

Abstract
Myostatin is a signaling molecule produced by skeletal muscle cells (myokine) that inhibits muscle hypertrophy and has further paracrine and endocrine effects in other organs including bone. Myostatin binds to activin receptor type 2B which forms a complex with transforming growth factor-β type I receptor (TGF-βRI) and induces intracellular p38MAPK and NFκB signaling. Fibroblast growth factor 23 (FGF23) is a paracrine and endocrine mediator produced by bone cells and regulates phosphate and vitamin D metabolism in the kidney. P38MAPK and NFκB-dependent store-operated Ca2+ entry (SOCE) are positive regulators of FGF23 production. Here, we explored whether myostatin influences the synthesis of FGF23. Fgf23 gene expression was determined by qRT-PCR and FGF23 protein by ELISA in UMR106 osteoblast–like cells. UMR106 cells expressed activin receptor type 2A and B. Myostatin upregulated Fgf23 gene expression and protein production. The myostatin effect on Fgf23 was significantly attenuated by TGF-βRI inhibitor SB431542, p38MAPK inhibitor SB202190, and NFκB inhibitor withaferin A. Moreover, SOCE inhibitor 2-APB blunted the myostatin effect on Fgf23. Taken together, myostatin is a stimulator of Fgf23 expression in UMR106 cells, an effect at least partially mediated by downstream TGF-βRI/p38MAPK signaling as well as NFκB-dependent SOCE.
Keywords: TGF-β, p38MAPK, Ca2+, Vitamin D, Phosphate
Introduction
Myostatin is part of a group of signaling molecules produced by skeletal muscle cells (myocytes) that are known under the name “myokines” in analogy to “cytokines” [25]. It was discovered in 1997 as a member of the TGF-β superfamily and first named “growth differentiation factor 8 (GDF-8)” [33]. Myostatin counteracts muscle hypertrophy as mice deficient in myostatin exhibit 2–3 times more muscle mass than wild-type animals [33]. Also the Belgian Blue, cattle with a loss of function mutation in the gene encoding myostatin, is characterized by enormous muscle mass [16, 25]. Finally, mutations in the human gene encoding myostatin, which result in muscle hypertrophy, are similarly described [25, 41].
In addition to myocytes, myostatin also affects bone metabolism including bone formation and osteoclastogenesis [25]. In osteocytes, myostatin upregulates sclerostin and dickkopf-related protein 1 (Dkk1) [25]. Thus, the myokine myostatin plays a role as regulatory molecule in the cross-talk between muscle and bone [25, 29].
Activin receptor type 2A and B (ACVR2A/B) are the membrane receptors for myostatin. Upon binding of myostatin, transcription factor activity of SMAD2/3 and forkhead box O (FOXO) is induced, ultimately resulting in the degradation of skeletal muscle proteins [17]. Further downstream effectors of myostatin also include p38MAPK [35].
Fibroblast growth factor 23 (FGF23) can be considered as an osteokine, a hormone mainly synthesized in the bone [25, 43]. Having classical endocrine effects, its main target organ is the kidney where it binds to a membrane receptor, which is dependent on transmembrane protein αKlotho [19], and inhibits the formation of calcitriol (1,25(OH)2D3, biologically active vitamin D3) by suppressing 25-hydroxyvitamin d-1α-hydroxylase (encoded by CYP27B1), the renal key enzyme for 1,25(OH)2D3 synthesis. Moreover, FGF23 fosters the internalization of membrane NaPiIIa (SLC34A1), the major Na+-dependent phosphate cotransporter in the proximal tubule of the kidney, thereby increasing the urinary excretion of phosphate [20, 39].
FGF23 may also be produced locally by other cells including hepatocytes [32] or cardiomyocytes thus exerting additional paracrine effects [28]. Thus, it was shown that FGF23 affects cardiac muscle and induces left ventricular hypertrophy [12], which underlines that this bone-derived molecule participates in the cross-talk between bone and muscle [25].
Moreover, the plasma concentration of FGF23 goes up in many acute and chronic diseases, notably renal and cardiovascular disorders [3, 21, 42]. Interestingly, myostatin is also upregulated in an early stage of chronic kidney diseases (CKD) [46]. Whether FGF23 is only a disease biomarker or actively drives pathophysiological processes is not entirely clear, yet [42].
The regulation of the production of FGF23 is subject to current research. Well-established regulators of FGF23 include 1,25(OH)2D3 [31], dietary phosphate [44], parathyroid hormone [27], pro-inflammatory cytokines and pathways [8, 14], interleukin-6 (IL-6) [7], erythropoietin [5, 18], iron metabolism [6], transforming growth factor (TGF)-β (TGF-β) [13], peroxisome proliferator-activated receptor α [11], and intracellular signaling cascades including AMP-dependent protein kinase (AMPK) [15] and insulin/IGF-1-dependent phosphoinositide 3-kinase/Akt/FOXO signaling [2]. Remarkably, myostatin downstream signaling effector p38MAPK also controls Fgf23 gene expression [10].
