

Abstract
The effect of rigidification of the n-butyl linker region of tetrahydroisoquinoline-containing D3R ligands via inclusion of an o-xylenyl motif was examined in this study. Generally, rigidification with an o-xylenyl linker group reduces D3R affinity and negatively impacts selectivity versus D2R for compounds possessing a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol primary pharmacophore group. However, D3R affinity appears to be regulated by the primary pharmacophore group and high affinity D3R ligands with 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline primary pharmacophore groups were identified. The results of this study also indicate that D3R selectivity versus the σ2R is dictated by the benzamide secondary pharmacophore group, this being facilitated with 4-substituted benzamides. Compounds 5s and 5t were identified as high affinity (Ki < 4 nM) D3R ligands. Docking studies revealed that the added phenyl ring moiety interacts with the Cys181 in D3R which partially accounts for the strong D3R affinity of the ligands.
Keywords: tetrahydroisoquinoline, dopamine receptor, D3R, σ2R, docking
Graphical abstract:

There are five subtypes of dopamine receptor (D1R-D5R) and these are divided based on structural similarity and pharmacological properties into two subgroups – D1-like which comprises D1R and D5R and D2-like which includes D2R, D3R and D4R. All dopamine receptors are G-protein-receptor coupled and recognize the endogenous neurotransmitter dopamine. D1-like receptors signal via activation of Gs pathways whereas D2-like receptors couple to Gi inhibitory proteins.1–3 Activation of dopamine receptors endogenously is known to play a role in a myriad of physiological effects including movement, cognition and addiction-related behaviors.4, 5 There is a high density of D3R in the mesolimbic region of the brain, an area associated with motivation and drug seeking behaviors.6 Thus, the D3R has received considerable attention, especially over the 2 past decades as a potential target for the treatment of psychostimulant addiction.7 In that regard, antagonism or partial agonism by selective D3R ligands is desirable.8 D3R antagonists have also been shown to increase cognitive performance and reverse cognitive deficits in animal studies and are thus promising as antipsychotic agents.9–11
Over years of research, there have been significant challenges with obtaining D3R ligands that are suitable for translational research. Selectivity, especially versus the closely related D2R and D4R is one such issue, although there are now available several D3R subtype selective ligands.12 However, several D3R ligands have other drawbacks such as poor oral bioavailability which limits their utility as therapeutics.
Several D3R ligands have been obtained that conform to a classical D3R pharmacophore which consists of three main regions: (i) an amine-containing primary pharmacophore region, (ii) a linker region – typically an n-butyl chain and (iii) an arylamide secondary pharmacophore moiety (Fig. 1). Various amine-containing primary pharmacophore groups have been used, among which the phenylpiperazine moiety has been fairly common (e.g. BP 897 and NGB2904, Fig. 1). A number of D3R ligands have been discovered that contain a 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline primary pharmacophore group. However, in addition to binding to D3R, compounds with this motif have also been reported to possess significant affinity for the σ2 receptor (σ2R)13. In fact, a number of these 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-containing compounds have been explored as σ2R selective ligands and as positron emission tomography (PET) cancer imaging agents.14–18 We recently reported new D3R ligands that contain a slight variation of this template in containing a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol motif.19 We found that compounds with this motif (e.g. compound 1, Fig. 2) exhibited good selectivity for D3R over σ2R (see ref 19 and Table S1 in Supporting Information) and may thus be advantageous as compared to compounds with the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline moiety with regards to retaining D3R selectivity.
Figure 1.

Structures of the D3R antagonist pharmacophore and selected D3R antagonist ligands
Figure 2.

Structures of typical tetrahydroisoquinoline-containing D3R and σ2R ligands - RHM-1–8620 and compound 119
Previous studies have investigated rigidification of the flexible n-butyl linker region of other D3R scaffolds (particularly those containing phenyl piperazine-based primary pharmacophore groups) with moieties including cis-alkenyl, trans-alkenyl, alkynyl, xylenyl and cycloalkyl motifs with varying results.21–24 We were curious to find out the extent to which rigidification of the n-butyl linker unit of a D3R scaffold containing a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol and related tetrahydroisoquinolyl primary pharmacophore groups may lead to improvements in D3R affinity and potentially increased selectivity versus σ2R. We considered rigidification with an o-xylenyl motif as this would: i) preserve the 4-atom connectivity between the amine of the primary pharmacophore group and the nitrogen of the arylamide group and ii) preserve the basicity of the nitrogen in the tetrahydroisoquinoline region which is deemed to be necessary towards formation of a critical salt bridge interaction with D3R.19 The synthesized compounds were pharmacologically characterized at dopamine D1R – D5R and at the σ2R. Data on these evaluations as well as receptor docking studies of the ligands at D3R are discussed herein.
