

Abstract
Background
The expansion of GGC repeat in the 5' untranslated region of the NOTCH2NLC has been associated with various neurogenerative disorders of the central nervous system and, more recently, oculopharyngodistal myopathy. This study aimed to report patients with distal weakness with both neuropathic and myopathic features on electrophysiology and pathology who present GGC repeat expansions in the NOTCH2NLC.
Methods
Whole‐exome sequencing (WES) and long‐read sequencing were implemented to identify the candidate genes. In addition, the available clinical data and the pathological changes associated with peripheral nerve and muscle biopsies were reviewed and studied.
Results
We identified and validated GGC repeat expansions of NOTCH2NLC in three unrelated patients who presented with progressive weakness predominantly affecting distal lower limb muscles, following negative results in an initial WES. We found intranuclear inclusions with multiple proteins deposits in the nuclei of both myofibers and Schwann cells. The clinical features of these patients are compatible with the diagnosis of distal motor neuropathy and rimmed vacuolar myopathy.
Interpretation
These phenotypes enrich the class of features associated with NOTCH2NLC‐related repeat expansion disorders (NRED), and provide further evidence that the neurological symptoms of NRED include not only brain, spinal cord, and peripheral nerves damage, but also myopathy, and that overlapping symptoms might exist.
Introduction
The number of disease‐associated short tandem repeat (STR) has been rapidly increasing with technological advances in genetic screening. Recently, GGC repeat expansion in the 5’untranslated region (5’UTR) of the NOTCH2NLC has been identified as the causative mutation of neuronal intranuclear inclusion disease (NIID). 1 , 2 , 3 , 4 NIID is a progressive degenerative disease with intricate clinical symptoms affecting multiple systems and pathological evidence of widespread eosinophilic intranuclear inclusions in a variety of organ tissues. 5 , 6 , 7 , 8 , 9 , 10 , 11 As neuroimaging abnormalities, 12 skin biopsy 13 and genetic analysis 1 , 2 , 3 , 4 became widely available for the diagnosis of NIID in recent years, the number of diagnosed NIID cases increased quickly. To date, GGC repeat expansion of NOTCH2NLC has been reported to not only be responsible for typical NIID, 1 , 2 , 3 , 4 but also associated with a group of NOTCH2NLC‐related repeat expansion disorders (NRED). 14 These disorders include Alzheimer’s disease (AD), 3 frontotemporal dementia, 15 parkinsonism‐related disorders, 3 , 16 , 17 multiple system atrophy (MSA), 18 essential tremor (ET), 19 , 20 adult leukoencephalopathy, 21 amyotrophic lateral sclerosis (ALS), 22 and oculopharyngodistal myopathy (OPDM), 23 , 24
Studies so far showed an indefinite tendency between the length of GGC repeat expansion in the NOTCH2NLC and the variable NRED phenotypes: intermediate‐length from 41 to 130 repeats (median 47 repeats) are potentially associated with Parkinson disease, 16 , 17 length from 44 to 143 repeats (median 75 repeats) are related to ALS, 22 around 100 repeats usually dominantly associated with dementia‐type NIID, and long expansions with more than 200 repeats may be associated with muscle‐type NIID or OPDM. 14 , 23 , 24 This indicates that the spectrum of NRED is highly complicated and may be closely related to the number of expanded GGC repeats, whereby it is worthy to further investigate whether there are new clinical subtypes of NRED and understand the relationship between the GGC repeat numbers and the heterogeneity of NRED.
Hereditary distal motor neuropathy and myopathy is a group of genetically heterogenous disorders. Mutations in a heat‐shock protein beta‐8 (HSPB8) and a heat‐shock protein beta‐1 (HSPB1), which are chaperon proteins involved in chaperone‐assisted selective autophagy and the degradation of misfolded proteins, are responsible for some patients with hereditary distal motor neuropathy and myopathy. 25 , 26 There are some clinical and pathological overlaps between NIID and the hereditary distal motor neuropathy and myopathy, including distal muscle weakness, rimmed vacuole with p62‐positve deposits. 6 , 27 However, the GGC repeat expansion of NOTCH2NLC has not been previously associated with a hereditary distal motor neuropathy and rimmed vacuolar myopathy.
In this study, we report two familial and one sporadic patients exhibiting adult‐onset hereditary distal motor neuropathy and rimmed vacuolar myopathy by clinical and pathological examination. Long‐read sequencing (LRS) identified the GGC repeat expansion in the NOTCH2NLC as a candidate mutation in these patients, following negative results after initial whole‐exome next generation sequencing (WES) screening.
Materials and methods
Study participants
We recruited three unrelated mainland Chinese patients, including two index patients from two autosomal dominant inherited families and one sporadic case from Peking University First Hospital in order to determine the underlying disease‐causing gene(s). Clinical features, laboratory findings, electrophysiological studies, muscle MRI, pathology of muscle, and peripheral nerve were reviewed and studied. We subsequently conducted Mini‐Mental State Examination (MMSE) and/or Montreal Cognitive Assessment (MoCA) evaluations, along with brain MRIs. Genomic DNA of peripheral blood leukocytes was obtained from the three patients and some of their family members. All participants provided their written informed consent. This study was approved by the Local Ethics Committee of Peking University First Hospital.
