

Abstract
Objective
Identification of brain regions susceptible to quantifiable atrophy in sporadic Creutzfeldt‐Jakob disease (sCJD) should allow for improved understanding of disease pathophysiology and development of structural biomarkers that might be useful in future treatment trials. Although brain atrophy is not usually present by visual assessment of MRIs in sCJD, we assessed whether using voxel‐based morphometry (VBM) can detect group‐wise brain atrophy in sCJD.
Methods
3T brain MRI data were analyzed with VBM in 22 sCJD participants and 26 age‐matched controls. Analyses included relationships of regional brain volumes with major clinical variables and dichotomization of the cohort according to expected disease duration based on prion molecular classification (i.e., short‐duration/Fast‐progressors (MM1, MV1, and VV2) vs. long‐duration/Slow‐progressors (MV2, VV1, and MM2)). Structural equation modeling (SEM) was used to assess network‐level interactions of atrophy between specific brain regions.
Results
sCJD showed selective atrophy in cortical and subcortical regions overlapping with all but one region of the default mode network (DMN) and the insulae, thalami, and right occipital lobe. SEM showed that the effective connectivity model fit in sCJD but not controls. The presence of visual hallucinations correlated with right fusiform, bilateral thalami, and medial orbitofrontal atrophy. Interestingly, brain atrophy was present in both Fast‐ and Slow‐progressors. Worse cognition was associated with bilateral mesial frontal, insular, temporal pole, thalamus, and cerebellum atrophy.
Interpretation
Brain atrophy in sCJD preferentially affects specific cortical and subcortical regions, with an effective connectivity model showing strength and directionality between regions. Brain atrophy is present in Fast‐ and Slow‐progressors, correlates with clinical findings, and is a potential biomarker in sCJD.
Introduction
Sporadic Creutzfeldt‐Jakob disease (sCJD) is a rapidly progressive dementia with underlying neurodegeneration, gliosis, and vacuolation. 1 Although early observations reported cerebral atrophy in sCJD through clinical neuroimaging, this was usually associated with very late disease course and/or long duration. 2 , 3 , 4 Currently, cortical atrophy in sCJD is not considered as a salient feature on visual inspection of MRI, except for cases of long duration. Studies on objective atrophy quantification in sCJD are scant, and there are no data on the presence of atrophy in Slow‐ versus Fast‐progressors based on sCJD molecular classification subtyping 5 or on brain atrophy patterns associated with some common clinical characteristics of sCJD. Previous morphometric studies have focused primarily on isolated brain regions and regions of interests, 6 , 7 diffusion tensor imaging metrics in the gray and white matter, 8 , 9 or cohorts which combined sporadic and genetic forms of prion disease. 10 , 11 Determination of atrophy patterns in sCJD might provide insights into the mechanisms of disease progression and identify vulnerable regions to track disease progression in both Fast‐ and Slow‐progressors, which could be targets of future network‐level‐based therapies. The primary goal of this cross‐sectional study is to quantify cerebral atrophy in sCJD using 3T brain magnetic resonance imaging (MRI) and voxel‐based morphometry (VBM). Based on the proposed mechanisms of transsynaptic propagation of prion particles, 12 our hypothesis is that neurodegeneration in sCJD causes selective brain network degeneration in anatomically connected regions, as opposed to indiscriminate global injury that might happen if prion disease was spreading within the brain either randomly or to adjacent regions. Structural equation modeling (SEM) is a statistical technique used in functional MRI and positron emission tomography (PET) studies to test whether activity in brain regions correlated with each other indiscriminately or if activity in one region influences that in another. 13 , 14 We therefore tested this hypothesis‐driven approach (SEM) to further investigate the relationship between brain regions vulnerable to sCJD pathology beyond simple correlations.
Participants and Methods
Patient selection
Participants or caregivers provided informed consent for participation in this study, which was approved by the University of California, San Francisco (UCSF) institutional review board. Participants were evaluated between January 2010 and August 2013 at the UCSF Memory and Aging Center (MAC) rapidly progressive dementia research program. Consecutive sCJD participants who ultimately either met 2007 UCSF clinical criteria for probable sCJD 15 (n = 8, 20%) or who had definite sCJD (pathology‐proven, n = 32, 80%), 16 and who had the same standardized MRI protocol of adequate quality (on the same scanner) from their first UCSF visit were included in the study. Forty sCJD participants were identified prior to MRI quality assessment. For the VBM analysis, 18 of these 40 participants were excluded due to severe motion artifact (n = 14) or lack of appropriate magnetization‐prepared rapid gradient‐echo (MPRAGE) T1‐weighted images (n = 4), leaving 22 participants with sufficient quality scan for VBM analyses. All 40 sCJD participants, however, had sufficient quality diffusion‐weighted imaging (DWI) MRIs for visual assessment (e.g., determination of pattern of involvement by DWI) and were used for analyses not requiring T1 sequences. Although we had initially intended to also examine longitudinal volume change, of the 14 participants with serial T1 scans, too few (n = 4) were of sufficient quality for cohort longitudinal analysis, so this was not performed. Twenty‐six healthy age‐ and gender‐matched participants, from the UCSF MAC Hillblom Healthy Aging Network project, who had MRIs performed with the same protocol on the same scanner were used as healthy controls (Controls). For some analyses, sCJD participants were grouped based on their DWI lesion patterns by visual inspection into 1) cortical‐subcortical, 2) cortical‐only, and 3) subcortical‐only cohorts. 17
Clinical and cognitive evaluation
sCJD participants had a standardized clinical evaluation including: neurological history and examination; Mini‐Mental State Examination (MMSE) 18 ; the Barthel index (measuring activities of daily living function) 19 ; a neuropsychological battery (although we only used MMSE); the neuropsychiatric inventory (NPI, 12‐item version) 20 to assess for behavioral symptoms and the presence of hallucinations; motor evaluation with the Unified Parkinson’s Disease Rating Scale – Motor (UPDRS‐Motor); and routine electroencephalogram (EEG). Controls underwent neurological history and exam, the clinical dementia rating scale, and a neuropsychological battery. Signs and symptoms were recorded from patient records and UCSF research visit summaries until the time of the research brain MRI. EEGs were classified into three categories: normal, slowing, or periodic sharp wave complexes, with the latter two categorized as abnormal for this study. 21 CSF total tau (t‐tau; performed at the U.S. National Prion Disease Pathology Surveillance Center (NPDPSC), Cleveland, OH) and neuron‐specific enolase (NSE; performed at Mayo Laboratories, Rochester, MN) levels, determined with enzyme‐linked immunosorbent assay, and protein 14‐3‐3 determined by Western Blot (NPDPSC; reported as either positive, inconclusive, or negative, with inconclusive considered as negative for this study) were available in 17, 13, and 19 sCJD participants, respectively. EEG and CSF biomarkers were obtained within 24 hours of the brain MRI. We also examined the effect of the prion protein gene (PRNP) codon 129 polymorphism on other clinical variables, including t‐tau, NSE, protein 14‐3‐3, EEG, and clinical scales. None of the 40 sCJD participants had a PRNP mutation, and all had codon 129 polymorphism analysis (NPDPSC, Case Western Reserve University, Cleveland, OH). Pathological confirmation was performed at either or both UCSF and NPDSPC, and prion typing was performed through the NPDPSC. The 32 pathology‐proven sCJD participants consisted of 18 of 22 patients included in the VBM analysis and 14 of 18 in the group excluded from the VMB analysis. One pathology‐proven subject without VBM had variably protease‐sensitive prionopathy and therefore had no prion typing. 22 , 23 , 24
Image acquisition
Participants underwent brain MRI at UCSF on a 3T scanner (Siemens TrioTim syngo, Erlangen, Germany) equipped with an eight‐channel transmit and receive head coil using an MPRAGE sequence with the following parameters: 160 sagittal slices; slice thickness = 1 mm; field of view = 256 mm; matrix = 256 × 240; voxel size 1.0 × 1.0 × 1.0 mm3; TR/TE/TI = 2300/2.98/900 ms, flip angle = 9°, and a HARDI dataset acquired using a single‐shot spin‐echo echo‐planar imaging (EPI) sequence including 55 contiguous axial slices acquired in an interleaved order with the following parameters: TR/TE = 8000/109 ms; flip angle = 90°; matrix = 100 × 100; in‐plane resolution = 2.2 mm2; slice thickness = 2.2 mm; 64 noncollinear diffusion sensitization directions at b = 2000 s/mm2, 1 at b = 0; and integrated parallel acquisition technique acceleration (IPAT) factor = 2.
Image processing
Image preprocessing for VBM analyses
All T1‐weighted images were visually inspected, and images with excessive motion or artifact were excluded. Processing for T1‐weighted sequences was performed using Statistical Parametric Mapping (SPM12, Welcome Trust Center for Neuroimaging, London, UK) running under MATLAB R2014b (MathWorks). Images were bias field‐corrected using N3 algorithm and segmented into gray matter, white matter, and CSF compartments using the unified segmentation algorithm from SPM12. A custom template was created from the whole cohort population (sCJD participants and Controls) by nonlinear registration template generation using Large Deformation Diffeomorphic Metric Mapping framework cite. 25 The images were then spatially normalized to the custom template and then modulated by multiplying the voxel values by the Jacobian determinant derived from the spatial normalization to preserve the relative volumes of gray matter. Finally, the images were smoothed with an 8‐mm full‐width‐at‐half‐maximum Gaussian kernel. This parcellation process created raw volumetric values for gray matter, white matter, and CSF compartments. The Anatomical Automated Labeling parcellation method was used to generate gray matter volumes, which were used in the SEM analysis and some other analyses to compare with CSF biomarkers (14‐3‐3, NSE, and total tau). The gray matter images then underwent VBM processing and analysis as discussed below.