Some human diseases with enhanced myostatin plasma levels including dermatomyositis and CKD are characterized by enhanced FGF23 production [23, 46]. This and the well-established muscle and bone cross-talk [25] prompted us to investigate whether myostatin directly impacts on FGF23 formation and to uncover the underlying mechanism. A direct effect could be of high relevance given that pharmacological manipulation of myostatin in disease may be a future option [25].
Methods
Cell culture and treatments
UMR106 rat osteoblast-like cells (CRL-1661; ATCC, Manassas, VA, USA) were cultured under standard conditions [13]. Per se, basal Fgf23 expression is low in UMR106 cells [40]. Therefore, they first have to be treated for 24 h with 100 nM 1,25(OH)2D3 (Tocris, Bristol, UK), which strongly enhances Fgf23 expression [40]. Next, cells were treated for further 24 h with recombinant myostatin protein (5–100 ng/mL, PeproTech, Rocky Hill, NJ, USA) in the presence or absence of TGF-β type I receptor (TGF-βRI) inhibitor SB431542 (10 µM, Sigma-Aldrich, Schnelldorf, Germany), p38MAPK inhibitor SB202190 (10 µM, Tocris), NFκB inhibitor withaferin A (500 nM, Tocris), or SOCE inhibitor 2-APB (150 µM, Sigma).
IDG-SW3 mouse osteocytes (CVCL_0P23; Kerafast, Boston, MA, USA) were plated on rat tail type I collagen–coated 12-well plates (150,000 cells per well) in α-Minimum Essential Medium (α-MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 U/mL interferon (IFN)-γ (all reagents from Gibco, Life Technologies, Darmstadt, Germany). Cells were grown for 24 h at 33 °C and 5% CO2. Next, differentiation was induced by replacing IFN-γ with 50 μg/ml ascorbic acid (Sigma-Aldrich) and 4 mM β-glycerophosphate (AppliChem, Darmstadt, Germany) and further incubation at 37 °C and 5% CO2. The medium was changed every 2nd to 3rd day. At day 28, cells were incubated with 100 ng/mL recombinant myostatin or vehicle in duplicate for 8 h.
MC3T3-E1 Subclone 4 mouse pre-osteoblast cells (CRL-2593; ATCC) were cultured in α‐MEM with 2 mM l-glutamine, nucleosides (Gibco, Life Technologies), 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. They were studied from passages 23 to 27. To this end, cells were seeded on rat tail type I collagen–coated 12-well plates (80,000 cells per well) for 24 h and incubated with 50 μg/mL ascorbic acid, and 4 mM β-glycerophosphate for 6 days. Then, 100 ng/mL myostatin or vehicle only was added in the presence of 1,25(OH)2D3 (10 nM) 24 h before harvesting the cells.
Quantitative real-time polymerase chain reaction
Total RNA was isolated with TriFast reagent (Peqlab, Erlangen, Germany), and 1.2 µg was used along with random primers and the GoScript™ Reverse Transcription System (Promega, Mannheim, Germany) for cDNA synthesis (program: 25 °C for 5 min, 42 °C for 1 h, and 70 °C for 15 min). Quantitative real-time polymerase chain reaction (qRT-PCR) using the Rotor-Gene Q cycler (Qiagen, Hilden, Germany) and GoTaq qPCR Master Mix (Promega) was performed. The amplification conditions for analysis of Fgf23 and TATA box-binding protein (Tbp) were 95 °C for 3 min, 40 cycles of 95 °C for 10 s, 57 °C for 30 s, 72 °C for 30 s (in UMR106 cells), and 95 °C for 3 min, 40 cycles of 95 °C for 10 s, 58 °C for 30 s (Fgf23), and 60 °C for 30 s (Tbp), 72 °C for 30 s (in IDG-SW3 and MC3T3-E1 cells). QRT-PCR conditions for analysis of Acvr2a and Acvr2b expression in UMR106, IDG-SW3, and MC3T3-E1 cells were 95 °C for 3 min, 40 cycles of 95 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s. The calculated Fgf23, Acvr2a, and Acvr2b mRNA transcript levels in UMR106, IDG-SW3, and MC3T3-E1 cells were normalized to the transcript levels of Tbp.
The Acvr2a and Acvr2b qRT-PCR products of the UMR106 cells were loaded on a 1.5% agarose gel and visualized by Midori Green.
