

Abstract
Background:
Tissue factor pathway inhibitor (TFPI) is an anticoagulant protein required for murine embryonic development. Intrauterine lethality of Tfpi−/− mice occurs at mid- and late gestation, the latter of which is associated with severe cerebrovascular defects. Megakaryocytes produce only the TFPIα isoform, which is stored within platelets and released upon activation.
Objectives:
To examine biological activities of platelet TFPIα (pTFPIα) by characterizing effects of pTFPIα overexpression in Tfpi−/− mice.
Methods:
Transgenic mice overexpressing pTFPIα were generated and crossed onto the Tfpi−/− background. Genetic and histological analyses of embryos were performed to investigate the function of pTFPIα during embryogenesis.
Results:
The transgene (Tg) increased pTFPIα 4- to 5- fold without altering plasma TFPI in adult Tfpi+/+ and Tfpi+/− mice but did not rescue Tfpi−/− mice to wean. Analyses of the impact of pTFPIα overexpression on Tfpi−/− survival, however, were complicated by linkage between the Tg integration site and the endogenous Tfpi locus on chromosome 2. Strain-specific genetic interactions also modulated Tfpi−/− embryonic survival. After accounting for these underlying genetic factors, pTFPIα overexpression completely suppressed mid-gestational lethality of Tfpi−/− embryos but had no effect on development of cerebrovascular defects during late gestation resulting in their lack of survival to wean.
Conclusions:
pTFPIα overexpression rescued Tfpi−/− embryos from mid-gestational but not late gestational lethality. The prevalence of underlying genetic factors complicating analyses within our study illustrates the importance of meticulously characterizing transgenic mouse models to avoid spurious interpretation of results.
Keywords: TFPI, platelets, transgene, embryogenesis
1 |. INTRODUCTION
Tissue factor pathway inhibitor (TFPI) is an essential anticoagulant protein1 that decreases the propensity for formation of intra-vascular thrombi2 and is a key modulator of bleeding severity in people with hemophilia.3,4 TFPI is an alternatively spliced Kunitz-type protease inhibitor with multiple anticoagulant activities that is present in plasma,5 on endothelium,6–8 and within platelets.9,10 It is expressed as two isoforms in humans: TFPIα and TFPIβ. In addition, mice express a third isoform, TFPIγ.11 In mice, TFPIα is the major isoform expressed by megakaryocytes and present in platelets,10 TFPIβ is the major isoform on endothelium in tissue vascular beds,12 and TFPIγ is the major isoform in plasma.13 In addition to its location in platelets, TFPIα is a heparin-releasable protein and is detectable in mouse plasma (~20% of total plasma TFPI) following heparin infusion.12 All isoforms are capable of inhibiting the tissue factor-factor VIIa (TF-FVIIa) catalytic complex and factor Xa (FXa) via the first (K1) and second (K2) Kunitz domains, respectively.11,14–16 TFPIα differs from the other isoforms because it has a C-terminal region encoding a third Kunitz domain (K3), which binds to protein S,17 and basic C-terminus, which is necessary for rapid inhibition of FXa by K2.18–20 The TFPIα basic C-terminus also binds to partially activated forms of factor V (FV) allowing for rapid binding of the FXa active site by K2 in forms of the FXa-FVa catalytic complex (prothrombinase) that assemble early in the procoagulant response, before thrombin generation.21,22 Thus, TFPIα is unique among the TFPI isoforms in that it is present within platelets and can inhibit prothrombinase.
Platelet TFPIα (pTFPIα) is an anticoagulant molecule within a procoagulant cell that is delivered directly to sites of vascular injury.23 In humans, 7% to 10% of TFPI in whole blood is in platelets.9,10 In a mouse vascular injury model, pTFPIα dampened thrombus growth by limiting platelet accumulation.24 The functional importance of pTFPIα is further demonstrated in mice with hemophilia, where its absence resulted in decreased blood loss following tail clip and increased accumulation of both platelets and fibrin following vascular injury.25
TFPI is required for successful embryogenesis. Mice lacking the TFPI K1 domain (Tfpitm1Gjb;Tfpi−/−) on a mixed 129-C57BL/6 genetic background succumb to embryonic lethality in two stages with about 60% dying during mid-gestation (E9.5-E11.5), and the remaining 40% during late gestation (E12.5–E18.5).26 Our laboratory recently described abnormal cerebrovascular development during late gestation in Tfpi−/− mice.27 Interestingly, cerebrovascular development is normal in Tfpi−/− embryos lacking endogenous FV, indicating that regulation of FV-dependent procoagulant activity by TFPI is necessary for proper cerebrovascular development.27
A transgenic mouse model expressing TFPIα under control of the platelet-specific Gp1ba promoter was generated to further define biological activities of pTFPIα. The transgene (Tg) was bred onto the Tfpitm1Gjb genetic background to examine how overexpression of pTFPIα alters embryonic survival of mice lacking functional TFPI.
2 |. METHODS
2.1 |. Generation of platelet-specific TFPIα transgenic mice and breeding strategy
The Institutional Animal Care and Use Committee at Medical College of Wisconsin approved all mouse experiments. The megakaryocyte-specific expression vector possessing the murine Gp1ba proximal promoter28,29 (gift from Mortimer Poncz, Children’s Hospital of Philadelphia) was digested with NotI and complementary DNA (cDNA) of the murine TFPIα coding sequence (Refseq: NM_011576.1) was subcloned into the vector. The 3.9-kb partial fragment containing the Gp1ba-Tfpi(α) Tg construct was isolated from the vector backbone by SalI digestion. The DNA was purified, sequenced, and injected into C57BL/6NCrl fertilized oocytes (strain code: 027; Charles River) at the Medical College of Wisconsin Transgenic Animal Model Core to generate G0 transgenic mice. Tg positive (Tg+) and negative (Tg0) genotypes were determined using TgF/TgR primers (Figure 1A; Table S1). Hemizygous Tfpi+/+ Tg+ G0 founders were bred to B6.129-Tfpitm1Gjb/AMast mice that had been backcrossed >10 generations (>N10) with C57BL/6 J (B6 J; stock #000664; The Jackson Laboratory). The Tg was maintained in a hemizygous state. The Tg and endogenous Tfpi+ and Tfpitm1Gjb (Tfpi−) alleles were differentiated using custom K1F, K1R1, and K1R2 primers (Figure 1B; Table S1).