VBM processing and analysis
For the MRIs with sufficient quality scans to undergo VBM processing, a general linear model was fit at each voxel using FMRIB Software Library v6.0 (FSL, Created by the Analysis Group, FMRIB, Oxford, UK). All comparisons included correction for age and total intracranial volume (TIV; calculated as the sum of gray matter, white matter, and CSF volumes). For comparisons in which we wanted to capture the effect of the speed of disease progression (based on molecular classification dichotomized into Fast‐progressors vs. Slow‐progressors), we controlled for disease severity by the Barthel index at the time of the MRI. This ensures that the findings are not due to the presence of more advanced disease in one group compared to the other. For comparisons that included clinical severity of symptoms (global cognition based on MMSE, motor symptoms based on UPDRS, and behavioral symptoms based on the NPI), we controlled for the MRI time‐ratio (the time the MRI was done relative to the total disease duration) to ensure that the findings are not simply due to being either early or late in the total disease process. For comparisons between those subjects with or without certain clinical symptoms (myoclonus, visual hallucinations, and ataxia), we controlled for both disease severity (Barthel index) and MRI time‐ratio in order to capture the brain regions associated with these symptoms. For these categorical VBM comparisons, a t‐test was performed for each voxel, with accepted threshold p value < .05 after correction for multiple comparisons with the permutation method used by the FMRIB Software Library (FSL), and the number of permutations was set at 5,000 using threshold‐free cluster enhancement. 26 , 27 Resulting statistical maps were normalized to Montreal Neurological Institute space for display. 28
We ran VBM analyses based on the following clinical variables:
sCJD versus Controls.
Fast‐progressors versus Slow‐progressors based on molecular classification (PRNP codon 129 genotype [MM, MV, or VV] and prion typing [type 1 or type 2]). 5 As the median disease duration of three sCJD molecular subtypes (MM1, MV1, and VV2) is less than 7 months (Fast‐progressors) and for the remaining three molecular subtypes (MM2, MV2, and VV1) typically is greater than 11 months (Slow‐progressors), 5 , 29 we conducted an analysis dichotomizing the sCJD VBM group into expected Fast‐progressors versus Slow‐progressors. For this analysis, we did not include sCJD cases with mixed prion types (i.e., 1‐2), as such cases often present more heterogeneously, varying along a spectrum between type 1 and type 2, 29 , 30 nor did we include the single variably protease‐sensitive prionopathy case, who by definition had no prion typing. 31 We also compared MM with MV cases (insufficient numbers for VV). Addtionally, we ran a VBM analysis comparing MM2 to MV2 participants (both Slow‐progressors) given their clinical and pathological differences, 5 and we had sufficient samples sizes of these two sCJD subtypes.
Global cognitive function dichotomized based on the median MMSE score of 20/30. Higher MMSE group for participants with score > 20/30 and lower MMSE group for participants with MMSE score ≤ 20/30.
The presence or absence of visual hallucinations captured on the NPI based on having visual hallucination at any point in the clinical course up until the time of brain MRI.
The presence or absence of ataxia.
The presence of absence of myoclonus.
Motor symptoms measured by the UPDRS‐Motor score and dichotomized based on the median UPDRS‐Motor score (lower ≤ 11, higher > 11; higher score indicates more motor impairment).
Behavioral changes dichotomized based on the median NPI FrequencyxSeverity product score (lower ≤ 20, higher > 20; higher score indicates more impairment/symptoms).
Statistical analyses
Tests of normality for all continuous data in sCJD and Controls were conducted with the Shapiro‐Wilk test. Means for continuous clinical variables were compared with the Student’s t or the Mann‐Whitney U tests, where appropriate. Because clinical and biomarker variables were not normally distributed, correlations were performed with the Spearman test. False discovery rate (FDR; at p < 0.05) was used to correct for multiple comparisons for all analyses, including non‐VBM (demographics, disease duration, clinical symptoms, cognitive and functional scales, CSF biomarkers, EEG pattern, DWI involvement pattern, codon 129, molecular classification, MRI‐based volume, and comparison of features between the sCJD subgroups included vs. those not included in VBM analysis in Table 1) and VBM comparisons. Data analysis was performed with SPSS (version 23, SPSS/IBM, Chicago, IL).
Table 1.
Clinical features of sporadic Creutzfeldt‐Jakob patients and Controls.
| Controls n = 26 | sCJD all n = 40 | sCJD included in VBM n = 22 | sCJD excluded from VBM* n = 18 | |
|---|---|---|---|---|
| Age at first evaluation, years, mean ± SD (median, range) | 66 ± 10 (67, 50‐77) | 63 ± 9 (66, 43‐80) | 64 ± 10 (68, 43‐80) | 63 ± 8 (64, 46‐75) |
| Sex, female (%) | 42 | 45 | 36 | 56 |
| Right‐handed (%) | 83 | 94 | 95 | 94 |
| Disease duration at the time of MRI, months, M ± SD (Md, r) | 8 ± 6 (7, 1‐32) | 8 ± 5 (8, 2‐23) | 7 ± 8 (6, 1‐32) | |
| Total disease duration, months, M ± SD (Md, r) | 14 ± 9 (14, 1‐32) | 16 ± 7 (18, 4‐28) 5 | 12 ± 10 (8, 3‐38) | |
| Interval from neuroimaging to death, months, M ± SD (Md, r) | 6 ± 2 (3, 0‐21) | 8 ± 2 (5, 0‐19) | 5 ± 2 (2, 2‐38) | |
| Clinical characteristics 1 (%) | ||||
| Cognitive difficulties | 90 | 86 | 94 | |
| Visual disturbance | 44 | 45 | 41 | |
| Ataxia | 31 | 45 6 | 12 | |
| Hallucinations | 36 | 31 | 41 | |
| Myoclonus | 44 | 31 | 59 | |
| MMSE score, M ± SD (Md, r, n) | 14 ± 10 (16, 0‐29, 38) | 18 ± 8 (19, 1‐29, 22) 7 | 8 ± 9 (5, 0‐29, 16) | |
| NPI score, M ± SD (Md, r, n) | 33 ± 24 (29, 0‐93, 33) | 27 ± 22 (20, 0‐79, 18) | 42 ± 25 (35, 8‐93, 15) | |
| UPDRS motor, M ± SD (Md, r, n) | 17 ± 16 (18, 0‐63, 29) | 16 ± 13 (11, 0‐42, 15) | 26 ± 17 (20, 0‐63, 14) | |
| Barthel index M ± SD (Md, r, n) | 66 ± 37 (80, 0‐100, 33) | 86 ± 23 (95, 15‐100, 19) 7 | 40 ± 38 (25, 0‐100, 14) | |
| CSF t‐tau (pg/mL) M ± SD (Md, r, n) 2 | 3870 ± 4172 (1800, 326‐15308, 29) | 2720 ± 3748 (1429, 326‐15308, 17) 6 | 5497 ± 4517 (4408, 1022‐13597, 12) | |
| CSF NSE (ng/mL) M ± SD (Md, r, n) 3 | 48 ± 47 (31, 4‐180, 26) | 46 ± 47 (31,18‐178, 13) | 49 ± 48 (31,4‐180, 13) | |
| CSF protein 14‐3‐3 | n = 34 | n = 19 | n = 15 | |
| Positive (%) | 44 | 25 6 | 67 | |
| Negative (%) | 18 | 35 6 | 0 | |
| Inconclusive (%) | 38 | 40 | 33 | |
| EEG | n = 36 | n = 21 | n = 15 | |
| Periodic epileptiform discharges (PED) (%) | 25 | 19 | 33 | |
| Slowing without PEDs (%) | 53 | 47 | 60 | |
| Normal (%) | 22 | 34 | 7 | |
| Diffusion‐weighted image pattern (%) | ||||
| Cortical‐subcortical | 55 | 45 | 67 | |
| Cortical‐only | 28 | 41 | 11 | |
| Subcortical‐only | 18 | 14 | 22 | |
| PRNP gene codon 129 genotype (n (%)) | 40 (100) | 22 (100) | 18 (100) | |
| MM (%) | 11 (28) | 6 (27) | 5 (28) | |
| MV (%) | 22 (55) | 14 (64) | 8 (44) | |
| VV (%) | 7 (17) | 2 (9) | 5 (28) | |
| Pathologically confirmed cases (n (%)) | 32 (80) | 18 (82) | 14 (77) | |
| Molecular Classification | ||||
| Prion typing not available (n (%)) | 10 (25)# | 4 (18) | 6 (33)# | |
| Prion typing available (n (%)) | 30 (75)# | 18 (81) | 12 (66) | |
| Fast‐progressors (n (%)) | 11 (35) (2 MM1, 5 MV1, 4 VV2) | 5 (27) (3 MV1, 2 VV2) | 6 (46) (2 MM1, 2 MV1, 2 VV2) | |
| Total disease duration, months, M ± SD, (Md, r) | 7 ± 4 (7, 3‐13) | 8 ± 6 (7, 4‐13) | 6 ± 3 (5.5, 3‐9) | |
| Slow‐progressors (n (%)) | 12 (38) (5 MM2, 6 MV2, 1 VV1) | 9 (50) (4 MM2, 5 MV2) | 3 (23) (1 MM2, 1 MV2, 1 VV1) | |
| Total disease duration, months, M ± SD (Md, r) | 19 ± 6 (20, 7‐27) | 20 ± 5 (20, 9‐27) | 18 ± 5 (23, 7‐24) | |
| Mixed Classification type (n (%)) | 7 (22) (3 MM1‐2, 4 MV1‐2) | 4 (22) (2 MM1‐2, 2 MV1‐2) | 3 (23) (1 MM1‐2, 2 MV1‐2 ) | |
| Total disease duration, months, M ± SD (Md, r) | 9 ± 7 (12, 3‐28) | 18 ± 9 (18, 10‐28) | 14 ± 7 (12, 3‐28) | |
| MRI‐based volume (corrected for TIV) | ||||
| Whole brain (mm 3 × 10 5 ) M (±SD) | 4.7 (±0.6) | 4.2 (±0.4) | ||
| Gray matter (mm 3 × 10 5 ) M (±SD) | 1.8 (±0.1) | 1.7 (±0.1) 4 | ||
| White matter (mm 3 × 10 5 ) M (±SD) | 1.4 (±0.2) | 1.2 (±0.1) | ||
| CSF (mm 3 × 10 5 ) M (±SD) | 1.4 (±0.2) | 1.2 (±0.1) |
CSF, cerebrospinal fluid; TIV, Total Intracranial Volume; MMSE, Mini‐mental state examination; M, Mean; d, median; t‐tau, total tau; NSE, neuronal‐specific enolase; r, range, NPI, Neuropsychiatric inventory; UPDRS, Unified Parkinson’s Disease Rating Scale motor; * = excluded due to poor‐quality MRI. Percentages might not sum to 100% due to rounding. # = 1 pathology‐proven patient had variably protease‐sensitive prion disease which by definition has no prion type identified.
