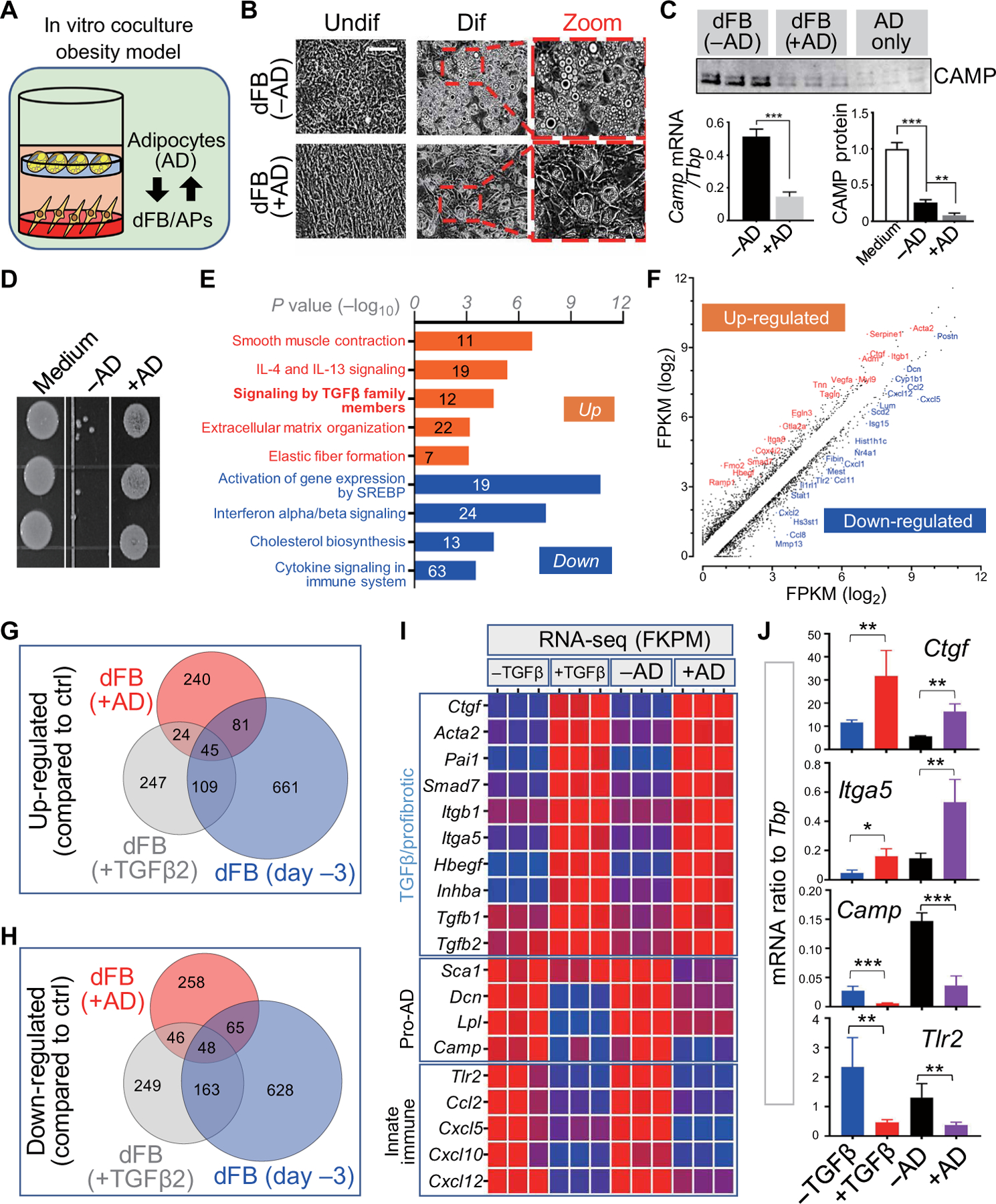
(A) Schematic for the in vitro coculture obesity model in which adipogenic dFB (pAD) seeded on a dish were cocultured with adipocytes (AD) seeded on a transwell insert on top. (B to D) dFBs seeded with or without AD coculture were subjected to adipocyte differentiation. (B) Representative phase contrast images (n = 3 per group) showing AD coculture triggered fibrotic cell morphology in undifferentiated dFBs and inhibited the subsequent adipocyte formation during differentiation. Scale bar, 100 µm. (C) Camp mRNA or CAMP protein expression in differentiating dFBs treated without or with AD coculture (n = 3 per group). (D) In vitro antimicrobial assay against S. aureus (n = 3 per group). (E and F) Reactome pathway analyses (E) or FPKM (fragments per kilobase of transcript per million mapped reads) correlation plots (F) of up- or down-regulated genes in dFB compared to controls upon AD coculture identified by bulk RNA-seq. IL, interleukin. (G and H) Venn diagram comparing up- or down-regulated genes in dFB with AD coculture, dFBs treated with TGFβ2 and s.c. dFBs (day −3, 3 days before confluency). (I) Heatmap of the FPKM of indicated genes showing a similar up-regulation of TGFβ/profibrotic genes and down-regulation of proadipogenic and innate immune genes in dFBs treated with TGFβ2 and dFBs seeded with AD coculture. (J) qRT-PCR validating the expression of select genes as indicated (n = 3 per group). All error bars indicate mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA).
To understand the mechanism of this inhibition, we performed reactome pathway analysis of the transcriptomes of dFB/AP cocultured with mature adipocytes, which revealed several pathways that were influenced by exposure to the soluble products of mature adipocytes (Fig. 4, E and F). Four of the top five up-regulated pathways were related to transforming growth factor–β (TGFβ): smooth muscle contraction, signaling by TGFβ, extracellular matrix organization, and elastic fiber formation. In contrast, the top pathways down-regulated in dFB/AP by mature adipocytes were those associated with early adipocyte development such as lipid biosynthesis [sterol regulatory element binding protein (SREBP) and cholesterol pathways] and innate immune responses (interferon and cytokine signaling). Because these features were similar to the response of dFB/AP when treated with TGFβ2 (fig. S4B) (11) or the s.c. day-3 fibrotic dFB (as shown in Fig. 3, E and F), we next compared the transcriptomes of these dFB/AP cells cultured under the three conditions. Venn diagram analyses revealed similarities between the responses of dFBs to mature adipocytes and TGFβ2 (Fig. 4, G and H). Specific transcripts induced such as Acta2, Ctgf, and Itga5 were downstream of TGFβ/profibrotic pathways, whereas down-regulated transcripts were associated with adipogenesis and innate immunity, for example, Sca1, Dcn, Camp, and Tlr2 (Fig. 4, I and J, and fig. S4C). Together, these results suggested that TGFβ could drive the loss of adipogenic and antimicrobial activity associated with obesity.