FIGURE 1.
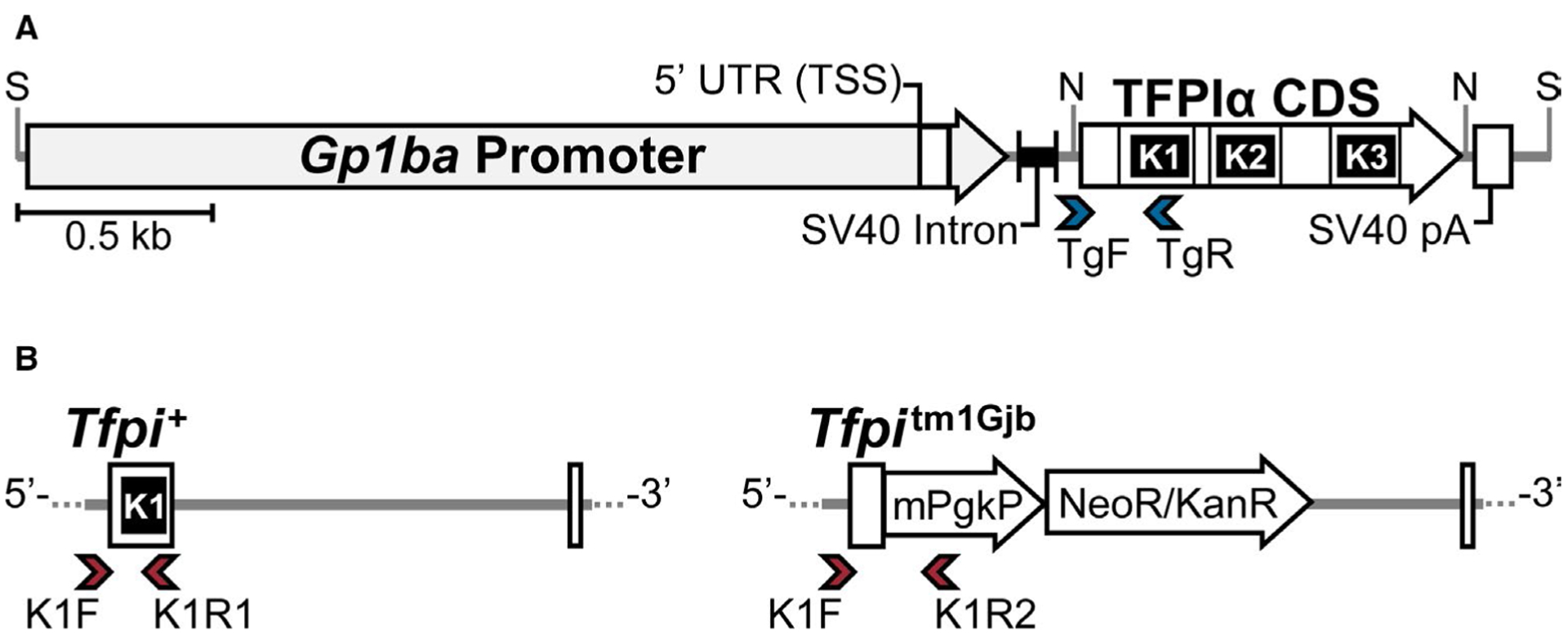
Schematic of the linearized Gp1ba-Tfpi(α) construct and methods to discriminate between Tfpi sequences within the genome. (A) The construct containing the murine Gp1ba proximal promoter sequence (2.3 kb) and TFPIα coding sequence (CDS; 918 bp) is drawn to scale with the positions of SalI (S) and NotI (N) restriction sites indicated. Location of the transcription start site (TSS) is shown by the left boundary of the Gp1ba 5′ untranslated region (UTR). The exon–exon junctions of the included TFPIα sequence and relative locations of the three Kunitz domains are shown. The Tg primer set (blue arrowheads) used for polymerase chain reaction genotyping amplifies a 300-bp product with the TgF primer located within the vector backbone downstream of the SV40 intronic sequence and the TgR primer located within the region encoding the K1 domain of the Tfpi sequence. SV40 pA indicates the Simian virus 40 polyadenylation signal. (B) Alleles were genotyped using a three-primer strategy (red arrowheads) to differentiate the Tg from the endogenous Tfpi locus with the common forward primer (K1F) located within the intron upstream of the exon encoding K1 that is absent in the Tg, and two reverse primers. The first (K1R1) is specific for the Tfpi+ wild-type allele (214 bp amplicon) and the second (K1R2) is specific for the mPgk/neo cassette insertion of the Tfpitm1Gjb (Tfpi−) allele (334 bp amplicon). TFPI, tissue factor pathway inhibitor; Tg, transgene
2.2 |. Plasma and platelet isolation
Whole blood was collected from the inferior vena cava into 3.2% citrate (10% vol/vol). Platelet poor plasma was prepared by consecutive 10-min centrifugation at 3000 and 9000g. Platelet lysates were prepared from platelet rich plasma from two genetically identical mice and pooled by collecting the upper 1/3 of whole blood after centrifugation (100g, 5 min), followed by centrifugation of each platelet-enriched plasma fraction (100g, 5 min). Platelets were pelleted (700g, 10 min), washed 3x with phosphate buffered saline and lysed by repeated freeze thaw (5x) followed by sonication (Bioruptor Pico sonicator; Diagenode Inc).
2.3 |. Tissue factor/factor VIIa initiated factor Xa activity assays
Assays were performed on plasma (1:200) and platelet lysates standardized by bicinchoninic acid assay (ThermoScientific) to 750 μg total protein/ml. Standard curves were generated using murine rTFPIα for platelet samples and B6 J wild-type plasma diluted with TFPI-deficient plasma obtained from Tfpi−/− F2rl3−/− mice30 for plasma samples. Samples were incubated for 20 min with 1:500 rabbit brain cephalin (Pel-Freez Biologicals), 0.001 nM human FVIIa (Novo Nordisk A/S, Bagsvaerd, DK), and 1:4000 human TF (Siemens). Spectrozyme FXa (0.5 mM, Sekisui Diagnostics) was added and reactions initiated with 20 nM human FX (Enzyme Research Labs). FXa production (Vmax) was measured at 405 nm for 1 h using softMAX pro ver4.3.1. TFPI inhibitory activity as fmoles TFPI/mg total platelet protein or percent B6 J wild-type plasma activity were interpolated using GraphPad Prism 8 ver8.0.0 using nonlinear regression parameters.