Includes signs and symptoms up until around the time of UCSF MRI.
Abnormal value ≥ 1150 ng/mL.
Abnormal value > 30 ng/mL. Comparisons between all sCJD, sCJD included in VBM, sCJD excluded from VBM, and Controls were done for all the variables in the tables and significant results are noted as below
Compared to Controls, P < 0.01
Compared to sCJD excluded from VBM analysis, P < 0.001
Compared to sCJD excluded from VBM analysis, P < 0.05
Compared to sCJD excluded from VBM analysis, P < 0.01
Volumetric correlations and structural equation modeling
To investigate the general correlation between the volumetric data in the brain regions that showed atrophy in sCJD compared with Controls, Pearson correlations were performed after correction for age and TIV. We then used SEM to investigate model fit of regional VBM volumetric data. Our hypothesis was that if region atrophy is not random, some regions will influence volumetric changes in another region or regions, and a model that includes the regions most commonly involved in sCJD will pass the strict significance thresholds of SEM only in sCJD but not in regions not commonly involved in sCJD or in Controls. Because previous research that used hypothesis‐driven SEM approaches recommends starting with parsimonious models, we tested SEM models in four canonical networks in sCJD participants and Controls first and then added regions commonly affected in sCJD.
SEM is a multivariate technique that combines “path analysis” (a statistic used to evaluate causal models) 32 and multiple regression to estimate linear relationships between specific variables in an analysis of the covariance among these variables. One might expect cortical volumes of certain brain regions in sCJD to correlate with each other. This correlation of volumes could occur for at least three reasons: 1.) regional vulnerability; 2.) prions spreading transynaptically to functionally connected regions; or 3.) prions spreading to adjacent areas. SEM extends beyond simple correlations to evaluate the presence of any directional relationship among brain regions, and it examines the strength as well as the directionality of relationships within a network. 14 SEM incorporates the residual variance of the independent variables to estimate the dependences among the observed variables allowing for the validation of a hypothetical model by empirical data using strict goodness‐of‐fit thresholds. 13 The fundamental concept for the application of SEM for brain network analysis is effective connectivity, which is defined as the influence that one neural system exerts over another and vice versa. It differs from functional connectivity in that it is not solely defined by statistical dependencies between remote events but takes into account the activity‐dependent, explicit, and directional coupling between them. Effective connectivity can be estimated from SEM by testing whether a theoretical connectivity model seeking to explain a network of influences can actually fit the influences estimated from observed data. 14 In our case, the theoretical models that we tested were based on regions known to be commonly clinically affected in sCJD (i.e., network of cortico‐subcortical regions typically affected by restricted diffusion, including thalamus and striatum) 11 , 17 , 33 or on known anatomical connectivity in healthy participants (i.e., default mode, executive, salience, and motor networks). The DMN analysis was based on Buckner et al. 2008, and it included the precuneus, angular gyrus (AG), anterior cingulate cortex (ACC), and mesial and lateral temporal cortical regions. 34 The data used as input for the SEM model were age‐ and TIV‐corrected cortical volumes derived from the VBM analysis. 35 The effective connectivity is described by beta coefficients (called “path coefficients” in SEM), which were generated from interregional volume correlations via a process of iterative data fitting using IBM SPSS Amos graphics software. Based on the evidence that prions propagate transynaptically (i.e., via paths), 12 it was hypothesized that atrophy in sCJD occurs selectively and that volumes in brain regions affected would show a directional relationship (i.e., one brain region driving atrophy in another brain region). Nonetheless, given that SEM is a causal modeling statistical tool, we wanted to avoid drawing causative biological conclusion, and hence we only focus on the general model fit rather than on the beta coefficients. Theoretical models being tested were graphically represented by nodes (i.e., brain regions) exerting trophic influences through anatomical pathways (arrows), with the direction of a trophic effect represented by the arrow direction and the connectivity strength (beta coefficient) represented by arrow thickness. Models for right, left, and whole brain were tested separately. Models with optimal data fit have low error (root mean square error of approximation (RMSEA) < .05 and goodness‐of‐fit index (GFI) > .90). 14
Results
Fluid, clinical, and neuroimaging biomarkers
The demographics, basic clinical features (including CSF biomarker, EEG, and MRI findings), and scores of our sCJD cohort (n = 40), its various subcohorts, and the Controls cohort (n = 26) are shown in Table 1 and in the Supplementary Material. The relationship among CSF results and other outcomes or variables is presented in Supplementary Material.
Comparing volumetric values, derived from image processing prior to VBM, between the 22 VBM sCJD cases and Controls showed significantly less total gray matter volume in sCJD (p < 0.01, Student’s t‐test) but no differences in total white matter or CSF (Table 1).
There was significant difference in t‐tau levels using dichotomization of sCJD as Fast‐ versus Slow‐progressors (based on the molecular classification). Among the cases with available t‐tau and molecular classification (n = 21 of 40 total sCJD participants), the majority of participants with nonelevated t‐tau were Slow‐progressors (total nine patients with normal tau: six (66%) Slow‐progressors, one (11%) Fast‐progressor, and two (22%) mixed prion type), whereas the majority of the participants with elevated t‐tau were Fast‐progressors (total 12 patients with elevated tau: nine (75%) Fast‐progressors, one (8%) Slow‐progressor, and two (16%) mixed) (χ2 = 10, p = 0.008).
In the total sCJD cohort (n = 40), there were no differences in clinical biomarkers (i.e., CSF biomarkers, signs/symptoms, clinical scales, and MMSE; see Methods) based on the three common DWI MRI patterns (cortical‐only vs. subcortical‐only vs. cortico‐subcortical). Because in our experience, patients with cortico‐subcortical DWI involvement tend to have greater functional impairment, in order to increase the possibility of finding an effect of the DWI MRI pattern of involvement, we also compared this group to a combined group of subcortical‐only and cortical‐only involvement. This subcohort with cortical and subcortical involvement on DWI had higher t‐tau (p = 0.014; Supplemental Figure 1D), NSE (p = 0.005; Supplemental Figure 1E), NPI scores (p = 0.021; not shown), and UPDRS‐Motor scores (p = 0.027; not shown), and they were more likely to have a positive 14‐3‐3 (63% vs. 20%, χ2 = 6, p = 0.017; not shown) than the combined cortical‐only and subcortical‐only group. There were no statistically significant differences in clinical symptoms, protein 14‐3‐3, EEG, clinical scales, or biomarkers based on the PRNP codon 129 polymorphism (data not shown).
The same analyses were performed for the 22 participants included in the VBM analysis and showed relatively similar results (Supplementary Material). In comparing the subgroup with VBM analysis to the subgroup without VBM analysis, the VBM‐excluded participants had shorter disease duration, lower MMSE scores, lower Barthel scores, higher CSF t‐tau concentrations, higher prevalence of positive CSF protein 14‐3‐3, and lower prevalence of ataxia (Table 1 and Supplementary Material). Most of these findings suggest that the VBM‐excluded group had poorer quality MRIs (unable to be used for VBM analysis) due to a higher degree of clinical impairment or disease severity. Interestingly, the VBM group (less impaired) appeared to be much more likely to have only cortical ribboning, whereas the impaired had subcortical involvement, either with or without cortical ribboning. This is consistent with our clinical experience.
Atrophy patterns and brain‐phenotype relationships
VBM analysis results of the comparisons between 22 sCJD participants and 26 Controls are shown in 3D brain views and in axial views in Figure 1A,F. VBM analysis revealed gray matter reduction in sCJD compared to Controls in multiple cortical regions, including the bilateral frontopolar, mesial and inferior frontal lobes, mesial and lateral parietal lobes, bilateral lateral temporal, left mesial temporal regions, bilateral insulae, and inferior posterior right occipital regions, as well as bilateral thalami (permutations‐based correction for multiple comparisons P < 0.05). Some areas of relative sparing of atrophy compared to Controls included the motor, much of the occipital, and some dorsolateral and prefrontal cortices as well as bilateral striatum and globus pallidi. Of note, no regions showed higher volumes in sCJD compared to Controls. Interestingly, the regions with atrophy in sCJD included all areas of the functionally defined default mode and salience, possibly some of the executive control (lateral parietal), but none of the motor network hubs. 34 , 36 , 37
Figure 1.

Regional gray matter atrophy in sporadic Creutzfeldt‐Jakob disease. A‐E show a 3D rendering, whereas F‐J show the same data rendered in axial view. All results shown in color passed permutations‐based correction for multiple comparisons p < 0.05. Orientation is neurological (e.g., left side is left brain). Redder colors (A‐E) signify higher level of significance (higher t‐stat). For F‐J (axial views), color bar represents various t‐scores. Only regions of t‐scores > 2 (i.e., > 2 SD away from the mean) are shown; blue regions color have significantly greater atrophy than the comparison group. Clusters with volume reductions in sCJD compared to Controls were found in the bilateral frontopolar, mesial and inferior frontal, mesial and lateral parietal, bilateral lateral temporal and left mesial temporal, and inferior posterior right occipital regions (A, F). sCJD participants with visual hallucinations had significant volume loss in the bilateral thalami, medial orbitofrontal, rectus gyri, and right fusiform compared to participants without visual hallucinations (B, G). The sCJD group with more severe cognitive impairment (based on dichotomization by the median MMSE score) showed volume reduction in the bilateral mesial and inferior frontal, cerebellum, left orbitofrontal, and right mesial temporal regions compared to the group with less cognitive impairment (C, H). Volume differences between Slow‐progressors (based on molecular classification subtype) and Controls were present in the bilateral mesial and lateral frontal, bilateral precuneal, middle temporal, postcentral, and occipitoparietal regions (Slow‐progressors = 4 MM2, 5 MV2) (D, J). Volume differences in Fast‐progressors, based on molecular classification, and Controls were found in bilateral mesial and lateral frontal, bilateral precuneal, middle temporal, postcentral, and occipitoparietal regions as well as occipital and temporal (Fast‐progressors = 3 MV1, 2 VV2) (E, I). No volume differences were found between comparison of Fast‐progressors versus Slow‐progressors (not shown; see text).