Mature adipocytes inhibit innate immune function of dFBs through the TGFβ-TGFBR-SMAD2/3 pathway
To further investigate the role of TGFβ in the inhibition of the innate immune defense function of adipogenesis, we measured the production of TGFβ1 and TGFβ2 during adipocyte maturation. Mature adipocytes secreted significantly (P < 0.01) more of these cytokines than immature pAD (Fig. 5A). We also observed that when dFBs were exposed to the products of mature adipocytes, there was rapid nuclear translocation of SMAD2/3 and accumulation of phospho-SMAD2/3 (pSMAD2/3) in the nucleus of the immature dFBs (Fig. 5B). These findings were consistent with activation of the TGFβ receptor (TGFBR). To confirm whether TGFβ released from mature adipocytes was indeed responsible for this effect, dFB was pretreated with a TGFBR inhibitor (SB431542) before coculture with mature adipocytes. Inhibition of TGFBR effectively blocked the capacity of mature adipocytes to promote a fibrotic morphology in dFBs and partially restored the adipogenic potential of these cells when exposed to differentiation conditions in the presence of mature adipocytes (Fig. 5C). Inhibition of TGFBR also restored expression of genes associated with reactive adipogenesis (Sca1 and Camp) and blocked induction of profibrotic genes (Ctgf and Acta2) (Fig. 5D). The TGFBR inhibitor also blocked the elevated protein expression of the profibrotic marker smooth muscle actin (SMA) on PDGFRA+ dFBs that was promoted by mature adipocytes as shown by FACS analysis of SMA expression on gated PDGFRA+ dFBs (Fig. 5, E and F, and fig. S5A).
Fig. 5. Adipocytes inhibit the adipogenic function of dFBs through the TGFβ-TGFBR-SMAD2/3 pathway.
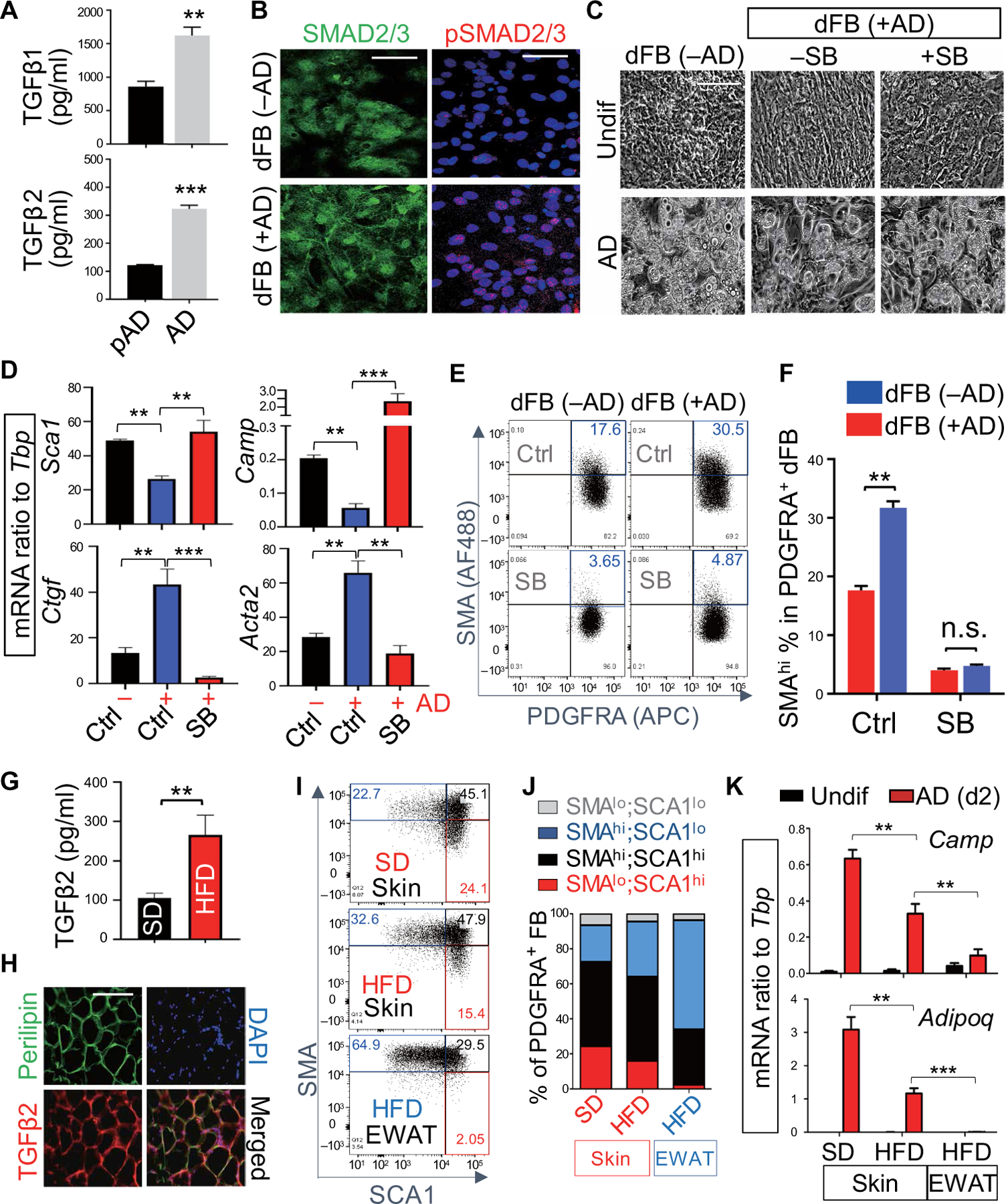
(A) Measurement of TGFβ1 and TGFβ2 concentrations in the culture supernatant collected from pAD or AD cells (n = 3 per group). (B) SMAD2/3 (green) and pSMAD2/3 (red) immunostaining of dFBs seeded with or without AD coculture. Nuclei were stained by DAPI (blue). Scale bars, 50 µm. (C to F) dFBs were cocultured with AD with or without SB431542 (SB; TGFBR inhibitor). (C) Phase-contrast images of undifferentiated or AD-differentiated cells. Scale bar, 100 µm. (D) qRT-PCR of the indicated adipogenic genes (Sca1 and Camp) or profibrotic genes (Ctgf and Acta2) (n = 3 per group). (E) Representative FACS plots of SMA (AF488) and PDGFRA [allophycocyanin (APC)] (n = 3 per group). The percentage of SMAhi PDGFRA+ dFBs is highlighted in blue. (F) Quantification of the percentage of SMAhi cells in PDGFRA+ dFB as shown in (E) (n = 3 per group). n.s., nonsignificant. (G) Measurement of TGFβ2 concentration in the serum of SD and HFD mice (n = 5 per group). (H) Immunostaining of perilipin (green) and TGFβ2 (red) in dermal fat of obese skin. Nuclei were stained by DAPI (blue). Scale bar, 100 µm. (I) Representative FACS plots of SMA and Sca1 in cultured Pdgfra+ dFBs isolated from skin or EWAT of SD or HFD mice as indicated (n = 3 per group). Red, black, and blue numbers indicate the percentage of indicated cell fractions. (J) Bar charts showing the percentage of indicated FB population within cultured primary PDGFRA+ FB from skin or EWAT. (K) Fibroblasts isolated from skin or EWAT of SD or HFD mice were treated with adipocyte differentiation medium, and mRNA expression of Camp or Adipoq in day 2 differentiated adipocytes (red bars) or undifferentiated cells (black bars) was measured by qRT-PCR as indicated (n = 3 per group). All error bars indicate mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA).