2.4 |. Mouse plasma TFPI enzyme-linked immunosorbent assay
Mouse total plasma TFPI (1:4000) was measured by enzyme-linked immunosorbent assay (ELISA) as previously described.24
2.5 |. RNA extraction, cDNA synthesis, and reverse transcriptase-polymerase chain reaction
Bone marrow and phosphate buffered saline–perfused tissues were harvested from Tfpi+/− Tg+ mice and stored in RNAlater (Sigma-Aldrich) at −80°C until homogenization in Trizol reagent (Invitrogen/Thermo Fisher Scientific) followed by phase separation. Total RNA was extracted from the resulting supernatants using the RNeasy Mini Kit (Qiagen) with the inclusion of genomic DNA (gDNA) digestion using the RNase-Free DNase Set (Qiagen). The concentration and purity of gDNA-free total RNA were determined by Nanodrop (ThermoFisher Scientific) and 1 μg was reverse transcribed using SuperScript VILO cDNA Synthesis Kit (Life Technologies). After first strand cDNA synthesis, transcripts were amplified using rt-Tg and rt-Tfpi primers listed in Table S1.
2.6 |. Determination of the Tg integration site
The gDNA was extracted from Tfpi+/− Tg+ mouse spleen by phenol:chloroform precipitation. The Tg integration site was determined using custom GSP1/GSP2 primers (Table S1) and the Universal GenomeWalker 2.0 kit (Takara Bio USA, Inc.). Bidirectional Sanger sequencing was performed by Retrogen Inc.. Sequences were aligned to the Tg sequence and the GRCm38 reference assembly using the National Center for Biotechnology Information BLASTn suite and SnapGene ver4.3.10 (GSL Biotech LLC). The Tg integration site was confirmed by polymerase chain reaction (PCR) using TgChr2 and Cpxm1 primer sets (Table S1). The physical position (Mb) of the Tg and Tfpi loci were converted to genetic position (cM) by linear interpolation on a standard genetic map for laboratory mice with male-specific parameters using the Mouse Map Converter tool (Jackson Laboratory, http://cgd.jax.org/mousemapconverter/)31,32
2.7 |. Histology and immunohistochemistry
Embryos from timed matings were aged as described previously.27 Embryos were fixed in 10% formalin, sectioned, paraffin embedded, and stained with Gomori’s trichrome. Immunohistochemistry was performed using anti-laminin-1 primary antibody (Sigma-Aldrich). For each genotype, three to seven embryos were randomly selected and provided to the pathologist who was blinded to genotype.
2.8 |. Statistical analyses
Statistical differences among mouse progeny were determined using Fisher exact test or goodness-of-fit tests (χ2 or exact multinomial and binomial tests using the method of small p values), as appropriate. For quantitative assays, Kruskal-Wallis test and post hoc analyses using the Wilcoxon signed-rank test were performed. The p values were adjusted for multiple comparisons using the Benjamini and Hochberg false discovery rate (FDR) with significance reported as the adjusted q value. Statistical analyses were performed in R ver3.6.3 using the base and XNomial packages with an alpha level of 0.05. Calculations for expected mouse progeny distributions are provided within the Supporting Information.
3 |. RESULTS
3.1 |. Generation of pTFPIα transgenic mice
The Gp1ba-Tfpi(α) linearized construct was microinjected into the pronuclei of fertilized eggs and transferred into pseudopregnant surrogates producing 23 G0 mice. Four of these founder mice (two males and two females) were positive for Tg integration (Tg+) by PCR genotyping. Independent breeding lines were initiated by outcrossing the hemizygous G0 founders to heterozygous Tfpitm1Gjb/AMast (Tfpi+/−) mice to produce F1 hybrids. Germline transmission did not occur in line 2 progeny. The Tg+ F1 progeny from lines 1, 3, and 4 were backcrossed by mating Tfpi+/+ Tg+ or Tfpi+/− Tg+ mice to Tfpi+/+ and Tfpi+/− mice. Paternal transmission of the Tg+ allele was maintained throughout line 3 and 4 pedigrees that were initiated by male founders. Distortion of sex ratios in lines 1 and 4 became apparent by generations N3–4. In line 1, approximately two males for every female were observed (confidence interval, 56.2%–71.6%; p = .0004). In line 4, approximately six females for every male were observed (confidence interval, 4.0%–32.7%; p = .0002). Equal frequencies of males and females were observed in line 3 progeny (Table S2), and further analyses were conducted with this line.
3.2 |. The pTFPIα Tg did not rescue the Tfpi−/− embryonic lethal phenotype
Homozygosity of the Tfpi− allele results in complete embryonic lethality on mixed and congenic C57BL/6 genetic backgrounds.25,26,33 Of 684 offspring from line 3, 386 were derived by backcrossing Tfpi+/− Tg+ males to Tfpi+/− females for 11 generations. The remaining 298 mice were derived from Tfpi+/− Tg0 × Tfpi+/− Tg+ N6 intercrosses (21 total) or were noninformative (e.g., the 26 F1 hybrids or the 251 progeny derived from backcrosses involving a parental Tfpi+/+ genotype). Of the 407 informative progeny from breeding heterozygous Tfpi females to Tfpi+/− Tg+ males, no Tfpi−/− Tg+ or Tfpi−/− Tg0 mice were observed at weaning (p = .3119; Table 1).
TABLE 1.