The following primers (5′ → 3′ orientation) were used:
Rat Acvr2a:
F: CAATATCTCACAGGGACATC,
R: TTTGGAAGTTTATAGCACCC;
Rat Acvr2b:
F: AACATCATCACGTGGAAC,
R: AACATTCTTGCTTTTGAAGTC;
Rat Fgf23:
F: TAGAGCCTATTCAGACACTTC,
R: CATCAGGGCACTGTAGATAG;
Rat Tbp:
F: ACTCCTGCCACACCAGCC,
R: GGTCAAGTTTACAGCCAAGATTCA;
Mouse Acvr2a:
F: GGTCTCTTGGAATGAACTTTG,
R: TTACTTTTGATGTCCCTGTG;
Mouse Acvr2b:
F: ATTACCTCAAGGGGAACATC,
R: CATTCTTGCTTTTGAAGTCC;
Mouse Fgf23:
F: TCGAAGGTTCCTTTGTATGGA,
R: AGTGATGCTTCTGCGACAAGT;
Mouse Tbp:
F: CCAGACCCCACAACTCTTCC,
R: CAGTTGTCCGTGGCTCTCTT.
Enzyme-linked immunosorbent assay
UMR106 cells were cultured as described and treated with 100 ng/mL myostatin for 24 h. The cell culture supernatant was stored at − 80 °C. C-terminal FGF23 was determined by an ELISA kit (Mouse/Rat FGF-23 (C-Term), Immuntopics, San Clemente, CA, USA) according to the manufacturer’s protocol. Intact FGF23 was determined in the supernatant after its concentration by means of Sartorius Vivaspin 6 Centrifugal Concentrators (Sartorius, Göttingen, Germany), with an ELISA kit (Mouse/Rat FGF-23 (Intact), Immuntopics).
Statistics
The data are shown as arithmetic means ± SEM, and n represents the number of independent experiments. Normal distribution was tested by Shapiro–Wilk normality test. Two groups were compared by unpaired Student’s t test (if necessary with Welch’s correction) or with Mann–Whitney-U-test for data not normally distributed. More than two groups were tested for significance by one-way ANOVA followed by Bonferroni’s multiple comparisons test (if necessary with Welch’s ANOVA followed by Dunnett’s T3 multiple comparisons test). Differences were considered significant if p < 0.05.
Results
Expression of the activin type 2 receptors in UMR106 cells
The impact of myostatin on Fgf23 expression was studied in UMR106 osteoblast–like cells. Employing RT-PCR, we first investigated the expression of myostatin receptors activin type 2A and B (Acvr2a and Acvr2b). As illustrated in Fig. 1, mRNA specific for receptor Acvr2b and to a markedly lesser extent for Acvr2a could be detected in UMR106 cells. Other cell lines used to study Fgf23 include IDG-SW3 and MC3T3-E1 cells. We used quantitative RT-PCR to compare Acvr2a and Acvr2b expression in all three cell lines. As a result, expression of Acvr2a relative to Tbp was 0.0008 ± 0.0000 in UMR106 cells, an expression level significantly (p < 0.001) lower than in IDG-SW3 cells (0.6014 ± 0.0615) and MC3T3-E1 cells (0.2768 ± 0.0162; for all n = 7). In contrast, expression of Acvr2b relative to Tbp was 0.5797 ± 0.0465 in UMR106 cells, an expression level significantly (p < 0.001) higher than in IDG-SW3 cells (0.0054 ± 0.0009) and MC3T3-E1 cells (0.0170 ± 0.0017; for all n = 7).
Fig. 1.