In vivo, serum concentrations of TGFβ2, but not TGFβ1, were significantly (P < 0.01) elevated in obese mice compared to lean mice (Fig. 5G and fig. S5B). TGFβ2 protein was also detectable in perilipin+ dermal mature adipocytes in dWAT of obese mice (Fig. 5H) and Tgfb2 mRNA expression was significantly (P < 0.01) elevated in the skin or epidydimal WAT (EWAT) from obese HFD mice compared to lean SD controls (fig. S5C). Cultured primary fibroblasts isolated from the skin dermis or EWAT tissue of mice fed SD and HFD were gated by PDGFRA+ and showed differential expression of SMA and Sca1. HFD promoted an increase in SMAhiSca1lo cells and a decrease in the proadipogenic SMAloSca1hi cells both in the dermis and EWAT (Fig. 5, I and J). These SMAhiSca1lo cells were profibrotic and had higher Acta2 expression but low Pref1 expression (fig. S5D), low adipogenic potential (fig. S5E), and expressed low amounts of Camp and Adipoq during differentiation (Fig. 5K). These findings of a proadipogenic to profibrotic shift in the dFB populations after HFD were consistent with the shift promoted by TGFβ during skin aging (11).
Rosiglitazone or inhibition of TGFβ signaling increases resistance of mice to S. aureus infection
Our observations suggested that mature adipocytes inhibit the antimicrobial function of dermal adipogenesis by producing TGFβ. To test this hypothesis, we used two independent methods to interfere or override TGFβ signaling in obese mice. The peroxisome proliferator–activated receptor–γ (PPARγ) agonist rosiglitazone was of interest as TGFβ suppresses the expression of PPARγ, a key transcription factor driving adipogenesis, and TGFβ-mediated suppression of PPARγ in fibroblasts has been linked with skin and lung fibrosis (27, 28). In addition, the PPARγ agonist has been shown to attenuate TGFβ-mediated fibrogenesis in lung fibroblasts (28). Here, we found that rosiglitazone pretreatment protected dFBs from TGFβ2-mediated suppression of adipogenic function in vitro (Fig. 6A and fig. S6A). Rosiglitazone also restored the adipogenic potential of cells exposed to TGFβ2 as seen by increased capacity to secrete FABP4 (fig. S6B) and express Pparg2, Adipoq, Camp, and Fabp4 (Fig. 6B and fig. S6C). Furthermore, combined rosiglitazone and TGFBR inhibitor SB431542 restored the adipogenic function of aged dFB (Fig. 6C), inhibited the expression of profibrotic genes (Fig. 6D), and induced proadipogenic genes (Fig. 6E). The combination of rosiglitazone and SB431542 worked the best to reverse the negative effects of coculture with mature adipocytes by decreasing expression of genes associated with fibrosis (Acta2 and Col1a1) while increasing genes associated with adipogenesis (Pparg2, Adipoq, and Camp) (Fig. 6, F and G, and fig. S6D). In vivo, administration of rosiglitazone and the TGFBR inhibitor also improved resistance to S. aureus infection in HFD obese mice, and the effect of both treatments combined was comparable to the improvement seen after treatment with either alone (Fig. 6H). Infected HFD mice treated with rosiglitazone and TGFBR inhibitor also had improved dermal reactive adipogenesis, including activation of C/EBPβ and increased cathelicidin AMP production in the dermal fat layer (Fig. 6, I and J).
Fig. 6. Rosiglitazone protects dFBs from TGFβ and increases resistance of mice to SA infection.
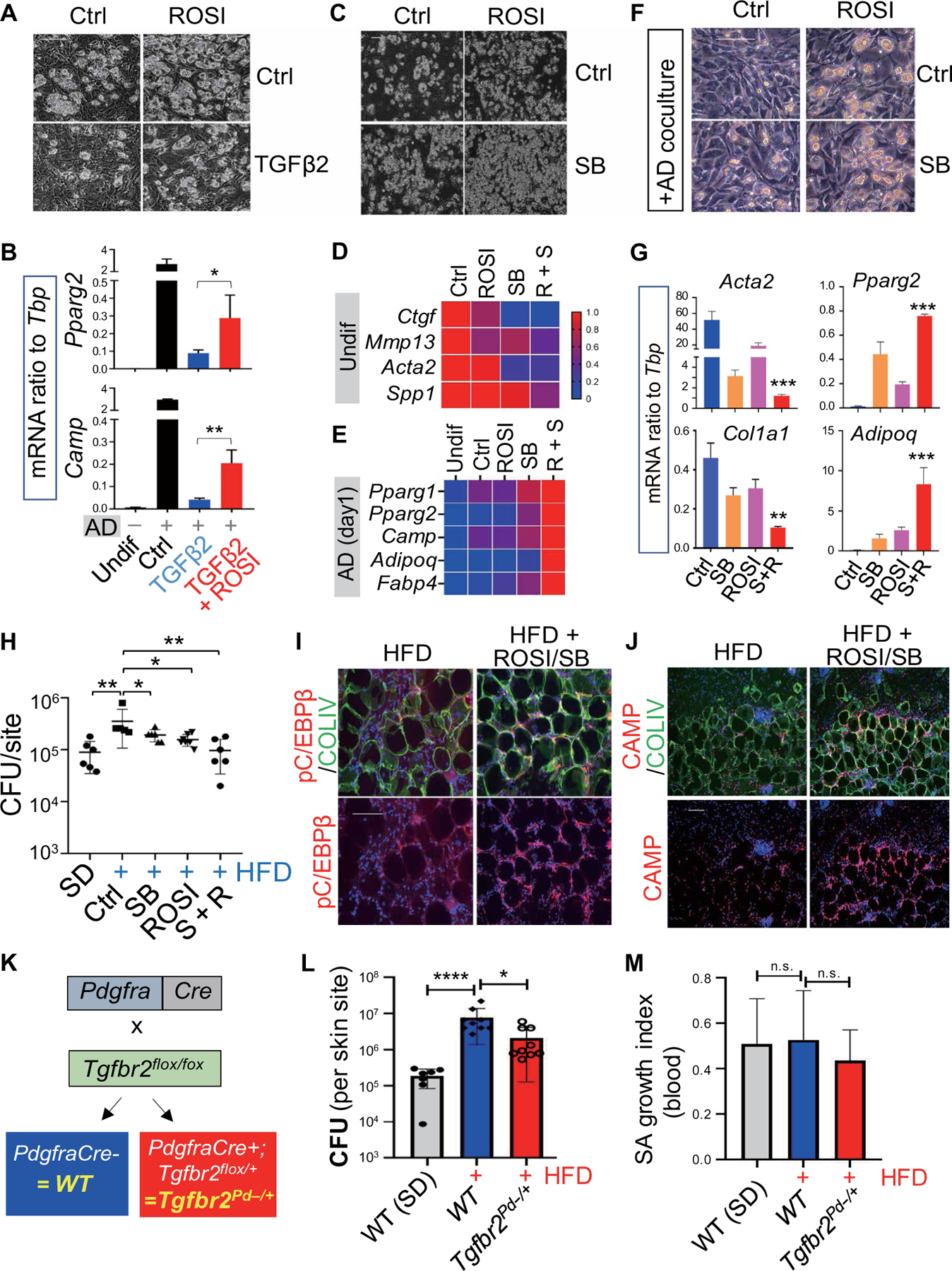
(A and B) Primary neonatal dFBs were pretreated with or without rosiglitazone (ROSI; 20 µM), followed by treatment with or without TGFβ2 (0.1 ng/ml) and then subjected to adipocyte differentiation. (A) Representative phase-contrast images showing adipocyte formation (n = 3 per group). (B) qRT-PCR analyses of Pparg2, Camp, Adipoq, and Fabp4 as indicated (n = 3 per group). (C and D) Primary neonatal dFBs were treated with ROSI with or without SB431542 (SB; TGFBR inhibitor) and then subjected to adipocyte differentiation conditions. (C) Representative phase contrast images showing adipocyte formation (n = 3 per group). (D and E) Heatmap showing relative mRNA expression (quantified by qRT-PCR) of selected fibrotic genes in undifferentiated cells and selected adipocyte genes in day 1 differentiated cells. (F and G) Neonatal dFB cocultured