

Abstract
Type I/III interferons (IFN) induce expression of hundreds of interferon stimulated genes (ISGs) through the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway to combat viral infections. Although JAK/STAT signaling is seemingly straightforward, it is nevertheless subjected to complex cellular regulation. Herein we show that an ubiquitination regulatory X (UBX) domain containing protein, UBXN6, positively regulates JAK-STAT1/2 signaling. Overexpression of UBXN6 enhanced type I/III IFNs-induced ISGs expression, while deletion of UBXN6 inhibited their expression. RNA viral replication was increased in human UBXN6-deficient cell lines, accompanied by a reduction in both type I/III IFN expression, when compared to UBXN6-sufficient cell lines. Mechanistically, UBXN6 interacted with tyrosine kinase 2 (TYK2) and inhibited IFN-β-induced degradation of both TYK2 and type I IFN receptor. These results suggest that UBXN6 maintains normal JAK-STAT1/2 signaling by stabilizing key signaling components during viral infection.
Keywords: Interferon, IFN, UBXN6, JAK-STAT, ubiquitination regulatory X, UBX
Introduction
The pattern recognition receptors (PRRs) sense invading pathogens and initiate innate immune responses. Retinoic acid inducible gene 1 (RIG-I) like receptors (RLRs) are primary viral RNA sensors that activate type I/III interferons (IFN) through a mitochondrial adaptor, mitochondrial antiviral signaling protein (MAVS) (1). On the other hand, the double stranded DNA sensor, cyclic GMP-AMP synthase (cGAS), induces stimulator-of-interferon-genes (STING)-dependent immune responses (2). The type I/III IFNs are crucial antiviral defense cytokines that induce rapid expression of several hundreds of effector molecules, termed interferon-stimulated genes (ISGs). These effectors directly interfere with viral life cycle (3), enhance endothelial/epithelial integrity (4), activate immune cells and/or shape adaptive immunity (5). The type I/III IFNs bind their respective heterodimeric receptors on the cell surface, leading to a conformational change. The receptor complex then recruits and activates Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), which undergo trans-autophosphorylation and phosphorylate specific tyrosine of the interferon receptors. This action creates a docking site for signal transducers and activators of transcription (STAT1/2), which are phosphorylated by JAK1/TYK2. The phosphorylated STAT1/2 dimerizes, forms a complex with interferon regulatory factor 9 (IRF9), translocates to nucleus, binds to interferon-stimulated response DNA element (ISRE), and initiate transcription of antiviral effectors (ISGs) (6). The JAK-STAT pathways are tightly regulated primarily by phosphorylation / dephosphorylation of receptors, kinases and STATs (6). K48-linked ubiquitination down-regulates JAK-STAT signaling by targeting STAT and IFN receptors to proteasome-mediated degradation (7). The suppressors of cytokine signaling (SOCS), classical ISGs, are direct inhibitors of JAK (8, 9). STAT-DNA binding can be inhibited by and/or modified by a family of small ubiquitin-like modifier (SUMO) E3 ligases, known as protein inhibitor of activated STATs (PIAS) (10) (11, 12). In addition, JAK-STAT signaling may be regulated by protein methyl transferases. Upon interleukin 6 (IL-6) stimulation, STAT3 is methylated on K140 by SET9 and de-methylated by LSD1 (13). The methylation of STAT1 on R31 by protein arginine methyl-transferase PRMT1 could be important for the function of tyrosine-phosphorylated STAT1 dimers (14), but this work has been controversial (15) (16).
The ubiquitin regulatory X (UBX) domain-containing protein family is comprised of 13 members. Although there is weak homology at the protein level, the UBX domain of all the members assumes an ubiquitin-like 3-dimentional structure. They are likely involved in many cellular processes, including endoplasmic reticulum (ER)-associated protein degradation (ERAD), lipid metabolism, vesicle fusion, cancer and cell cycle probably as adaptors bridging an ATPase p97 (also known as VCP/CDC48) and E3 ligases (17, 18) (19, 20). Some UBXNs have been recently shown to regulate immune signaling pathways including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (21, 22), Fas cell surface death receptor (Fas) (23, 24), interferon regulatory factor 3 (IRF3) (25), RIG-I-MAVS (26–28) (29) and STING (30). In the current study, we identify UBXN6 as a novel positive regulator of JAK1-STAT1/2 signaling. UBXN6-deficient cells are defective in ISG expression stimulated by type I/III IFNs, and are more prone to viral infection, when compared to UBXN6-sufficient cells. Mechanistically, UBXN6 interacted with tyrosine kinase 2 (TYK2) and inhibited degradation of both TYK2 and type I IFN receptor (IFNAR1) triggered by IFN-β.
Materials and Methods
Major reagents
The rabbit Actin (Cat# 8456), mouse anti-tubulin (Cat# 3873), rabbit anti- STAT1 (Cat#9172), phospho-STAT1 (Cat # 9167), rabbit anti-phospho-JAK1 (Cat # 74129S), rabbit anti-JAK1 (Cat # 3344S), rabbit anti-phosphor-TYK2 (Cast # 68790S), rabbit anti-TYK2 (Cat # 9312S), and rabbit anti-RIG-I (Cat #3743S) were purchased from Cell Signaling Technology (Danvers, MA 02241, United States). The rabbit anti-IFNAR1 antibody (Cat #A304–289A) used for western blots was purchased from Bethyl Laboratories, Inc (Montgomery, TX 77365, United States). M2 (anti-FLAG) magnetic beads (Cat# A2220), mouse anti-FLAG antibody (Cat# F3165), rabbit anti-GAPDH (Cat # G9545) and 3× FLAG peptide (Cat# F4799) were available at Sigma-Aldrich (St. Louis, MO 63103, United States). Lipofectamine 2000 (Cat# 11668019) was obtained from Thermo Fisher Scientific (Waltham, MA, United States). The heavy molecule weight polyinosinic-polycytidylic acid (poly I:C-H) was purchased from InvivoGen (San Diego, CA 92121, United States). The Dual-Luciferase Reporter Assay (Cat# E1910) was available from Promega (Madison, WI 53711, United States). The recombinant human IFN-β (Cat#3 00–02BC), human IFN-λ1 (Cat # 300–02L) and rabbit anti-UBXN6 (Cat # 14706-1-AP) were from Proteintech (Rosemont, IL 60018, United States). The IFN-λ2 (Cat # RHF333CK) and IFN-β (Cat# RHF842CK ELISA kit were available from Antigenix America, INC (Huntington Station, NY 11746, United States). The PCR array kit (Cat# PAHS-016Z) was from QIAGEN (Germantown, MD 20874, United States). The siRNA against human UBXN6 (Cat# SR312922), FLAG-Myc-UBXN2A (Cat# MR203340), −2B (Cat# MR204884) were purchased from Origene Technologies (Rockville, MD 20850, USA). The rest of FLAG-UBXNs (UBXN1, UBXN3A, UBXN3B, UBXN4, UBXN6, UBXN7, UBXN8, UBXN9, UBXN10, UBXN11 and P47) were reported previously (18) (31).
Cell lines and viruses
Human embryonic kidney (HEK) 293 T (Cat# CRL-3216), Vero cells (monkey kidney epithelial cells, Cat# CCL-81), placental trophoblast (Cat# CRL-3271), THP-1 monocytic cell line (Cat# TIB202), and the Zika virus FLR strain (Cat# VR-1844) were from American Type Tissue Culture (ATCC) (Manassas, VA 20110, United States). Green fluorescence protein (GFP) tagged vesicular stomatitis virus (VSV) was derived from the Indiana strain of VSV by Dr. Rose at Yale University (31, 32).These cell lines are not listed in the database of commonly misidentified cell lines maintained by ICLAC and have not been authenticated in our hands. They are routinely treated with MycoZAP (Lonza) and tested for mycoplasma contamination in our hands.
Cell culture
HEK293T, Vero cells and 2fGTH cells were grown in Dulbecco’s Modified Eagle Medium (DMEM); trophoblast and THP-1 were grown in Roswell Park Memorial Institute (RPMI)1640 supplemented with 10% FBS, glutamine, antibiotics (ThermoFisher Scientific/Gibco), and Mycozap (Lonza Group, Basel, Switzerland) at 37°C, 5% CO2.
For viral infection, viruses were allowed to infect cells for two hours in the serum-free medium; the cells were then washed with 1× phosphate-buffered saline (PBS) once and incubated with pre-warmed complete medium. The multiplicity of infection (MOI) and infection conditions were specified for each experiment in the figure legend. When appropriate, total integrated density of GFP fluorescence signal was quantified using ImageJ. In brief, total fluorescence signal was strengthened using the ‘Measure’ function to include all of the GFP+ cells while minimizing signal from the background. Five fields of view were taken from each biological replicate (n=4) and averaged for a mean integrated density measurement.
Transfection
Transfection of plasmid DNA/siRNA/poly(I:C) into cells with Lipofectamine 2000 was performed exactly in the product manual. One day before transfection, cells were seeded in a culture plate at ~40–50% confluence. For preparation of transfection mix, briefly, appropriate amount of DNA/siRNA and Lipofectamine 2000 was suspended in serum-free DMEM separately, incubated for 5 minutes, and then combined, incubated for 20 minutes at room temperature. The complete culture medium was replaced with pre-warmed serum free medium, and the transfection mix was added to cells (~60–80% confluence) in a dropwise manner. After 6–10 hours, the culture medium was changed to complete medium.
Calcium phosphate transfection was sometimes employed for delivery of plasmid DNA into HEK293T cells. Briefly, for one transfection (24-well plate), 10–200ng of plasmid DNA was diluted in 50μL of 0.25M CaCl2 and incubated at room temperature for 15 minutes. Then 50μL 2× HBSS (Hank’s Balanced Salt Solution) was added dropwise with agitation. The mix was incubated at room temperature for 20 minutes and then added to ~50–70% confluent cell culture dropwise.