Histology and electron microscopic study
We performed muscle and peripheral nerve biopsies following routine histological and histochemistry staining. Specifically, we collected samples of the gastrocnemius muscle and the sural nerve from patients F1‐III‐5 and F2‐IV‐5, and from the tibialis anterior muscle and superficial peroneal nerve from sporadic patient 1. Muscle samples were frozen in liquid nitrogen. Sections of all samples (8 μm) were stained by hematoxylin & eosin (H&E), modified Gomori trichrome (mGT), NADH‐tetrazolium reductase (NADH‐TR), and adenosine triphosphatase (ATPase), as previously reported. 19 Nerve samples were fixed in 2‐3% glutaraldehyde and postfixed in 1% osmium tetroxide. Semithin sections for light microscopy were stained with toluidine blue. 28 The samples used for electron microscopy were prepared as previously described. 29
WES by next generation sequencing
WES was performed in these three patients in a commercial company (RunningGene Inc, Beijing, China). For short‐read sequencing, a DNA sequencing library was constructed using an MGIEasy DNA Library Prep Kit following the manufacturer’s instructions (BGI, China) in order to generate DNA nanoballs (DNB). This was implemented by using the exact same methodology as that employed in a previous study. 29
LRS and STR‐Scoring framework
LRS was performed on the three patients on a joint research platform in our lab and Grandomics Biosciences (Beijing, China). 24 , 29 Library preparation was carried out using a 1D Genomic DNA ligation kit (SQKLSK109) according to the manufacturer’s protocol. Each individual used one PRO‐002 (R9.4.1) flowcell. PromethION data base‐calling was performed using guppy v3.3.0 (Oxford Nanopore Technologies). Only pass reads (qscore ≥ 7) were used for subsequent analysis. STR‐Scoring framework was performed and some updates were made based on the previous reported methods. 29 Firstly, a gene‐associated STR list spanning all genes from hg38 RefSeq database (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/refGene.txt.gz) was prepared based on the repeatmasker file from UCSC Genome Browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/rmsk.txt.gz). Genes upstream and downstream of the 10 kb region and the STR repeat unit ranged in length from 3 to 12 bp, so that the number of STR was increased to 228,281. Then, the pass reads from PromethION were aligned to the reference genome hg38 using ngmlr. 30 For each repeat, the repeat count of each read that aligned with the STR locus was detected using repeatHMM 31 without the peak calling step.
Reads with the longer insertion size was defined as the individual’s estimated repeat count (ERC) as in a previous study. 16 To better screen the disease‐associated STR, we performed STR‐Scoring framework as previously reported, 29 which can offset the impact of error bias and population common repeat expansion to prioritize the STR.
According to the above‐mentioned description, we constructed ERC matrixes for the healthy individuals (Hm×n) and affected individuals (Am×s), where m, n, and s denote the number of STRs, and the numbers of healthy, and affected individuals, respectively. For example, Hi,j denotes the jth healthy individual’s ERC at ith STR locus for i = 1···m, j = 1···n.
The scoring procedure was implemented as follows:
Repeat count change: for each repeat, we used cubic mean 32 ci = to represent the STR repeat count in healthy individuals, and used di,j = max(Aij‐ci, 0)/ci to represent the difference between each affected individual and all healthy individuals.
Population proportion: the proportion of healthy individuals with an ERC greater than the one affected individual was defined as the population proportion of the affected individual for every repeat, pi, j = sum(Hi·≥Ai,j)/n; for i = 1··· m, j = 1··· s. In addition, the number of affected individuals whose population proportion was less than 5% was normalized by the total number of affected individuals as a weighted score, qi = sum(pi, j < 0.05)/s for i = 1··· m, j = 1··· s.
Gene function: based on the previous method, 32 for each repeat, we made a slight change, fi = 5 for exon, 2 for UTR, 1.5 for promoter, 1.5 for exon of non‐protein‐coding RNA, 0.5 for intron, 0.2 for upstream, 0.2 for downstream.
STR Score: for each repeat, we calculated the integrative scores as: STR_Score=avg(di,j·(1‐pi,j))qi·fi and the STRs were ranked in order of STR_Score to determine the priority.
RP‐PCR verification
The genomic DNA from the three patients and their available family members were analyzed by repeat‐primed polymerase chain reaction (RP‐PCR) as described in a previous study. 4
Immunofluorescence study
Immunofluorescence staining was conducted on serially frozen muscle sections (8 μm) with an anti‐p62 antibody (Abcam, ab56416), and an anti‐NOTCH2NL antibody (Abcam, ab233287) which was reported to target NOTCH2NLC protein, 17 as previously described. 29 The nuclei were counter‐stained with DAPI. The images were acquired with a confocal microscopy (Nikon A1MP) at 60× magnification.
Data availability
The data supporting the results of this study are available from the corresponding author upon request.
Results
Clinical features
Family 1
The index patient (F1‐III‐5) (Fig. 1) is a 45‐year‐old female who noticed fatigue and pain in her distal lower limbs after walking 300 m at the age of 35. Gradually, she started having increased difficulty in climbing stairs, squatting up, and walking on her tiptoes. She also noticed tremor in both hands, fasciculation in proximal lower limbs. At the age of 45, physical examination revealed she had steppage gait. Her muscle strength (Medical Research Council) were grades 5 in neck flexors, 5 in the proximal upper limbs, 4 in the distal upper limbs, 3‐4 in the hip flexion, and 3‐4 in the extension and flexion of ankle and toes. The weakness was a bit more severe in her right limbs. No ocular, facial, and bulbar muscle weakness was detected. Interosseous muscle atrophy of her left hand was observed. Deep tendon reflexes were extensively diminished. Sensation examination showed mild hyperesthesia in her feet. She had tremors both at resting and position. However, there were no other signs of cerebellar dysfunction. Cognitive assessments showed 30 points in the MMSE and 29 points in the MoCA.
Figure 1.
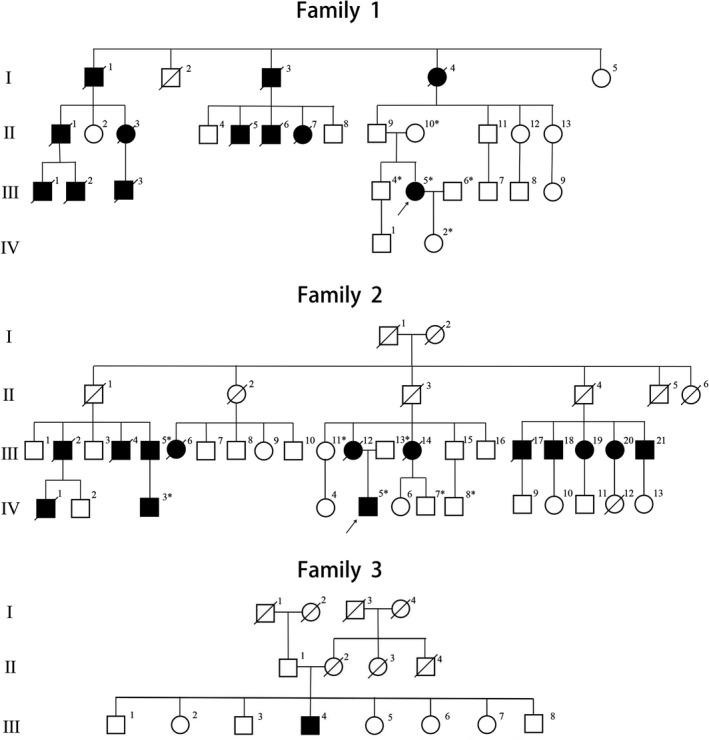
Pedigrees of three unrelated families with the neuromyopathy phenotype. The squares represent males and the circles represent females. Unaffected and affected individuals are marked by white and black symbols, respectively. A diagonal line across a symbol indicates a deceased individual. Asterisks indicate patients with available genomic DNA obtained from peripheral blood leukocytes.