with AD with or without SB or ROSI as indicated. (F) Representative phase images showing adipocyte formation (n = 3 per group). (G) qRT-PCR analyses of selected fibrotic genes or adipocyte genes (n = 3 per group). (H to J) SD or HFD mice were treated with SB or ROSI then infected intradermally with S. aureus. (H) Measurement of bacterial CFU from the infection edge area of the skin (n = 5 to 6 per group). (I and J) Skin sections were stained with pC/EBPβ (red) and COLIV (green) as shown in (I) or CAMP (red) and COLIV (green) as shown in (J). Nuclei were stained by DAPI (blue). Scale bars (A, C, F, I, and J), 100 µm. (K) Conditional heterozygous Tgfbr2 deletion in Pdgfra+ fibroblasts, Tgfbr2flox/+;Pdgfra-cre = Tgfbr2Pd−/+, was achieved through the breeding of Tgfbr2flox/flox mice with Pdgfra-cre mice. (L to M) WT (Tgfbr2flox/flox) or Tgfbr2Pd−/+ mice were infected intradermally with S. aureus. (L) Effect of heterozygous deletion of Tgfbr2 in Pdgfra+ dFBs on resistance of obese HFD mice to S. aureus intradermal infection as shown by measurement of bacterial CFU count in infected skin sites (n = 5 to 8 per group). (M) S. aureus killing activity of whole mouse blood (n = 5 per group). All error bars indicate mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (one-way ANOVA).
To further test whether TGFβ drives the loss of host dermal defense against infection in obesity, we deleted expression of the TGFBR in dFBs. To achieve targeted deletion of Tgfbr2 specifically in Pdgfra+ dFBs, Tgfbr2flox/flox mice were bred with Pdgfra-Cre mice (Fig. 6K). As we have described in a previous study (11), no homozygous Tgfbr2flox/flox;Pdgfra-Cre offspring were generated despite extensive breeding, and thus, experiments were conducted only with heterozygous Tgfbr2 deletion mice (Tgfbr2flox/+;Pdgfra-Cre, Tgfbr2Pd−/+) offspring and wild-type (WT) littermates (Tgfbr2flox/flox). Tgfbr2Pd−/+ and WT littermates were fed with HFD for 6 months and then subjected to S. aureus intradermal infection. Heterozygous deletion of Tgfbr2 in dFBs significantly (P < 0.05) increased the resistance of HFD mice compared to WT controls (Fig. 6L). The increased resistance to S. aureus in Tgfbr2Pd−/+ mice was not caused by an increased ability of blood neutrophils to kill S. aureus (Fig. 6M). These observations confirm the likely critical role of TGFβ signaling in suppressing innate antimicrobial defense against S. aureus infection seen in obese mice.
Human skin from obese subjects also shows a loss of antimicrobial pADs
To determine whether observations in mice are also relevant to humans, we evaluated changes in adiposity and fibrotic features in the dermis of human skin samples collected from lean and obese Caucasian donors. Lipid (Bodipy) staining showed that whereas Bodipy+ adipocytes were only detected in the lower dermis from lean subjects, Bodipy+ cells were abundant through the upper dermis of the obese skin (Fig. 7A). Similar observations were also found in skin dermis collected from obese Asian skin donors (fig. S7A). Perilipin confirmed that adipocytes were present in the dermis of obese subjects (fig. S7B). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses found that expression of CAMP and pAD marker PREF1 was significantly (P < 0.01) down-regulated in obese skin dermis compared to lean controls, whereas expression of TGFB2 as well as profibrotic genes including SPP1, FN1, COL3A1, COL1A1, and TIMP1 was elevated in obese dermis compared to lean controls (Fig. 7, B to E, and fig. S7, C to F). Together, these results in humans compare closely to our observations in obese mice.
Fig. 7. Obesity promotes dermal adiposity, fibrosis, and loss of AMP expression in human skin.
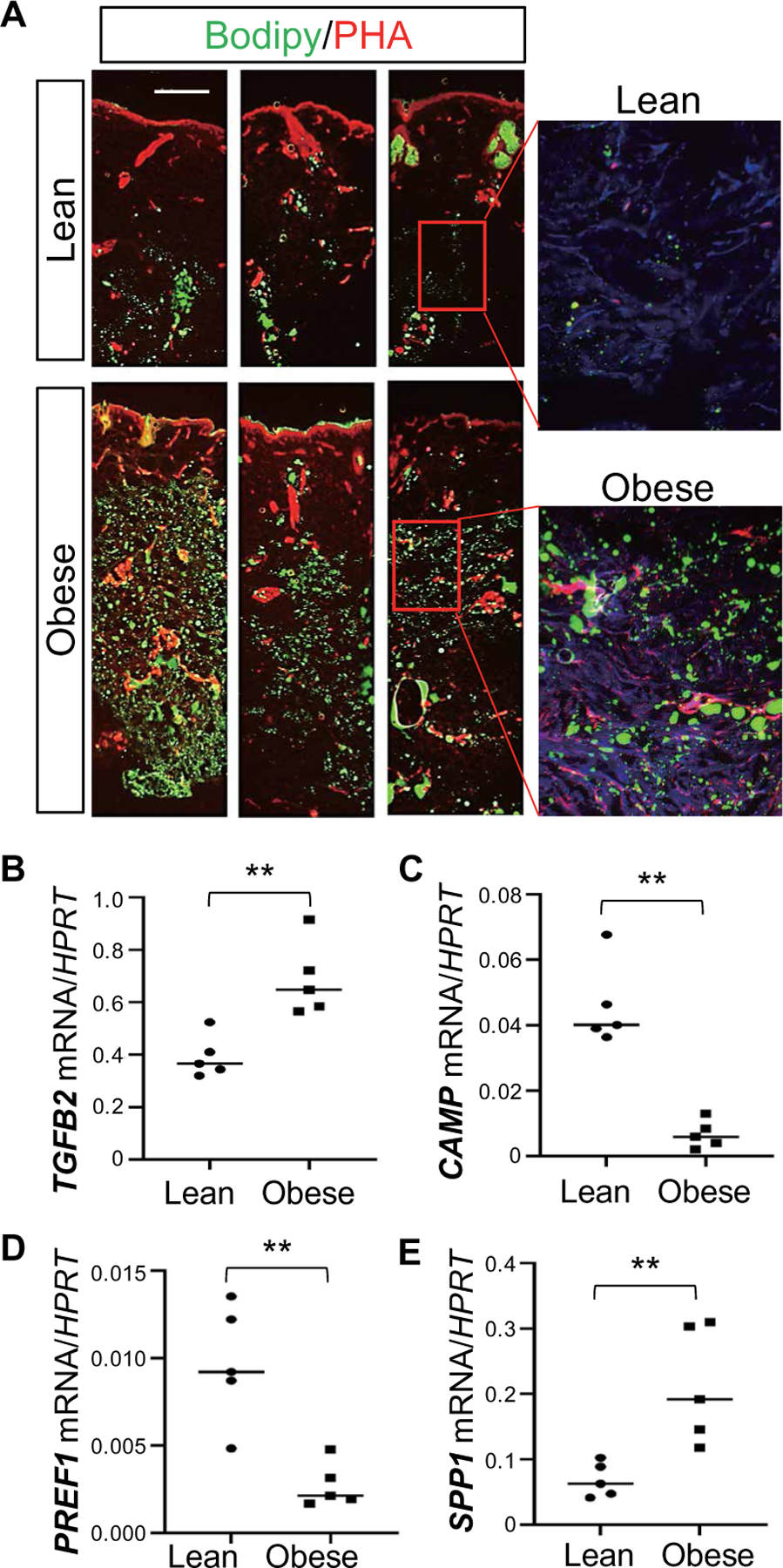
(A) Representative Bodipy (green) and phalloidin (PHA) staining of human back skin sections from lean (BMI <23) and obese individuals (BMI >29). All individuals who participated in the study are white Caucasians (n = 3 to 6 per group). Scale bar, 1 mm. (B to E) qRT-PCR analysis of indicated genes (ratio to housekeeping gene HPRT) in lean or obese human skin (n = 4 to 5 per group). All error bars indicate mean ± SEM. **P < 0.01; n.s., nonspecific (unpaired Student’s t test).