Transfection protocol for UBXN6-specific siRNA into THP-1 cells was completed as described above, with minor modifications to improve knockdown efficiency. First, THP-1 cells were seeded at a 60% confluency and differentiated into macrophages in the presence of 10 ng ml−1 phorbol 12-myristate 13-acetate (PMA) (Fisher Scientific, Hampton, NH, Cat # BP685–1) for 3 days. After 3 days, the medium containing PMA was removed and replaced with complete RPMI. Cells were then transfected with either UBXN6-specific siRNA or scrambled control siRNA and incubated for 48-hours before experimentation.
Immunoblotting
Whole-cell extracts were separated by 4–20% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a nitrocellulose membrane, which was followed by blocking with 5% non-fat milk in Tris-buffered saline (TBST) with Tween 20 [(Tris: 20 mM, NaCl: 150 mM, Tween 20 detergent: 0.1% (w/v)) for one-hour. Membranes were probed with primary antibodies. Each antibody was diluted in 5% non-fat milk in TBST at the manufacturers’ suggested concentration. After washing, the membrane was probed with a horseradish peroxidase (HRP)-conjugated secondary antibody and developed using an enhanced chemiluminescent (ECL) substrate (ThermoFisher, Cat# 32106).
Co-immunoprecipitation (IP)
HEK293T cells were seeded at a ~50% confluency in a 6-well plate. The next day, cells were transfected with plasmid DNA using Lipofectamine 2000. Whole-cell extracts were prepared from transfected cells in IP lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 1 mM EDTA, 0.5% NP40, 10% glycerol), centrifuged at 7000 × g for 15-minutes to remove cell debris and nuclei, and the resulting supernatant was incubated with 50μl of anti-FLAG magnetic beads overnight at 4°C. Co-immunoprecipitation was performed according to the manufacturers’ instructions [M2 (anti-FLAG) magnetic beads (Cat# A2220, Sigma Aldrich)].
Luciferase reporter assay
50–70% confluent cells were transfected with 3 plasmids including 100 ng of pGL3-ISRE luciferase reporter (firefly luciferase; experimental reporter) plasmid, 10ng of Actin-renilla reporter (renilla luciferase; internal control), and 20–100 ng of individual test gene expressing plasmid. Twenty-four hours after transfection, the cells were treated with 10ng mL−1 of recombinant hIFN-β or hIFN-λ. Sixteen hours after treatment, luciferase activity was measured using a Promega Dual-Glow Luciferase assay system. Briefly, cells were washed with 500μL ice-cold 1X PBS and lysed in 50μL of 1× passive lysis buffer. Five microliters of cell lysate was added to 30μL Dual-Glo Luciferase Reagent, and the firefly luciferase activity was measured immediately on a GloMax 20/20 Luminometer (Promega). Immediately after the first reading, 30μL STOP-Glo Luciferase Reagent was added to the sample and mixed thoroughly, and the Renilla luciferase activity was measured. The result was calculated as a ratio of firefly luciferase/Renilla, and expressed as fold change over control/unstimulated. For the reporter assay involving RNA Interference, 1× 105 HEK293T cells were plated into polyornithine-coated 24-well cell culture plates. Sixteen hours later, cells were transfected with 40pmol of siRNA mix directed against human UBXN6 (three 27-mer siRNAs from Origene) or scramble control, by Lipofectamine 2000. Cells were transfected again with luciferase reporter plasmids (100ng ISRE-Luc and 10ng Act-Renilla per well) together with a RLR pathway gene expressing plasmid 48hrs after siRNA transfection. Luciferase was then quantitated 16-hours after the second transfection.
PCR array
Total RNA was reverse transcribed using a cDNA kit. The cDNA was used for the real-time RT Profiler PCR Array (QIAGEN, Cat. No. PAHS-016Z) in combination with SYBR Green qPCR Mastermix (Cat. No. 330529). The PCR results were analyzed through a web portal at http://www.qiagen.com/geneglobe. Samples were assigned to controls and test groups. The threshold cycle (Ct) values were normalized based on a panel of reference genes (HKG). Briefly, delta Ci is calculated between the gene of interest (GOI) and an average of reference genes (HKG), followed by delta-delta Ct calculations [delta Ct (UBXN6 knockout)-delta Ct (WT)]. Fold change was then calculated using 2^ (-delta delta Ct) formula.
Generation of gene knockout by CRISPR-Cas9
The guide RNA was UBXN6 KO-1: 5’-TCGGATG GTGTCCTGCGATG-3’ and KO-2: 5’-GCTGGTT GGGCTTCTCTTTG-3’. The guide RNA was cloned into Lenti-CRISPR V2 plasmid separately (33), and validated by sequencing using the LKO.1 primer 5’-GAC TATCATATGCTTACCGT-3’. The gRNA construct was transfected into HEK293T cells with the packaging plasmids pCMV-VSV-G (Addgene # 8545) and psPAX2 (Addgene # 12260) in a molar ratio 1:2:2. The lentiviral particles in the cell culture medium were collected between 48–96-hours after transfection. For transduction, lentiviral culture and a final concentration of 8μg mL−1 of polybrene was added to target cells (HEK293T, trophoblasts) at a 30–50% confluence. 48-hours later, the transduction medium was replaced with fresh growth medium with 1μg mL−1 of puromycin. The wild type (WT) control was lentiCRISPRv2 vector only. A well of cells that were not treated with lentiviruses served as a negative control for antibiotic selection. Antibiotic selection was considered complete, usually within 3–5 days, when all non-transduced control cells died. The antibiotic-resistant cells were subjected to immunoblotting to confirm gene deletion.
Type I/III IFN bioavailability assay
Type I/III IFN concentrations in the cell culture supernatant was measured on a 2fGTH reporter cell line. This cell line stably expresses an ISRE-luciferase reporter, which is activated by Type I/III IFN (31). Briefly, 1 × 105 2fGTH cells were seeded in each well of a 24-well plate and grown in complete DMEM medium at 37°C, 5% CO2 overnight. Two-hundred microlitters of samples (dilution may be necessary if the IFN concentration is too high) were added to the 2fGTH-ISRE-Luc cells and incubated at 37°C, 5% CO2 for 10-hours. A serial two-fold dilution of recombinant human IFN-β served as a positive control and was used for plotting a standard curve. Unstimulated HEK293T cell culture medium served as a negative control. After the 10-hour treatment, cells were washed with 500μL ice-cold 1x PBS and lysed in 50μL of 1× passive lysis buffer. Then, 5μL of cell lysate was added to 30μL Dual-Glo Luciferase Reagent, and the luciferase activity was measured immediately on a GloMax 20/20 Luminometer (Promega).
ELISA (enzyme-linked immunosorbent assay)
A commercial ELISA kit was used to measure the interferon levels in the cell culture supernatants. The levels of human IFN-λ2 (Cat # RHF333CK) and IFN-β (Cat# RHF842CK) were purchased from Antigenix America, INC. Briefly, a microtiter plate was coated with capture antibody overnight at 4°C. After washing with 300μL of wash buffer four times, the plate was coated with 200μL of a coating stabilizer (Cat # EA150) for 60-minutes at room temperature. The samples and standards were added to each well in triplicate and incubated at room temperature for 2-hours with agitation. The wells were aspirated and washed four times. One-hundred microliters of diluted detection antibody (0.2μg mL−1) was added and incubated at room temperature for 1-hour. The plate was aspirated and washed four times. One-hundred microliters of HRP conjugate (1:1000) was added and incubated for 30-minutes at room temperature. The plate was aspirated and washed four times. Then, 100μL of substrate solution was added and incubated for 10-minutes at room temperature. Finally, 100μL of stop solution was added to stop color development and the plate was read at λ 450 nm on a spectrophotometer.
Quantification of infectious viral particles by plaque-forming assay
Vero cells (3×105 cells well−1) were seeded onto a 6-well plate and grown to a confluent monolayer overnight. Serial dilutions (in serum-free DMEM) of viral culture supernatants were applied to the Vero cells at 37 °C for 2-hours. The inoculum was then removed and replaced with 2-ml of DMEM complete medium containing 1% SeaPlaque agarose (Cat# 50100, Lonza). Plaques were visualized using Neutral red (Sigma-Aldrich) after 3-days of incubation at 37 °C, 5% CO2.
Reverse transcription and quantitative PCR (qPCR)
Approximately 105 cells were collected in 350μL of RLT buffer (QIAGEN RNeasy mini kit). RNA was extracted following the QIAGEN manual exactly, reverse-transcribed into cDNA using the BIO-RAD iScriptTM cDNA Synthesis Kit. qPCR was performed with gene-specific primers and SYBR Green master mix. Results were calculated using the −ΔΔCt method and beta actin gene used as an internal control. The qPCR primers are listed in Table 1.
Table 1.
qPCR primers.
| Gene | Forward | Reverse |
|---|---|---|
| hOAS1 | AGGAAAGGTGCTTCCGAGGTAG | GGACTGAGGAAGACAACCAGGT |
| hIFIT1 | GCCTTGCTGAAGTGTGGAGGAA | ATCCAGGCGATAGGCAGAGATC |
| hISG15 | CTCTGAGCATCCTGGTGAGGAA | AAGGTCAGCCAGAACAGGTCGT |
| hIL28 | TCGCTTCTGCTGAAGGACTGCA | CCTCCAGAACCTTCAGCGTCAG |
| hIL29 | AACTGGGAAGGGCTGCCACATT | GGAAGACAGGAGAGCTGCAACT |
| hTNFA | CTCTTCTGCCTGCTGCACTTTG | ATGGGCTACAGGCTTGTCACTC |
| hIFNB1 | CAGCAATTTTCAGTGTCAGAAGCT | TCATCCTGTCCTTGAGGCAGT |
| hACTB | ATCCTGGCCTCGCTGTCCAC | GGGCCGGACTCGTCATAC |
| hMDA-5 | GCTGAAGTAGGAGTCAAAGCCC | CCACTGTGGTAGCGATAAGCAG |
| VSV | TGATACAGTACAATTATTTTGGGAC | GAGACTTTCTGTTACGGGATCTGG |
| ZIKV | CCGCTGCCCAACACAAG | CCACTAACGTTCTTTTGCAGACAT |
| EMCV | CCTCTTAATTCGACGCTTGAA | GGCAAGCATAGTGATCGAG |
h-homo sapiens
Graphing and Statistics
All data were analyzed with a GraphPad Prism software. Specific statistical analyses used are described in the legend of each figure and include two-tailed Student’s t-test and two-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test. P values of ≤0.05 were considered statistically significant.