As for laboratory examinations, the serum creatine kinase (CK) level was 256 IU/L (normal range: 22‐269 IU/L). Nerve conduction studies (NCS) revealed a remarkable reduction of motor nerve conduction velocity predominantly affecting the lower limbs, while the sensory nerve conduction velocity was only mildly decreased (Table 1). EMG findings showed neurogenic change and myotonic discharge. Brain MRI showed mildly high signal on the splenium of the corpus callosum on T2‐FLAIR (Fig. 2A‐D). Muscle MRI showed selective fatty infiltration in the posterior muscle group of the lower limbs (Fig. 2L,M).
Table 1.
Electrophysiological findings of three patients.
| Motor nerve | Patient1 (F1‐III‐5) | Patient2 (F2‐IV‐5) | Patient3 (S1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCV (m/s) | dL (ms) | CMAP (mV) | MCV (m/s) | dL (ms) | CMAP (mV) | MCV (m/s) | dL (ms) | CMAP (mV) | ||
| L Median | E‐W | 54 | 3.9 | 56.7 | 9.6 | 42.4 | 13.0 | |||
| W‐APB | 4.1 | 4.1 | 3.1 | 10.0 | 5.3 | 14.2 | ||||
| R Median | E‐W | 50.8 | 5.2 | 50.9 | 10,4 | 43.7 | 6.9 | |||
| W‐APB | 4.1 | 6.2 | 3.5 | 11.6 | 4.8 | 8.3 | ||||
| L Peroneal | FH‐A | 37 | 2.9 | 36.9 | 0.9 | 28.7 | 0.8 | |||
| A‐EBD | 5.0 | 3.2 | 5.9 | 1.3 | 6.6 | 0.8 | ||||
| R Peroneal | FH‐A | 33.5 | 2.2 | 37.4 | 0.9 | ND | ||||
| A‐EBD | 5.4 | 2.0 | 6.3 | 3.4 | ||||||
| L Tibial | PF‐A | 35.4 | 1.6 | 37.3 | 8.2 | ND | ||||
| A‐AA | 3.8 | 3.2 | 3.9 | 10.4 | ||||||
| R Tibial | PF‐A | 38.0 | 2.4 | 37.2 | 4.1 | 33.5 | 4.3 | |||
| A‐AA | 5.1 | 3.2 | 5.1 | 6.9 | 7.3 | 4.7 | ||||
| Sensory nerve# | SCV (m/s) | SNAP (μV) | SCV (m/s) | SNAP (μV) | SCV (m/s) | SNAP (μV) | |
|---|---|---|---|---|---|---|---|
| L‐Median | IIIF‐W | 42.3 | 4.9 | 56.9 | 13.5 | 34.4 | 7.3 |
| R‐Median | IIIF‐W | 40.5 | 5.4 | 57.8 | 12.2 | 37.9 | 8.2 |
| L Ulnar | VF‐W | 40.7 | 5.0 | 52.3 | 12.1 | 35.7 | 8.6 |
| R Ulnar | VF‐W | 38.5 | 5.1 | 49.2 | 10.5 | 36.3 | 4.6 |
| L‐Medial Plantar | Pl‐MM | 44.8 | 6.1 | ND | 33.5 | 6.0 | |
| R‐Medial Plantar | Pl‐MM | 58.5 | 7.6 | 49.1 | 11.0 | 37.8 | 7.0 |
| L‐Sural | A‐MLL | ND | ND | 51.3 | 2.5 | 31.5 | 2.5 |
| R‐Sural | A‐MLL | ND | ND | ND | ND | \ |
For each nerve, data from both sides are shown. MCV, motor nerve conduction velocity; CMAP, compound motor action potential; dL, distal motor latency; SCV, sensory nerve conduction velocity; SNAP, sensory nerve action potential; ND, not done; E, elbow; W, wrist; Ab. E–Bel. E, above elbow–below elbow; APB, abductor pollicis brevis; ADM, abductor digiti minimi; PF, popliteal fossa; A, ankle; MLL, middle lower leg; FH, fibula head; TA, tibialis anterior; EDB, extensor digitorum brevis; AA, abductor allucis; IIIF, third finger; VF, fifth finger; Pl, plantae; MM, malleolus medialis; R, right; L, left; # Near‐nerve recording.
Figure 2.