DISCUSSION
The recent observation that dermal adipogenesis is an important source of antimicrobial defense against infection (11, 12) appeared in conflict with many observations that obese individuals have an increased risk for bacterial skin infections (29–32). Here, we found that obesity impaired dWAT innate immune functions by at least three mechanisms. First, HFD feeding triggered hyperplastic growth of adipocytes from dermal progenitors, leading to depletion of dermal APs and accumulation of mature adipocytes. Second, mature adipocytes lost the capacity to produce AMP. Third, mature adipocytes also indirectly suppressed the adipogenic and antimicrobial potential of their progenitors by secreting TGFβ. Therefore, we have uncovered a pathological feature of obesity. This observation may also provide a critical link to explain associations between obesity and other systemic immune dysfunctions associated with AMP dysregulation (33–37).
Adipose tissue expansion results from an increase in adipocyte size (hypertrophy) or through differentiation of APs into mature, lipid-laden adipocytes, a process known as adipogenesis (hyperplasia) (1, 2). We have shown that dermal fat expansion triggered by S. aureus infection involves both hyperplastic and hypertrophic modes (12) and termed this S. aureus–triggered adipocyte expansion as dermal reactive adipogenesis (13). During this event in immune defense, PDGFRA+THY1+ APs proliferate locally and differentiate into new lipid-laden adipocytes that produce the AMP cathelicidin, conferring innate antimicrobial protection against bacteria (11, 12). It was not previously clear why excessive gain of fat during obesity would enable increased infection.
Adipose tissue expands by hypertrophy with or without hyperplasia in a depot-specific manner during HFD feeding of mice. It has been shown that DIO promotes the expansion of subcutaneous WAT primarily via hypertrophic growth of adipocytes, whereas it induces a transient proliferation of adipocyte precursors followed by adipocyte hypertrophy in perigonadal visceral WAT (2, 3). However, how dermal fat expands during DIO has not been elucidated. Here, using Adipoq-CreER lineage-tracing mice, we show that dermal fat expansion in response to HFD resulted from hyperplasia of adipocytes from dermal adipogenic fibroblasts. This was then followed by hypertrophic growth of mature adipocytes during the chronic phase of DIO. The adipogenic potential of dermal fat relies on the existence of a large pool of adipogenic progenitors residing in the RET layer of the dermis (38). These fibroblasts normally respond to S. aureus infection by locally differentiating into adipocytes and producing AMPs (11, 12). Our data now show that the dermal AP pool is depleted by DIO, leading to loss of defense protection from these APs.
To study the influence of adipocyte hypertrophy on antimicrobial function, we established an in vitro primary culture system in which Sca1+ dermal APs were sequentially differentiated into pADs, early adipocytes, and then mature adipocytes. We found that upon commitment of APs to pADs and during early stages of adipocyte differentiation, pADs down-regulated the expression of genes associated with fibrosis but transiently up-regulated innate immune receptors including Tlr2 and Il1r as well as the AMP Camp. These differentiating early adipocytes are an effective mechanism for immune defense as the conditioned medium from these early adipocytes potently inhibited the growth of S. aureus. However, fully mature adipocytes lost this ability to limit bacterial growth. Therefore, pADs or early adipocytes are active in innate immune defense, but DIO leads to accumulation of mature adipocytes that do not fight against bacterial infection.
Activation of the TGFβ signaling pathway results in the loss of adipogenic precursors. TGFβ plays an important role in regulating many aspects of cellular processes such as proliferation, extracellular matrix production, and differentiation in adipose tissue (11, 26, 39, 40). Here, using a coculture model, we found that mature adipocytes not only lost the ability to suppress bacterial growth but also acted in a feedback loop to suppress the adipogenic and antimicrobial potential of their progenitors. The factors responsible for this negative feedback were TGFβ1 and TGFβ2. In vivo, obese mice showed increased serum concentrations of TGFβ2, but not TGFβ1. This observation suggests that adipocytes may be the major cellular source of TGFβ2, whereas TGFβ1 is also produced by immune cells such as regulatory T cells (41) and macrophages (42). A recent study has identified TGFβ2 as an exercise-induced adipokine that regulates metabolism and attenuates adipose tissue inflammation in HFD-fed mice (39). In the current study, we found that TGFβ2 treatment or adipocyte coculture not only suppressed the adipogenic potential of dFBs but also led to innate immune suppression characterized by decreased expression of Tlr2, Il1r1, and inflammatory cytokines/chemokines. TGFβ2-mediated immunosuppression may help to attenuate obesity-associated adipose tissue inflammation but is detrimental to the production of AMPs during reactive adipogenesis, which is necessary for optimal resistance to bacterial infection.