Results
UBXN6 potentiates ISG induction by type I/III IFNs
We have recently demonstrated that a subset of UBXNs regulate MAVS and STING signaling (26, 30). We now performed a screening in human embryonic kidney 293 cells transformed with T antigen of SV40 (HEK293T) cells for UBXNs that enhanced type I/III IFN- stimulated response element (ISRE) reporter activity. We observed that forced expression of UBXN6 could increase basal ISRE by ~2.5-fold over the empty vector control (Fig.1A). Both recombinant IFN-β and IFN-λ stimulated robust ISRE activity (Vector+IFN versus Vector alone), which was enhanced by ~2.5–3.4 fold following UBXN6 overexpression (Fig.1B,C). The ability of UBXN6 to activate ISRE was dose-dependent (Fig.1D). We further confirmed the ISRE reporter assay by quantitative reverse transcription PCR (qPCR). UBXN6 potentiated the mRNA expression of well-established ISGs (Fig. 1E) These results demonstrate that UBXN6 is a unique UBXN that regulates optimal JAK-STAT activity.
Figure 1. Ectopic expression of UBXN6 potentiates Type I/III IFN responses.
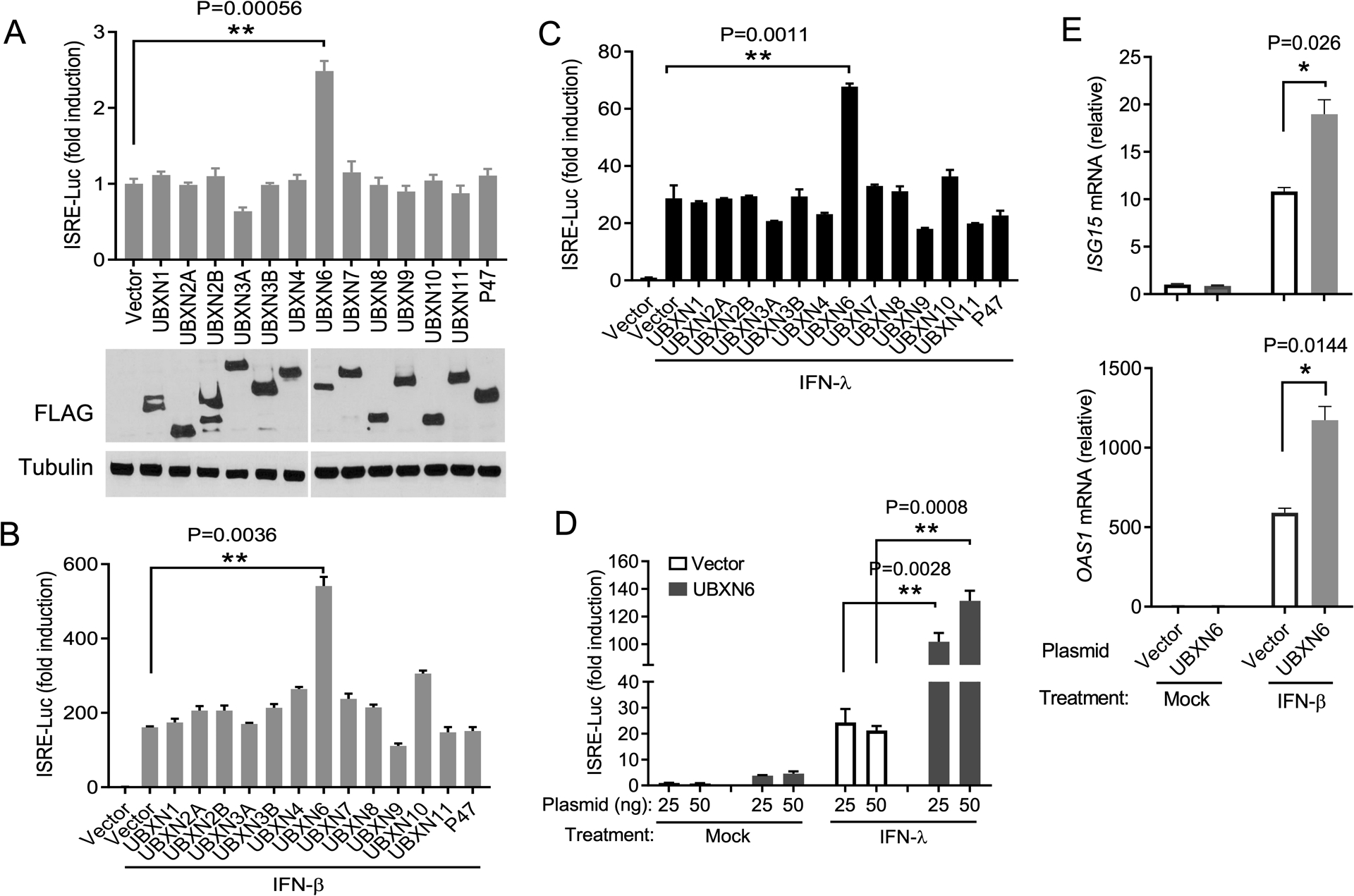
(A) Quantification of ISRE promoter activity by a dual-luciferase assay in HEK293T cells transfected with either 100ng of empty vector or individual FLAG-UBXN plasmids. The immunoblot depicts the expression levels of FLAG-UBXN proteins and α-tubulin as a housekeeping protein control. (B-D) Measurement of ISRE reporter after plasmid transfection as in (A) and 16-hours after stimulation with 10ng mL−1 of recombinant human (B) IFN-β and (C, D) IFN-λ1. The results are presented as fold change over non-stimulated vector control. (E) qPCR analysis of mRNA expression of interferon-stimulated genes ISG15 and OAS1A 16-hours after IFN-β stimulation. Mock: no IFN stimulation. Bar: the mean ± SEM (n = 3), *p < 0.05; ** p < 0.01 (two-tailed Student’s t-test).
To validate the results from the UBXN6 overexpression studies, we next generated UBXN6 knockout HEK293T and human trophoblasts cell lines using CRISPR-Cas9 technology (Clustered Regularly Interspaced Short Palindromic Repeats and its associated proteins). Western blotting shows successful deletion of UBXN6 protein expression (Fig.2A). We then analyzed expression of 84 genes related to type I/III IFN responses by a PCR array in UBXN6+/+ and UBXN6−/− trophoblasts. Expression of ~20 ISGs including well-characterized IFIT1, OAS1/2, IFITM1, ISG15 and DDX58 (encoding RIG-I) were >1.5-fold lower in UBXN6−/− than UBXN6+/+ cells treated with recombinant IFN-λ for 6 hours. Three genes IL6, VEGF and IFNE that are not conventional ISGs were upregulated (Fig.2B,C). The reduction in IFIT1 and OAS1 expression in two UBXN6 knockout cell lines was further validated by qPCR (Fig.2D,E). This reduced expression of ISGs was consistent with an apparent decrease in STAT1 phosphorylation (p-STAT1) after 30-minutes of IFN-β treatment, which activates identical JAK/STAT components as IFN-λ does (Fig.2F). We next asked whether the regulation of ISG induction by UBXN6 is consistent in immune cells. In THP-1 macrophages, transient knockdown of UBXN6 using 2 unique siRNAs significantly compromised the expression of ISG15 and MX2 (Fig.2G,H). Of note, IFIT1 and OAS1A were not induced in THP-1 cells, regardless of the dose of IFN-β (data not shown), suggesting these genes are not a component of the ISG signature in this cell line. Therefore, UBXN6 appears to regulate ISG responses in both epithelial and immune cell types.
Figure 2. UBXN6 deficiency impairs ISG induction by type I and III IFNs.