Progeny distribution from Tfpi+/− × Tfpi+/− Tg+ (female × male) crosses at wean
| Genotype | % Exp Zygotes | % Exp at Weaning | No. Obs | % Obs |
|---|---|---|---|---|
| Tfpi+/+ Tg0 | 12.5 | 16.7 | 73 | 17.9 |
| Tfpi+/+ Tg+ | 12.5 | 16.7 | 49 | 12.0 |
| Tfpi+/− Tg0 | 25.0 | 33.3 | 144 | 35.4 |
| Tfpi+/− Tg+ | 25.0 | 33.3 | 141 | 34.6 |
| Tfpi−/− Tg0 | 12.5 | 0 | 0 | 0 |
| Tfpi−/− Tg+ | 12.5 | 0 | 0 | 0 |
Abbreviations: Exp, Expected; Obs, Observed; Tg0, transgene negative; Tg+, transgene positive.
3.3 |. pTFPIα transgenic mice had increased platelet TFPI inhibitory activity and normal plasma TFPI
Tg expression was examined by tissue reverse transcriptase-PCR analysis. The pTFPIα transgenic message was present only in bone marrow (Figure S1), indicating megakaryocyte-specific transcription consistent with previous studies using the Gp1ba promoter-driven expression vector.28 Tg function was examined by measurement of TFPI in platelets and plasma of line 3 mice. Tfpi+/+ Tg+ mice had 4- to 5- fold increased TFPI inhibitory activity with 55.9 ± 14.1 fmoles TFPI/mg total platelet protein compared with 13.8 ± 4.6 in Tfpi+/+ Tg0 littermates (q = 1.6 × 10−4; Figure 2A). Similarly, Tfpi+/− Tg+ mice had 40.8 ± 16.5 fmoles TFPI/mg total platelet protein compared with 8.0 ± 1.6 in Tfpi+/− Tg0 littermates (q = 1.7 × 10−4). Plasma TFPI activity (Figure 2B) and total antigen (Figure 2C) were unaltered in Tg+ mice regardless of Tfpi zygosity. Thus, TFPI produced in platelets did not alter TFPI in mouse plasma.
FIGURE 2.

pTFPIα transgenic mice have increased platelet TFPI inhibitory activity and unaltered plasma TFPI. (A,B) Tissue factor/factor VIIa initiated factor Xa generation assays. (A) TFPI inhibitory activity in platelet lysates from N7 mice expressed as fmoles TFPI/mg total platelet protein using recombinant mouse TFPIα as standard. (B) TFPI inhibitory activity in plasma from N2–6 mice expressed as % wild-type (WT) plasma activity using B6 J plasma diluted with Tfpi−/−F2rl3−/− plasma as standard. (C) TFPI antigen enzyme-linked immunosorbent assay using plasma from N2–6 mice. (A-C) Plots are displayed as the mean ± standard deviation. *q < 0.001. pTFPIα, platelet TFPIα; TFPI, tissue factor pathway inhibitor
3.4 |. Genetic interactions modulated the Tfpi−/− embryonic lethal phenotype
Embryos (line 3, N7–9) from 17 Tfpi+/− × Tfpi+/− Tg+ matings were harvested between E14.5 and E18 (Table 2). Tfpi alleles from these 120 late-gestation embryos displayed normal Mendelian distribution with 28% Tfpi+/+, 49% Tfpi+/−, and 23% Tfpi−/−. The high percentage of Tfpi−/− embryos was unexpected because it was reported that only ~5% survive to late gestation when on a mixed 129 × C57BL/6 genetic background.26 Because the line 3 embryos analyzed here were ≥99% C57BL/6 J, we hypothesized that mouse strain effects as well as the Tg influenced Tfpi−/− gestational viability. To investigate these hypotheses, we first examined embryos derived from Tfpi+/− × Tfpi+/− intercrosses on the congenic C57BL/6 J background (Table 3). Of 359 embryos harvested between E14.5 and E16.5, 14% were Tfpi−/−. This is significantly fewer than the expected 25% Mendelian frequency (q = 1.2 × 10−6) and significantly more than the 5% frequency on the mixed background (q = 8.4 × 10−11). Thus, Tfpi−/− embryos were more likely to survive to late-stage gestation on the C57BL/6 J background than on the mixed 129 × C57BL/6 background. Additionally, it appeared that pTFPIα overexpression further ameliorated Tfpi−/− mid-stage embryonic lethality, which was investigated next.
TABLE 2.
Progeny distribution Tfpi+/− × Tfpi+/− Tg+ (female × male) crosses at E14.5–18
| Genotype | % Exp zygotes | % Exp E14.5–18 | No. Obs | % Obs | 95% CIa | q Valuea |
|---|---|---|---|---|---|---|
| Tfpi+/+ Tg0 | 18.6 | 21.3 | 26 | 21.7 | 14.8–30.1 | 0.999 |
| Tfpi+/+ Tg+ | 6.4 | 7.4 | 7 | 5.8 | 2.4–11.6 | 0.999 |
| Tfpi+/− Tg0 | 25.0 | 28.7 | 34 | 28.3 | 20.5–37.3 | 0.999 |
| Tfpi+/− Tg+ | 25.0 | 28.7 | 25 | 20.8 | 14.0–29.2 | 0.206 |
| Tfpi−/− Tg0 | 6.4 | 3.6 | 4 | 3.3 | 0.9–8.3 | 0.999 |
| Tfpi−/− Tg+ | 18.6 | 10.4 | 24 | 20.0 | 13.3–28.3 | 0.009b |
Abbreviations: CI, confidence interval; Exp, Expected; Obs, Observed; Tg0, transgene negative; Tg+, transgene positive.
Post hoc exact binomial test of individual genotypes at E14.5 to E18 and false discovery rate–adjusted probabilities.
Significant.
TABLE 3.