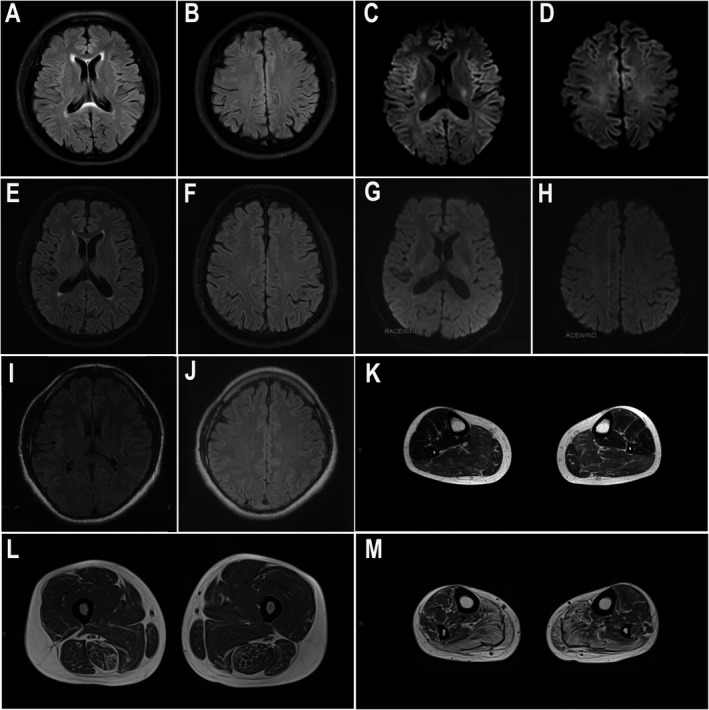
Brain and muscle MRI findings. (A‐J) Brain MRIs of F1‐III‐5 (A‐D), S1 (E‐H), and F2‐IV‐5 (I, J). (A, B, E, F) showed mildly high signal on the splenium of corpus callosum on T2‐FLAIR. (C, D) showed no obvious abnormalities on DWI. (G‐J) showed no obvious abnormalities on DWI (G, H) or T2‐FLAIR (I, J). (K‐M) Muscle MRIs on T2‐FLAIR were evaluated at 8 and 4 years after the onset of symptoms in patients F2‐IV‐5 (K) and F1‐III‐5 (L, M), respectively. Remarkable fatty degeneration and mild amyotrophy were observed. The distal muscles (M, calf level) were more severely affected than the proximal muscles (L, thigh level).
Her family history showed an incomplete dominant inheritance pattern. Specifically, her grandmother (F1‐I‐4) walked with the assistance of crutches in her early 20s. She presented generalized muscle weakness and wasting in her fourth decade. She died at 42 years of age. Two of her grandmother’s siblings (F1‐I‐1 and F1‐I‐3) also developed weakness and pain in distal limb muscles in their early 20s and died in their 30s. There were another eight individuals in her family (F1‐II‐1, F1‐II‐3, F1‐II‐5, F1‐II‐6, F1‐II‐7, F1‐III‐1, F1‐III‐2, F1‐III‐3 and F1‐III‐5) with similar symptoms, including weakness and pain in distal limb muscles. Four of these individuals (F1‐II‐1, F1‐II‐5, F1‐II‐6, and F1‐II‐7) died in their 30s and one individual (F1‐III‐3) died in his 50s. A detailed history and the cause of death among affected Family 1 members were not available.
Family 2
The index patient (F2‐IV‐5) (Fig. 1) is a 31‐year‐old male who noticed hand tremor at the age of 26. At age 28, he developed distal lower limb weakness and had difficulty walking on his tiptoes. Physical examination at the age of 30 revealed steppage gait and mild weakness in bilateral ankle and toe movements (MRC 4). Otherwise, the patient showed normal strength levels in all other limb muscles. Tendon reflexes were reduced in the four limbs. There were no sensory deficits. He presented both resting and postural tremors, without additional cerebellar signatures. No cranial nerve sign was observed. Cognitive assessment revealed 30 points in the MMSE.
The laboratory examinations showed that the CK level was 989 IU/L (normal range: 24‐194 IU/L). NCS revealed a reduction in motor nerve conduction velocity and prolonged distal latency in the lower limbs. Motor nerve conduction studies in the upper limbs and sensory nerve conduction studies in the four limbs were normal. Brain MRI was normal (Fig. 2I, J) and muscle MRI showed selective fatty infiltration in bilateral soleus muscles (Fig. 2K).
His family’s pedigree was also consistent with an incomplete dominance inheritance pattern (Fig. 1). The index patient’s mother (F2‐III‐12) had weakness on her distal lower limbs that was accompanied by static tremor at 34 years old. At the age of 38, she started to develop upper muscle weakness and recurrent upper abdominal distension, vomiting, and constipation. The digestive symptoms worsened and she was diagnosed with psychogenic nervosa at 40 years old. The symptoms were partly alleviated after the administration of chlorpromazine. Her limb muscles weakness and wasting progressed insidiously. She was not able to independently climb stairs at the age of 42. She began to use a wheelchair and could not eat meals by herself at the age of 48. Memory loss and reading difficulty were noticed at the age of 56. She died of pneumonia when she was 57 years old.
As for other family members, the index patient’s maternal aunt (F2‐III‐14) presented with weakness and wasting in her distal limbs and became dependent on a wheelchair at 44 years of age. She died of pneumonia when she was 53 years old. There were another 11 members (F2‐III‐2, F2‐III‐4, F2‐III‐5, F2‐III‐6, F2‐III‐17, F2‐III‐18, F2‐III‐19, F2‐III‐20 F2‐III‐21 F2‐IV‐1, and F2‐IV‐3) with similar muscle weakness symptoms affecting the distal limbs. Unfortunately, the detailed histories of these family members for other patients were not available because of follow‐up loss.
Sporadic patient 1
Sporadic patient 1 is a 56‐year‐old male who had weakness and wasting in bilateral legs at 53 years old. The symptoms developed into an inability to climb stairs or squat up within half a year, and were accompanied by fasciculation in the proximal upper limbs. He was wheelchair‐bound from the age of 54 onwards. At the age of 55, physical examination revealed that muscle strength indices were graded 4 in neck flexors, 5 for upper limbs, 3 for hip flexion, 4 for knees flexion and extension, 2‐3 for ankle dorsiflexion, and 1 for toe extension. The weakness was a bit more severe in his right limbs. Deep tendon reflexes were extensively diminished. Sensory examinations were normal. No cranial nerve sign was noted. Cognitive assessment revealed 28 points in the MMSE. His family history showed no signs of symptomatic individuals (Fig. 1).
The laboratory examinations showed that the serum CK level was 311 IU/L (normal range: 25‐195 IU/L). NCS revealed reduced conduction velocity in both motor and sensory nerves, but the motor nerve and the lower limbs were more severely affected. EMG findings showed neurogenic change. Brain MRI showed mildly high signal on the splenium of the corpus callosum on T2‐FLAIR and no abnormalities on DWI (Fig. 2E‐H).