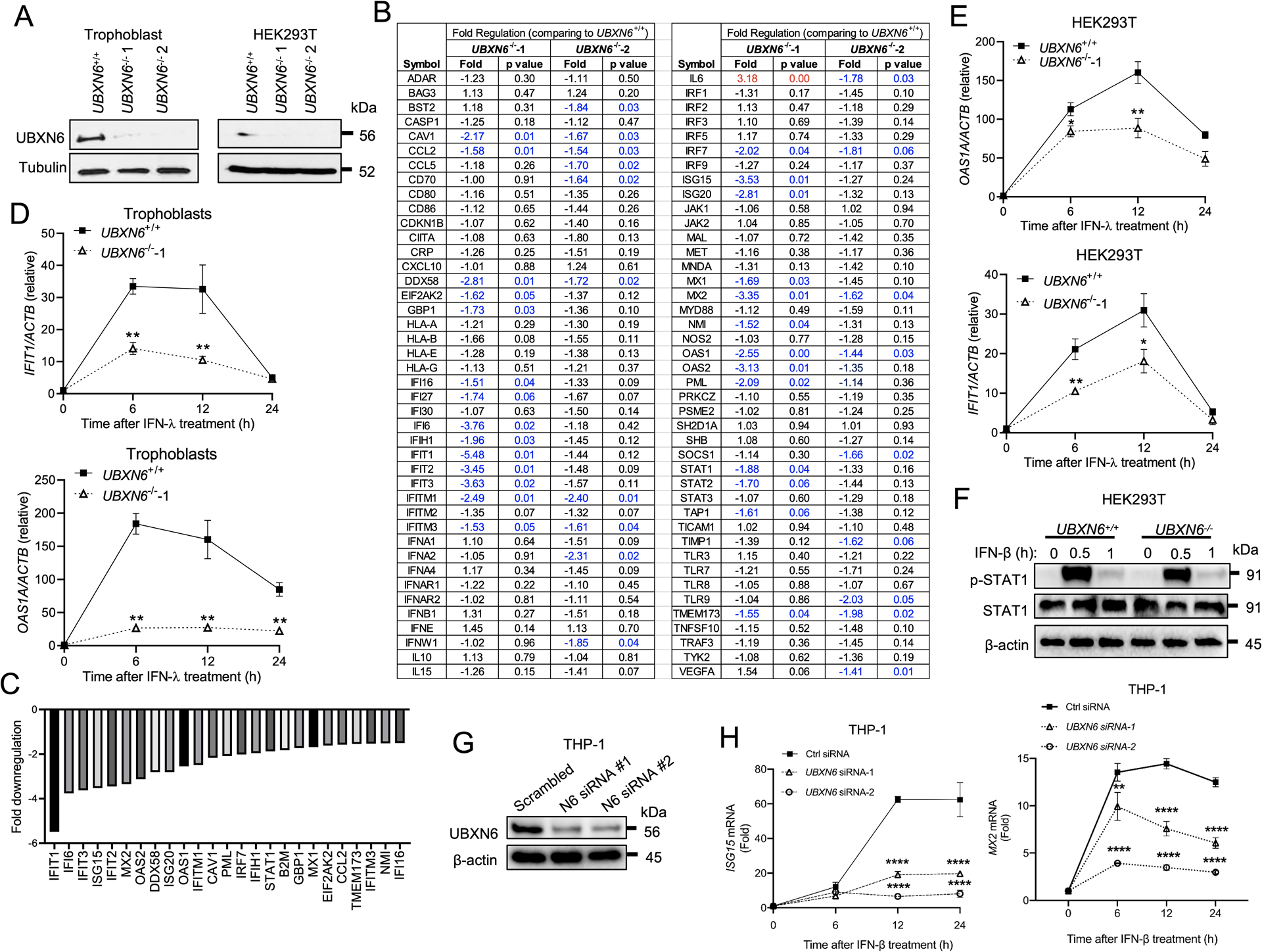
(A) Immunoblots showing UBXN6 and tubulin protein levels in UBXN6+/+ and UBXN6−/− human trophoblasts and HEK293T cells. (B) The relative fold changes in gene expression, mainly ISGs, of the type I/III IFN pathway by PCR array in two knockout trophoblasts 6-hours after treatment with 10ng mL−1 of IFN-λ1 (n=3). The significantly down-regulated genes (p<0.05) are highlighted in blue. (C) A bar graph summarizing all the genes with significantly altered expression in UBXN6−/−−1 compared to UBXN6+/+. (D) qPCR analysis of mRNA expression of canonical interferon-stimulated genes (ISGs) at 0, 6, 12, 24-hours after IFN-λ1 treatment in (D) trophoblasts and (E) HEK293T. (F) Immunoblots showing the phosphorylation status of STAT1 (p-STAT1) at 0, 30 and 60-minutes following IFN-β treatment in the same UBXN6+/+ and UBXN6−/− HEK293T cells as in (E). (G) Immunoblot analysis depicting knockdown efficiency of UBXN6 in THP-1 macrophages treated with control siRNA or UBXN6 siRNA-1 and 2. (H) qPCR analysis of mRNA expression of ISGs in THP-1 macrophages stimulated with 500 pg ml−1 IFN-β for 24-hours. Data are representative of two (B-E) and three (F-H) independent experiments. Data points represent the mean ± SEM (n =4–6 biological replicates) For (D,E,H), *P < 0.05; ** P < 0.01, ****P<0.0001 (two-way ANOVA with Dunnett’s multiple comparisons test).
UBXN6 potentiates type I IFN induction by poly(I:C) and viral infection
The aforementioned data altogether suggest that UBXN6 regulates JAK-STAT1/2 signaling. We next examined the role of UBXN6 in antiviral immune responses. We first employed a viral double-stranded RNA analogue, poly(I:C), to stimulate the ISRE reporter activity primarily through RIG-I like receptors [including melanoma differentiation-associated gene 5 (MDA5) and retinoic acid-inducible gene-1, (RIG-I)] in HEK293T cells. Forced UBXN6 expression potentiated poly(I:C)-induced ISRE-Luc and IFN-I protein expression in a dose-dependent manner (Fig.3A,C). As a control, UBXN11 failed to do so (Fig.3B). In agreement with overexpression results, IFNB1 and TNFA mRNA expression was lower at 6 and 12-hours, but not 24-hours, in UBXN6−/− than UBXN6+/+ cells (Fig.3D) after poly(I:C) treatment. Moreover, immune responses were induced rapidly, reaching the peak at 12 hours and decreasing by 24-hours in UBXN6+/+ cells; however, in UBXN6−/− cells, the peak was noticeably delayed by 12-hours (Fig.3D). Given some UBXN genes can be induced after IFN treatment—albeit at lower levels than well characterized ISGs (26, 30)— and have been implicated in RNA virus sensing pathways, we investigated whether UBXN6 may be an ISG itself, hence efficient IFNB1 expression may be dependent on a higher threshold of protein than present at basal levels. However, neither IFN-β nor poly(I:C) treatments could significantly enhance the protein expression of UBXN6 over baseline, as compared to a canonical ISG, RIG-I (Fig.3E).
Figure 3. UBXN6 positively regulates IFN responses induced by a dsRNA analogue, poly (I:C).
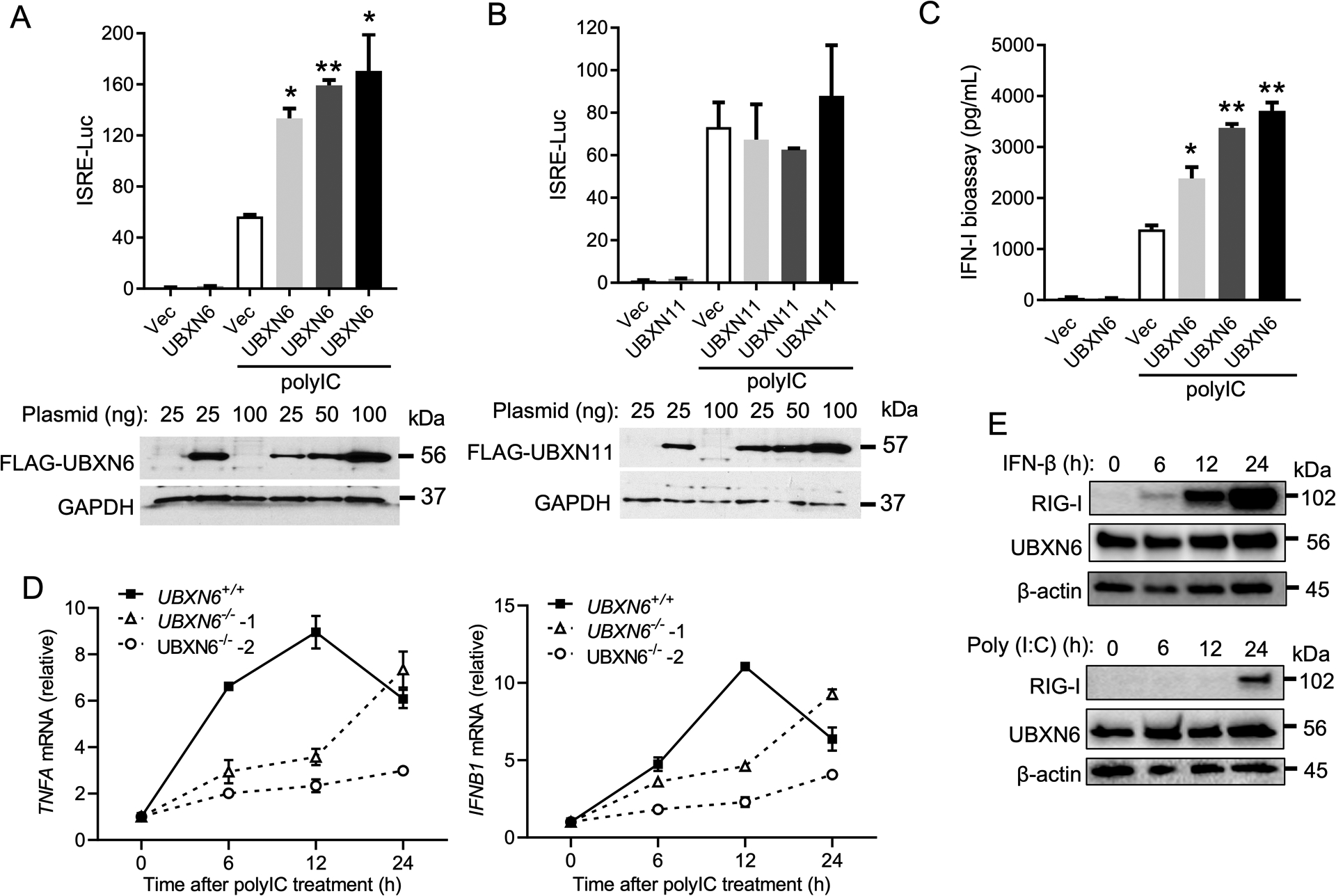
Quantification of ISRE promoter activity by a dual-luciferase assay in HEK293T cells transfected with an empty vector, (A) FLAG-UBXN6 or (B) FLAG-UBXN11 plasmid, respectively, and 24-hours later transfected with 4μg ml−1 poly I:C for an additional 16-hours. Immunoblots show the expression levels of FLAG-tagged proteins and GAPDH as a housekeeping protein control. (C) Quantification of type-I IFN bioavailability in the culture supernatants from (A) using a stable 2fGTH-ISRE reporter cell line. (D) qPCR analysis of TNFA and IFNB1 mRNA expressions at 0, 6, 12, and 24-hours after poly I:C stimulation in UBXN6+/+, and UBXN6−/− trophoblast cells. (E) Immunoblots of RIG-I and UBXN6 in HEK293T cells treated with 500 pg ml−1 IFN-β or 4μg ml−1 poly I:C for 24-hours. β-actin served as a housekeeping loading control. Data represent the mean ± SEM (n = 3), *P < 0.05; ** P < 0.01 (two-tailed Student’s t-test). For (E), western blots represent one biological replicate from a total of n=3.