In terms of structural diversity of the designed analogues, we targeted the synthesis of compounds with the following structural variations: i) primary pharmacophore group - either a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol, 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline or 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline motif, ii) linker – o-xylene unit and iii) secondary pharmacophore region – a variety of arylamide units, including some found in previously identified D3R ligands. The compounds were synthesized as outlined in Scheme 1.
Scheme 1.

Synthesis of analogues 5a-t and 6a-c
Reagents and conditions: (a) Appropriate aldehyde, NaBH(OAc)3, DCM, 12 h, rt, 69–72%; (b) LiAlH4, THF, 1h, rt, 60–63%; (c) HBTU, appropriate carboxylic acid, TEA, 12 h, rt; (d) conc. HCl, CH3COOH, 12 h, 40 °C, 55–63% (over two steps); (e) BBr3, DCM, 0°C, 2h, 87–90%
To commence the synthesis of compounds containing a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol motif in tandem with the o-xylenyl linker unit, readily available compound 2a was subjected to reductive amination conditions with 2-formylbenzonitrile yielding 3a. Compound 3a was subsequently reduced with lithium aluminium hydride to afford the amine 4a. Compound 4a in turn underwent acid-amine coupling with various carboxylic acid groups to afford intermediate amides which were debenzylated under acidic conditions (without purification of the intermediate amide) to give the target analogues 5a – r. Analogues 5s and 5t with the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline motif were prepared from 2b in an analogous route to that described for analogues 5a – r (except for the acidic cleavage step). Compounds 5g, 5s and 5t were treated with BBr3 to effect O-demethylation, thus yielding catechol analogues 6a, 6b and 6c respectively.
The results of radioligand binding evaluations on analogues containing the o-xylenyl linker unit are compiled in Table 1. In general, compounds in this series lacked affinity for D1R and D5R. Compound 5a which contains a 4-fluorophenyl arylamide motif was devoid of affinity at all receptors tested. The 2-naphthylamide-containing 5b had only moderate affinity for D3R (Ki =780 nM); affinity was still moderate but approximately twice as high at D2R. No affinity was detected for the σ2R. Compounds with a 2-chloro, 2-bromo and 2-methoxy mono-substituted benzamide motif (i.e. 5c – 5e) displayed moderate affinity for D3R (Ki = 120 – 540 nM). In all cases these compounds were slightly more selective for D2R. Affinities generally increased in the order D4R<D3R<D2R. These analogues also had moderate affinity for the σ2R. Compounds 5f – 5h with the 3-chloro, 3-bromo and 3-methoxy benzamide motifs, displayed a similar binding profile and selectivity trends at D2R, D3R, D4R and σ2R as their previously described 2-substituted congeners. Compound 5i, with a 3-cyano substituent however displayed strong affinity for D3R (Ki = 26 nM) with comparable affinity for D2R and good selectivity versus D4R (>100-fold). This compound lacked any appreciable affinity for the σ2R. Compounds 5j – 5m with a 4-halophenyl moiety in the benzamide motif, had similar binding profiles in that they all lacked affinity for D4R and the σ2R. Compounds 5j – 5m showed good affinity for D2R and D3R; compound 5m had good D2R affinity (84 nM) but lacked D3R affinity and was the compound with the best D2R selectivity identified in this study. Replacement of the 4-halo substituent groups with 4-methoxy or 4-cyano groups (compounds 5n and 5o respectively) resulted in a rebound in (albeit weak: Ki = 1040 – 2800 nM) D4R affinity. The 2,3-dichloro benzamide analogue 5p displayed a binding profile that was reminiscent of the 2- and 3-substituted analogues described above. Interestingly, the 2,3-dimethoxy benzamide analogue (5q) showed complete selectivity for D3R (Ki = 57 nM) with no affinity at the other receptors evaluated. This compound has the best D3R selectivity profile of all compounds evaluated in this study. The sole 3,4-disubstituted benzamide tested (5r) showed strong affinity at D3R (Ki = 24 nM), comparable to that at D2R and with low D1R affinity (Ki = 1970 nM).