Pathological changes
Peripheral nerve biopsies revealed that the density of myelinated fibers was mildly to moderately decreased in Patient F1‐III‐5 and sporadic Patient 1 (4500/mm2 and 5000/mm2, respectively), and that the density was relatively normal in Patient F2‐IV‐5 (Fig. 3). In all three patients, we observed a few thinly myelinated fibers but no onion‐bulb, axonal degeneration, or clustering of regenerated fibers was found. EM revealed mild to moderate loss of unmyelinated nerve fibers, many collagen pockets, and denervated Schwann cell units. Ultrastructural examination of peripheral nerve specimens demonstrated typical intranuclear inclusions in the Schwann cells, capillary pericytes, and fibroblasts.
Figure 3.
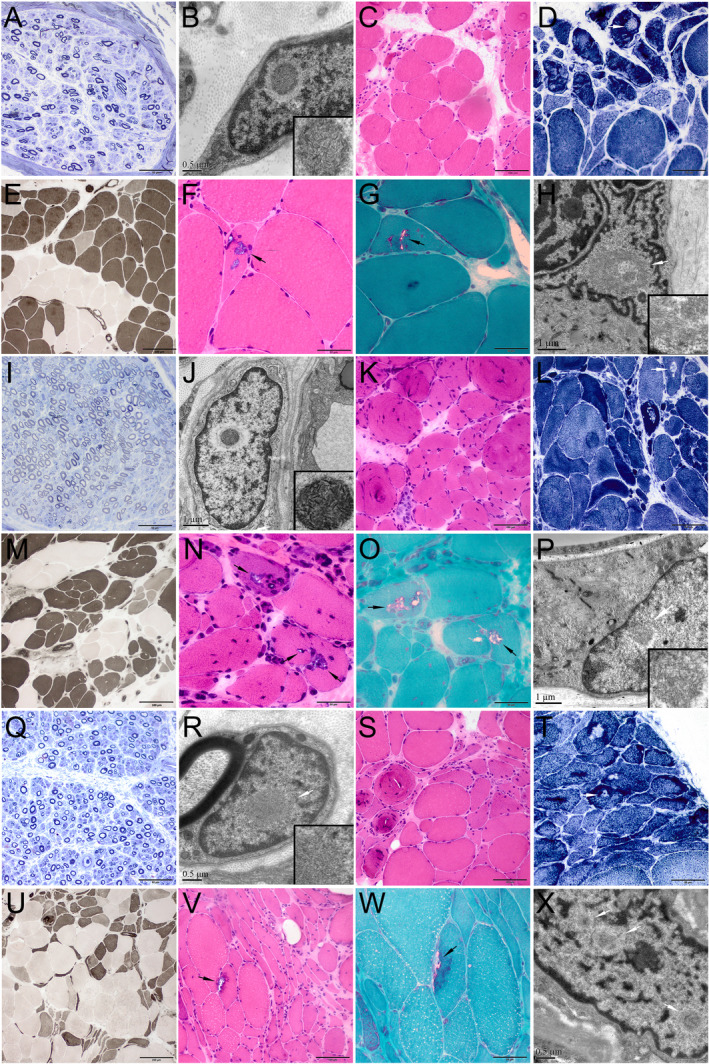
Pathological changes of nerve and muscle biopsies from patients F1‐III‐5 (A‐H), F2‐IV‐5 (I‐P), and S1 (Q‐X). (A, I, Q) Toluidine blue staining of nerve sections showed a reduced density of myelinated nerve fibers. A few thin‐myelinated nerve fibers were present. Ultrastructural examination of peripheral nerve tissues (B, J, R) demonstrated that the intranuclear inclusions containing filamentous aggregates (B, fibroblast; J, capillary pericyte; R, Schwann cell). (C‐G, K‐O, S‐W) Muscle biopsies showed both neurogenic changes (groups of small angular atrophic muscle fibers, type grouping, and target or targetoid fibers) and myopathic changes (rimmed vacuoles fibers, swirling fibers, endomysial fibrosis). (H, P, X) Ultrastructural examination of muscle tissues demonstrated that the intranuclear inclusions containing filamentous aggregates. (C, F, K, N, S, V) H&E staining; (D, L, T) NADH staining; (E, M, U) ATPase staining (pH 10.7, pH 4.4, pH 10.5, respectively); (G, O, W) mGT staining. Scale bars = 50 μm in A, D, F, G, I, N, O, Q, T, W; 100 μm in C, E, K, L, M, S, V; 200 μm in U; 1 μm in H, J, P; 0.5 μm in B, R, X.
Muscle biopsies in the three patients showed both neuropathic and myopathic changes (Fig. 3). The findings of angular fiber clustering, target fibers, and fiber grouping indicated neuropathic changes. At the same time, the presence of chronic myopathic changes was also obvious, including many small round fibers, endomysial fibrosis, and swirling fibers. Moreover, we identified some typical rimmed vacuoles in variable proportions of muscle fibers on H&E and mGT staining in all three patients. Finally, ultrastructural examination revealed typical intranuclear inclusions in the muscle fibers of these patients.
Identification of GGC repeat expansion in NOTCH2NLC
WES was initially conducted in the three index patients, but failed to identify any likely disease‐causing mutations. Subsequently, we performed LRS on a PromethION sequencing machine in order to screen the underlying STR variants. An STR‐Scoring method was used to analyze the sequencing data, and identify nucleotide repeat expansions in the long‐read data based on comparisons between healthy and affected individuals. Among the 228,281 STRs obtained by STR‐Scoring analysis from the long‐read whole genome sequencing data, the GGC repeat expansion in the 5’UTR of NOTCH2NLC had the highest score (4.77) in the top 5 candidate genes (Fig. 4, Supplementary Table 1). The number of GGC repeats in NOTCH2NLC of patient F1‐III‐5, F2‐IV‐5, and S1 was 203, 306, and 68, respectively (Fig. 4, Supplementary Figure 1), which were in the pathogenic range of GGC repeat (>40 repeats). 22 Hence, these results suggested that the GGC repeat expansion in the 5’UTR of NOTCH2NLC is associated with the clinical manifestations observed in these patients.
Figure 4.