Genotypic distribution of embryonic progeny derived from Tfpi+/− × Tfpi+/− crosses on the C57BL/6 J genetic background
| Day of gestation | No. of embryos | Tfpi genotypes Obs (% of Total) | |||||
|---|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | |||||
| 14.5 | 240 | 74 | (31%) | 132 | (55%) | 34 | (14%) |
| 15.5 | 69 | 23 | (33%) | 36 | (52%) | 10 | (14%) |
| 16.5 | 50 | 20 | (40%) | 23 | (46%) | 7 | (14%) |
| Total | 359 | 117 | (33%) | 191 | (53%) | 51 | (14%)a,b |
Abbreviations: FDR, false discovery rate; Obs, Observed.
q-value = 1.2 × 10−6. FDR-adjusted probability from exact binomial test comparing Tfpi−/− genotypes observed at E14.5 to E16.5 to the 25% Mendelian probability.
q value = 8.4 × 10−11. FDR-adjusted probability from exact binomial test comparing Tfpi−/− genotypes observed at E14.5 to E16.5 to the 5% probability reported in Huang et al.26
3.5 |. The pTFPIα Tg and the endogenous Tfpi locus were linked in line 3
Of 28 Tfpi−/− embryos, 86% were Tg+ and 14% were Tg0, which is consistent with the Tg promoting viability to late gestation. Unexpectedly, only 21% of the 33 Tfpi+/+ embryos were Tg+ (50% expected), which suggested the Tg caused lethality in Tfpi+/+ embryos. This was perplexing because the overall transmission of the Tg was not distorted (Table 2), the Tfpi genotypes were in Mendelian proportions (Table 2), and this cross yielded expected numbers of Tfpi+/+ Tg+ weaning-age progeny (Table 1). We hypothesized the Tg integration site and the endogenous Tfpi locus on chromosome 2 were linked resulting in nonindependent assortment between the Tg and Tfpi loci. The location of the Tg integration site was determined using PCR-based DNA walking (Figure S2). Analysis of genomic DNA from an N9 Tfpi+/− Tg+ mouse revealed that the 3′ end of the Tg randomly integrated into a 227-kb intergenic region on mouse chromosome 2 at 47 996 318 bp (Figure 3A,B). Integration at this site was confirmed via PCR amplification over the Tg-genome junction (Figure 3C). This site is approximately 36.4 Mb upstream of the Tfpi gene, and the genetic distance between these two loci is approximately 21.4 cm.31,32 Thus, the DNA walking experiments revealed that the endogenous Tfpi gene and the pTFPIα Tg were linked with cross-over events predicted to occur in 21.4% of male gametes. Therefore, line 3 progeny genotype probabilities were non-Mendelian and dependent upon the recombination rate and whether the Tg+ allele in male sires was in phase with the Tfpi+ or the Tfpi− allele.
FIGURE 3.

The Tg integrated into an intergenic region on mouse chromosome 2. (A) Ideogram of mouse chromosome (Chr) 2. Lower scale represents physical map positions (Mb) with the upper hashmarks indicating the endogenous Tfpi locus at 84.4 Mb and the integration site (X) for the 3′ end of the Tg at 48.0 Mb. (B) RefSeq genes in the 1 Mb region (Chr2:47481416–484881416) surrounding the Tg integration site (X) on the GRCm38 reference assembly. This integration site is flanked in closest proximity by predicted genes Gm52576 located 162 kb upstream and Gm13481 located approximately 65 kb downstream. Transcription start sites for the nearest known protein-coding genes Zeb2 and Acvr2a are approximately 2.9 Mb upstream and 0.8 Mb downstream, respectively (not shown). (C) Upper panel: schematic displaying primers used to amplify the 698-bp region spanning the junction between Chr 2 and the final truncated SV40 polyA site of the Tg concatemer (pA*; see Figure S2). TgChr2F is an exon–exon junction spanning primer between the exons encoding K2 and the linker region before K3 of the TFPIα coding sequence. The TgChr2R primer is located within the endogenous intergenic region, 143-bp distal to the Tg-Chr2 junction at 47996318 bp on Chr 2. Lower panel: PCR amplified products from Tfpi+/− Tg+ and Tfpi+/+ Tg0 gDNA obtained at wean and the N9 mouse spleen genomic DNA isolated for PCR-based DNA walking. Amplicons for the Tg-Chr2 junction and the 397-bp internal amplification control using Cpxm1 primers are indicated. NTC, no template control; PCR, polymerase chain reaction; Tg, transgene
3.6 |. Overexpression of platelet TFPIα completely suppressed Tfpi−/− mid-gestation lethality
The maternal/paternal haplotype of each Tfpi+/− Tg+ male breeder was phased within the line 3 pedigree based on progeny genotype frequencies. Of the 407 weaning-age offspring (Table 1), 51% were sired by Tfpi−Tg0/Tfpi+Tg+ males and 49% were sired by Tfpi+Tg0/Tfpi−Tg+ males (Table S3). This equal distribution along with the Tfpi−/− intrauterine lethality made evidence of the linkage nearly undetectable in the weaning-age dataset. In stark contrast and by chance, 93% of the embryos in the timed mating studies were sired by males with the Tg in phase with the Tfpi− allele. Given this finding and the 21.4% recombination rate between the Tg and Tfpi loci, two separate analyses were performed to examine how the Tg impacted mid-gestational survival. First, the percent expected line 3 zygote genotypes at conception were calculated (Supporting Information) after adjusting for the phase of the transgene in the breeding males (Table 2). In this analysis, the percent expected Tfpi−/− Tg+ zygotes (18.6%) did not differ from the percent Tfpi−/− Tg+ embryos observed between E14.5 and E18, indicating that the Tg completely rescued mid-gestational lethality of Tfpi−/− embryos (q = 0.870, Table 2). Second, the percent expected embryos at E14.5 to E18 were calculated (Supporting Information), accounting for both the phase of the transgene in the breeding males and the expected late gestational survival of Tfpi−/− embryos on the C57/BL/6 J genetic background (Table 3) and are shown in Table 2. The overall genotype distribution at E14.5 to E18 was significantly different from expected at late gestation (p = .045, Table 2). This deviation was solely due to the difference in frequencies of the Tfpi−/− Tg+ genotype (20.0% vs. 10.4%; confidence interval, 13.3%–28.3%; q = 0.009). Although this second analysis used controls from a separate cross with potential for residual differences in genetic backgrounds and environmental effects between the two mouse colonies to skew results, it again indicated complete suppression of mid-gestation lethality in Tfpi−/− embryos by the Tg expression.