VBM analyses revealed atrophy clusters that survived permutations‐based correction for multiple comparisons within sCJD subgroups when dichotomizing based on the dementia severity (MMSE) or the presence of visual hallucinations (Fig. 1B,C,G,H). sCJD participants with visual hallucinations (n = 6; 1 MM1, 1 MV1, 2 MV2, 1 VV2, and 1 MV without typing available) displayed right more than left thalamus, bilateral medial orbitofrontal, rectus gyri, and right fusiform atrophy compared to those without visual hallucinations (n = 15; 1 MM1, 3 MV1, 4 MM2, 2 MV2, 1 VV2, and 4 MV without typing available, one subject excluded because no Barthel score was available to adjust for disease severity – see Methods – VBM processing and analysis). Participants with more severe dementia (MMSE score ≤ 20, n = 12; 1 MM1, 4 MV1, 3 MM2, 2 MV2, 1 VV2, and 1 MV without typing available) had significant regional volume loss in several areas including bilateral mesial and inferior frontal, bilateral insula, thalamus, cerebellum, left orbitofrontal, and right mesial temporal regions compared to those with less severe dementia (MMSE score > 20, n = 7; 1 MM1, 1 MM2, 2 MV2, 1 VV2, and 2 MV without typing available). No effects were observed when MMSE was used as a continuous variable; comparing these groups separately against Controls, however, showed significant differences. No volumetric differences were found when comparing codon 129 MM2 to MV2 (the sample size was small, however: 4 MM2 and 5 MV2) or MM to MV genotypes. Compared to Controls, Fast‐progressors (n = 5; 3 MV1 and 2 VV2) had atrophy in bilateral mesial and lateral frontal, bilateral precuneal, middle temporal, postcentral, and occipitoparietal regions as well as bilateral thalamic atrophy. Slow‐progressors (n = 9; 4 MM2 and 5 MV2) compared to Controls showed a similar pattern of atrophy as the Fast‐progressors compared to Controls, but did not have atrophy of cerebellum, occipital, and most temporal regions (Fig. 1D, E, I, J). Surprisingly, comparing Fast‐progressors directly to Slow‐progressors showed no significant differences in regional atrophy (not shown). Participants with ataxia (n = 10; 1 MM1, 3 MV1, 2 MM2, 2 MV2, and 2 VV2) displayed a trend of posterior (left more than right) cerebellar atrophy compared to participants without ataxia (n = 10; 1 MM1, 1 MV1, 2 MM2, 2 MV2, and 4 MV without available type) (uncorrected, p < 0.001; not shown), but did not survive multiple correction. There were no statistically significant differences when comparing participants with myoclonus to participants without myoclonus. There were also no statistically significant differences when comparing participants with higher UPDRS score (n = 11; 1 MM1, 2 MV1, 2 MV2, 2 VV2, 1 MM2, 1 MV1‐2, and 2 MV without typing available) to those with lower UPDRS score (n = 10; 1 MV1, 3 MM2, 3 MV2, 2 MV1‐2, and 1 MV without typing available) or when comparing participants with higher NPI score (n = 9; 1 MM1, 2 MV2, 3 MM2, 1 VV2, 1 MV1‐2, and 1 MV without typing available) to those with lower NPI score (n = 8; 1 MV1, 1 MM2, 3 MV2, 1 MM1‐2, 1 MV1‐2, and 1 MV without typing available). Some comparisons did not include all 22 sCJD participants if subjects were missing a variable being controlled for or relevant to that analysis (e.g., Barthel or sCJD molecular classification).
Modeling atrophy covariance
Based on our VBM analysis, the atrophy patterns in sCJD involved all areas of the default mode network (DMN), except minimal hippocampal involvement, but also involved other regions outside of the DMN. The DMN is a topographically distant but highly connected network of functionally connected hubs. 34 Although we found high correlations of the volumetric data between these regions, we could not deduce whether these volumetric changes are happening simultaneously or if one region is influencing the atrophy in another (Supplementary Table 1 shows the Pearson correlation between some of the brain regions). Because of this and that many of these same areas are preferentially affected with abnormal diffusion in sCJD, 11 , 17 , 38 we investigated whether a structural equation model would fit the data and, if so, whether these simultaneously implicated atrophic regions are affected due to intrinsic network vulnerability independent of connectivity or if one brain region could be mathematically, although not necessarily biologically, influencing atrophy in another region. To test this hypothesis, we evaluated volumetric interactions in brain connectivity network models using SEM. Models with optimal data fit (RMSEA < .05 and GFI > .90) should fit only in CJD but not Controls, and the beta coefficient from region A to region B in the SEM model would mathematically indicate a directional relationship. 14 Tests of data fitness were done first in four functional networks: 1) DMN 34 ; 2) motor 36 ; 3) executive control 37 ; and 4) salience networks. 37 None of these SEM models met the significance and goodness‐of‐fit thresholds in either sCJD or Controls (RMSEA > 0.05 and GFI < 0.90). A model, however, based on the DMN model plus the addition of deep nuclei regions (striatum and thalamus) – which commonly show restricted diffusion in sCJD – resulted in optimal data fit in sCJD but not the Controls (Fig. 2). For sCJD and Control groups, left, right, and whole brain volumetric data were tested separately in the model, with only sCJD showing a significant effect (Fig. 2) but not the Controls (all RMSEA > 0.05 and all GFI < 0.90). The most meaningful model clinically, however, is the whole brain model, as it allows connectivity between hemispheres. The whole brain model showed that volumetric changes in the precuneus influenced the volumetric changes in the anterior cingulate cortex, angular gyrus, and mesial and lateral temporal cortex regions, whereas the connectivity was commensurate between the precuneus and the thalamus. Furthermore, striatal atrophy appeared to cause precuneus atrophy but not vice versa (as RMSEA > .05 and GFI < .90, not shown). Thus, coupled (connected) regions within the network changed their volumes together (i.e., high covariance), and these changes were directional within the network. Thus, there was SEM effective connectivity present in sCJD in a model including the DMN plus the striatum and thalamus, with the precuneus being the main influencer of directionality.
Figure 2.
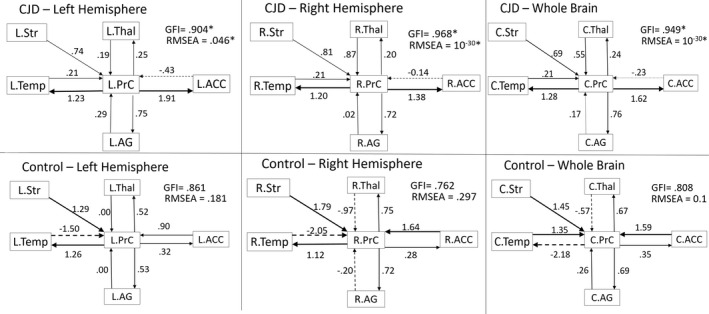
Sporadic Creutzfeldt‐Jakob disease selectively changes the effective connectivity between specific cortical and subcortical brain regions that overlap with the default mode network nodes. The figure shows the models of brain effective connectivity when brain volume data are tested in a network of cortical and subcortical regions usually noted by the authors to be commonly affected clinically on diffusion imaging in sCJD, specifically the default mode network plus the striatum and thalamus. Two key take‐away points from this figure are (1) the model fit in sCJD but not in Controls, and (2) that the precuneus (PrC) seems to play a central role in influencing volumetric changes in other regions. In the following text, we explain the SEM model and the meaning of the arrows from a mathematical standpoint. The graphs represent anatomical nodes in boxes connected by paths of trophic influence (arrows) that determine the regional volumetric influence on the target nodes. The effective connectivity (i.e., direction of the trophic effect) is represented by the arrow direction. Connectivity strength (i.e., strength of an effect) is represented by path coefficients (i.e., beta coefficient) displayed by the number over each arrow, with higher numbers meaning stronger tropic influence. The thickness of the arrow is a visual representation of the strength of the correlation and the dashed lines representing a negative correlation. Positive values indicate induction of atrophy in the direction of the arrow, whereas negative values indicate induction of increased volume. Goodness‐of‐fit statistics (GFIs) > .900 are considered significant with the p value equivalent shown by root mean square error of approximation (RMSEA)—only the models in sCJD, and none of the models in Controls, were significant (significant results are indicated with an *). In the whole brain and the right hemisphere models, and partially in the left hemisphere model, the precuneus exerts a large and disproportionate effect on the anterior cingulate (ACC), angular gyrus (AG), and temporal lobe (Temp). For example, in the whole brain model, one‐unit change in the precuneus volume results in 1.62, .76, and 1.28 points change in the ACC, AG, and Temp, respectively. Conversely, changes in the ACC, AG, and Temp volumes results in −.23, .17, and .21 unit change, respectively, in the precuneus. Interestingly, compared to the tropic influence of the precuneus, the effects were more balanced between the thalamus (Thal) and the precuneus and were unidirectional from the striatum (Str) to the precuneus. Models that included bidirectional effect between the precuneus and the Str did not meet the goodness‐of‐fit and the statistical significance parameters. This suggests that the striatum influenced atrophy of the precuneus, but not the reverse. L = left hemisphere, R = right hemisphere, C = combined or bilateral structure.