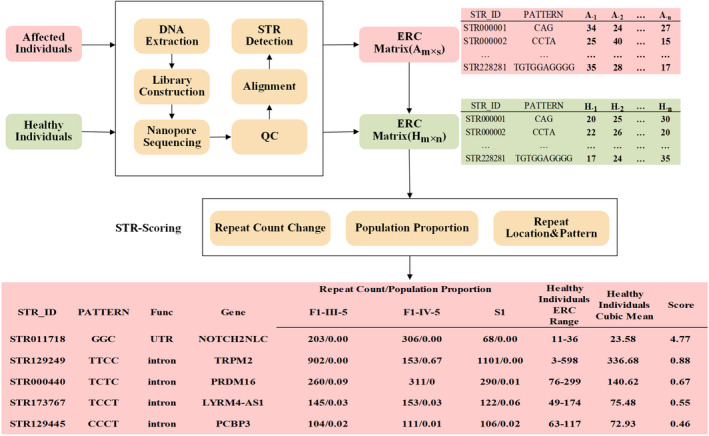
Detection of expanded repeats by LRS and STR‐Scoring method. LRS data for individuals affected with neuromyopathy (red boxes) and healthy individuals (green boxes) was obtained by ONT PromethION sequencing and analyzed using the STR‐Scoring method. A total of 228,281 STRs were screened in genic regions. The Top 5 candidate genes (bottom box) showed that the GGC repeats in the 5’UTR region of the NOTCH2NLC was expanded in three index patients and got a considerable higher score (4.77) than the other candidate genes.
Validation of the GGC repeat expansion in NOTCH2NLC by RP‐PCR
We performed RP‐PCR in order to validate the GGC repeat expansion in the three index patients and some of the family members of patients F1‐III‐5 and F2‐IV‐5. Typical saw‐tooth patterns that were consistent with GGC repeat expansions in the NOTCH2NLC were identified in the three index patients and another two affected individuals in Family 2, but not in the unaffected individuals (Fig. 5).
Figure 5.
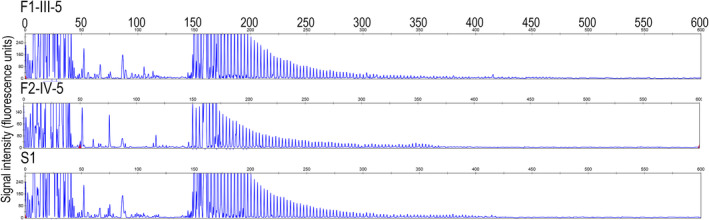
Validation of GGC repeat expansions in the NOTCH2NLC gene among three index patients by RP‐PCR. Three index patients showed a saw‐tooth pattern of the repeat expansion in the NOTCH2NLC.
Verification of intranuclear inclusions by immunofluorescence
To elucidate the pathological changes associated with NOTCH2NLC‐related repeat expansion disorders, immunofluorescent studies were further conducted to observe the p62‐positive intranuclear inclusions. We were able to identify, in all three index patients, p62‐positive protein deposits in the intranuclear inclusions and in the rimmed vacuoles (Fig. 6). Interestingly, the NOTCH2NLC proteins were partially co‐localized with p62 protein deposits, which was consistent with our recent observation in NOTCH2NLC‐related OPDM patients. The GGC repeat expansions located in the coding region of the transcript isoform 2 of NOTCH2NLC might result in the translation of a toxic NOTCH2NLC‐polyGlycine protein, which is co‐localized with p62. 24 It suggested that toxic protein gain‐of‐function mechanism could be involved in the pathogenesis of NRED.
Figure 6.

Immunofluorescence staining reveals the distribution of intranuclear inclusions in both muscle fibers and nerve cells. (A) NOTCH2NLC protein partially co‐localized with p62‐positive intranuclear inclusions (marked with arrows and shown at a higher magnification) and p62‐positive inclusions in the rimmed vacuole (marked with arrow heads and shown at a higher magnification) in muscle fibers. (B) NOTCH2NLC protein partially co‐localized with p62‐positive intranuclear inclusions in nerve cells (marked with arrows and shown at a higher magnification). Nuclei were counterstained with DAPI. Scale bars = 25 μm.
Discussion
The prominent manifestations in these patients were a gradually progressive distal muscle weakness and wasting sparing of obvious sensory disturbance. The electrophysiological changes and peripheral nerve biopsy suggested that the distal weakness might be associated with length‐dependent motor neuropathy, with predominant demyelinating changes. Additionally, the muscle MRI and myopathological features indicated that primary muscle involvement, including the presence of rimmed vacuoles might be also responsible for the distal muscle weakness. We implemented a comprehensive gene screening and identified GGC repeat expansions in NOTCH2NLC in these three index patients. This suggested that these patients were presented with a new phenotype of distal hereditary demyelinating motor neuropathy and rimmed vacuolar myopathy associated with the GGC repeat expansions of the NOTCH2NLC. It is not unexpected that both peripheral nerve and muscle tissue were simultaneously involved in the pathological process of a NOTCH2NLC‐related repeat expansion disorder, because the clinical spectrum included length‐dependent motor–sensory neuropathy and distal myopathy, and the eosinophilic intranuclear inclusions can exist in both nerve and muscle tissues. 6 , 11
The clinical manifestations in these patients were initially and predominantly characterized as muscle weakness in the distal lower limbs. Subsequently, mild involvements of proximal limbs were observed in some patients with a disease duration between 10 and 20 years. Although some subclinical abnormalities of the sensory nerves were identified in the NCS examinations, the dominant symptoms in these patients resembled a type of distal hereditary motor neuropathy with genetic uncertainness. Similarly, previous studies have described a subtype of muscle weakness‐dominant NIID ascribed to neurogenic weakness. 6 , 33 However, the elevated CK level in patients 2 and 3 initially provided us with an indication of possible muscle involvement, which was further supported by the selective fatty infiltration indicating myopathic involvement. Finally, the pathological changes observed in the muscle showed a chronic myopathic features. The clinical spectrum of NRED has been known to affect and cause widespread multiple system symptoms across the body. In particular, the neurological symptoms of NRED include the brain and spinal cord, peripheral nerves, and myopathy. However, a phenotype of predominant distal weakness ascribed to both length‐dependent neuropathy and distal myopathy has not yet been described or highlighted. Therefore, we propose that the patients with typical muscle weakness‐dominant NIID should be retrospectively investigated to assess whether their symptoms have a myogenic origin.