3.7 |. Overexpression of platelet TFPIα did not prevent the cerebrovascular defects associated with Tfpi−/− late-gestation lethality
We recently reported that Tfpi−/− late-gestation embryos display stunted brain growth, delayed development of the meninges, and a severe vascular pathology with formation of glomeruloid bodies surrounding areas of cellular death, fibrin deposition, and hemorrhage.27 Microscopic examination of brains from the line 3 N7–9 embryos revealed pathology of Tfpi−/− Tg0 and Tfpi−/− Tg+ embryos consistent with our previous findings (Figure 4A). In seven of seven randomly selected Tfpi−/− Tg+ embryos, regions of cellular death and hemorrhage surrounded by glomeruloid bodies were present within the forebrain, midbrain, hindbrain, and spinal cord. Immunohistochemistry for laminin clearly delineated the glomeruloid bodies within the E17.0 Tfpi−/− Tg0 and E16.5 Tfpi−/− Tg+ brain parenchyma (Figure 4A,B). Thus, overexpression of pTFPIα did not restore normal cerebrovascular development in Tfpi−/− embryos explaining why the Tg did not prolong survival to wean.
FIGURE 4.

The Tg did not prevent the embryonic cerebrovascular defect associated with Tfpi−/− late gestation lethality. (A) Trichrome staining (upper panel) and laminin immunohistochemistry (lower panel) of brain sagittal sections from E16.5 Tfpi+/+ Tg0, E17.0 Tfpi−/− Tg0, and E16.5 Tfpi−/− Tg+ embryos at the level of the diencephalon. Hemorrhage was depicted with trichrome staining while laminin antibody staining clearly outlined the glomeruloid bodies seen in the Tfpi−/− embryos with or without the Tg. No glomeruloid bodies, hemorrhage, or cell death was seen in the Tfpi+/+ Tg0 embryos. Arrows/arrow brackets indicate glomeruloid bodies. *hemorrhage. Bar = 200 μm. cp, choroid plexus; v, ventricle. (B) Laminin immunohistochemistry of glomeruloid bodies within Tfpi−/− Tg0 and Tfpi−/− Tg+ insets in (A). Bar = 25 μm. Tg, transgene
4 |. DISCUSSION
A transgenic mouse model overexpressing TFPIα in platelets was generated and crossed with Tfpi+/− mice. The Tg increased pTFPIα inhibitory activity 4- to 5- fold without altering plasma TFPI indicating storage of transgenic TFPIα within circulating platelets. The Tg did not rescue Tfpi−/− mice to wean or restore late gestation cerebrovascular development. However, it completely suppressed embryonic lethality at mid-gestation indicating a functional role for TFPIα within fetal platelets during this developmental stage. We encountered several pitfalls in developing the pTFPIα transgenic mice that were identified and addressed to reach these conclusions. These included the distorted male:female ratios within two of four transgenic founder lines, which suggested that Tg integration altered traits beyond TFPIα platelet-specific expression in these two lines. Further, the Tg randomly integrated into chromosome 2 and was linked to the endogenous Tfpi locus in the founder line with normal progeny sex ratios that was selected for characterization. This required extensive genetic and statistical analyses to determine the effect of pTFPIα overexpression on embryonic survival of Tfpi−/− mice.
Previous studies have shown that the Tfpi−/− lethal phenotype can be at least partially rescued by genetic deficiencies in TF,33 FVII,34 PAR4,30 or FV.35 Further, isoform-specific pools of TFPI may play pivotal roles during embryonic development as mice with the TFPI K1 domain conditionally removed with Tek/Tie2-cre and Meox2-cre Tgs survive to wean.36,37 Here, we found that the pTFPIα Tg failed to rescue any Tfpi−/− mice to wean. This result was complicated by linkage between the Tg and the endogenous Tfpi locus but was not dependent on it because 46 Tfpi−/− Tg+ zygotes from our informative crosses were expected if the Tg completely rescued to wean. Death of the Tfpi−/− mice overexpressing pTFPIα may have occurred from the cerebral vascular defects that develop in Tfpi−/− embryos during late gestation.27 The absence of an effect of the Tg on this phenotype indicates that either pTFPIα does not modulate cerebrovascular development or that the Tg did not produce sufficient TFPIα to overcome the effect.
Megakaryocytes are detected in the yolk sac at E9.5 and circulating platelets have been identified in murine embryonic vasculature at E10.5.38 CD42b+ cells have also been identified in the yolk sac at E10.5, indicating expression of GPIbα at this time point.39 The ability of the pTFPIα Tg to rescue lethality during mid-gestation suggests a potential role for TFPIα that is specific for fetal platelets because the Tg was not present in the Tfpi+/− maternal genome. However, p45NF-E2 knockout mice with severe thrombocytopenia survive to birth,40 indicating that fetal platelets are dispensable for placentation.41 Thus, fetal pTFPIα overexpression may have a compensatory role at mid-gestation that serves to circumvent the deleterious impact of global TFPI deficiency. Approximately 68% of knockout mouse lines that are lethal between E9.5 and 14.5 exhibit placental malformations,42 and mouse strain genetic backgrounds influence mid-gestation survival.43,44 Although the specific phenotypic effects of Tfpi−/− at mid-gestation remain to be defined, we did find that genetic modifiers between the C57BL/6 and 129 substrains influence the Tfpi−/− mid-gestation lethal phenotype with lower penetrance observed within embryos on the C57BL/6 J background. This finding confirms a previous report by Pedersen et al.,33 in a much larger cohort. Because penetrance of lethality differed on different backgrounds at mid-gestation but not at late gestation, it is possible that that the mechanisms leading to death in Tfpi−/− embryos at these two time points are divergent.
Random Tg integration is widely reported to distort mouse phenotypic analyses. For example, a recent study analyzing 40 highly used transgenic mouse lines found that transgenic alleles commonly produce aberrant genetic events, such as coding sequence disruption and large genomic structural variations at the integration site, which could potentially confound experimental interpretations and limit their reproducibility.45 Our study was a testament to the presence and frequency of these effects. Our observation of the sex ratio distortion in lines 1 and 4 was likely explicitly the result of integration effects because the male:female ratio differed between these two lines, whereas normal Mendelian sex ratios were observed in line 3. Because line 3 overexpressed pTFPIα 4- to 5- fold, pTFPIα overexpression itself did not affect sex ratios, a notion echoed within human studies that found no effect of sex on platelet TFPIα expression.46,47
Our description of the complexity involved in analyses when combining unknown linked alleles is a cautionary tale and a reminder that this is not a rare phenomenon. Within this report, we describe several encounters with notorious, yet often underestimated factors known to confound the phenotypic evaluation of genetic mouse models. The detail provided regarding breeding schemes, genetic analyses, and progeny mapping strategies, serves to not only reiterate the impact that Tg integration effects and strain-dependent penetrance can have on study outcomes, but also describes the course for their identification, characterization, and management needed to derive proper conclusions regarding the effects of pTFPIα overexpression in this model. In doing so, our findings have provided evidence regarding important physiological functions of TFPIα specifically within fetal platelets and new insights into the roles of this anticoagulant platelet protein in mouse embryonic development.