Table 1.
Binding affinity of o-xylenyl ring rigidified analogues at dopamine and σ2 receptors
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ki ± SEM (nM)a | |||||||||
| Cmpd. | R1 | R2 | R3 | D1Rb | D2Rc | D3Rc | D4Rc | D5Rb | σ2d |
| 5a | Me | H |  |
nae | na | na | na | na | na |
| 5b | Me | H |  |
na | 310 ± 40 | 780 ± 100 | na | na | na |
| 5c | Me | H |  |
na | 190 ± 25 | 380 ± 49 | 3900 ± 500 | na | 440 ± 57 |
| 5d | Me | H |  |
na | 420 ± 54 | 540 ± 70 | 880 ± 110 | na | 360 ± 46 |
| 5e | Me | H |  |
na | 78 ± 10 | 120 ± 15 | 950 ± 120 | na | 140 ± 18 |
| 5f | Me | H |  |
na | 100 ± 13 | 140 ± 18 | 950 ± 110 | na | 240 ± 31 |
| 5g | Me | H |  |
na | 150 ± 19 | 170 ± 22 | na | na | 290 ± 37 |
| 5h | Me | H |  |
na | 140 ± 18 | 280 ± 36 | 1070 ± 1 40 | na | 610 ± 79 |
| 5i | Me | H |  |
na | 39 ± 5 | 26 ± 3.4 | 3360 ± 430 | na | >10000 |
| 5j | Me | H |  |
na | 52 ± 6.7 | 140 ± 18 | na | na | na |
| 5k | Me | H |  |
na | 47 ± 6.1 | 55 ± 7.1 | na | na | na |
| 5l | Me | H |  |
na | 76 ± 9.8 | 160 ± 21 | na | na | na |
| 5m | Me | H |  |
na | 84 ± 11 | na | na | na | na |
| 5n | Me | H |  |
na | 74 ± 9.5 | 180 ± 23 | 1040 ± 130 | na | 1400 ± 180 |
| 5o | Me | H |  |
na | 190 ± 25 | 310 ± 40 | 2800 ± 360 | na | na |
| 5p | Me | H |  |
na | 280 ± 36 | 460 ± 5.9 | 1150 ± 150 | na | 480 ± 62 |
| 5q | Me | H |  |
na | na | 57 ± 7.4 | na | na | na |
| 5r | Me | H |  |
1970 ± 250 | 27 ± 3.5 | 24 ± 3.1 | na | na | na |
| 5s | Me | Me |  |
510 ± 66 | 46 ± 5.9 | 1.2 ± 0.2 | 57 ± 7.4 | 300 ± 39 | na |
| 5t | Me | Me |  |
58 ± 7.5 | 50 ± 6.5 | 3.4 ± 0.4 | 140 ± 18 | 340 ± 44 | na |
| 6a | H | H |  |
91 ± 12 | 56 ± 7.2 | 2.0 ± 0.2 | 101 ± 13 | na | na |
| 6b | H | H |  |
na | 900 ± 120 | 370 ± 48 | na | na | na |
| 6c | H | H |  |
na | 83 ± 11 | 28 ± 3.6 | - | na | na |
| (+)-butac1amo1 | 2.84 ± 0.05 | ||||||||
| Haloperidol | 2.61 ± 0.03 | 7.21 ± 0.9 | |||||||
| Nemonapride | 0.86 ± 0.04 | 0.75 ± 0.06 | |||||||
| SKF 83566 | 1.75 ± 0.1 | ||||||||
Experiments conducted in triplicate;
[3H]-SCH23390 used as radioligand;
[3H]N-methylspiperone used as radioligand;
[3H]DTG used as radioligand;
na- not active- ligands displayed <10% inhibition in a primary assay at a ligand concentration of 10 μM
Two compounds with a 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline motif were evaluated – the 3-cyano and 4-cyano benzamide derivatives 5s and 5t respectively. These compounds displayed very high affinity for D3R (Ki = 1.2 and 3.4 nM; among the most potent D3R ligands in this study) and exhibited selectivities ranging from 15- to 420-fold versus the other dopamine receptors tested. Interestingly, both compounds lacked σ2R affinity.