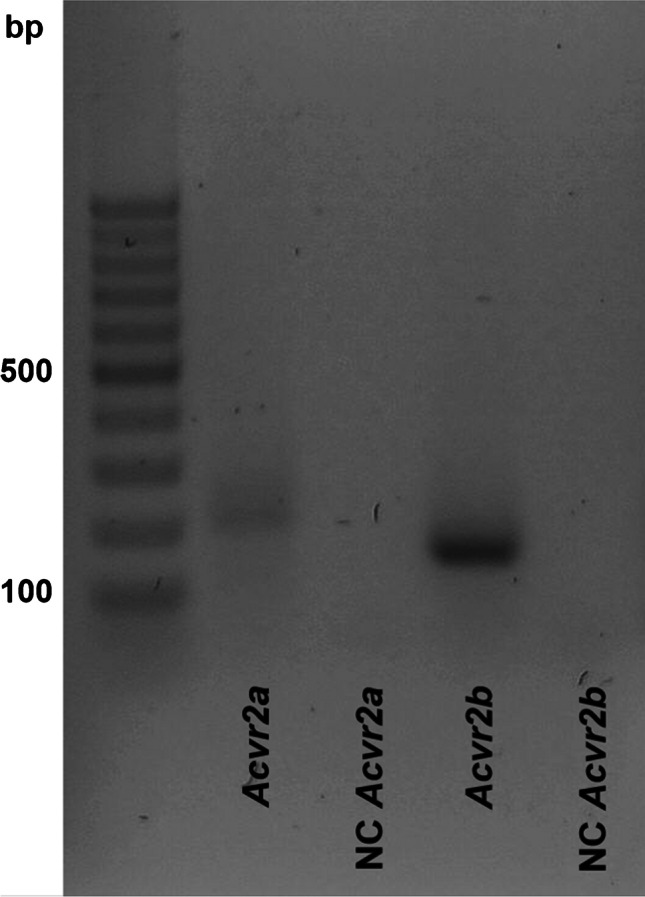
Expression of activin type 2 receptor isoforms in rat UMR106 osteoblast–like cells. Original agarose gel photo showing cDNA specific for activin type 2 receptor A (Acvr2a) and activin type 2 receptor B (Acvr2b) in UMR106 cells. NC, non-template control
Myostatin induces Fgf23 expression in UMR106 cells
Next, we examined whether myostatin influences Fgf23 expression. To this end, UMR106 cells were incubated without or with different concentrations of myostatin for 24 h and Fgf23 gene expression was analyzed by quantitative RT-PCR. As shown in Fig. 2a, myostatin significantly increased Fgf23 gene expression in a dose-dependent manner. Using ELISA, we tested whether the stimulatory effect of myostatin on Fgf23 gene expression is translated into enhanced FGF23 protein secretion into the cell culture supernatant. Indeed, 24 h treatment with myostatin also upregulated C-terminal FGF23 (Fig. 2b) and intact FGF23 (Fig. 2c) production. In IDG-SW3 cells, an 8 h treatment with 100 ng/mL myostatin resulted in a relative Fgf23 expression of 0.0006 ± 0.0004 (n = 4), a level not significantly different from that in control cells (0.0006 ± 0.0002; n = 4). Similarly, a 24 h treatment with 100 ng/mL myostatin led to a relative Fgf23 expression of 0.0382 ± 0.0025 (n = 7) in MC3T3-E1 cells, a level not significantly different from that in control-treated cells (0.0401 ± 0.0042; n = 7).
Fig. 2.

Myostatin induces Fgf23 expression in UMR106 cells, a Arithmetic means ± SEM of relative (rel.) Fgf23 mRNA abundance or b C-terminal or c intact FGF23 protein concentration in the cell culture supernatant of UMR106 osteoblast–like cells incubated without (white bars) or with (black bars) myostatin (a indicated concentrations, n = 7; and b, c 100 ng/mL, n = 10) for 24 h. *p < 0.05, **p < 0.01, ***p < 0.001 indicate significant difference from control. AU, arbitrary units; ctr, control (a one-way ANOVA; b unpaired Student’s t test with Welch’s correction; c Mann–Whitney-U-test)
Transforming growth factor-β type I receptor is necessary for myostatin-induced Fgf23 expression
The next experiments explored the signaling of the myostatin effect on Fgf23. The binding of myostatin to ACVR2B induces complex formation with TGF-βRI [38]. In order to study whether this process is involved, we incubated UMR106 cells with 100 ng/mL myostatin in the presence or absence of TGF-βRI inhibitor SB431542 (10 µM) for 24 h. SB431542 treatment significantly abrogated myostatin-induced Fgf23 expression in UMR106 cells (Fig. 3).
Fig. 3.