Discussion
Summary of major findings
This high‐resolution 3T MRI VBM study of 22 sCJD cases provides evidence that regional, but not global, atrophy is a feature of sCJD. Selective regional atrophy was observed in a composite of cortical and deep nuclei regions, including DMN hubs, 34 salience network (SN) hubs, 37 the thalami as well as other regions. Specifically, selective atrophy in sCJD (compared with Controls) was predominant in multimodal association regions, including the bilateral precunei, bilateral lateral parietal, bilateral mesial prefrontal cortices, and bilateral lateral temporal (all DMN regions, with the hippocampus as the only DMN region being only minimally involved), and bilateral anterior cingulate and insula (SN regions), in addition to bilateral lateral and inferior prefrontal cortices, and bilateral thalami (Fig. 1). Notably, in sCJD there was relative sparing of atrophy in the dorsolateral prefrontal cortex (part of the executive control network) and no atrophy in the primary motor, sensory, and visual cortices, as well as the striatum and cerebellum. We explored whether certain clinical variables and symptomatology were associated with regions of atrophy. Both Slow‐ and Fast‐progressors (defined based on sCJD molecular classification) 5 , 29 had significant atrophy compared to Controls but, to our surprise, were not statistically significantly different from each other. The sCJD subgroup with visual hallucinations had significantly greater atrophy in the right fusiform gyrus, right more than left thalamus, and bilateral orbitofrontal areas compared to the subgroup without visual hallucinations. Although MMSE as a continuous variable did not correlate with atrophy, participants with lower MMSE scores (dichotomized by median MMSE of 20) had more frontal, temporal, insula, thalamus, and cerebellum atrophy compared with participants with higher MMSE scores. Finally, SEM of volumetric brain data revealed significant covariance and trophic (directional) effects among key functional nodes of the DMN (precuneus, anterior cingulate cortex, angular gyrus, and mesial and lateral temporal cortex) 34 as well as the thalamus and striatum in sCJD but not in Controls. This model of effective connectivity showed trophic influence of subcortical regions on the precuneus and suggests a central role for the precuneus in driving volumetric changes in other cortical regions.
Atrophy in sCJD (vs. Controls) and comparison to literature
The pattern of selective brain volume reduction in sCJD overlaps with its typical DWI‐restricted diffusion pattern, which involves the same multimodal association regions including the cingulate, precuneus, angular gyrus, and superior and middle frontal gyri, with relative sparing of primary motor and sensory cortices. 11 , 17 , 33 We might expect that areas first involved in sCJD would be the areas to show greatest atrophy later on in the disease. We found five case reports of neurologically normal persons who had brain MRIs for reasons unrelated to sCJD (e.g., carotid bulb tumor, studies assessing utility of annual MRI for standard of clinical care, etc) and who became symptomatic with sCJD between 3 and 14 months later. Overall, these cases showed DWI abnormalities (reduced diffusion) in the presymptomatic phase of disease in association cortices including the bilateral temporo‐parietal‐occipital junction, lateral parietal, precunei, and mesial frontal regions, 39 , 40 , 41 , 42 , 43 similar to regions where we detected atrophy with VBM. Furthermore, our findings are consistent with studies in human genetic prion disease showing atrophy in cortical association regions. 10 , 11 Alner et al. 2011, using 1.5T MRI and FreeSurfer imaging analysis, described cortical thickness reduction in 10 patients with a genetic prion disease (gPrD; 6‐OPRI PRNP mutation), predominantly in the precuneus, supramarginal, parietal lobule, and lingual regions. 10 In a follow‐up study also with 1.5T MRI, but using SPM8 VBM analysis, De Vita et al. 2013 described precuneus, perisylvian, lingula, and basal ganglia volume reduction in nine symptomatic patients with the same 6‐OPRI PRNP mutation. 11 Similarly, using 3T VBM analysis in 30 symptomatic predominantly gPrD subjects (10% sCJD), Caine et al. described volume reductions in frontal and parietal gray matter volumes which correlated with predominant frontoparietal dysfunction on neurocognitive testing. 44 These three studies all showed atrophy in regions for which we also found atrophy in our sCJD cohort (Fig. 1). In a study using 3T VBM analysis, Grau‐Rivera et al. 2015 found significant gray matter loss in 15 CJD cases (13 sCJD and 2 genetic, E200K) specifically in the bilateral thalami, putamen, fusiform gyrus, cerebellum, and left perirolandic cortex. 45 Although we had similar thalamic and fusiform findings, we did not observe basal ganglia (striatum and globus pallidus), perirolandic, or cerebellar volume loss in sCJD vs. Controls. It is interesting that we did not find striatal volume loss despite visible striatal diffusion reductions present in 60% of our sCJD participants (Table 1). This parallels findings in a study by Seror et al. 2010 with 12 patients with E200K genetic prion disease who had striatal‐restricted diffusion but did not show quantifiable volume reduction. 46 In our experience, for most sCJD cases, striatal involvement on DWI usually occurs after extensive cortical ribboning appears (except in MV2 and VV2 cases which usually have isolated deep nuclei involvement). The fact that we found overlapping but slightly different cortical areas involved than these other studies might be due to other studies using mostly genetic cases and few sporadic cases or to smaller sample sizes in Navid et al. 7 (n = 11) and Grau‐Rivera et al. 45 (n = 13) compared with 22 sCJD subjects in our study.
Atrophy in sCJD subgroups and comparison to literature
As noted above, we predicted that Fast‐progressors might not have sufficient time to develop detectable atrophy compared to Slow‐progressors (Fast and Slow based on sCJD molecular classification). Contrary to our prediction, however, each of these subgroups showed significant atrophy when compared to Controls. The atrophy pattern in Fast‐progressors compared to Controls and Slow‐progressors compared to Controls was relatively similar (Fig. 1). On visual inspection of the VBM data, Fast‐progressors had more atrophic areas than Slow‐progressors, including cerebellum, occipital, and temporal regions. These differences, however, did not reach statistical significance, which might be due to the relatively small sample size. Nevertheless, these findings suggest that brain atrophy is present, even in Fast‐progressors, and although usually not detectable by visual assessment, it can be quantified using VBM. To our knowledge, VBM analysis of the Fast‐ versus Slow‐progressors in sCJD based on molecular classification has not been reported previously.
We also identified a number of brain‐behavior relationships with VBM in sCJD. Participants with visual hallucinations showed more significant right fusiform, right more than left thalamus, and bilateral medial orbitofrontal volume loss compared with participants without visual hallucinations (Fig. 1). The involvement of the fusiform gyrus is consistent with previous studies showing atrophy on VBM and functional (fMRI) abnormalities in visual association areas in patients with neurodegenerative conditions such as Parkinson’s disease. 47 Similarly, orbitofrontal cortex involvement in Parkinson’s disease dementia patients experiencing visual hallucinations has been reported in multiple studies, including VBM, 48 , 49 selective serotonin 2A receptor ligand F‐18 PET, 50 and diffusion tensor imaging studies. 51 Moreover, fluorodeoxyglucose‐PET (FDG‐PET) hypometabolism in the orbitofrontal cortex has been found in patients with schizophrenia who have visual hallucinations. 52 Furthermore, recent studies suggest a role for thalamic‐DMN decoupling as the mechanism for visual hallucinations in synuclinopathies. 53
The sCJD group with more severe cognitive impairment, based on dichotomization of MMSE scores had scattered volume loss in the bilateral mesial orbitofrontal, inferior frontal, posterior central, bilateral middle temporal, and bilateral medial precuneus gyri, in addition to more confluent volume loss in the bilateral insula, thalamus, and cerebellum, even after adjusting for the time point of MRI in the disease course (ratio of disease duration at the time of MRI over total disease duration). When we corrected for Barthel, however, this effect went away, probably because Barthel and MMSE were correlated. This atrophy pattern is consistent with the role of the cerebellar‐thalamic‐cortical connections in a wide range of cognitive functions. 54
Although it did not survive correction for multiple comparisons, sCJD participants with ataxia compared with those without ataxia showed a trend toward greater left posterior cerebellar atrophy (not shown), which is consistent with a previous finding of a correlation between cerebellar atrophy and ataxia in E200K genetic prion disease. 6 Perhaps a larger sample size is needed to show this effect in sCJD.