In the case of poly(I:C) stimulation, RLRs, which basal protein expression levels are barely detectable (Fig.3E), need to be upregulated by a functional JAK1-STAT1/2 pathway for optimal IFNB1 expression. To evaluate the contribution of UBXN6 in the primary IFN-I/III pathways, we utilized a MAVS and TBK1 expression plasmid (pMAVS and pTBK1, respectively) of the RLR pathway which can stimulate IFNB1 transcription directly without JAK1-STAT1/2. Overexpression of pMAVS/pTBK1 in UBXN6 knockdown cells (Fig.4A) reduced ISRE-luciferase activity, corroborating our previous results indicating a defect in proper JAK-STAT signaling (Fig.4B). Furthermore, we observed a modest defect in IFNB1 transcription in UBXN6 knockdown cells transfected with pMAVS, but not cells receiving pTBK1 (Fig.4C).
Figure 4. UBXN6 controls viral infection independently of MAVS signaling.
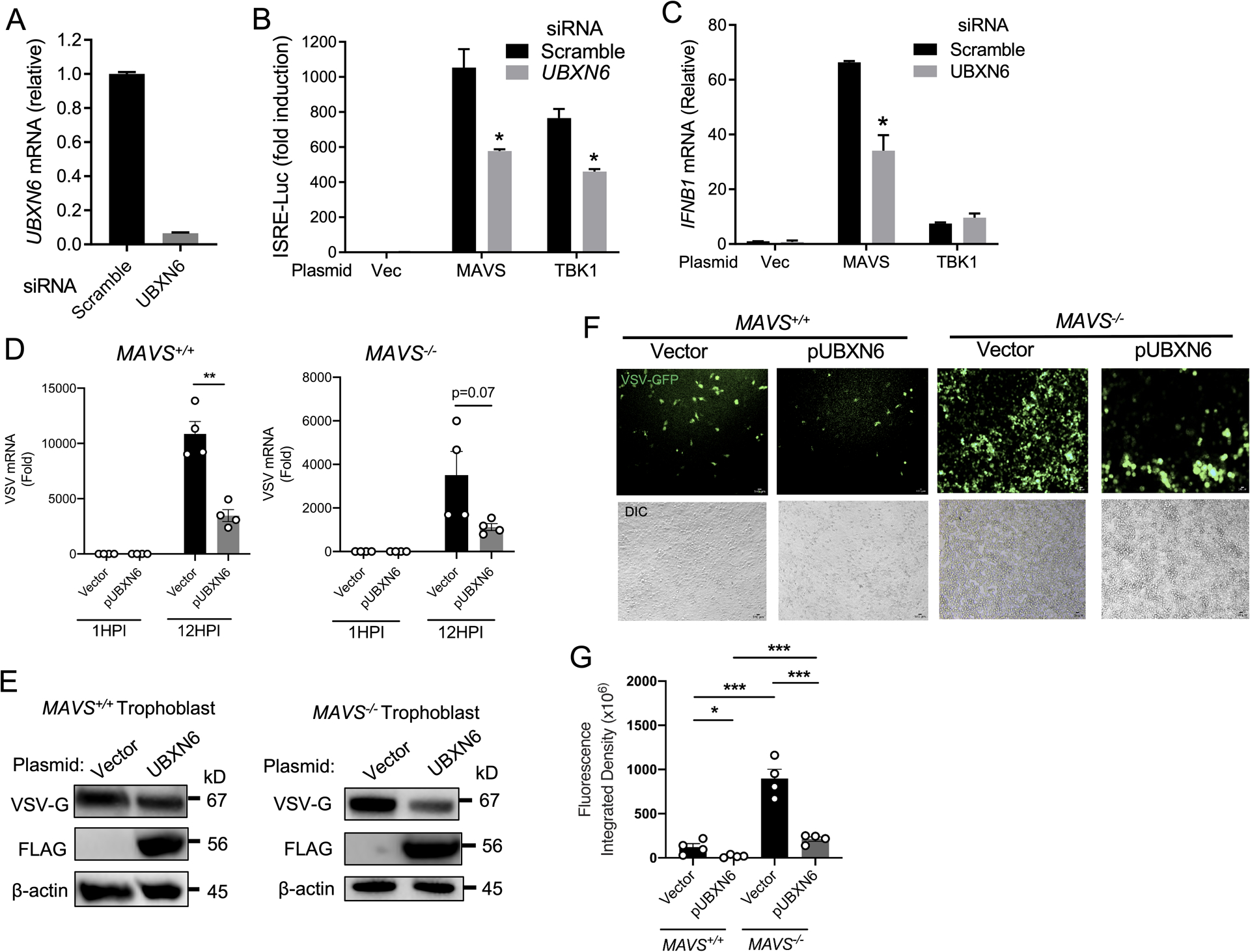
qPCR analysis of (A) UBXN6 mRNA expression 48-hours after transfection with scrambled and UBXN6-specific siRNA. (B) Quantification of ISRE promoter activity by dual-luciferase assay in HEK293T cells 48-hours after siRNA transfection as in (A). After 24-hours, vector, MAVS, or TBK1 plasmids were transfected and ISRE promoter activity assessed 16-hours later. (C) qPCR analysis of IFNB1 mRNA in HEK293T cells that had been transfected with siRNA and plasmid DNA as detailed in (B). (D) qPCR analysis of VSV mRNA expression in MAVS+/+ and MAVS−/− trophoblast cells 24-hours after vector or FLAG-UBXN6 plasmid transfection. Multiplicity of infection (MOI):1; hours post infection (HPI). (E) Immunoblots of the surface glycoprotein of vesicular stomatitis virus (VSV-G) protein expression in MAVS+/+ and MAVS−/− trophoblast cells treated as in (D). The image development time for the VSV-G blot of MAVS−/− was shorter than for MAVS+/+, to avoid overexposure. Blots were subsequently probed for FLAG expression. β-actin used as a housekeeping loading control. (F) Fluorescent microscopic images of VSV-GFP (MOI:1) 12-hours after infection in MAVS+/+ and MAVS−/− trophoblast cells transfected with indicated plasmids as in (D). The chart shows quantification of GFP fluorescence signal displayed as integrated density from 4 biological replicates. Objective: 10x; Green: VSV-GFP; scale bar: 100μM. For (B-D,G), bar: the mean ± SEM (n = 3). *P < 0.05 (two-tailed Student’s t-test). Data are representative of three (D-E) and two (F-G) independent experiment.
These data demonstrate UBXN6 can also promote primary IFN-I responses, specifically at the MAVS adaptor level. We next asked whether the reciprocal phenotype was true: amplified IFN-I responses from UBXN6 are dependent on MAVS. To this end, we utilized a previously generated MAVS−/− trophoblast cell line (31) and overexpressed FLAG-UBXN6 prior to infection with vesicular stomatitis virus (VSV)-GFP. A transcriptional unit encoding green fluorescent protein (GFP) is incorporated into the VSV genome upstream of the L polymerase to facilitate viral detection by fluorescence microscopy (32). The overexpression of UBXN6 in MAVS+/+ cells significantly decreased VSV replication as compared to cells which received a vector control plasmid (Fig.4D). This apparent control of viral replication in UBXN6-overexpressing cells was consistent with diminished VSV-G protein and GFP signal (Fig.4D–G). Although MAVS−/− cells were observationally overwhelmed by VSV infection (Fig.4F, right panel, vector), UBXN6 overexpression could decrease VSV mRNA (P=0.07), VSV-G protein and GFP fluorescent signal (Fig.4D–G). Nevertheless, these data suggest that UBXN6 serves two crucial roles in the regulation of type-I IFN responses: 1) it promotes efficient JAK/STAT signaling and heightened ISG transcription important for responding to virus infection, and 2) it inhibits virus replication independently of MAVS signaling.
Next, we employed three models of RNA viruses: VSV, encephalomyocarditis virus (EMCV) and Zika virus (ZIKV), to further elucidate whether UBXN6 is a broad antiviral effector and not restricted solely to responding to VSV pathogen associated molecular patterns (PAMPs). All of these viruses induce IFN-I/III and ISGs primarily via RLRs (34) (31). In accordance with abovementioned results, overexpression of UBXN6 repressed VSV replication most dramatically among all of the UBXNs (Fig.5A). Consistently, intracellular VSV-GFP was much increased in UBXN6−/− than UBXN6+/+ cells, as was VSV mRNA and infectious virus (Fig.5B–D). Similar results were noted for EMCV infection (Fig.5E–F). Additionally, IFN-β and IFN-λ concentrations produced by UBXN6−/− cells were much lower than UBXN6+/+ cells at 12- and 24-hours post infection (Fig.5G–H). Trophoblasts act as a primary barrier of the placenta to congenital viral transmission, such as ZIKV, by constitutively secreting type III IFNs (35). We then examined if UBXN6 is important for limiting ZIKV in trophoblasts. Indeed, ZIKV load was 10-fold higher in UBXN6−/− than UBXN6+/+ cells at 12- and 24-hours after infection (Fig.5I). Thus, UBXN6 appears to have a positive influence on IFN signaling pathways in response to a breadth of RNA viruses.