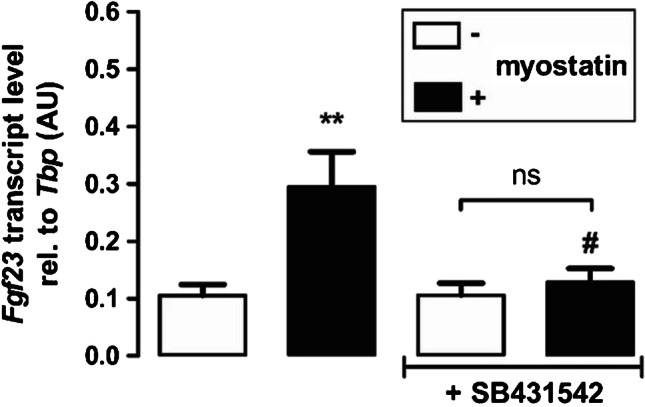
Transforming growth factor-β type I receptor (TGF-βRI) is necessary for myostatin-induced Fgf23 gene expression. Arithmetic means ± SEM of relative (rel.) Fgf23 mRNA abundance in UMR106 osteoblast–like cells incubated without (white bars) or with (black bars) myostatin (100 ng/mL, 24 h, n = 7) in the presence or absence of TGF-βRI inhibitor SB431542 (10 µM, 24 h). **p < 0.01 indicates significant difference from control. #p < 0.05 indicates significant difference from the absence of SB431542 (2nd vs 4th bar). AU, arbitrary units (one-way ANOVA)
The myostatin effect on Fgf23 is at least in part dependent on p38MAPK and NFκB-mediated store-operated Ca2+ entry (SOCE)
P38MAPK is a downstream target of myostatin [45] and also a potent regulator of FGF23 production [10]. To test whether p38MAPK contributes to the myostatin effect on Fgf23 gene expression, UMR106 cells were incubated with or without 100 ng/mL myostatin in the presence or absence of 10 µM p38MAPK inhibitor SB202190 for 24 h. As demonstrated in Fig. 4a, SB202190 significantly blunted myostatin-mediated upregulation of Fgf23 gene expression. Nevertheless, myostatin was capable of significantly enhancing Fgf23 gene expression even in the presence of SB202190, pointing to the involvement of further effectors. Since pro-inflammatory transcription factor complex NFκB is a powerful regulator of FGF23 and myostatin induces NFκB activity [1], we performed further experiments to elucidate an involvement of NFκB. To this end, UMR106 cells were treated with and without 100 ng/mL myostatin in the presence or absence of NFκB inhibitor withaferin A (500 nM) for 24 h. As shown in Fig. 4b, myostatin did not significantly alter Fgf23 gene expression in the presence of withaferin A.
Fig. 4.

The myostatin effect on Fgf23 is at least in part dependent on p38MAPK and on NFκB-mediated store-operated Ca2+ entry (SOCE). Arithmetic means ± SEM of relative (rel.) Fgf23 mRNA abundance in UMR106 cells treated without (white bars) or with (black bars) myostatin (100 ng/mL, 24 h) in the presence or absence of p38MAPK inhibitor SB202190 (a 10 µM, 24 h, n = 16) or NFκB inhibitor withaferin A (b 500 nM, 24 h, n = 6), or SOCE inhibitor 2-APB (c 150 µM, 24 h, n = 6). *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significant difference from control. ##p < 0.01 and ###p < 0.001 indicate significant difference from the absence of a SB202190; b withaferin A, or c 2-APB (2nd vs 4th bar). AU, arbitrary units (a Welch’s ANOVA; b, c one-way ANOVA)
NFκB enhances Fgf23 gene expression by upregulating SOCE [47]. Hence, we sought to determine whether the myostatin effect is also dependent on SOCE. To this end, UMR106 cells were treated with or without myostatin in the presence or absence of SOCE inhibitor 2-APB (150 µM) for 24 h. No significant effect of myostatin on Fgf23 gene expression was observed in UMR106 cells treated with 2-APB (Fig. 4c).
Discussion
This study provides evidence that myostatin, a signaling molecule produced by skeletal muscle cells, is a potent regulator of the production of FGF23, a hormone produced by bone cells. According to our results, myostatin upregulated Fgf23 gene expression and secretion of FGF23 protein in UMR106 osteoblast–like cells. Both, production of C-terminal and intact FGF23 were enhanced upon treatment with myostatin, suggesting that the myokine indeed regulates biologically active FGF23.
Myostatin was discovered as a myokine with mainly paracrine effects in skeletal muscle, i.e., the inhibition of skeletal muscle growth [16]. Our study adds to the growing concept of a muscle-bone cross-talk and further supports the notion of myostatin having paracrine and endocrine effects [25]. In this respect, it shares similarity with FGF23 which is also characterized by both paracrine and endocrine effects in different tissues and cells [28]. Other factors involved in this cross-talk are, among others, irisin, receptor activator of NF-κB ligand (RANKL), osteocalcin, sclerostin, or TGF-β [25, 29]. The regulation of bone-derived FGF23 through muscle-derived myostatin according to our study again underlines the mutual influence of bone and muscle. Interestingly, also IL-6 and TGF-β, other factors involved in bone-muscle cross-talk, are potent regulators of FGF23 [7, 13]. Conversely, bone-derived FGF23 also acts on skeleton muscle, inducing muscle atrophy [24, 29]
Our experiments also addressed the cellular mechanisms through which myostatin exerts its stimulatory effect on FGF23. The main membrane receptor for myostatin, ACVR2B [25, 38], was strongly expressed in UMR106 cells, whereas the expression level of ACVR2A was markedly lower. Conversely, expression of ACVR2B was low and that of ACVR2A was high in IDG-SW3 and MC3T3-E1 cells. Interestingly, myostatin failed to significantly affect Fgf23 expression in these cells, a result in line with ACVR2B being the major mediator of the myostatin effect on FGF23. Moreover, our results suggest that TGF-βRI is involved, as TGF-βRI inhibitor SB431542 significantly attenuated the myostatin effect on Fgf23. In line with a decisive role of TGF-βRI signaling for the production of FGF23, an earlier study identified TGF-β as a major trigger of FGF23 formation [13].