We showed the ability of rosiglitazone and a TGFBR inhibitor to restore normal antimicrobial functions in obese mice. However, multiple cell types participate in antimicrobial defense, and innate AMPs are secreted by other cells in the skin, such as keratinocytes, neutrophils, and mast cells (8, 9). Furthermore, other host defense processes including the action of reactive oxygen species, complement binding, and phagocytosis are essential for antimicrobial function. A limitation of our study is that only a mouse DIO model was used, and further studies are required to determine whether a similar defective reactive adipogenesis is present in humans and genetic models of obesity such as the db/db and ob/ob mice. Another limitation of this study is that observations of improved antimicrobial function in obese mice may also be due to influence on other cells, either directly by rosiglitazone or the TGFBR inhibitor or indirectly by effects on adipogenesis that enhance cell-mediated immune defense. The expression of cathelicidin has potent indirect activity on cell recruitment, cytokine release, and angiogenesis (33, 43, 44). Thus, although reactive adipogenesis and the production of AMPs by the differentiating pADs are important elements in host defense against S. aureus, it is likely that the direct antimicrobial function observed in vitro is not the only mechanism that explains the in vivo response.
Adipose tissue fibrosis is considered as a hallmark of adipose malfunction that is linked to insulin resistance (1, 45–47). Adipose fibrosis occurs when PDGFRA+ APs become activated and shift from adipogenesis to extracellular matrix–synthesizing profibrotic cells. However, how adipocyte hypertrophy triggers a shift in progenitor cell plasticity remains unclear. Here, we have shown that mature adipocytes secreted TGFβ ligands that promote a shift of dFBs from proadipogenic to profibrogenic. In DIO mice, dFBs failed to initiate a reactive adipogenesis program but, in turn, exhibited a profibrotic expression profile upon S. aureus skin infection. Whereas DIO promoted a notable profibrotic phenotype in mouse dermal fat upon infection, obesity-triggered dermal fibrosis was apparent in human skin even in the absence of infection. In human skin, increased dermal adiposity was associated with an increase in TGFβ2 expression and a shift from adipogenesis to fibrosis characterized by a decrease in AMP gene expression and a gain of dermal fibrosis shown by thickened skin dermis and increased profibrotic gene expression.
Collectively, our data reveal how DIO impairs innate immune antimicrobial function and highlights a central role of TGFβ as an immunosuppressive adipokine. Our observations that infection by S. aureus in mice can be treated by inhibiting TGFβ or activating PPARγ may suggest a therapeutic approach to assist in resisting infections in the obese population.
MATERIALS AND METHODS
Study design
The main objective of this study is to determine whether DIO impairs the beneficial antimicrobial defense function of dermal fat against S. aureus and to determine the underlying mechanisms. A DIO mouse model (60% HFD) was used to induce obesity in mice, and mice fed with standard or LFD were used as control. Intradermal infection with S. aureus was used as infection model as described previously (11, 12). In vivo phenotypes were characterized by analyzing neutrophil recruitment and dermal reactive adipogenesis on skin from HFD or SD mice infected with S. aureus. scRNA-seq was used to determine how HFD influences the skin immune response against S. aureus at a single-cell level. To determine how DIO affects dermal adipogenesis, a dual-color adipocyte-specific lineage-tracing mouse (AdipoqERT2Cre;mTmG) was used to characterize hyperplastic and hypertrophic growth of dermal adipocytes during HFD feeding. In vitro adipocyte differentiation from primary dermal pADs was established to determine how adipocytes change their antimicrobial function during maturation. In vitro coculture between mature adipocytes and undifferentiated pADs was used to determine whether the accumulation of mature adipocytes feedback to inhibit the adipogenic potential of pADs, and RNA-seq was performed to identify key upstream pathways driving the phenotype. TGFβ was identified as a key factor secreting by mature adipocytes to inhibit reactive adipogenesis. TGFβ is known to inhibit PPARγ, a key transcription factor for adipogenesis. Therefore, we used pharmacological inhibitor of TGFBR inhibitor and PPARγ agonist to determine whether the combination of these two treatments could restore reactive adipogenesis in the in vitro coculture model or in the obese mice in the in vivo infection model. Last, human skin samples were collected from lean and obese subjects to validate our key observations from mice in human clinical samples. All mice were randomly assigned to treatment groups, and all experiments were conducted with at least three technical replicates per group.
Animals and animal care
All animal experiments were approved by the University of California, San Diego (UCSD) and the University of California, Irvine (UCI), Institutional Animal Care and Use Committee. C57BL/6 WT mice (Jackson Laboratory/JAX) were originally purchased from the Jackson laboratory and then bred and maintained in the animal facility at UCSD. C57BL/6 male mice were fed with 60% HFD (Research Diets Inc.) from 8 weeks of age for 6 months. In some studies, an LFD (LFD = 10% calories from fat; Research Diets Inc.) was used as nutrient- and calorie-matched control diet. Adipoq-CreER (JAX, 025124) and mTmG (JAX, 007576) mice were originally purchased from the Jackson laboratory and then bred, maintained, and fed with 60% HFD in the animal facility at the UCI. For all animal studies, animals were randomly selected without formal prerandomization, and quantitative measurements were done without the opportunity for introduction of bias.
Mouse model of S. aureus skin infection
Skin infection experiments were done as described (11, 12). S. aureus strain USA300/methicillin-resistant S. aureus was used for in vivo infection. In brief, the backs of sex- and age-matched adult WT were shaved, and hair was removed by chemical depilation (Nair). Mice were then injected intradermally with 100 µl of a midlogarithmic growth phase of S. aureus (1 × 107 CFU of bacteria) in phosphate-buffered saline (PBS). Mice were sacrificed after day 3, and skin biopsies covering the infection edge or center area were harvested as described previously (11). Skin biopsies were homogenized in 1 ml of TRIzol (for RNA) or PBS (for CFU counting) with 2-mm zirconia beads in a Mini-Beadbeater 16 (Biospect). To count CFUs, homogenized skin samples were serially diluted, plated onto Tryptic Soy Agar, and enumerated after overnight culture to quantify the CFU per site of infection. For some experiments, the PPARγ agonist rosiglitazone (10 mg/kg in PBS) or vehicle control was administered by daily intraperitoneal injection from 1 day before infection to day 2 of infection, and administration of TGFBR inhibitor SB431542 (0.125 mg in 100 µl of 2% dimethyl sulfoxide + 30% polyethylene glycol 300 + PBS) or vehicle control was performed through intradermal injection within the infection site at 24 and 48 hours after infection. Skin biopsies were collected at day 3 after infection for RNA extraction or CFU counting as described above. For BrdU experiments, mice were injected intraperitoneally with BrdU (50 mg/g) 4 hours before being sacrificed for skin section collection.
Human skin sample collection
Fresh adult human (Caucasian) full-thickness skin biopsies, from the back of lean [body mass index (BMI) <23] and obese (BMI >29) male donors between 50 and 65 years of age, were collected by the UCSD dermatology clinics. Fresh adult human (Chinese) full-thickness skin biopsies were collected by the Hospital for Skin Disease, Institute of Dermatology, Chinese Academy of Medical Science and Peking Union Medical College. All sample acquisitions were approved and regulated by the UCSD Institutional Review Board (reference number 140144) or by the Institute of Dermatology, Chinese Academy of Medical Science Medical Ethics Committee (reference number 2012003). Informed consent was obtained from all subjects before skin biopsies. All human biopsies were taken from the back skin, where it is relatively protected from sun exposure compared to other regions such as face and neck of the body. Upon collection, these samples were directly fixed with paraformaldehyde and then either paraffin embedded or optimum cutting temperature embedded for histological or immunofluorescent analyses.