Figure 5. UBXN6 deficiency enhances RNA virus infection and impairs type I/III IFN responses.
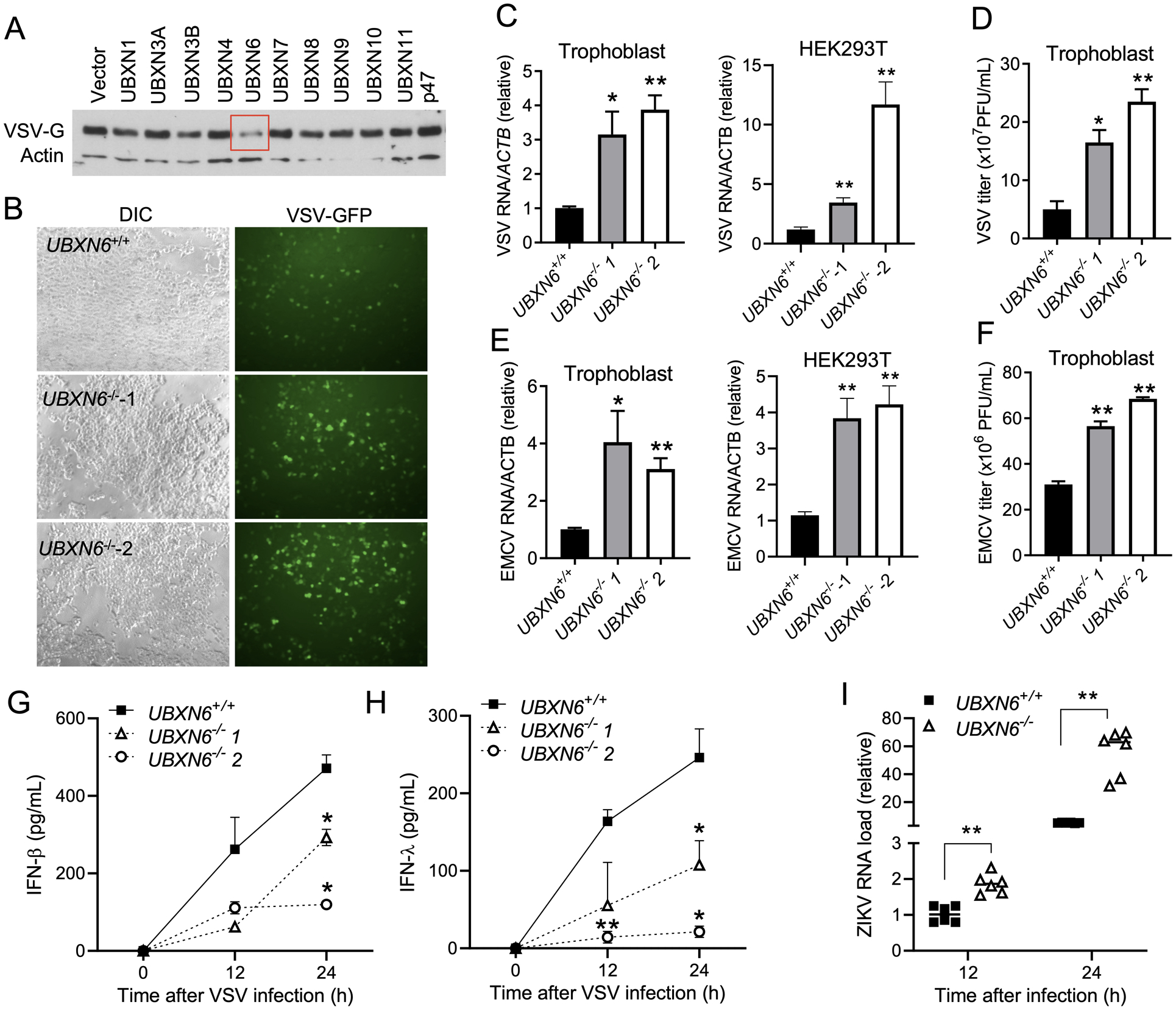
(A) Immunoblot showing protein expression of VSV-G 12-hours after infection (MOI=0.1) in HEK293T cells transfected with individual UBXN plasmids. Actin serves as a housekeeping protein control. (B) Fluorescent microscopic images of VSV-GFP (MOI=0.1) in UBXN6+/+ and UBXN6−/− trophoblast cells 12-hours after infection. Objective: 5x. qPCR analysis of intracellular (C) VSV, (E) encephalomyocarditis virus (EMCV) RNA 12-hours after infection in UBXN6+/+ and UBXN6−/− trophoblast cells (VSV MOI=0.1, EMCV MOI=0.01), (n=6). (D, F) Extracellular viral titers of VSV (D) and EMCV (F) from UBXN6+/+ and UBXN6−/− trophoblast cells quantified by plaque forming assay 12-hours after infection, n=3. The concentrations of (G) IFN-β and (H) IFN-λ2 quantified by ELISA in the culture supernatants of UBXN6+/+ and UBXN6−/− trophoblasts after VSV infection (MOI=0.1), n=3. The data represent the mean ± SEM, *P < 0.05; ** P < 0.01 (two-tailed Student’s t-test). (I) qPCR analysis of Zika virus mRNA expression in UBXN6+/+ and UBXN6−/− trophoblasts (MOI=5), n=6. The data represent the mean ± SEM, *P < 0.05; ** P < 0.01 (two-tailed Student’s t-test).
UBXN6 maintains normal JAK-STAT signaling by stabilizing TYK2 and IFNAR1 during IFN-β stimulation
In order to understand the underlying mechanism by which UBXN6 positively regulates JAK-STAT signaling, we next sought to observe the activation kinetics of JAK-STAT components in either UBXN6 overexpression or knockout cells. Overexpression of FLAG-UBXN6 enhanced the phosphorylation of STAT1 from 2 through 6-hours post IFN-β treatment, compared to the vector control (Fig.6A). UBXN6 has been shown to regulate endolysosomal sorting of ubiquitylated caveolin-1, a surface receptor (36–38). We reasoned if UBXN6 regulates JAK-STAT signaling at the IFN receptor level. Indeed, the IFNAR1 protein level was increased in UBXN6-overexpressing cells, delaying the beginning stages of degradation appearing as early as 4-hours post-IFN-β stimuli in vector control cells (Fig.6A). Consistently, IFN-β-stimulated STAT1, JAK1 and TYK2 phosphorylation was reduced in UBXN6−/− cells. Furthermore, IFN-β induced obvious degradation of IFNAR1 and TYK2 proteins in UBXN6+/+ cells by 12-hours post treatment, serving as a negative feedback mechanism to prevent prolonged activation of this pathway, which may be detrimental to tissues. However, this degradation happened as early as 2 through 12-hours post treatment in UBXN6−/− cells (Fig.6B). These new results suggest that UBXN6 helps maintain normal JAK-STAT signaling by stabilizing TYK2 and IFNAR1 proteins.
Figure 6. UBXN6 maintains normal JAK-STAT signaling by stabilizing TYK2 and IFNAR1 during IFN-β stimulation.