Downstream intracellular effectors of myostatin include p38MAPK [45]. Using p38MAPK inhibitor SB202190, we could demonstrate that also the myostatin effect on FGF23 is, at least in part, dependent on p38MAPK. This finding corroborates another study showing that p38MAPK signaling is a regulator of FGF23, in part through pro-inflammatory transcription factor complex NFκB [10]. Importantly, NFκB is a downstream target of myostatin [1] and itself is an important mediator of the stimulatory effect of inflammation and pro-inflammatory cytokines on FGF23 synthesis [22, 47]. NFκB is effective through inducing SOCE [47]. In line with this, both, NFκB and SOCE inhibition, significantly prevented myostatin from upregulating Fgf23 gene expression. A summary of the putative signaling is presented in Fig. 5.
Fig. 5.
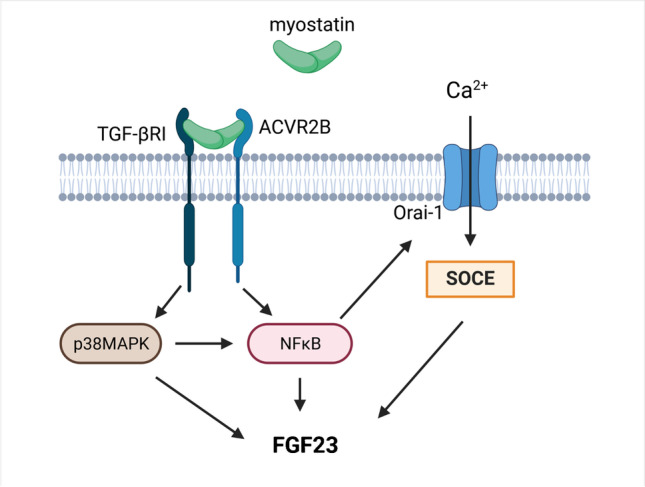
Schematic illustration of myostatin-induced FGF23 production in UMR106 cells. Myostatin binding to ACVR2B and following partnering with TGF-βRI activates p38MAPK and NFκB. NFκB induces SOCE, resulting in induction of Fgf23 gene expression. Created with BioRender.com. Activin type 2 receptor B (ACVR2B); fibroblast growth factor 23 (FGF23); nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB); p38 mitogen–activated protein kinase (p38MAPK); store-operated Ca2 + entry (SOCE); transforming growth factor-β type I receptor (TGF-βRI)
Myostatin not only prevents muscle hypertrophy but also an increase in bone mass as myostatin deficiency results in higher bone mass [9], bone mineral density and mineral content [34]. In line with this, myostatin inhibits osteoblast differentiation [37]. FGF23 induces a reduction of the plasma phosphate and 1,25(OH)2D3 concentration. Both effects are expected to favor a reduction in bone mineralization and mass [3]. Hence, the stimulatory effect of myostatin on FGF23 fits well into the concept of myostatin limiting skeletal muscle and bone mass.
In dermatomyositis, an inflammatory condition, the plasma myostatin concentration is elevated [23]. Importantly, the same study also found higher FGF23 levels in patients with dermatomyositis [23]. Moreover, CKD, another disease associated with elevated FGF23 levels [26], is characterized by a higher myostatin plasma concentration [46] and enhanced myostatin expression in muscle of mice [48]. These results are in line with our major finding, i.e., myostatin-dependent stimulation of FGF23 production.
Pharmacological manipulation of myostatin has already been tested as a therapeutic approach [25]: Myostatin inhibition may theoretically be beneficial in diseases with muscle weakness such as Duchenne muscular dystrophy or rheumatoid arthritis and was already tested [25]. According to our study, myostatin inhibition could result in lower FGF23, which may indeed be beneficial with regard to the reduced bone mass typical of both Duchenne muscular atrophy [4] and rheumatoid arthritis [30].
Data on the in vivo relevance of our in vitro results are sparse thus far: No gross differences in serum Ca2+ or phosphate were reported in Holstein Friesian calves and Belgian Blue calves [36]. Clearly, further in vivo studies exploring FGF23 and phosphate metabolism in myostatin-deficient mice or in Belgian Blue cattle are needed to confirm the significance of our findings.