Among the compounds with a 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline unit, compound 6a and 6c displayed the highest D3R affinity. In fact, compound 6a had one of the highest D3R affinities (2 nM) of all compounds evaluated and had no affinity for D5R and σ2R. Selectivity of 6a versus D1R, D2R and D4R was modest (<50-fold). In comparing the catechol-containing analogues 6b and 6c with their dimethoxy counterparts (5s and 5t respectively) and 7-hydroxy-6-methoxy congeners (5i and 5o respectively), it is apparent that D3R affinity is better tolerated with the dimethoxy functionality than either the catechol or 7-hydroxy-6-methoxy motifs.
On a whole, it appears that 4-substituents on the benzamide moiety are less likely to promote binding to the σ2R as compared to their 2- or 3-substituted benzamide counterparts. However, compounds with a 2,3-disubstituted benzamide motif do not necessarily follow the same trend as for 2- and 3-monosubstitued benzamides. In that regard, compound 5q stands out in maintaining selectivity versus all other receptors including the σ2R.
A comparison of the previously evaluated 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol – containing ligands with a flexible linker group (e.g. 1) with their congeners containing the rigidifying o-xylenyl motif in the present study, also allows some general conclusions to be drawn about the impact of the introduced o-xylenyl sub-structure on D3R affinity and selectivity. In our previous study the compounds with flexible linker groups displayed strong D3R affinity (ranging from 2– 28 nM) with moderate or no affinity for other dopamine (See Table S1 in Supporting Information for data on comparator compounds from our previous work).19 However, in this study the highest D3R affinity seen for a rigidified compound with the 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol motif was 26 nM (compound 5i); most such compounds in the present study had affinities >100 nM at the D3R. Generally then, introduction of the o-xylenyl linker group resulted in a reduction in D3R affinity receptors (see Table S1). It appears that the inclusion of the o-xylenyl ring improves D2R affinity as most compounds with flexible linker groups (i.e. from our previous study) had low or no affinity unlike analogous rigidified compounds in the present study. Taken together, the data indicate that the rigidifying o-xylenyl ring motif is detrimental towards D3R versus D2R selectivity. One area where the SAR of the flexible and rigid compounds diverged in a positive sense was with regards to D4R affinity of the 4-halo-substituted benzamide derivatives. With the flexible analogues we found moderate D4R affinity (95 – 1500 nM) for this sub-group of compounds; for the analogous rigidified compounds there was no D4R affinity.19 Therefore, the inclusion of the o-xylenyl ring in this instance is beneficial in promoting D3R selectivity versus D4R. There does not seem to be any major impact on D1R or D5R affinity as both the flexible and rigidified compounds generally lacked D1R and D5R affinity.
A comparison of the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-containing compound 5t with its flexible counterpart from our previous study (see Table in Supporting Information), indicates that the presence of the rigidifying phenyl unit is beneficial for D3R affinity (Ki at D3R = 3.4 and 410 nM for 5t and its more flexible congener respectively). Further investigations are required in order to determine the extent to which this SAR trend holds for other 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-containing compounds.
The compounds with the highest D3R affinity contained either a 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline or 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline moiety. This suggests that the presence of an o-xylenyl ring linker motif in tandem with a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol moiety is less favorable for strong D3R affinity.
A molecular docking investigation was conducted to attempt to provide a structural interpretation of the effects of linker rigidification and methylation of substituents on the tetrahydroisoquinoline primary pharmacophore moiety that have been observed in this study. Our previous study showed that, with a flexible linker, compounds with the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline primary pharmacophore moiety displayed less favorable affinity to D3R relative to 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol-containing analogues. Based on previous molecular modeling results, this behavior was attributed to the ability of the latter group of compounds to form two hydrogen bond interactions between the primary pharmacophore group and Ser192 of the receptor compared to only one hydrogen bond in the case of the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-containing compounds.19, 25 A similar molecular docking study conducted here indicates that rigidification of the linker induces a mode of binding distinct from the one observed earlier and consistent with the observed SAR. As shown in Figure 3, the three most potent compounds (5s, 5t, and 6a) dock as generally expected with the primary pharmacophore group in the orthosteric pocket and forming the key salt bridge between the alkyl protonated nitrogen and Asp110.