Supplementary Material
Essentials.
Platelets contain the anticoagulant protein Tissue Factor Pathway Inhibitor (pTFPIα).
pTFPIα transgenic mice were produced and effects on lethality of Tfpi−/− mice were examined.
Overexpression of pTFPIα completely suppressed Tfpi−/− lethality at mid-, but not late-gestation.
Tg chromosome 2 integration and linkage with endogenous Tfpi gene complicated phenotype analyses.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (HL068835 [A.E.M.]). A.E.S. was supported by training grant HL007209.
Footnotes
CONFLICT OF INTEREST
A.E.M. receives research funding from Novo Nordisk and has received honoraria for serving on Novo Nordisk Advisory Boards. The other authors have declared that no conflict of interest exists.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Rao LV, Rapaport SI. Studies of a mechanism inhibiting the initiation of the extrinsic pathway of coagulation. Blood. 1987;69:645–651. [PubMed] [Google Scholar]
- 2.Dahm A, Van Hylckama VA, Bendz B, Rosendaal F, Bertina RM, Sandset PM. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood. 2003;101:4387–4392. 10.1182/blood-2002-10-3188 [DOI] [PubMed] [Google Scholar]
- 3.Nordfang O, Valentin S, Beck TC, Hedner U. Inhibition of extrinsic pathway inhibitor shortens the coagulation time of normal plasma and of hemophilia plasma. Thromb Haemost. 1991;66:464–467. [PubMed] [Google Scholar]
- 4.Shapiro AD, Angchaisuksiri P, Astermark J, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 2019;134:1973–1982. 10.1182/blood.2019001542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novotny WF, Girard TJ, Miletich JP, Broze GJ Jr. Purification and characterization of the lipoprotein-associated coagulation inhibitor from human plasma. J Biol Chem. 1989;264:18832–18837. [PubMed] [Google Scholar]
- 6.Ameri A, Kuppuswamy MN, Basu S, Bajaj SP. Expression of tissue factor pathway inhibitor by cultured endothelial cells in response to inflammatory mediators. Blood. 1992;79:3219–3226. [PubMed] [Google Scholar]
- 7.Ott I, Miyagi Y, Miyazaki K, et al. Reversible regulation of tissue factor-induced coagulation by glycosyl phosphatidylinositol-anchored tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2000;20:874–882. 10.1161/01.atv.20.3.874 [DOI] [PubMed] [Google Scholar]
- 8.Mast AE, Acharya N, Malecha MJ, Hall CL, Dietzen DJ. Characterization of the association of tissue factor pathway inhibitor with human placenta. Arterioscler Thromb Vasc Biol. 2002;22:2099–2104. 10.1161/01.atv.0000042456.84190.f0 [DOI] [PubMed] [Google Scholar]
- 9.Novotny WF, Girard TJ, Miletich JP, Broze GJ Jr. Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood. 1988;72:2020–2025. [PubMed] [Google Scholar]
- 10.Maroney SA, Haberichter SL, Friese P, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109:1931–1937. 10.1182/blood-2006-07-037283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroney SA, Ferrel JP, Collins ML, Mast AE. Tissue factor pathway inhibitor-gamma is an active alternatively spliced form of tissue factor pathway inhibitor present in mice but not in humans. J Thromb Haemost. 2008;6:1344–1351. 10.1111/j.1538-7836.2008.03033.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maroney SA, Ferrel JP, Pan S, et al. Temporal expression of alternatively spliced forms of tissue factor pathway inhibitor in mice. J Thromb Haemost. 2009;7:1106–1113. 10.1111/j.1538-7836.2009.03454.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girard TJ, Grunz K, Lasky NM, Malone JP, Broze GJ Jr. Re-evaluation of mouse tissue factor pathway inhibitor and comparison of mouse and human tissue factor pathway inhibitor physiology. J Thromb Haemost. 2018;16:2246–2257. 10.1111/jth.14288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girard TJ, Warren LA, Novotny WF, et al. Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338:518–520. 10.1038/338518a0 [DOI] [PubMed] [Google Scholar]
- 15.Petersen LC, Bjorn SE, Olsen OH, Nordfang O, Norris F, Norris K. Inhibitory properties of separate recombinant Kunitz-type-protease-inhibitor domains from tissue-factor-pathway inhibitor. Eur J Biochem. 1996;235:310–316. 10.1111/j.1432-1033.1996.0310f.x [DOI] [PubMed] [Google Scholar]
- 16.Chang JY, Monroe DM, Oliver JA, Roberts HR. TFPIbeta, a second product from the mouse tissue factor pathway inhibitor (TFPI) gene. Thromb Haemost. 1999;81:45–49. [PubMed] [Google Scholar]
- 17.Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103:3106–3111. 10.1073/pnas.0504240103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wesselschmidt R, Likert K, Girard T, Wun TC, Broze GJ Jr. Tissue factor pathway inhibitor: the carboxy-terminus is required for optimal inhibition of factor Xa. Blood. 1992;79:2004–2010. [PubMed] [Google Scholar]
- 19.Lockett JM, Mast AE. Contribution of regions distal to glycine-160 to the anticoagulant activity of tissue factor pathway inhibitor. Biochemistry. 2002;41:4989–4997. 10.1021/bi016058n [DOI] [PubMed] [Google Scholar]