Taken together, this study found a direct stimulatory effect of myostatin on Fgf23 gene expression and protein production in UMR106 cells. Moreover, it uncovered that this effect is, at least in part, mediated by p38MAPK and NFκB. These findings may contribute to higher FGF23 levels in some diseases with enhanced myostatin production and may be relevant for future therapeutic approaches involving pharmacological manipulation of myostatin.
Acknowledgements
The authors thank S. Ross, F. Reipsch, S. Münz, and H. Froß for technical help.
Author contribution
F. Ewendt and M. Föller designed the research; F. Ewendt analyzed the data; F. Ewendt and M. Feger performed the research; and M. Föller, F. Ewendt, and M. Feger wrote the paper.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the Deutsche Forschungsgemeinschaft.
Data Availability.
Data and material will be shared.
Code availability.
Not applicable.
Declarations
Ethics approval
Not applicable.
Consent to participate.
Not applicable.
Consent for publication.
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aravena J, Abrigo J, Gonzalez F, et al. Angiotensin (1–7) Decreases myostatin-induced NF-κB signaling and skeletal muscle atrophy. Int J MolSci. 2020;21:1167. doi: 10.3390/ijms21031167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bär L, Feger M, Fajol A, et al. Insulin suppresses the production of fibroblast growth factor 23 (FGF23) Proc Natl Acad Sci U S A. 2018;115:5804–5809. doi: 10.1073/pnas.1800160115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bär L, Stournaras C, Lang F, et al. Regulation of fibroblast growth factor 23 (FGF23) in health and disease. FEBS Lett. 2019;593:1879–1900. doi: 10.1002/1873-3468.13494. [DOI] [PubMed] [Google Scholar]
- 4.Buckner JL, Bowden SA, Mahan JD. Optimizing bone health in Duchenne muscular dystrophy. Int J Endocrinol. 2015;2015:928385. doi: 10.1155/2015/928385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daryadel A, Bettoni C, Haider T, et al. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflugers Arch. 2018;470:1569–1582. doi: 10.1007/s00424-018-2171-7. [DOI] [PubMed] [Google Scholar]
- 6.David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89:135–146. doi: 10.1038/ki.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durlacher-Betzer K, Hassan A, Levi R, et al. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018;94:315–325. doi: 10.1016/j.kint.2018.02.026. [DOI] [PubMed] [Google Scholar]
- 8.Egli-Spichtig D, Imenez Silva PH, Glaudemans B, et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019;96:890–905. doi: 10.1016/j.kint.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Elkasrawy MN, Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact. 2010;10:56–63. [PMC free article] [PubMed] [Google Scholar]
- 10.Ewendt F, Föller M. p38MAPK controls fibroblast growth factor 23 (FGF23) synthesis in UMR106-osteoblast-like cells and in IDG-SW3 osteocytes. J Endocrinol Invest. 2019;42:1477–1483. doi: 10.1007/s40618-019-01073-y. [DOI] [PubMed] [Google Scholar]
- 11.Ewendt F, Hirche F, Feger M, et al. Peroxisome proliferator-activated receptor α (PPARα)-dependent regulation of fibroblast growth factor 23 (FGF23) Pflugers Arch. 2020;472:503–511. doi: 10.1007/s00424-020-02363-8. [DOI] [PubMed] [Google Scholar]
- 12.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feger M, Hase P, Zhang B, et al. The production of fibroblast growth factor 23 is controlled by TGF-β2. Sci Rep. 2017;7:4982. doi: 10.1038/s41598-017-05226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis C, David V. Inflammation regulates fibroblast growth factor 23 production. Curr Opin Nephrol Hypertens. 2016;25:325–332. doi: 10.1097/MNH.0000000000000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glosse P, Feger M, Mutig K, et al. AMP-activated kinase is a regulator of fibroblast growth factor 23 production. Kidney Int. 2018;94:491–501. doi: 10.1016/j.kint.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Grobet L, Martin LJ, Poncelet D, et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- 17.Han HQ, Zhou X, Mitch WE, et al. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol. 2013;45:2333–2347. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Hanudel MR, Eisenga MF, Rappaport M, et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol Dial Transplant. 2019;34:2057–2065. doi: 10.1093/ndt/gfy189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu MC, Shi M, Moe OW. Role of αKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflugers Arch. 2019;471:99–108. doi: 10.1007/s00424-018-2238-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Indirli R, Guabello G, Longhi M, et al. FGF23-related hypophosphatemia in patients with low bone mineral density and fragility fractures: challenges in diagnosis and management. J Endocrinol Invest. 2020;43:787–798. doi: 10.1007/s40618-019-01165-9. [DOI] [PubMed] [Google Scholar]