Recently Ogasawara et al 23 reported NOTCH2NLC‐related repeat expansions were also associated with patients suffering from OPDM, after CGG repeat expansions in LRP12 and GGC repeat expansions in GIPC1 were identified as causative genes of OPDM. 1 , 29 , 34 OPDM is another condition presenting with prominent distal muscle wasting, but also with dysphagia/dysarthria and ptosis/ophthalmoplegia. Some of OPDM patients have even been reported to be complicated with peripheral neuropathy. 23 , 29 However, in the present study, the patients did not exhibit oculo‐pharyngo‐facial weakness, which is not compatible with OPDM. The NOTCH2NLC‐related OPDM patients by Ogasawara et al not only showed primary myopathy, but also manifestations in various extra‐skeletal muscle organs, including the central nervous systems. 23 Similarly, two out of the three index patients investigated in this study also presented with tremor and mild signal abnormalities on the splenium of the corpus callosum. However, typical central nervous system symptoms, such as episodic encephalopathy, dementia, cerebellar ataxia, or autonomic dysfunction were not observed in our study. Nevertheless, symptoms affecting the extra‐muscle might be useful the diagnostic indicators for distal neuromyopathy associated with NOTCH2NLC repeat expansions. In particular, some family members showed typical symptoms of NIID similar to those observed in F2‐III‐12, which indicates that the clinical heterogeneity of NIID also exists in certain families. It should be note that the positive family history was very useful for this diagnosis.
The characteristic pathological changes of NRED are widespread eosinophilic intranuclear inclusions that distribute across all organs of the body. 11 In this study, the affected nerves and muscle presented with numerous intranuclear inclusions which settled a reasonable pathological correlation with the phenotype of both length‐dependent neuropathy and distal myopathy. In similar scenarios, some chaperonopathies also presented with a phenotype of distal myopathy and motor neuropathy associated with HSPB1 25 or HSPB8 26 which are simultaneously expressed in both nerves and muscles, and thus share with a common pathological basis. Additionally, we found that rimmed vacuoles as an invariable pathological phenomenon of distal myopathy, not only appeared in the distal neuromyopathy associated with NOTCH2NLC repeat expansion, but were also the characteristic of the distal neuromyopathy associated with HSPB1 or HSPB8. Therefore, muscle pathological studies might play an important role in delimiting the phenotypical borderlines associated with NOTCH2NLC repeat expansion.
The genetic anticipation was not clearly seen in the present study, which is consistent with a previous study. 3 Considering the number of patients in this study was very small, more NRED‐affected families are needed to explore the genetic pattern, onset of age and severity effect over multiple generations. However, some correlations have been previously observed between the length of the GGC repeat and the phenotype. For example, the average number of GGC repeat was different between Parkinson disease (< 80 repeats in 91.7% patients), dementia‐type NIID (around 100 repeats), and muscle‐type NIID (around 200 repeats). 14 In this study, the two familial patients, F1‐III‐5 and F2‐IV‐5, had a GGC repeat range of nearly 200, which is consistent with the muscle‐weakness phenotype observed. 14 Interestingly, the number of GGC repeats in the sporadic patient was less than 100, indicating other factors including genetic modifiers might have some effects on the phenotype. More patients are needed to explore the correlation between genotype and phenotype in this disease.
Conclusion
In summary, the GGC repeat expansion in the NOTCH2NLC is associated with a new type of hereditary distal neuromyopathy. The widespread eosinophilic intranuclear inclusions in both nerves and muscles results from a common pathological basis. It is important to emphasize that myopathy is involved the pathogenesis of the distal muscle weakness besides peripheral neuropathy, because this will enable more therapeutic targets, and provide more precise evaluations for the prognosis of this disease. Additionally, we suggest screening of GGC repeat expansions in the NOTCH2NLC in those patients presenting with distal motor neuropathy and rimmed vacuoles with yet undetermined genetic causes.
Authors’ Contributions
JWD, DJH, and ZXW conceived the idea, designed studies, and supervised the project; JXY and XHL recruited designed and carried out experiments, analyzed data; MY, WZ, HL, LC, LCM, MZ, BBZ, XRW, QG, JL, XS, WL, SY, ZRJ, YY, DJH, and ZXW contributed to the clinical diagnosis and biopsy of hereditary distal neuromyopathy individuals; PDL carried out the bioinformatic analysis on LRS data. JXY, JWD, DJH, and ZXW wrote and edited the manuscript. All authors were involved in revising the article critically for intellectual content. All authors read and approved the final manuscript.
Conflict of Interest
The authors claim no conflict of interest.
Supporting information
Supplementary Figure. Consensus sequences of the GGC repeat expansions of NOTCH2NLC in three index patients.
Supplementary Table. Top 30 candidate genes in three index patients by LRS and STR‐Scoring method.
Acknowledgments
We are indebted to the cooperation of the individuals and their families. Professor Ichizo Nishino (National Center of Neurology and Psychiatry, Japan) for helpful comments. We thank Mr. Lijun Chai (Peking University First Hospital) for his work in taking electron microscopy pictures, Ms. Yuehuan Zuo and Ms. Qiurong Zhang (Peking University First Hospital) for their work in preparations for pathological sections.
Funding Information
Double thousand talents program of Jiangxi province; National Natural Science Foundation of China : 81571219 : 81801243 : 82071409 : U20A20356; Peking University Medicine Fund of Fostering Young Scholars’Scientific & Technological Innovation.
Funding Statement
This work was funded by Double thousand talents program of Jiangxi province grant ; National Natural Science Foundation of China grants 81571219, 81801243, 82071409, and U20A20356; Peking University Medicine Fund of Fostering Young Scholars’Scientific & Technological Innovation grant .
Contributor Information
Jianwen Deng, Email: raywen1@126.com.
Daojun Hong, Email: hongdaojun@hotmail.com.