Statistical analysis
Experiments were repeated at least three times with similar results and were statistically analyzed by GraphPad Prism 8 software. For experiments with two groups, statistical significance was determined using Student’s unpaired two-tailed t test. Normality was tested using the Shapiro-Wilk test, and for datasets that were not normally distributed, nonparametric tests were used to determine statistical significance. For experiments with more than two groups, one-way analysis of variance (ANOVA) multiple comparison test was performed as indicated in the legend. A P value of <0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).
Supplementary Material
Fig. S1. Dermal reactive adipogenesis response upon S. aureus infection is lost in DIO.
Fig. S2. Identification of cell communities and cell cycle metrics on skin cells.
Fig. S3. DIO promotes expansion of dermal fat, thinning of skin dermis, and depletion of dermal cells.
Fig. S4. Adipocytes inhibit the adipogenic function of dFBs.
Fig. S5. Adipocytes inhibit the adipogenic function of dFBs through the TGFβ-TGFBR-SMAD2/3 pathway.
Fig. S6. Rosiglitazone protects dFBs from TGFβ in vitro.
Fig. S7. Obesity promotes dermal adiposity.
Table S1. Primers used for RT-qPCR of mouse genes.
Table S2. Primers used for RT-qPCR of human genes.
References (48–56)
Acknowledgments:
We thank the UCSD flow cytometry core for FACS studies and the Xiamen University central biomedical instruments core for imaging studies.
Funding:
L-.j.Z. was supported by Chinese NSFC grants (81971551 and K2918001) and Xiamen University grant X2123303. R.L.G. was supported by NIH R01 AR076082, R37 AI052453, and U19 AI117673. M.V.P. was supported by a Pew Charitable Trust Grant, LEO Foundation Award, NIH grants R01-AR067273 and R01-AR069653, P30-AR075047, NSF grant DMS1763272, and Simons Foundation grant (594598 to Q. Nie). This work was also supported by U01-AR073159 (to Q. Nie, M.V.P., and X. Dai). C.F.G.-J. was supported by UC Irvine Chancellor’s ADVANCE Postdoctoral Fellowship Program, NSF-Simons Postdoctoral Fellowship, NSF grant DMS1763272, Simons Foundation grant (594598 to Q.N.), and a gift from the Howard Hughes Medical Institute Hanna H. Gray Postdoctoral Fellowship Program.
Footnotes
Competing interests: R.L.G. is a cofounder, scientific advisor, and consultant; has equity in MatriSys Biosciences and is a consultant; receives income; and has equity in Sente. The other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. scRNA-seq data are deposited at the NCBI GEO (http://ncbi.nlm.nih.gov/geo/) under accession GSE150729.
REFERENCES AND NOTES
- 1.Ghaben AL, Scherer PE, Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol 20, 242–258 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS, Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat. Cell Biol 17, 376–385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang QA, Tao C, Gupta RK, Scherer PE, Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med 19, 1338–1344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, Treloar AE, Archer S, Brown WA, Muller M, Watt MJ, Ohara O, McLean CA, Tiganis T, Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 175, 1289–1306.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR, TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell 172, 731–743.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryu J, Loza CA, Xu H, Zhou M, Hadley JT, Wu J, You H, Wang H, Yang J, Bai J, Liu F, Bialowas C, Dong LQ, Potential roles of adiponectin isoforms in human obesity with delayed wound healing. Cell 8, 1134 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu CL, Kimmerling KA, Little D, Guilak F, Serum and synovial fluid lipidomic profiles predict obesity-associated osteoarthritis, synovitis, and wound repair. Sci. Rep 7, 44315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L-J, Gallo RL, Antimicrobial peptides. Curr. Biol 26, R14–R19 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Gallo RL, Hooper LV, Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol 12, 503–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL, Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Zhang L-J, Chen SX, Guerrero-Juarez CF, Li F, Tong Y, Liang Y, Liggins M, Chen X, Chen H, Li M, Hata T, Zheng Y, Plikus MV, Gallo RL, Age-related loss of innate immune antimicrobial function of dermal fat is mediated by transforming growth factor beta. Immunity 50, 121–136.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, Gallo RL, Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347, 67–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen SX, Zhang L-J, Gallo RL, Dermal white adipose tissue: A newly recognized layer of skin innate defense. J. Invest. Dermatol 139, 1002–1009 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Guerrero-Juarez CF, Plikus MV, Emerging nonmetabolic functions of skin fat. Nat. Rev. Endocrinol 14, 163–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt BA, Horsley V, Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development 140, 1517–1527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zwick RK, Guerrero-Juarez CF, Horsley V, Plikus MV, Anatomical, physiological, and functional diversity of adipose tissue. Cell Metab 27, 68–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Festa E, Fretz J, Berry R, Schmidt B, Rodeheffer M, Horowitz M, Horsley V, Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell 146, 761–771 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Shao M, Hepler C, Zi Z, Zhao S, An YA, Zhu Y, Ghaben AL, Wang M-Y, Li N, Onodera T, Joffin N, Crewe C, Zhu Q, Vishvanath L, Kumar A, Xing C, Wang QA, Gautron L, Deng Y, Gordillo R, Kruglikov I, Kusminski CM, Gupta RK, Scherer PE, Dermal adipose tissue has high plasticity and undergoes reversible dedifferentiation in mice. J. Clin. Invest 129, 5327–5342 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrero-Juarez CF, Dedhia PH, Jin S, Ruiz-Vega R, Ma D, Liu Y, Yamaga K, Shestova O, Gay DL, Yang Z, Kessenbrock K, Nie Q, Pear WS, Cotsarelis G, Plikus MV, Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun 10, 650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plikus MV, Guerrero-Juarez CF, Ito M, Li YR, Dedhia PH, Zheng Y, Shao M, Gay DL, Ramos R, Hsi T-C, Oh JW, Wang X, Ramirez A, Konopelski SE, Elzein A, Wang A, Supapannachart RJ, Lee H-L, Lim CH, Nace A, Guo A, Treffeisen E, Andl T, Ramirez RN, Murad R, Offermanns S, Metzger D, Chambon P, Widgerow AD, Tuan T-L, Mortazavi A, Gupta RK, Hamilton BA, Millar SE, Seale P, Pear WS, Lazar MA, Cotsarelis G, Regeneration of fat cells from myofibroblasts during wound healing. Science 355, 748–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shook BA, Wasko RR, Mano O, Rutenberg-Schoenberg M, Rudolph MC, Zirak B, Rivera-Gonzalez GC, López-Giráldez F, Zarini S, Rezza A, Clark DA, Rendl M, Rosenblum MD, Gerstein MB, Horsley V, Dermal adipocyte lipolysis and myofibroblast conversion are required for efficient skin repair. Cell Stem Cell 26, 880–895.