Cortico‐subcortical regions as the epicenter in sCJD
From a brain network perspective, our VBM data support the idea that frontoparietal and temporal regions, including but not limited these areas within the DMN, 55 as well as several subcortical structures, are at the epicenter of neurodegenerative changes in sCJD. The selective volume reduction we observed in sCJD within most of the DMN (only minimal hippocampal atrophy) and other regions may be explained by both distinctive regional neurometabolic profiles and network connectivity properties in this disease. For example, parietal regions such as the precuneus in particular, but also certain occipital and temporal regions, display the highest state of oxidative neuronal metabolism in the brain. 56 , 57 In such regions with high baseline metabolism, oxidative stress may play a crucial role in prion protein misfolding. 58 These results are in line with an FDG‐PET study of sCJD showing hypometabolism to be common particularly in parietal and temporal regions. 59 Networks, such as the DMN, often feature a number of heteromodal association cortices that operate as hub nodes. Such networks are resistant to random node failure but become vulnerable when hub failure occurs, thus facilitating transsynaptic degeneration or other forms of network pathological spread. 60 The biological vulnerability of these hubs is supported by its implication as a pathogenic substrate in various neurodegenerative diseases. For example, Alzheimer’s disease (AD), which like prion disease involves protein misfolding, has prominent posterior DMN hypometabolism and atrophy. 61 Interestingly, the pattern of cortical atrophy in sCJD, in part targeting DMN hubs, is reminiscent of the involvment of the DMN in AD. 61 , 62 This also is consistent with literature suggesting shared pathogenic mechanisms and overlap between AD and sCJD. 63 , 64 , 65 , 66 Furthermore, selective vulnerability of networks containing high‐degree hubs is also characteristic of other neurodegenerative disorders such as behavioral variant frontotemporal dementia, semantic dementia, progressive supranuclear palsy, and corticobasal syndrome. 37 , 67 , 68
VBM results implicating the involvement of cortical and subcortical regions in sCJD (through atrophy) were supported using SEM in connectivity models that were specifically tested for the presence of network‐level involvement in sCJD. The cortico‐subcortical effective connectivity in sCJD can be interpreted as a common trophic fate (i.e., volume reduction) for involved cortical and subcortical regions. A structural connectivity model that involved DMN regions plus subcortical regions consisting of the thalamus and striatum fit in the sCJD group but not in the Controls (Fig. 2). In both sCJD and Controls, alternative models involving either the DMN without the addition of subcortical structures, as well as the executive, salience, or motor networks did not fit the model. In the cortico‐subcortical network model that met the strict significance thresholds of SEM (GFIs>.90 and RMSEA<.05), the strongest path effect was from the precuneus to the anterior cingulate cortex, mesial and lateral temporal, and angular gyrus regions in the left, right, and combined hemispheres of the sCJD group but not in the Controls. These SEM findings potentially point to a central role for the precuneus in sCJD connectivity‐mediated neurodegeneration. This finding is consistent with our clinical experience and literature suggesting that the precuneus and the adjacent posterior cingulate cortex are very commonly involved by visual assessment of the DWI/ADC maps and quantitative mean diffusivity measurement in sCJD. 17 , 33 The precuneus exhibited trophic influences on other cortical regions, whereas its trophic influence on the thalamus was bidirectionally equivalent, and the striatum exhibited a unidirectional trophic influence on the precuneus. We speculate that the bidirectional effects on a group‐wise level could be due to the high connectivity between the DMN hubs – once one region is affected it begins influencing atrophy in other highly connected regions. Thus, regardless of where a patient's disease begins, involvement eventually converges on this highly connected network. Some bidirectionality, however, was positive in one direction and negative in another, meaning that atrophy in one region is influencing increased volume in another region (negative arrow in Fig. 2). We specultate that this might occur because astrocytosis, neuroinflammation, and/or possibly edema involved in neurodegeneration might produce volumetric changes that can be interpreted by SEM as increased volumes within the sCJD group, which we simply were not able to detect in our VBM analyses.
Despite the striatum not showing atrophy on VBM, the SEM showed a volumetric trophic effect of the striatum on the precuneus in sCJD, which was not found in Controls. We suspect that, despite the lack of detectable striatal changes on VBM in sCJD, the striatum likely plays an important role on the network‐level pathogenesis of sCJD, along a continuum of diffusivity changes preceding atrophy. These findings point to the importance of subcortical regions, such as the striatum and thalamus, in the pathophysiology of sCJD, as similar models including only cortical regions were not solvable and only models including both cortical and subcortical regions were. Furthermore, this SEM model provides mathematical evidence for the hypothesis that the selective atrophy pattern in sCJD is mediated by atrophy spread via anatomical connections, such as those used for the transsynaptic spread of prions, rather than selective atrophy due to intrinsic regional vulnerability alone, independent of connectivity and transsynaptic prion spread. In other words, if atrophy is mediated only by intrinsic regional vulnerability, then we would expect high volumetric correlations in atrophied regions but without a directional relationship as we found in our SEM analysis.
We also identified several associations between various biomarkers and clinical outcomes in both the entire cohort of 40 sCJD participants who had DWI and the subcohort of 22 sCJD participants who also had sufficient quality T1 MRI volumetric analysis. sCJD participants with both cortical and subcortical DWI involvement had higher CSF biomarkers (t‐tau and NSE) and more motor and behavioral dysfunction compared with the group combining cortical‐only and subcortical‐only involvement. This is consistent with our clinical experience in which we find that patients with both cortical and subcortical DWI involvement tend to decline more rapidly, particularly compared to those with cortical‐only involvement. Whole brain volumes inversely correlated with CSF t‐tau levels, which is generally consistent with the modestly high sensitivity and specificity of an elevated t‐tau in sCJD. 69 Similarly, t‐tau levels were inversely correlated with total disease duration and MMSE scores (i.e., participants with lower levels had longer disease durations and better cognitive performance), which parallels previously observed changes of t‐tau with disease duration 70 , 71 , 72 , 73 and cognitive function in prion disease. 74
This study has a number of limitations. Only cross‐sectional volumetric quantifications were performed, as we had too few cases with sufficient quality serial data. Longitudinal studies, which are often difficult to obtain in sCJD, particularly of sufficient quality for volumetric analysis, will be needed to better characterize atrophy progression in sCJD. Although our cohort was relatively large for a single‐site sCJD imaging cohort, our sample size nevertheless is relatively small, limiting our ability to identify more interactions of clinical variables and volumetric changes or to compare all six main sCJD molecular classification subgroups. The fact that even with our limited sample size, however, we still found any associations suggests the strength of our findings. Although our cohort with DWI (n = 40) included all molecular subtypes and had a plurality of Fast‐progressors (MM1, MV1, and VV2) consistent with the general sCJD population, 29 , 75 Fast‐progressors were underrepresented in the subcohort included in the VBM analysis because of difficulties obtaining sufficient quality volumetric scans due to motion artifact. This may represent sampling bias favoring cases with slower progression. Furthermore, we only compared cases whom could easily be classified as Fast‐ or Slow‐progressors based on their sCJD molecular classificaiton, excluding five cases (four with mixed prion types and one variably protease‐sensitive prionopathy). This dichotomization of sCJD into Fast‐ vs. Slow‐progressors based on the mean disease duration of each of the six major molecular subgroups has limitations. For example, even though on average a molecular classification subtype is rather homogenous, an individual patient might be more heterogenous. Additionally, the clinical and biomarker data of the VBM‐excluded participants suggest that these participants in general had more severe disease, which likely interfered with MRI acquisition and led to VBM analysis of the relatively less severely affected subjects. Lastly, although our cohort might appear to have longer survival than many other large published sCJD cohorts, our 2‐day research visit protocol attempts to identify the very first symptoms, which we commonly find to be several weeks and usually months earlier than the medical records or even the family had initially reported. This results in our patients' disease durations appearing longer than those reported in national surveillance cohorts that are largely based on retrospective external record review. 5 , 65 , 76 , 77
Our cohort in this study had extensive neurological, PRNP codon 129 genotyping, and CSF biomarker characterization and, to our knowledge, represents the first quantitative VBM study exclusively in sporadic prion disease. The results of this study add a valuable dimension to the neuroimaging characterization of sCJD and support the value of unbiased quantification of brain volume as a potential clinical biomarker in sCJD. Trophic SEM analyses of VBM data suggest that atrophy in sCJD might not just be due to regional vulnerability, but that there is a directionality of influence of atrophy in sCJD, which we hypothesize might be influenced by propagation of prions along synaptic pathways.
Conflict of Interest
MDG receives/received research support on prion disease from the NIH/NIA (R01‐AG031189; R56‐AG055619; R01AG062562), the Michael J. Homer Family Fund and Alliance Biosecure. He has consulted for Adept Field Consulting (Backbay consulting), Advanced Medical Inc., Anderson Boutwell Traylor, Acsel Health LLC, Best Doctors Inc., Blade Therapeutics, Biohaven Pharmaceuticals, Bioscience Pharma Partners, LLC (BPP), ClearView HealthCare Partners, Grand Rounds Inc./Second Opinion Inc., Gerson Lehrman Group (GLG) Inc., Guidepoint Global LLC, Market Plus, InThought Consulting, LifeSci Capital LLC, Maupin Cox Legoy, MEDACorp, Quest Diagnostics, 3M Communications (Microvention Terumo), Smith & Hennessey LLC, TeleDoc Health Inc., and Trinity Partners LLC. He has received speaking honoraria for various medical center lectures and from Oakstone Publishing. He has received past research support from CurePSP, the Tau Consortium, Quest Diagnostics, and NIH. Dr. Geschwind serves on the board of directors for San Francisco Bay Area Physicians for Social Responsibility and on the editorial board of Dementia & Neuropsychologia. JCR is a site PI for clinical trials supported by Eli Lilly and receives support from NIH. IIG is supported by the Rio Hortega grant (CM17/00074) from “Acción Estratégica en Salud 2013‐2016” and the Global Brain Health Institute.
Author Contributions
KY and JR drafted the manuscript and contributed data acquisition, analysis, and interpretation. AW, GY, MP, GT, EC, MM, IG, and YC contributed data acquisition, analysis and interpretation, and critical review of the manuscript. JHK contributed data acquisition, BLM contributed study concept and design, HR and MG contributed study concept and design, data interpretation, and critical review of the manuscript.
Supporting information
Supplementary Figure. The relationship between magnetic resonance imaging findings and clinical variables in sporadic Creutzfeldt‐Jakob disease. Regarding correlations with whole brain volume (A‐C), CSF t‐tau levels correlated negatively with whole brain volume (A), whereas NSE did not correlate with whole brain volume (B), and MMSE correlated positively with whole brain volume (C). For D and E, we compared CSF t‐tau and NSE in cases with both cortical‐ and subcortical‐restricted diffusion on DWI (cortical and subcortical: C‐S) against the combination of cases with cortical‐only involvement and subcortical‐only involvement (combined cortical‐only and subcortical‐only: Co‐S/C) (see text). This was because, our hypothesis was that those with cortical and subcortical diffusion would be more severely affected and more likely to have these biomarkers positive. As we suspected, sCJD participants with a cortical and subcortical (C‐S) pattern of restricted diffusion (gray) showed elevated t‐tau (C) and NSE (D) levels compared to the group of subjects of combined cortical‐only and subcortical‐only (Co‐S/C) restricted diffusion (white box).
Supplementary Material. Biofluid, clinical, and neuroimaging biomarker results including CSF biomarker results and comparison of CSF results and other outcomes or variables among entire sCJD cohort, those with and those without VBM.