- 21.Isakova T, Cai X, Lee J, et al. Longitudinal FGF23 trajectories and mortality in patients with CKD. J Am Soc Nephrol. 2018;29:579–590. doi: 10.1681/ASN.2017070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito N, Wijenayaka AR, Prideaux M, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cell Endocrinol. 2015;399:208–218. doi: 10.1016/j.mce.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Kerschan-Schindl K, Gruther W, Föger-Samwald U, et al. Myostatin and markers of bone metabolism in dermatomyositis. BMC Musculoskelet Disord. 2021;22:150. doi: 10.1186/s12891-021-04030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kido S, Hashimoto Y, Segawa H, et al. Muscle atrophy in patients wirh ckd results from fgf23/klotho-mediated supression of insulin/igf-i signaling. Kidney Research and Clinical Practice. 2012;31:A44. doi: 10.1016/j.krcp.2012.04.435. [DOI] [Google Scholar]
- 25.Lara-Castillo N, Johnson ML. Bone-muscle mutual interactions. Curr Osteoporos Rep. 2020 doi: 10.1007/s11914-020-00602-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larsson T, Nisbeth U, Ljunggren O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 27.Lavi-Moshayoff V, Wasserman G, Meir T, et al. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299:F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 28.Leifheit-Nestler M, Haffner D. Paracrine effects of FGF23 on the heart. Front Endocrinol (Lausanne) 2018;9:278. doi: 10.3389/fendo.2018.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G, Zhang L, Wang D, et al. Muscle-bone crosstalk and potential therapies for sarco-osteoporosis. J Cell Biochem. 2019;120:14262–14273. doi: 10.1002/jcb.28946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lodder MC, de Jong Z, Kostense PJ, et al. Bone mineral density in patients with rheumatoid arthritis: relation between disease severity and low bone mineral density. Ann Rheum Dis. 2004;63:1576–1580. doi: 10.1136/ard.2003.016253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masuyama R, Stockmans I, Torrekens S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattinzoli D, Ikehata M, Tsugawa K, et al. FGF23 and Fetuin-A interaction in the liver and in the circulation. Int J Biol Sci. 2018;14:586–598. doi: 10.7150/ijbs.23256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 34.Morissette MR, Stricker JC, Rosenberg MA, et al. Effects of myostatin deletion in aging mice. Aging Cell. 2009;8:573–583. doi: 10.1111/j.1474-9726.2009.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005;17:365–375. doi: 10.1016/j.cellsig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Probo M, Giordano A, Moretti P, et al. Serum biochemical profile in Holstein Friesian and Belgian blue calves in the first 48 hours of life. Ital J Anim Sci. 2019;18:657–662. doi: 10.1080/1828051X.2018.1551073. [DOI] [Google Scholar]
- 37.Qin Y, Peng Y, Zhao W, et al. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: a novel mechanism in muscle-bone communication. J Biol Chem. 2017;292:11021–11033. doi: 10.1074/jbc.M116.770941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rebbapragada A, Benchabane H, Wrana JL, et al. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/mcb.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez-Ortiz ME, Rodríguez M (2015) FGF23 as a calciotropic hormone. F1000Res 4:1472. 10.12688/f1000research.7189.1 [DOI] [PMC free article] [PubMed]
- 40.Saini RK, Kaneko I, Jurutka PW, et al. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcif Tissue Int. 2013;92:339–353. doi: 10.1007/s00223-012-9683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 42.Stöhr R, Schuh A, Heine GH, et al. FGF23 in cardiovascular disease: innocent bystander or active mediator? Front Endocrinol (Lausanne) 2018;9:351. doi: 10.3389/fendo.2018.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takashi Y, Fukumoto S. Phosphate-sensing and regulatory mechanism of FGF23 production. J Endocrinol Invest. 2020 doi: 10.1007/s40618-020-01205-9. [DOI] [PubMed] [Google Scholar]
- 44.Vervloet MG, van Ittersum FJ, Büttler RM, et al. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin J Am Soc Nephrol. 2011;6:383–389. doi: 10.2215/CJN.04730510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallner C, Jaurich H, Wagner JM, et al. Inhibition of GDF8 (Myostatin) accelerates bone regeneration in diabetes mellitus type 2. Sci Rep. 2017;7:9878. doi: 10.1038/s41598-017-10404-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yano S, Nagai A, Isomura M, et al. Relationship between blood myostatin levels and kidney function: Shimane CoHRE Study. PLoS ONE. 2015;10:e0141035. doi: 10.1371/journal.pone.0141035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang B, Yan J, Umbach AT, et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J Mol Med. 2016;94:557–566. doi: 10.1007/s00109-015-1370-3. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Rajan V, Lin E, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25:1653–1663. doi: 10.1096/fj.10-176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and material will be shared.
Not applicable.