- 20.Cunningham AC, Hasty KA, Enghild JJ, Mast AE. Structural and functional characterization of tissue factor pathway inhibitor following degradation by matrix metalloproteinase-8. Biochem J. 2002;367:451–458. 10.1042/BJ20020696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor-alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci USA. 2013;110:17838–17843. 10.1073/pnas.1310444110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood JP, Petersen HH, Yu B, Wu X, Hilden I, Mast AE. TFPIalpha interacts with FVa and FXa to inhibit prothrombinase during the initiation of coagulation. Blood Adv. 2017;1:2692–2702. 10.1182/bloodadvances.2017011098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siebert AE, Mast AE. Platelet anticoagulant proteins: modulators of thrombosis propensity within a procoagulant cell. J Thromb Haemost. 2020;18:2083–2086. 10.1111/jth.14995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maroney SA, Cooley BC, Ferrel JP, Bonesho CE, Mast AE. Murine hematopoietic cell tissue factor pathway inhibitor limits thrombus growth. Arterioscler Thromb Vasc Biol. 2011;31:821–826. 10.1161/ATVBAHA.110.220293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maroney SA, Cooley BC, Ferrel JP, et al. Absence of hematopoietic tissue factor pathway inhibitor mitigates bleeding in mice with hemophilia. Proc Natl Acad Sci USA. 2012;109:3927–3931. 10.1073/pnas.1119858109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang ZF, Higuchi D, Lasky N, Broze GJ Jr. Tissue factor pathway inhibitor gene disruption produces intrauterine lethality in mice. Blood. 1997;90:944–951. [PubMed] [Google Scholar]
- 27.Maroney SA, Westrick RJ, Cleuren AC, et al. Tissue factor pathway inhibitor is required for cerebrovascular development in mice. Blood. 2021;137:258–268. 10.1182/blood.2020006054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarovoi HV, Kufrin D, Eslin DE, et al. Factor VIII ectopically expressed in platelets: efficacy in hemophilia A treatment. Blood. 2003;102:4006–4013. 10.1182/blood-2003-05-1519 [DOI] [PubMed] [Google Scholar]
- 29.Gewirtz J, Thornton MA, Rauova L, Poncz M. Platelet-delivered factor VIII provides limited resistance to anti-factor VIII inhibitors. J Thromb Haemost. 2008;6:1160–1166. 10.1111/j.1538-7836.2008.02992.x [DOI] [PubMed] [Google Scholar]
- 30.Ellery PE, Maroney SA, Cooley BC, et al. A balance between TFPI and thrombin-mediated platelet activation is required for murine embryonic development. Blood. 2015;125:4078–4084. 10.1182/blood-2015-03-633958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox A, Ackert-Bicknell CL, Dumont BL, et al. A new standard genetic map for the laboratory mouse. Genetics. 2009;182:1335–1344. 10.1534/genetics.109.105486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu EY, Morgan AP, Chesler EJ, Wang W, Churchill GA, Pardo-Manuel de Villena F. High-resolution sex-specific linkage maps of the mouse reveal polarized distribution of crossovers in male germline. Genetics. 2014;197:91–106. 10.1534/genetics.114.161653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedersen B, Holscher T, Sato Y, Pawlinski R, Mackman N. A balance between tissue factor and tissue factor pathway inhibitor is required for embryonic development and hemostasis in adult mice. Blood. 2005;105:2777–2782. 10.1182/blood-2004-09-3724 [DOI] [PubMed] [Google Scholar]
- 34.Chan JC, Carmeliet P, Moons L, et al. Factor VII deficiency rescues the intrauterine lethality in mice associated with a tissue factor pathway inhibitor deficit. J Clin Invest. 1999;103:475–482. 10.1172/JCI5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westrick RJ, Cleuren A, Martinez ND, et al. Platelet FV deficiency restores survival of TFPI null mice independently of plasma FV. Blood. 2017;130:365. 10.1182/blood.V130.Suppl_1.365.365 [DOI] [Google Scholar]
- 36.White TA, Johnson T, Zarzhevsky N, et al. Endothelial-derived tissue factor pathway inhibitor regulates arterial thrombosis but is not required for development or hemostasis. Blood. 2010;116:1787–1794. 10.1182/blood-2009-10-250910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo MM, Yang Q, Zhan M, et al. Maintaining extraembryonic expression allows generation of mice with severe tissue factor pathway inhibitor deficiency. Blood Adv. 2019;3:489–498. 10.1182/bloodadvances.2018018853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tober J, Koniski A, McGrath KE, et al. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood. 2007;109:1433–1441. 10.1182/blood-2006-06-031898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juban G, Sakakini N, Chagraoui H, et al. Oncogenic Gata1 causes stage-specific megakaryocyte differentiation delay. Haematologica. 2020. 106(4):1106–1119. 10.3324/haematol.2019.244541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81:695–704. 10.1016/0092-8674(95)90531-6 [DOI] [PubMed] [Google Scholar]
- 41.Kashif M, Hellwig A, Kolleker A, et al. p45NF-E2 represses Gcm1 in trophoblast cells to regulate syncytium formation, placental vascularization and embryonic growth. Development. 2011;138:2235–2247. 10.1242/dev.059105 [DOI] [PubMed] [Google Scholar]
- 42.Perez-Garcia V, Fineberg E, Wilson R, et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature. 2018;555:463–468. 10.1038/nature26002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Threadgill DW, Dlugosz AA, Hansen LA, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. 10.1126/science.7618084 [DOI] [PubMed] [Google Scholar]
- 44.Strunk KE, Amann V, Threadgill DW. Phenotypic variation resulting from a deficiency of epidermal growth factor receptor in mice is caused by extensive genetic heterogeneity that can be genetically and molecularly partitioned. Genetics. 2004;167:1821–1832. 10.1534/genetics.103.020495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodwin LO, Splinter E, Davis TL, et al. Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 2019;29:494–505. 10.1101/gr.233866.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winckers K, Thomassen S, Ten Cate H, Hackeng TM. Platelet full length TFPI-alpha in healthy volunteers is not affected by sex or hormonal use. PLoS ONE. 2017;12:e0168273. 10.1371/journal.pone.0168273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellery PER, Hilden I, Sejling K, et al. Correlates of plasma and platelet tissue factor pathway inhibitor, factor V, and Protein S. Res Pract Thromb Haemost. 2018;2:93–104. 10.1002/rth2.12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.