Supplementary Table. Pearson correlations between the cortical and subcortical regions included in structural equation modeling (SEM). This table shows that high correlations (≥0.5 strong; moderate 0.3‐0.49; < 0.3 low) are present in sCJD in some of the brain regions included in the SEM model (significant correlations are shown in bold). This methodology, however, cannot discern if there is any effective connectivity and if this effective connectivity has any preferential directionality. Figure 2 in the main text, however, does show the effective connectivity and any directionality using SEM. * Correlation is significant at the 0.05 level (two‐tailed). ** Correlation is significant at the 0.01 level (two‐tailed). "Control Variables: Gender, Age, TIV, and Barthel"
Acknowledgments
NIH/NIA R01 AG031189 (MDG), P50 AG023501(BLM, HJR), T32 AG023481 (JCR), AG032289 (JHK), NIH/NCRR UCSF‐CTSI (UL1 RR024131), JCR is supported by NIA‐NIH: K23AG059888. Hellman Family Foundation, Michael J. Homer Family Fund (MDG), Larry L. Hillblom Network 2018‐A‐006‐NET (JHK), i‐PFIS grant (IF15/00060), Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (I.I‐G). Global Brain Health Institute Atlantic Fellow for Equity in Brain Health (I.I‐G). The authors thank Drs. Stephen J. DeArmond and Henry Sanchez at UCSF and the National Prion Disease Pathology Surveillance Center (NPDSPC) at Case Western Reserve University for their assistance in pathological confirmation of many of our sCJD cases. The authors also express their gratitude to the patients and their loved ones for participating in our research program.
Funding Statement
This work was funded by NIH/NIA grants AG031189 (MDG), P50AG023501 (BLM, HJR), AG032289 (JHK), K23AG059888 (JCR), and T32 AG023481 (BLM, HJR); NIH/NCRR UCSF‐CTSI UL1 grant RR024131; Hellman Family Foundation (JCR) grant ; Michael J. Homer Family Fund (MDG) grant ; Larry L. Hillblom Network 2018‐A‐006‐NET (JHK) grant ; i‐PFIS grant grant IF15/00060; Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (I.I‐G) grant ; Global Brain Health Institute Atlantic Fellow for Equity in Brain Health (I.I‐G) grant .
References
- 1. Geschwind MD. Prion diseases. Continuum (Minneap Minn) 2016;21(16 Neuroinfectious Disease); 1612–38. 10.1212/CON.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovanen J, Erkinjuntti T, Iivanainen M, et al. Cerebral MR and CT imaging in Creutzfeldt‐Jakob disease. J Comput Assist Tomogr 1985;9(1):125–128. 10.1097/00004728-198501000-00023. [DOI] [PubMed] [Google Scholar]
- 3. Hayashi R, Hanyu N, Kuwabara T, Moriyama S. Serial computed tomographic and electroencephalographic studies in Creutzfeldt‐Jakob disease. Acta Neurol Scand 1992;85(3):161–165. 10.1111/j.1600-0404.1992.tb04020.x. [DOI] [PubMed] [Google Scholar]
- 4. Finkenstaedt M, Szudra A, Zerr I, et al. MR imaging of Creutzfeldt‐Jakob disease. Radiology 1996;199(3):793–798. 10.1148/radiology.199.3.8638007. [DOI] [PubMed] [Google Scholar]
- 5. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46(2):224–233. https://www.ncbi.nlm.nih.gov/pubmed/10443888. [PubMed] [Google Scholar]
- 6. Cohen OS, Hoffmann C, Lee H, et al. MRI detection of the cerebellar syndrome in Creutzfeldt‐Jakob disease. Cerebellum 2009;8(3):373–381. 10.1007/s12311-009-0106-8. [DOI] [PubMed] [Google Scholar]
- 7. Navid J, Day GS, Strain J, et al. Structural signature of sporadic Creutzfeldt‐Jakob disease. Eur J Neurol 2019;26(8):1037–1043. 10.1111/ene.13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eisenmenger L, Porter MC, Carswell CJ, et al. Evolution of diffusion‐weighted magnetic resonance imaging signal abnormality in sporadic Creutzfeldt‐Jakob disease with histopathological correlation. JAMA Neurol 2016;73(1):76–84. 10.1001/jamaneurol.2015.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee H, Cohen OS, Rosenmann H, et al. Cerebral white matter disruption in Creutzfeldt‐Jakob disease. AJNR Am J Neuroradiol 2012;33(10):1945–1950. 10.3174/ajnr.A3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alner K, Hyare H, Mead S, et al. Distinct neuropsychological profiles correspond to distribution of cortical thinning in inherited prion disease caused by insertional mutation. J Neurol Neurosurg Psychiatry 2012;83(1):109–114. 10.1136/jnnp-2011-300167. [DOI] [PubMed] [Google Scholar]
- 11. De Vita E, Ridgway GR, Scahill RI, et al. Multiparameter MR imaging in the 6‐OPRI variant of inherited prion disease. AJNR Am J Neuroradiol 2013;34(9):1723–1730. 10.3174/ajnr.A3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stopschinski BE, Diamond MI. The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol 2017;16(4):323–332. 10.1016/S1474-4422(17)30037-6. [DOI] [PubMed] [Google Scholar]
- 13. Krause JB, Taylor JG, Schmidt D, et al. Imaging and neural modelling in episodic and working memory processes. Neural Netw 2000;13(8–9):847–859. 10.1016/s0893-6080(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 14. de Marco G, Vrignaud P, Destrieux C, et al. Principle of structural equation modeling for exploring functional interactivity within a putative network of interconnected brain areas. Magn Reson Imaging 2009;27(1):1–12. 10.1016/j.mri.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 15. Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin 2007;25(3):783–807. 10.1016/j.ncl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt‐Jakob disease. Arch Neurol 1996;53(9):913–920. http://www.ncbi.nlm.nih.gov/htbin‐post/Entrez/query?db=m&form=6&dopt=r&uid=8815857. [DOI] [PubMed] [Google Scholar]
- 17. Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion‐weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011;76(20):1711–1719. 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189–198. 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 19. Mahoney FI, Barthel DW. Functional evaluation: the barther index. Md State Med J 1965;14:61–65. https://www.ncbi.nlm.nih.gov/pubmed/14258950. [PubMed] [Google Scholar]
- 20. Cummings JL. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology 1997;48(5 Suppl 6):S10–S16. 10.1212/wnl.48.5_suppl_6.10s. [DOI] [PubMed] [Google Scholar]
- 21. Tartaglia MC, Johnson DY, Thai JN, et al. Clinical overlap between Jakob‐Creutzfeldt disease and Lewy body disease. Can J Neurol Sci 2012;39(3):304–310. 10.1017/s0317167100013421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zou WQ, Puoti G, Xiao X, et al. Variably protease‐sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68(2):162–172. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20695009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zou WQ, Gambetti P, Xiao X, et al. Prions in variably protease‐sensitive prionopathy: an update. Pathogens 2013;2(3):457–471. 10.3390/pathogens2030457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gambetti P, Cali I, Notari S, et al. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol 2011;121(1):79–90. 10.1007/s00401-010-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashburner J, Friston KJ. Diffeomorphic registration using geodesic shooting and Gauss‐Newton optimisation. NeuroImage 2011;55(3):954–967. 10.1016/j.neuroimage.2010.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jenkinson M, Beckmann CF, Behrens TEJ, et al. Fsl. Neuroimage 2012;62(2):782–790. 10.1016/j.neuroimage.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 27. Winkler AM, Ridgway GR, Webster MA, et al. Permutation inference for the general linear model. NeuroImage 2014;92:381–397. 10.1016/j.neuroimage.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fonov VS, Evans AC, McKinstry RC, et al. Unbiased nonlinear average age‐appropriate brain templates from birth to adulthood. NeuroImage 2009;47:S102. 10.1016/s1053-8119(09)70884-5. [DOI] [Google Scholar]
- 29. Collins SJ, Sanchez‐Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt‐Jakob disease. Brain 2006;129(Pt 9):2278–2287. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16816392. [DOI] [PubMed] [Google Scholar]
- 30. Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012;11(7):618–628. 10.1016/S1474-4422(12)70063-7. [DOI] [PubMed] [Google Scholar]
- 31. Gambetti P, Puoti G, Zou WQ. Variably protease‐sensitive prionopathy: a novel disease of the prion protein. J Mol Neurosci 2011;45(3):422–424. 10.1007/s12031-011-9543-1. [DOI] [PubMed] [Google Scholar]
- 32. Wright S. The method of path coefficients. Ann Math Stat 1934;5(3):161–215. 10.1214/aoms/1177732676. [DOI] [Google Scholar]
- 33. Caverzasi E, Mandelli ML, DeArmond SJ, et al. White matter involvement in sporadic Creutzfeldt‐Jakob disease. Brain 2014;137(Pt 12):3339–3354. 10.1093/brain/awu298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buckner RL, Andrews‐Hanna JR, Schacter DL. The brain’s default network: Anatomy, function, and relevance to disease. Ann N Y Acad Sci 2008;1124:1–38. 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 35. Collins DL, Zijdenbos AP, Kollokian V, et al. Design and construction of a realistic digital brain phantom. IEEE Trans Med Imaging 1998;17(3):463–468. 10.1109/42.712135. [DOI] [PubMed] [Google Scholar]
- 36. DeLong MR, Wichmann T. Basal ganglia circuits as targets for neuromodulation in Parkinson disease. JAMA Neurol 2015;72(11):1354–1360. 10.1001/jamaneurol.2015.2397. [DOI] [PubMed] [Google Scholar]
- 37. Seeley WW, Menon V, Schatzberg AF, et al. Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci 2007;27(9):2349–2356. 10.1523/JNEUROSCI.5587-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caverzasi E, Henry RG, Vitali P, et al. Application of quantitative DTI metrics in sporadic CJD. NeuroImage Clin 2014;4:426–435. 10.1016/j.nicl.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zanusso G, Camporese G, Ferrari S, et al. Long‐term preclinical magnetic resonance imaging alterations in sporadic Creutzfeldt‐Jakob disease. Ann Neurol 2016;80(4):629–632. 10.1002/ana.24757. [DOI] [PubMed] [Google Scholar]
- 40. Iwasaki Y, Mori K, Ito M, et al. An autopsied case of MM1 + MM2‐cortical with thalamic‐type sporadic Creutzfeldt‐Jakob disease presenting with hyperintensities on diffusion‐weighted MRI before clinical onset. Neuropathology 2017;37(1):78–85. 10.1111/neup.12327. [DOI] [PubMed] [Google Scholar]