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, Scherer PE, Tourtellotte WG, Varga J, Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol 67, 1062–1073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasza I, Suh Y, Wollny D, Clark RJ, Roopra A, Colman RJ, MacDougald OA, Shedd TA, Nelson DW, Yen M-I, Yen C-LE, Alexander CM, Syndecan-1 is required to maintain intradermal fat and prevent cold stress. PLOS Genet 10, e1004514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA, Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwalie PC, Dong H, Zachara M, Russeil J, Alpern D, Akchiche N, Caprara C, Sun W, Schlaudraff K-U, Soldati G, Wolfrum C, Deplancke B, A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559, 103–108 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, Seale P, Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364, eaav2501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang QQ, Jain M, Whitfield ML, Feghali-Bostwick C, Varga J, PPARγ downregulation by TGFß in fibroblast and impaired expression and function in systemic sclerosis: A novel mechanism for progressive fibrogenesis. PLOS ONE 5, e13778 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, Szibor M, Kosanovic D, Schwind F, Schermuly RT, Henneke I, MacKenzie BA, Quantius J, Herold S, Ntokou A, Ahlbrecht K, Braun T, Morty RE, Günther A, Seeger W, Bellusci S, Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 20, 571–571 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Karppelin M, Siljander T, Vuopio-Varkila J, Kere J, Huhtala H, Vuento R, Jussila T, Syrjanen J, Factors predisposing to acute and recurrent bacterial non-necrotizing cellulitis in hospitalized patients: A prospective case–control study. Clin. Microbiol. Infect 16, 729–734 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Bartholomeeusen S, Vandenbroucke J, Truyers C, Buntinx F, Epidemiology and comorbidity of erysipelas in primary care. Dermatology 215, 118–122 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Dupuy A, Benchikhi H, Roujeau JC, Bernard P, Vaillant L, Chosidow O, Sassolas B, Guillaume JC, Grob JJ, Bastuji-Garin S, Risk factors for erysipelas of the leg (cellulitis): Case-control study. BMJ 318, 1591–1594 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edmonds R, Cuschieri J, Minei J, Rosengart M, Maier R, Harbrecht B, Billiar T, Peitzman A, Moore E, Sperry J; Inflammation the Host Response to Injury Investigators, Body adipose content is independently associated with a higher risk of organ failure and nosocomial infection in the nonobese patient postinjury. J. Trauma 70, 292–298 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Zhang L-J, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, Adase C, Sanford JA, Gao N, Chensee M, Sato E, Fritz Y, Baliwag J, Williams MR, Hata T, Gallo RL, Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity 45, 119–130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DYM, Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med 347, 1151–1160 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, Zal T, Mellman I, Schröder J-M, Liu Y-J, Gilliet M, Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Mihailovic PM, Lio WM, Yano J, Zhao X, Zhou J, Chyu K-Y, Shah PK, Cercek B, Dimayuga PC, The cathelicidin protein CRAMP is a potential atherosclerosis self-antigen in ApoE(−/−) mice. PLOS ONE 12, e0187432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benachour H, Zaiou M, Samara A, Herbeth B, Pfister M, Lambert D, Siest G, Visvikis-Siest S, Association of human cathelicidin (hCAP-18/LL-37) gene expression with cardiovascular disease risk factors. Nutr. Metab. Cardiovasc. Dis 19, 720–728 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, Watt FM, Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi H, Alves CRR, Stanford KI, Middelbeek RJW, Nigro P, Ryan RE, Xue R, Sakaguchi M, Lynes MD, So K, Mul JD, Lee M-Y, Balan E, Pan H, Dreyfuss JM, Hirshman MF, Azhar M, Hannukainen JC, Nuutila P, Kalliokoski KK, Nielsen S, Pedersen BK, Kahn CR, Tseng Y-H, Goodyear LJ, TGF-β2 is an exercise-induced adipokine that regulates glucose and fatty acid metabolism. Nat. Metab 1, 291–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrus P, Mejhert N, Corrales P, Lecoutre S, Li Q, Maldonado E, Kulyte A, Lopez Y, Campbell M, Acosta JR, Laurencikiene J, Douagi I, Gao H, Martinez-Alvarez C, Heden P, Spalding KL, Vidal-Puig A, Medina-Gomez G, Arner P, Ryden M, Transforming growth factor-β3 regulates adipocyte number in subcutaneous white adipose tissue. Cell Rep 25, 551–560.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Bommireddy R, Doetschman T, TGFβ1 and Treg cells: Alliance for tolerance. Trends Mol. Med 13, 492–501 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keophiphath M, Achard V, Henegar C, Rouault C, Clement K, Lacasa D, Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol. Endocrinol 23, 11–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adase CA, Borkowski AW, Zhang L.-j., Williams MR, Sato E, Sanford JA, Gallo RL, Non-coding double-stranded RNA and antimicrobial peptide LL-37 induce growth factor expression from keratinocytes and endothelial cells. J. Biol. Chem 291, 11635–11646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi K-Y, Mookherjee N, Multiple immune-modulatory functions of cathelicidin host defense peptides. Front. Immunol 3, 149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hasegawa Y, Ikeda K, Chen Y, Alba DL, Stifler D, Shinoda K, Hosono T, Maretich P, Yang Y, Ishigaki Y, Chi J, Cohen P, Koliwad SK, Kajimura S, Repression of adipose tissue fibrosis through a PRDM16-GTF2IRD1 complex improves systemic glucose homeostasis. Cell Metab 27, 180–194 e186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, Henegar C, Hulot JS, Poitou C, Torcivia A, Nail-Barthelemy R, Bichet JC, Gautier EL, Clement K, A PDGFRα-mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab 25, 673–685 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, Olson LE, PDGFRα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev 29, 1106–1119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan J-B, Zhang K, Chun J, Kharchenko PV, Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L-J, Isolation, culture, and characterization of primary mouse epidermal keratinocytes. Methods Mol. Biol 1940, 205–215 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Li F, Adase CA, Zhang L-J, Isolation and culture of primary mouse keratinocytes from neonatal and adult mouse skin. J. Vis. Exp, 56027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hulsen T, de Vlieg J, Alkema W, BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu G, Wang L-G, Han Y, He Q-Y, clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rivera-Gonzalez GC, Shook BA, Andrae J, Holtrup B, Bollag K, Betsholtz C, Rodeheffer MS, Horsley V, Skin adipocyte stem cell self-renewal is regulated by a PDGFA/AKT-signaling axis. Cell Stem Cell 19, 738–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chia JJ, Zhu T, Chyou S, Dasoveanu DC, Carballo C, Tian S, Magro CM, Rodeo S, Spiera RF, Ruddle NH, McGraw TE, Browning JL, Lafyatis R, Gordon JK, Lu TT, Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis. J. Clin. Invest 126, 4331–4345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Dermal reactive adipogenesis response upon S. aureus infection is lost in DIO.
Fig. S2. Identification of cell communities and cell cycle metrics on skin cells.
Fig. S3. DIO promotes expansion of dermal fat, thinning of skin dermis, and depletion of dermal cells.
Fig. S4. Adipocytes inhibit the adipogenic function of dFBs.
Fig. S5. Adipocytes inhibit the adipogenic function of dFBs through the TGFβ-TGFBR-SMAD2/3 pathway.
Fig. S6. Rosiglitazone protects dFBs from TGFβ in vitro.
Fig. S7. Obesity promotes dermal adiposity.
Table S1. Primers used for RT-qPCR of mouse genes.
Table S2. Primers used for RT-qPCR of human genes.
References (48–56)