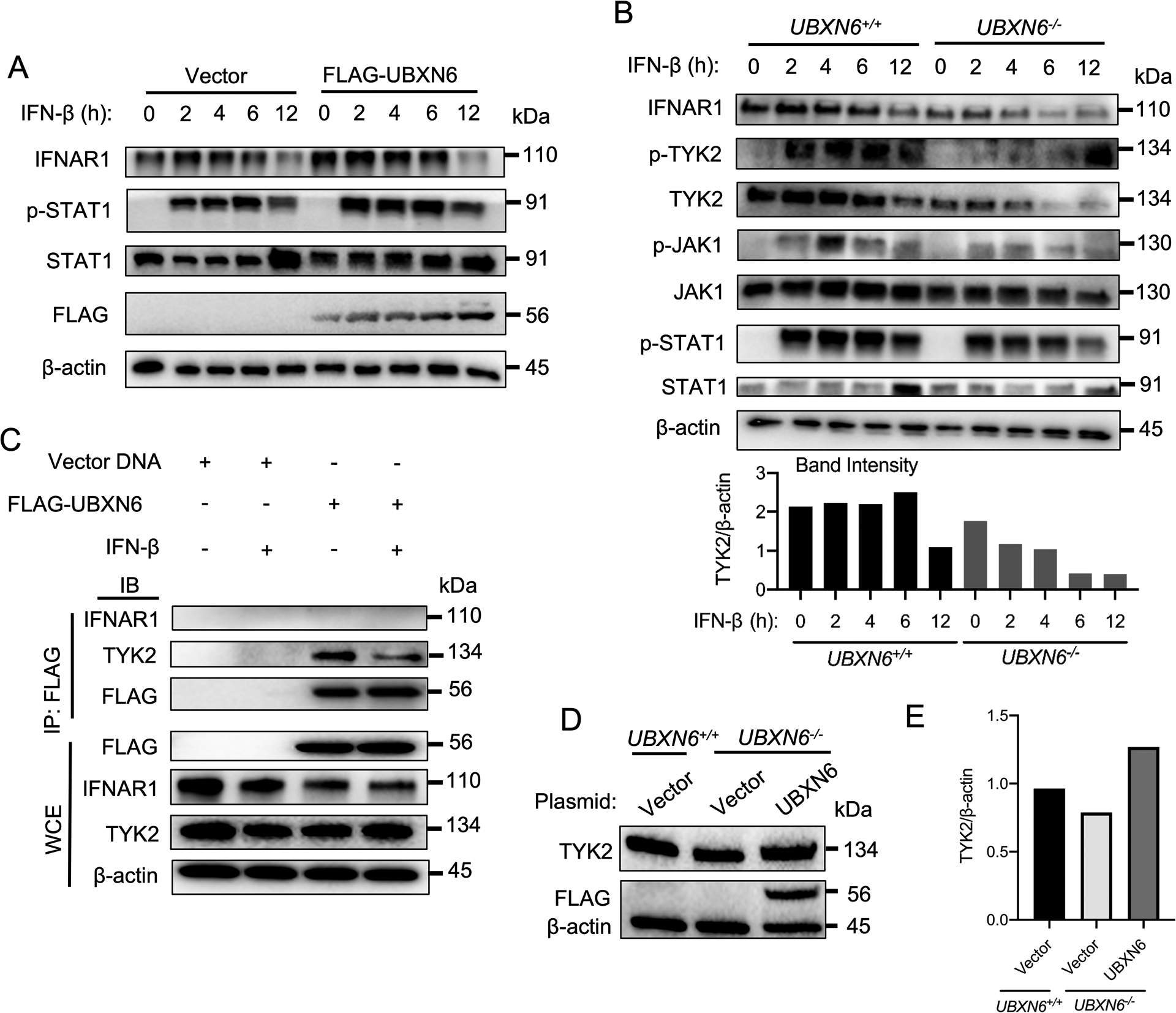
(A) The STAT1 phosphorylation and IFNAR1 expression kinetics in HEK293T cells transiently transfected with either vector or FLAG-UBXN6 plasmids followed by 500 pg ml−1 IFN-β treatment for 12-hours as assessed by immunoblotting. (B) Immunoblot of key JAK-STAT components in UBXN6+/+ and UBXN6−/− HEK293T cells treated with 500 pg ml−1 IFN-β. The bar graph shows the ratio of the band intensity of total TYK2 to their respective actin controls. Actin serves as a housekeeping protein control for (A-B). (C) Immunoprecipitates (IP:FLAG) and whole cell extracts (WCE) from HEK293T cells transfected with vector or FLAG-UBXN6 plasmids and analyzed by immunoblotting. Lysate was either collected before or after 1-hour of 500 pg ml−1 IFN-β treatment. FLAG serves as an input control for immunoprecipitation samples, whereas actin is used as a control in WCE. (D) TYK2 expression in UBXN6+/+ and UBXN6−/− HEK293T cells transfected with vector or FLAG-UBXN6 plasmids for 24-hours. Immunoblot depicts restored basal expression of TYK2 following UBXN6 expression. Bands in (D) are quantified in (E). Data are representative of two (D-E) and three (A-C) independent experiments.
TYK2 constitutively associates with IFNAR1, stabilizes IFNAR1 on the cell surface and delays its degradation that this is due, at least in part, to inhibition of IFNAR1 endocytosis (39). To determine whether UBXN6 is a member of the IFNAR1-TYK2 complex and thereby supports its stability, we overexpressed FLAG-UBXN6 in HEK293T cells before or after 1-hour IFN-β treatment and co-immunoprecipitated any binding partners. We observed a strong interaction between UBXN6 and TYK2 that was not present in vector-overexpressing cells indicating its specificity (Fig.6C). However, 1-hour IFN-β treatment diminished this interaction, possibly due to phosphorylation of TYK2 necessary for activation of downstream kinases (e.g., STAT1) (Fig.6C). Indeed, UBXN6 failed to interact with p-TYK2 (data not shown). Further probing of any subsequent interactions was negative, in particular IFNAR1, which could not be detected. Because a slight decrease in steady state TYK2 expression was observed in UBXN6−/− cells (Fig.6B) and UBXN6 appears to interact strongly with TYK2 prior to IFN stimuli, we further explored if exogenous UBXN6 could restore TYK2 expression. Indeed, overexpressing FLAG-UBXN6 rescued TYK2 levels to a greater extent than vector plasmid and those present in UBXN6+/+ cells (Fig.6D–E).
Discussion
UBXN6 belongs to a family of proteins that are putatively involved in p97-mediated and/or ubiquitination cellular processes (35). Recent studies suggest that UBXN6 participates in ERAD (36), protein trafficking (37–39), clearance of ruptured lysosomes by autophagy (40), and degradation of mitochondrial outer membrane protein myeloid cell leukemia 1 (MCL1) in a model of Huntington’s disease (41). However, the immunological function of UBXN6 remains largely unexplored. In this study, we present several pieces of evidence implicating UBXN6 as a novel positive regulator of the JAK-STAT1/2 pathway. First, UBXN6 overexpression potentiates STAT1 phosphorylation and ISG induction in response to recombinant IFN-β and IFN-λ, which activates JAK1-STAT1/2 signaling via IFNAR1/2 and IFNLR1/IL-10R2, respectively (Fig.1, Fig.6A). Second, UBXN6−/− cells are deficient in STAT1 phosphorylation and ISG transcription in response to IFN-β and IFN-λ treatment (Fig.2). Third, UBXN6 overexpression potentiates, while UBXN6 deficiency weakens immune responses to poly(I:C) (Fig.3). Fourth, UBXN6−/− cells are unable to control viral infection and produce sufficient IFN-β and IFN-λ, when compared to UBXN6+/+ cells (Fig.5). Although these data convincingly suggest that UBXN6 primarily regulates the JAK-STAT1/2 signaling, the observations that poly(I:C) and virus-induced type I/III expression was reduced suggest a role of UBXN6 in the RLR signaling as well. The two major poly(I:C)/viral RNA receptors are melanoma differentiation-associated gene 5, MDA5 (encoded by IFIH1), and retinoic acid-inducible gene-1, RIG-I (encoded by DDX58) (31), which are also well characterized ISGs. Their basal levels are generally very low and need to be induced via JAK-STAT1/2 in the context of poly(I:C)/virus stimulation. Indeed, we observed a ~1.8–2.8-fold reduction in MDA-5/RIG-I following IFN-λ treatment in UBXN6−/−, when compared to UBXN6+/+ cells (Fig.2B–C). Thus, a deficiency in JAK-STAT1/2 signaling may also negatively impact the primary IFN response. Indeed, a STAT1/2 deficiency in mice led to a significant reduction in type I IFNs in response to dengue virus infection (42). To further clarify this point, we overexpressed the major molecules (MAVS/TBK1) of the RLR pathway to activate IFNB1 transcription independently of JAK-STAT1/2. Intriguingly, MAVS/TBK1-activated ISRE activity and MAVS –activated IFNB1 transcription was impaired in UBXN6-knockdown cells, while TBK1-induced IFNB1 expression was not. These results further validate a positive role of UBXN6 in JAK1-STAT1/2 signaling and suggest that UBXN6 positively regulates MAVS. Under physiological conditions, endogenous UBXN6 may depend on RLR-MAVS to achieve a maximal antiviral effect because RLRs are ISGs downstream of JAK-STAT1/2. Nonetheless, the impact of UBXN6 deficiency on MAVS signaling is moderate (~50% reduction in IFNB1 expression, Fig.4C), when compared to that on JAK-STAT1/2 signaling (up to 4-fold reduction in some ISGs, Fig.2B–D). Moreover, overexpression of UBXN6 inhibited viral infection independently of MAVS (Fig.4D–G). Therefore, the primary target of UBXN6 may be JAK-STAT1/2 during viral infection.
The observations that UBXN6 influences ISG expression in response to both IFN-β and INF-λ suggest a role of UBXN6 downstream of their cognate receptors, as both IFNs utilize identical JAK-STAT1/2 members after receptor dimerization. Indeed, UBXN6 interacts with non-phosphorylated TYK2, but not other components of the JAK-STAT1/2 pathway, suggesting that UBXN6 targets TYK2. Of note, the UBXN6-TYK2 interaction is reduced in response to IFN-β, suggesting that this interaction is physiologically relevant to JAK-STAT1/2 signaling (Fig.6C). This reduction is likely because of TYK2 phosphorylation, which activates STAT1, and in the meantime, reduces the availability of non-phosphorylated TYK2 for UBXN6. Nonetheless, the interaction of UBXN6 with TYK2 may have an impact on TYK2 function. Indeed, TYK2 phosphorylation following IFN-β treatment is significantly impaired and delayed in UBXN6−/− compared to that in UBXN6+/+ cells (Fig.6B), suggesting that UBXN6 acts on the very early step of the JAK-STAT1/2 pathway. Of note, TYK2 protein is degraded in response to IFN-β treatment in UBXN6+/+ cells starting from 6 hours, and this is accelerated in UBXN6−/− cells (beginning from 2 hours) (Fig.6B), suggesting that UBXN6 stabilizes TYK2. To our surprise, IFNAR1 degradation following IFN-β treatment is also accelerated in the absence of UBXN6, though no interaction between them is detected (Fig.6B,C). This is likely because of degradation of TYK2, which constitutively associates with IFNAR1 (43), stabilizes IFNAR1 on the cell surface and delays its degradation that is due, at least in part, to inhibition of IFNAR1 endocytosis (44). Degradation of TYK2 and IFN receptors in response to stimuli is well known as a negative feedback mechanism to prevent prolonged activation of this pathway, which may be detrimental to tissues (43). Our results suggest that UBXN6 may bind and slow down TYK2 degradation in proteasomes (45), which in turn stabilizes IFNAR1, and this can explain the positive role of UBXN6 in both type I and III IFN signaling. Consistent with this notion, UBXN6, together with p97, regulates endolysosomal sorting of ubiquitylated caveolin-1, a surface receptor (37–39), and degradation of mitochondrial outer membrane protein myeloid cell leukemia 1 (MCL1) in a model of Huntington’s disease (41).
In summary, our studies identify UBXN6 as a positive regulator of the JAK-STAT1/2 pathway and demonstrate that UBXN6 stabilizes the TYK2 protein. Future work is required to pinpoint the molecular mechanism including the specific E3 ligase involved.