- 41. Satoh K, Nakaoke R, Nishiura Y, et al. Early detection of sporadic CJD by diffusion‐weighted MRI before the onset of symptoms. J Neurol Neurosurg Psychiatry 2011;82(8):942–943. 10.1136/jnnp.2008.155242. [DOI] [PubMed] [Google Scholar]
- 42. Maeda K, Sugihara Y, Shiraishi T, et al. Cortical hyperintensity on diffusion‐weighted images as the presymptomatic marker of sporadic creutzfeldt‐Jakob disease. Intern Med 2019;58(5):727–729. 10.2169/internalmedicine.1155-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verde F, Ticozzi N, Messina S, et al. MRI abnormalities found 1 year prior to symptom onset in a case of Creutzfeldt‐Jakob disease. J Neurol 2016;263(3):597–599. 10.1007/s00415-016-8022-6. [DOI] [PubMed] [Google Scholar]
- 44. Caine D, Tinelli RJ, Hyare H, et al. The cognitive profile of prion disease: a prospective clinical and imaging study. Ann Clin Transl Neurol 2015;2(5):548–558. 10.1002/acn3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grau‐Rivera O, Calvo A, Bargalló N, et al. Quantitative magnetic resonance abnormalities in creutzfeldt‐jakob disease and fatal insomnia. J Alzheimer’s Dis 2016;55(1):431–443. 10.3233/JAD-160750. [DOI] [PubMed] [Google Scholar]
- 46. Seror I, Lee H, Cohen OS, et al. Putaminal volume and diffusion in early familial Creutzfeldt‐Jakob disease. J Neurol Sci 2010;288(1–2):129–134. 10.1016/j.jns.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ibarretxe‐Bilbao N, Junque C, Marti MJ, Tolosa E. Cerebral basis of visual hallucinations in Parkinson’s disease: Structural and functional MRI studies. J Neurol Sci 2011;310(1–2):79–81. 10.1016/j.jns.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 48. Sanchez‐Castaneda C, Rene R, Ramirez‐Ruiz B, et al. Frontal and associative visual areas related to visual hallucinations in dementia with lewy bodies and Parkinson’s disease with dementia. Mov Disord 2010;25(5):615–622. 10.1002/mds.22873. [DOI] [PubMed] [Google Scholar]
- 49. Rollins CPE, Garrison JR, Simons JS, et al. Meta‐analytic evidence for the plurality of mechanisms in transdiagnostic structural MRI studies of hallucination status. EClinicalMedicine 2019;8:57–71. 10.1016/j.eclinm.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol 2010;67(4):416–421. 10.1001/archneurol.2010.35. [DOI] [PubMed] [Google Scholar]
- 51. Ohtani T, Bouix S, Hosokawa T, et al. Abnormalities in white matter connections between orbitofrontal cortex and anterior cingulate cortex and their associations with negative symptoms in schizophrenia: a DTI study. Schizophr Res 2014;157(1–3):190–197. 10.1016/j.schres.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Silbersweig D. A., Stern E., Frith C., et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature 1995;378(6553):176–179. 10.1038/378176a0. [DOI] [PubMed] [Google Scholar]
- 53. Onofrj M, Espay AJ, Bonanni, L , et al. Hallucinations, somatic‐functional disorders of PD‐DLB as expressions of thalamic dysfunction. Mov Disord. 2019;34(8), 1100–1111. 10.1002/mds.27781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmahmann J. The cerebellar cognitive affective syndrome. Brain 1998;121(4):561–579. 10.1093/brain/121.4.561. [DOI] [PubMed] [Google Scholar]
- 55. Buckner RL, Andrews‐Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci 2008;1124:1–38. 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 56. Li Y, Rinne JO, Mosconi L, et al. Regional analysis of FDG and PIB‐PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2008;35(12):2169–2181. 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shamchi SP, Khosravi M, Taghvaei R, et al. Normal patterns of regional brain18F‐FDG uptake in Normal aging. Hell J Nucl Med 2018;21(3):175–180. 10.1967/s002449910902. [DOI] [PubMed] [Google Scholar]
- 58. Yuan F, Yang L, Zhang Z, et al. Cellular prion protein (PrPC) of the neuron cell transformed to a PK‐resistant protein under oxidative stress, comprising main mitochondrial damage in prion diseases. J Mol Neurosci 2013;51(1):219–224. 10.1007/s12031-013-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Henkel K, Zerr I, Hertel A, et al. Positron emission tomography with [(18)F]FDG in the diagnosis of Creutzfeldt‐Jakob disease (CJD). J Neurol 2002;249(6):699–705. 10.1007/s00415-002-0695-3. [DOI] [PubMed] [Google Scholar]
- 60. Albert R, Jeong H, Barabási AL. Error and attack tolerance of complex networks. Struct Dyn Networks 2011;9781400841:503–506. 10.1515/9781400841356.503. [DOI] [PubMed] [Google Scholar]
- 61. Zhou J, Seeley WW. Network dysfunction in Alzheimer’s disease and frontotemporal dementia: implications for psychiatry. Biol Psychiatry 2014;75(7):565–573. 10.1016/j.biopsych.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 62. Greicius MD, Srivastava G, Reiss AL, Menon V. Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101(13):4637–42.http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15070770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science (80‐) 2012;336(6088):1511–1513. 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jucker M, Walker LC. Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013;501(7465):45–51. 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tousseyn T, Bajsarowicz K, Sanchez H, et al. Prion Disease Induces Alzheimer disease‐like neuropathologic changes. J Neuropathol Exp Neurol 2015;74(9):873–888. 10.1097/NEN.0000000000000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaunmuktane Z, Mead S, Ellis M, et al. Evidence for human transmission of amyloid‐beta pathology and cerebral amyloid angiopathy. Nature 2015;525(7568):247–250. 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- 67. Gardner RC, Boxer AL, Trujillo A, et al. Intrinsic connectivity network disruption in progressive supranuclear palsy. Ann Neurol 2013;73(5):603–616. 10.1002/ana.23844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fredericks CA, Brown JA, Deng J, et al. Intrinsic connectivity networks in posterior cortical atrophy: a role for the pulvinar? NeuroImage Clin 2019;21:101628. 10.1016/j.nicl.2018.101628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Forner SA, Takada LT, Bettcher BM, et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt‐Jakob disease. Neurol Clin Pract 2015;5(2):116–125. 10.1212/CPJ.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sanchez‐Juan P, Sanchez‐Valle R, Green A, et al. Influence of timing on CSF tests value for Creutzfeldt‐Jakob disease diagnosis. J Neurol 2007;254(7):901–906. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17385081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sanchez‐Juan P, Green A, Ladogana A, et al. CSF tests in the differential diagnosis of Creutzfeldt‐Jakob disease. Neurology 2006;67(4):637–643. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16924018. [DOI] [PubMed] [Google Scholar]
- 72. Skillback T, Rosen C, Asztely F, et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt‐Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol 2014;71(4):476–483. 10.1001/jamaneurol.2013.6455. [DOI] [PubMed] [Google Scholar]
- 73. Staffaroni AM, Kramer AO, Casey M, et al. Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic Creutzfeldt‐Jakob disease. JAMA Neurology. 2019;76(8), 969. 10.1001/jamaneurol.2019.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cohen OS, Chapman J, Korczyn AD, et al. CSF tau correlates with CJD disease severity and cognitive decline. Acta Neurol Scand 2016;133(2):119–123. 10.1111/ane.12441. [DOI] [PubMed] [Google Scholar]
- 75. Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt‐Jakob disease. Ann Neurol 1996;39(6):767–778. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8651649. [DOI] [PubMed] [Google Scholar]
- 76. Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt‐Jakob disease. Neurology 2006;66(2):286–287. 10.1212/01.wnl.0000196440.00297.67. [DOI] [PubMed] [Google Scholar]
- 77. Paterson RW, Torres‐Chae CC, Kuo AL, et al. Differential diagnosis of Jakob‐Creutzfeldt disease. Arch Neurol 2012;69(12):1578–1582. 10.1001/2013.jamaneurol.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. The relationship between magnetic resonance imaging findings and clinical variables in sporadic Creutzfeldt‐Jakob disease. Regarding correlations with whole brain volume (A‐C), CSF t‐tau levels correlated negatively with whole brain volume (A), whereas NSE did not correlate with whole brain volume (B), and MMSE correlated positively with whole brain volume (C). For D and E, we compared CSF t‐tau and NSE in cases with both cortical‐ and subcortical‐restricted diffusion on DWI (cortical and subcortical: C‐S) against the combination of cases with cortical‐only involvement and subcortical‐only involvement (combined cortical‐only and subcortical‐only: Co‐S/C) (see text). This was because, our hypothesis was that those with cortical and subcortical diffusion would be more severely affected and more likely to have these biomarkers positive. As we suspected, sCJD participants with a cortical and subcortical (C‐S) pattern of restricted diffusion (gray) showed elevated t‐tau (C) and NSE (D) levels compared to the group of subjects of combined cortical‐only and subcortical‐only (Co‐S/C) restricted diffusion (white box).
Supplementary Material. Biofluid, clinical, and neuroimaging biomarker results including CSF biomarker results and comparison of CSF results and other outcomes or variables among entire sCJD cohort, those with and those without VBM.
Supplementary Table. Pearson correlations between the cortical and subcortical regions included in structural equation modeling (SEM). This table shows that high correlations (≥0.5 strong; moderate 0.3‐0.49; < 0.3 low) are present in sCJD in some of the brain regions included in the SEM model (significant correlations are shown in bold). This methodology, however, cannot discern if there is any effective connectivity and if this effective connectivity has any preferential directionality. Figure 2 in the main text, however, does show the effective connectivity and any directionality using SEM. * Correlation is significant at the 0.05 level (two‐tailed). ** Correlation is significant at the 0.01 level (two‐tailed). "Control Variables: Gender, Age, TIV, and Barthel"