Figure 3.
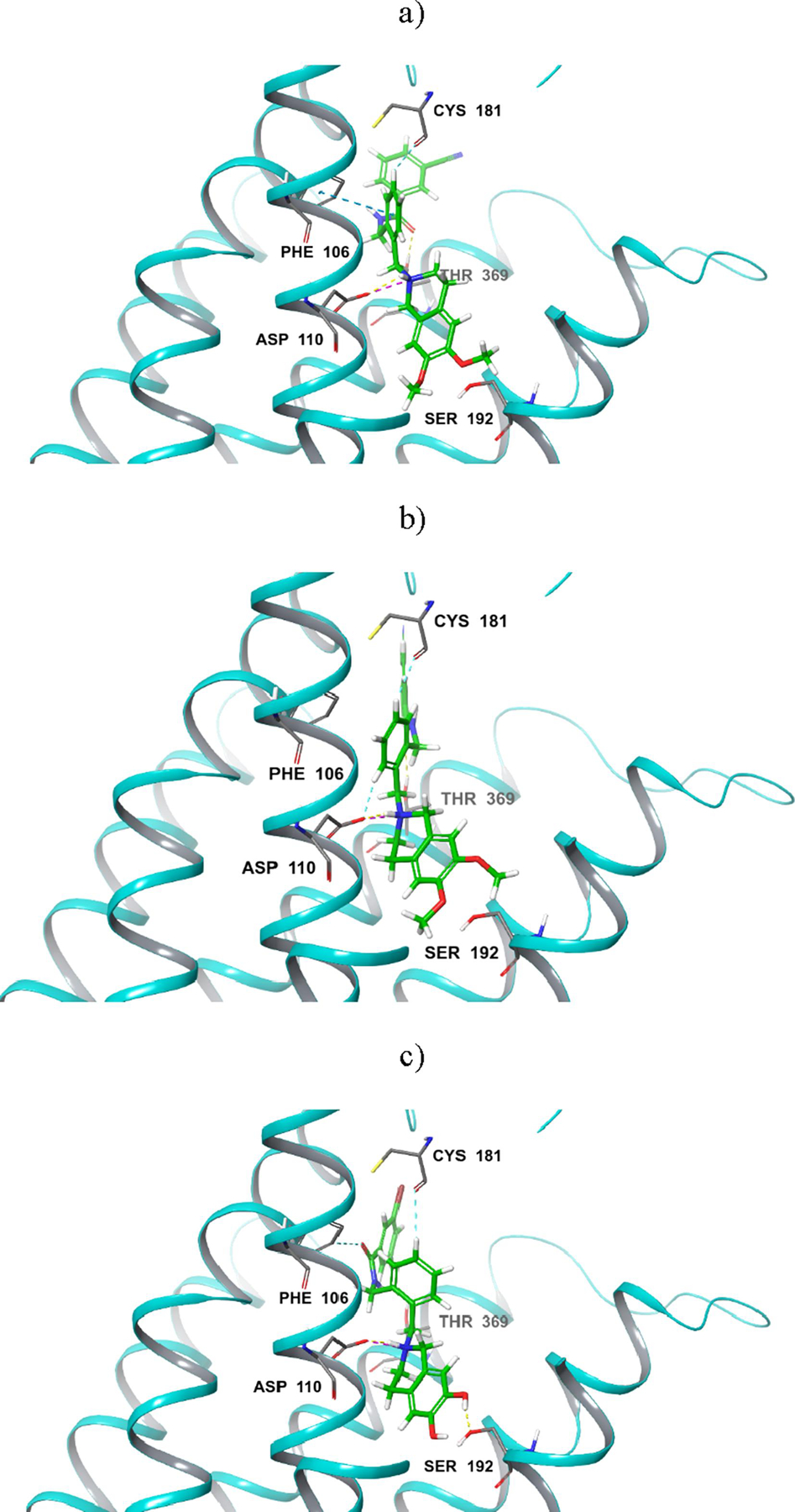
Docked poses of compounds 5s (a), 5t (b) and 6a (c) at D3R
However, while the unmethylated compound 6a makes a hydrogen-bond interaction with Ser192 as observed in the previous series, compounds 5s and 5t do not. Interestingly, these compounds offset the lack of hydrogen-bond interactions in the orthosteric pocket with an interaction between the linker and regions further up the D3R binding cavity. In particular, in 5s and 5t there is an interaction between Cys181 of D3R and the newly introduced phenyl ring of these analogues (via an aromatic H-bond interaction). A similar interaction with the introduced phenyl ring is also seen in 6a. In compound 5s, the phenyl ring also forms a π-π interaction with Phe106. In addition, the positioning of the phenyl ring observed for 5s and 5t enables additional hydrogen-bonds between the carbonyl of the aryl amide moiety and Thr369 (see Figure 3a and 3b). Such interactions do not occur with compound 6a probably because it is shifted deeper into the orthosteric pocket to establish the hydrogen bond with Ser192 as mentioned above.