Revision Summary.
UBXN6 is positive regulator of both primary and secondary IFN responses.
UBXN6 exhibits antiviral immune responses independent of MAVS signaling.
IFNAR1 and TYK2 protein stability are compromised in the absence of UBXN6.
Acknowledgements
We thankfully acknowledge both Dr. Antoine L. Menoret and Timofey Karginov of The University of Connecticut, Department of Immunology, for their excellent experimental and philosophical discussions pertaining to the data/results.
Financial Support
This work was supported in part by a National Institutes of Health grant R01AI132526 and R21AI155820 to P.W.
Footnotes
Conflict of interest
The authors declare no conflict of financial and non-financial interest.
References
- 1.Kell AM, and Gale M Jr. 2015. RIG-I in RNA virus recognition. Virology 479–480: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Q, Sun L, and Chen ZJ. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17: 1142–1149. [DOI] [PubMed] [Google Scholar]
- 3.Schneider WM, Chevillotte MD, and Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miner JJ, and Diamond MS. 2016. Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr Opin Immunol 38: 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Navajas JM, Lee J, David M, and Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hammaren HM, Virtanen AT, Raivola J, and Silvennoinen O. 2018. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. [DOI] [PubMed] [Google Scholar]
- 7.Chen K, Liu J, and Cao X. 2017. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J Autoimmun 83: 1–11. [DOI] [PubMed] [Google Scholar]
- 8.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, and Hilton DJ. 1997. A family of cytokine-inducible inhibitors of signalling. Nature 387: 917–921. [DOI] [PubMed] [Google Scholar]
- 9.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, and Nicola NA. 2012. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, and Shuai K. 1997. Specific inhibition of Stat3 signal transduction by PIAS3. Science 278: 1803–1805. [DOI] [PubMed] [Google Scholar]
- 11.Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, and Shuai K. 1998. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A 95: 10626–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shuai K 2006. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 16: 196–202. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, Chance MR, Chen X, Du Y, Wang Y, An L, Wang Q, Lu T, Zhang X, Wang Z, and Stark GR. 2010. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A 107: 21499–21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, and David M. 2001. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 104: 731–741. [DOI] [PubMed] [Google Scholar]
- 15.Meissner T, Krause E, Lodige I, and Vinkemeier U. 2004. Arginine methylation of STAT1: a reassessment. Cell 119: 587–589; discussion 589–590. [DOI] [PubMed] [Google Scholar]
- 16.Komyod W, Bauer UM, Heinrich PC, Haan S, and Behrmann I. 2005. Are STATS arginine-methylated? J Biol Chem 280: 21700–21705. [DOI] [PubMed] [Google Scholar]
- 17.Schuberth C, and Buchberger A. 2008. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell Mol Life Sci 65: 2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, and Deshaies RJ. 2008. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134: 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raman M, Sergeev M, Garnaas M, Lydeard JR, Huttlin EL, Goessling W, Shah JV, and Harper JW. 2015. Systematic proteomics of the VCP-UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nat Cell Biol 17: 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rezvani K 2016. UBXD Proteins: A Family of Proteins with Diverse Functions in Cancer. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park MY, Moon JH, Lee KS, Choi HI, Chung J, Hong HJ, and Kim E. 2007. FAF1 suppresses IkappaB kinase (IKK) activation by disrupting the IKK complex assembly. J Biol Chem 282: 27572–27577. [DOI] [PubMed] [Google Scholar]
- 22.Wang YB, Tan B, Mu R, Chang Y, Wu M, Tu HQ, Zhang YC, Guo SS, Qin XH, Li T, Li WH, Li AL, Zhang XM, and Li HY. 2015. Ubiquitin-associated domain-containing ubiquitin regulatory X (UBX) protein UBXN1 is a negative regulator of nuclear factor kappaB (NF-kappaB) signaling. J Biol Chem 290: 10395–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryu SW, Lee SJ, Park MY, Jun JI, Jung YK, and Kim E. 2003. Fas-associated factor 1, FAF1, is a member of Fas death-inducing signaling complex. J Biol Chem 278: 24003–24010. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, O’Boyle K, Auer J, Raju S, You F, Wang P, Fikrig E, and Sutton RE. 2017. Multiple UBXN family members inhibit retrovirus and lentivirus production and canonical NFkappaBeta signaling by stabilizing IkappaBalpha. PLoS Pathog 13: e1006187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song S, Lee JJ, Kim HJ, Lee JY, Chang J, and Lee KJ. 2016. Fas-Associated Factor 1 Negatively Regulates the Antiviral Immune Response by Inhibiting Translocation of Interferon Regulatory Factor 3 to the Nucleus. Mol Cell Biol 36: 1136–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang P, Yang L, Cheng G, Yang G, Xu Z, You F, Sun Q, Lin R, Fikrig E, and Sutton RE. 2013. UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Rep 3: 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JH, Park ME, Nikapitiya C, Kim TH, Uddin MB, Lee HC, Kim E, Ma JY, Jung JU, Kim CJ, and Lee JS. 2017. FAS-associated factor-1 positively regulates type I interferon response to RNA virus infection by targeting NLRX1. PLoS Pathog 13: e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu C, Kim BS, Park M, Do YJ, Kong YY, and Kim E. 2018. FAF1 mediates necrosis through JNK1-mediated mitochondrial dysfunction leading to retinal degeneration in the ganglion cell layer upon ischemic insult. Cell Commun Signal 16: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai T, Wu L, Wang S, Wang J, Xie F, Zhang Z, Fang X, Li J, Fang P, Li F, Jin K, Dai J, Yang B, Zhou F, van Dam H, Cai D, Huang H, and Zhang L. 2018. FAF1 Regulates Antiviral Immunity by Inhibiting MAVS but Is Antagonized by Phosphorylation upon Viral Infection. Cell Host Microbe 24: 776–790 e775. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Wang L, Ketkar H, Ma J, Yang G, Cui S, Geng T, Mordue DG, Fujimoto T, Cheng G, You F, Lin R, Fikrig E, and Wang P. 2018. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat Commun 9: 2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Ketkar H, Geng T, Lo E, Wang L, Xi J, Sun Q, Zhu Z, Cui Y, Yang L, and Wang P. 2018. Zika Virus Non-structural Protein 4A Blocks the RLR-MAVS Signaling. Front Microbiol 9: 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalton KP, and Rose JK. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279: 414–421. [DOI] [PubMed] [Google Scholar]
- 33.Sanjana NE, Shalem O, and Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, and Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441: 101–105. [DOI] [PubMed] [Google Scholar]
- 35.Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr., Cherry S, Sadovsky Y, and Coyne CB. 2016. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 19: 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritz D, Vuk M, Kirchner P, Bug M, Schutz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, and Meyer H. 2011. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 13: 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirchner P, Bug M, and Meyer H. 2013. Ubiquitination of the N-terminal region of caveolin-1 regulates endosomal sorting by the VCP/p97 AAA-ATPase. J Biol Chem 288: 7363–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haines DS, Lee JE, Beauparlant SL, Kyle DB, den Besten W, Sweredoski MJ, Graham RL, Hess S, and Deshaies RJ. 2012. Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Mol Cell Proteomics 11: M111 016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ragimbeau J, Dondi E, Alcover A, Eid P, Uze G, and Pellegrini S. 2003. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J 22: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madsen L, Andersen KM, Prag S, Moos T, Semple CA, Seeger M, and Hartmann-Petersen R. 2008. Ubxd1 is a novel co-factor of the human p97 ATPase. Int J Biochem Cell Biol 40: 2927–2942. [DOI] [PubMed] [Google Scholar]
- 41.Nagahama M, Ohnishi M, Kawate Y, Matsui T, Miyake H, Yuasa K, Tani K, Tagaya M, and Tsuji A. 2009. UBXD1 is a VCP-interacting protein that is involved in ER-associated degradation. Biochem Biophys Res Commun 382: 303–308. [DOI] [PubMed] [Google Scholar]
- 42.Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, Lux V, Ehrmann M, Weihl CC, and Meyer H. 2017. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36: 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X, and Qi X. 2017. VCP cooperates with UBXD1 to degrade mitochondrial outer membrane protein MCL1 in model of Huntington’s disease. Biochim Biophys Acta Mol Basis Dis 1863: 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perry ST, Buck MD, Lada SM, Schindler C, and Shresta S. 2011. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog 7: e1001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao W, Chen X, Liu L, Shu Y, Zhang M, and Zhong Y. 2019. Role of protein arginine methyltransferase 5 in human cancers. Biomed Pharmacother 114: 108790. [DOI] [PubMed] [Google Scholar]
- 46.Pollack BP, Kotenko SV, He W, Izotova LS, Barnoski BL, and Pestka S. 1999. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J Biol Chem 274: 31531–31542. [DOI] [PubMed] [Google Scholar]
- 47.Igarashi H, Kuwahara K, Yoshida M, Xing Y, Maeda K, Nakajima K, and Sakaguchi N. 2009. GANP suppresses the arginine methyltransferase PRMT5 regulating IL-4-mediated STAT6-signaling to IgE production in B cells. Mol Immunol 46: 1031–1041. [DOI] [PubMed] [Google Scholar]
- 48.Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, Deblasio A, Hatlen M, Menendez S, and Nimer SD. 2011. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 19: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar KG, Tang W, Ravindranath AK, Clark WA, Croze E, and Fuchs SY. 2003. SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22: 5480–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar KG, Krolewski JJ, and Fuchs SY. 2004. Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279: 46614–46620. [DOI] [PubMed] [Google Scholar]
- 51.Wack A, Terczynska-Dyla E, and Hartmann R. 2015. Guarding the frontiers: the biology of type III interferons. Nat Immunol 16: 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]