Zhaoxia Wang, Email: drwangzx@163.com.
References
- 1. Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 2019;51(8):1222–1232. [DOI] [PubMed] [Google Scholar]
- 2. Sone J, Mitsuhashi S, Fujita A, et al. Long‐read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019;51(8):1215–1221. [DOI] [PubMed] [Google Scholar]
- 3. Tian Y, Wang JL, Huang W, et al. Expansion of human‐specific GGC repeat in neuronal intranuclear inclusion disease‐related disorders. Am J Hum Genet 2019;105(1):166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deng J, Gu M, Miao Y, et al. Long‐read sequencing identified repeat expansions in the 5'UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet 2019;56(11):758–764. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi‐Fujigasaki J. Neuronal intranuclear hyaline inclusion disease. Neuropathology 2003;23(4):351–359. [DOI] [PubMed] [Google Scholar]
- 6. Sone J, Mori K, Inagaki T, et al. Clinicopathological features of adult‐onset neuronal intranuclear inclusion disease. Brain 2016;139(Pt 12):3170–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen H, Lu L, Wang B, et al. Re‐defining the clinicopathological spectrum of neuronal intranuclear inclusion disease. Ann Clin Transl Neurol 2020;7(10):1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindenberg R, Rubinstein LJ, Herman MM, Haydon GB. A light and electron microscopy study of an unusual widespread nuclear inclusion body disease. A possible residuum of an old herpesvirus infection. Acta Neuropathol 1968;10(1):54–73. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Wang B, Wang L, et al. Diagnostic indicators for adult‐onset neuronal intranuclear inclusion disease. Clin Neuropathol 2020;39(1):7–18. [DOI] [PubMed] [Google Scholar]
- 10. Liang H, Wang B, Li Q, et al. Clinical and pathological features in adult‐onset NIID patients with cortical enhancement. J Neurol 2020;267(11):3187–3198. [DOI] [PubMed] [Google Scholar]
- 11. Lu X, Hong D. Neuronal intranuclear inclusion disease: recognition and update. J Neural Transm (Vienna) 2021;128(3):295–303. [DOI] [PubMed] [Google Scholar]
- 12. Sone J, Kitagawa N, Sugawara E, et al. Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy. J Neurol Neurosurg Psychiatry 2014;85(3):354–356. [DOI] [PubMed] [Google Scholar]
- 13. Sone J, Tanaka F, Koike H, et al. Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease. Neurology 2011;76(16):1372–1376. [DOI] [PubMed] [Google Scholar]
- 14. Westenberger A, Klein C. Essential phenotypes of NOTCH2NLC‐related repeat expansion disorder. Brain 2020;143(1):5–8. [DOI] [PubMed] [Google Scholar]
- 15. Jiao B, Zhou L, Zhou Y, et al. Identification of expanded repeats in NOTCH2NLC in neurodegenerative dementias. Neurobiol Aging 2020;89(142):142.e1–142.e7. [DOI] [PubMed] [Google Scholar]
- 16. Ma D, Tan YJ, Ng ASL, et al. Association of NOTCH2NLC repeat expansions with Parkinson disease. JAMA Neurol 2020;77(12):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi CH, Fan Y, Yang J, et al. NOTCH2NLC intermediate‐length repeat expansions are associated with Parkinson disease. Ann Neurol 2021;89(1):182–187. [DOI] [PubMed] [Google Scholar]
- 18. Fang P, Yu Y, Yao S, et al. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann Clin Transl Neurol 2020;7(4):517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun QY, Xu Q, Tian Y, et al. Expansion of GGC repeat in the human‐specific NOTCH2NLC gene is associated with essential tremor. Brain 2020;143(1):222–233. [DOI] [PubMed] [Google Scholar]
- 20. Ng ASL, Lim WK, Xu Z, et al. NOTCH2NLC ggc repeat expansions are associated with sporadic essential tremor: variable disease expressivity on long‐term follow‐up. Ann Neurol 2020;88(3):614–618. [DOI] [PubMed] [Google Scholar]
- 21. Okubo M, Doi H, Fukai R, et al. GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann Neurol 2019;86(6):962‐968. [DOI] [PubMed] [Google Scholar]
- 22. Yuan Y, Liu Z, Hou X, et al. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology 2020;95(24):e3394–e3405. [DOI] [PubMed] [Google Scholar]
- 23. Ogasawara M, Iida A, Kumutpongpanich T, et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol Commun 2020;8(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu J, Deng J, Guo X, et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lewis‐Smith DJ, Duff J, Pyle A, et al. Novel HSPB1 mutation causes both motor neuronopathy and distal myopathy. Neurol Genet 2016;2(6):e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghaoui R, Palmio J, Brewer J, et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2016;86(4):391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kimber TE, Blumbergs PC, Rice JP, et al. Familial neuronal intranuclear inclusion disease with ubiquitin positive inclusions. J Neurol Sci 1998;160(1):33–40. [DOI] [PubMed] [Google Scholar]
- 28. Li J, Meng L, Wu R, et al. Sural nerve pathology in TFG‐associated motor neuron disease with sensory neuropathy. Neuropathology 2019;39(3):194–199. [DOI] [PubMed] [Google Scholar]
- 29. Deng J, Yu J, Li P, et al. Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am J Hum Genet 2020;106(6):793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single‐molecule sequencing. Nat Methods 2018;15(6):461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Q, Zhang P, Wang D, et al. Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long‐read sequencing. Genome Med 2017;9(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mitsuhashi S, Frith MC, Mizuguchi T, et al. Tandem‐genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol 2019;20(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sone J, Hishikawa N, Koike H, et al. Neuronal intranuclear hyaline inclusion disease showing motor‐sensory and autonomic neuropathy. Neurology 2005;65(10):1538–1543. [DOI] [PubMed] [Google Scholar]
- 34. Xi J, Wang X, Yue D, et al. 5' UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain 2021;144(2):601–614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. Consensus sequences of the GGC repeat expansions of NOTCH2NLC in three index patients.
Supplementary Table. Top 30 candidate genes in three index patients by LRS and STR‐Scoring method.
Data Availability Statement
The data supporting the results of this study are available from the corresponding author upon request.