Overall, the molecular docking models developed here offer useful structural interpretations of the observed affinity of the compounds with the rigid linkers. The drawback of having the o-xylenyl linker is the decrease in selectivity. The orthosteric binding sites of D2R and D3R are highly conserved, but the secondary binding pocket of these receptors differ significantly. Interactions with the D3R secondary binding pocket are important for achieving D3R versus D2R selectivity.26, 27 For compounds with both a 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline motif, the rigid o-xylenyl linker present in this new series does not allow the arylamide secondary pharmacophore of the molecules to make strong contacts with residues in the secondary binding pocket. This diminished binding of the secondary pharmacophore in the secondary binding pocket may be responsible for the lowered D3R versus D2R selectivity in this series.
In conclusion, we examined the effect of rigidification of the linker region of tetrahydroisoquinoline-containing D3R antagonist chemotypes via the inclusion of an o-xylenyl linker motif. Various oxygenated tetrahydroisoquinoline motifs and benzamide sub-structures were utilized for the primary and secondary pharmacophore regions respectively.
We found that in general, rigidification with an o-xylenyl ring is detrimental to D3R selectivity versus D2R. However, selectivity versus the σ2R seems to be dependent on the benzamide motif employed; 4-subsituted benzamide groups in particular afforded compounds with excellent selectivity versus σ2R (i.e. low or no affinity for σ2R). In compounds with a 6-methoxy-1,2,3,4,-tetrahydroisoquinolin-7-ol primary pharmacophore group, D3R affinity was negatively impacted. However, we identified compounds with 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline primary pharmacophore groups with high D3R affinity. Overall, in this series of o-xylenyl ring rigidified compounds, it appears that the choice of the primary pharmacophore group is critical for D3R affinity while the benzamide secondary pharmacophore region is important for maintaining D3R affinity as well as selectivity versus σ2R. Compounds 5s and 5t were among the most potent D3R ligands identified in this study. Docking studies indicate that the high D3R affinity of some compounds may be due to the extra interactions of the phenyl ring of the linker with, especially, Cys181.
Supplementary Material
Acknowledgments
This publication was made possible by Grant Number 1SC1DA049961 from the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions. Ki determinations, and receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. For experimental details please refer to the PDSP website http://pdsp.med.unc.edu/ and click on “Binding Assay” or “Functional Assay” on the menu bar. The authors thank Tom Kurtzman (Lehman College, CUNY) for computational support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Vallone D, Picetti R, Borrelli E. Structure and function of dopamine receptors. Neurosci Biobehav Rev. 2000;24(1): 125–132. [DOI] [PubMed] [Google Scholar]
- 2.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78(1): 189–225. [DOI] [PubMed] [Google Scholar]
- 3.Nishi A, Kuroiwa M, Shuto T. Mechanisms for the modulation of dopamine d(1) receptor signaling in striatal neurons. Front Neuroanat. 2011;5: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. 2010;68(5): 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cools R Dopaminergic control of the striatum for high-level cognition. Curr Opin Neurobiol. 2011;21(3): 402–407. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich EV, Joyce JN. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology. 1999;20(1): 60–80. [DOI] [PubMed] [Google Scholar]
- 7.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci. 2010;1187: 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maramai S, Gemma S, Brogi S, et al. Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front Neurosci. 2016;10: 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakajima S, Gerretsen P, Takeuchi H, et al. The potential role of dopamine D(3) receptor neurotransmission in cognition. Eur Neuropsychopharmacol. 2013;23(8): 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sokoloff P, Le Foll B. The dopamine D3 receptor, a quarter century later. Eur J Neurosci. 2017;45(1): 2–19. [DOI] [PubMed] [Google Scholar]
- 11.Sokoloff P, Diaz J, Le Foll B, et al. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord Drug Targets. 2006;5(1): 25–43. [DOI] [PubMed] [Google Scholar]
- 12.Micheli F Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem. 2011;6(7): 1152–1162. [DOI] [PubMed] [Google Scholar]
- 13.Mach RH, Huang Y, Freeman RA, Wu L, Vangveravong S, Luedtke RR. Conformationally-flexible benzamide analogues as dopamine D3 and sigma 2 receptor ligands. Bioorg Med Chem Lett. 2004;14(1): 195–202. [DOI] [PubMed] [Google Scholar]
- 14.Lee I, Lieberman BP, Li S, Hou C, Makvandi M, Mach RH. Comparative evaluation of 4 and 6-carbon spacer conformationally flexible tetrahydroisoquinolinyl benzamide analogues for imaging the sigma-2 receptor status of solid tumors. Nucl Med Biol. 2016;43(11): 721–731. [DOI] [PubMed] [Google Scholar]
- 15.Hou C, Hsieh CJ, Li S, et al. Development of a Positron Emission Tomography Radiotracer for Imaging Elevated Levels of Superoxide in Neuroinflammation. ACS Chem Neurosci. 2018;9(3): 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lever JR, Miller DK, Green CL, et al. A selective sigma-2 receptor ligand antagonizes cocaine-induced hyperlocomotion in mice. Synapse. 2014;68(2): 73–84. [DOI] [PubMed] [Google Scholar]
- 17.Hajipour AR, Guo LW, Pal A, Mavlyutov T, Ruoho AE. Electron-donating para-methoxy converts a benzamide-isoquinoline derivative into a highly Sigma-2 receptor selective ligand. Bioorg Med Chem. 2011;19(24): 7435–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan KH, Lever JR, Lever SZ. Effect of structural modification in the amine portion of substituted aminobutyl-benzamides as ligands for binding sigma1 and sigma2 receptors. Bioorg Med Chem. 2011;19(6): 1852–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadhiya S, Cordone P, Pal RK, et al. New Dopamine D3-Selective Receptor Ligands Containing a 6-Methoxy-1,2,3,4-tetrahydroisoquinolin-7-ol Motif. ACS Med Chem Lett. 2018;9(10): 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luedtke RR, Perez E, Yang SH, et al. Neuroprotective effects of high affinity Sigma1 receptor selective compounds. Brain Res. 2012;1441: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Ding K, Levant B, Wang S. Design of novel hexahydropyrazinoquinolines as potent and selective dopamine D3 receptor ligands with improved solubility. Bioorg Med Chem Lett. 2006;16(2): 443–446. [DOI] [PubMed] [Google Scholar]
- 22.Hackling A, Ghosh R, Perachon S, et al. N-(omega-(4-(2-methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as dopamine D2 and D3 receptor ligands. J Med Chem. 2003;46(18): 3883–3899. [DOI] [PubMed] [Google Scholar]
- 23.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. N-(4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg Med Chem Lett. 2003;13(13): 2179–2183. [DOI] [PubMed] [Google Scholar]
- 24.Grundt P, Carlson EE, Cao J, et al. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J Med Chem. 2005;48(3): 839–848. [DOI] [PubMed] [Google Scholar]
- 25.Pal RK, Gadhiya S, Ramsey S, et al. Inclusion of enclosed hydration effects in the binding free energy estimation of dopamine D3 receptor complexes. PLoS One. 2019;14(9): e0222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chien EY, Liu W, Zhao Q, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007): 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newman AH, Beuming T, Banala AK, et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem. 2012;55(15): 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.