

Abstract
ATPases Associated with Diverse Cellular Activity (AAA ATPase) are essential enzymes found in all organisms. They are involved in various processes such as DNA replication, protein degradation, membrane fusion, microtubule serving, peroxisome biogenesis, signal transduction, and the regulation of gene expression. Due to the importance of AAA ATPases, several researchers identified and developed small-molecule inhibitors against these enzymes. We discuss six AAA ATPases that are potential drug targets and have well-developed inhibitors. We compare available structures that suggest significant differences of the ATP binding pockets among the AAA ATPases with or without ligand. The distances from ADP to the His20 in the His-Ser-His motif and the Arg finger (Arg353 or Arg378) in both RUVBL1/2 complex structures bound with or without ADP have significant differences, suggesting dramatically different interactions of the binding site with ADP. Taken together, the inhibitors of six well-studied AAA ATPases and their structural information suggest further development of specific AAA ATPase inhibitors due to difference in their structures. Future chemical biology coupled with proteomic approaches could be employed to develop variant specific, complex specific, and pathway specific inhibitors or activators for AAA ATPase proteins.
Keywords: AAA ATPases, Small molecule inhibitors, p97, RUVBL1/2, ATAD2
Graphical Abstract

1. Introduction
ATPases Associated with Diverse Cellular Activities (AAA ATPase proteins) are a kind of enzymes sharing a common conserved ATP-binding domain of approximately 200 to 250 amino acid residues and oligomerizes into hexamers. These ATPases belong to a functionally diverse protein family of the AAA protein superfamily of ring-shaped P-loop NTPases that catalyze the hydrolysis of a phosphate bond in adenosine triphosphate (ATP) to form adenosine diphosphate (ADP) and release inorganic phosphate. They provide energy to facilitate a variety of cellular processes that are essential for life: including protein folding [1], intracellular transport [2], protein degradation [3], initiation of DNA replication [4], DNA repair [5], DNA remodeling [6], and ion transport [7]. To date, there are 53 members in the AAA ATPase family based on the HGNC database [8], some of which have been characterized and identified as druggable targets to explore as potential treatments for some diseases such as cancer [9–11] and Alzheimer’s disease [12, 13]. Most AAA ATPases form barrel-shaped hexameric rings, where each monomer consists of one or two ATPase domains (also known as D1 and D2) together with a regulatory N-terminal domain. Each ATPase domain contains a conserved Walker A domain, Arginine finger, and Walker B domain.
Recent reviews have summarized AAA ATPase inhibitors for p97 (14,15), ATAD2 (16,17), and dynein (18,19). Wipf and co-authors reviewed the discovery, optimization, characterization, and therapeutic potential of p97 inhibitors, which were classified into D2 ATP Site Inhibitors, D1-D2 Linker region allosteric Inhibitors, covalent inhibitors, and inhibitors with unknown mechanisms of action [14]. Sui and co-authors reviewed molecular mechanism, cofactors, and small-molecule inhibitors of p97 [15]. Hussain and co-authors reviewed the functions of ATAD2 in normal physiology and cancer-etiology and discussed technical challenges rendered by ATAD2’s BRD active site and the drug discovery efforts to find small molecule inhibitors against ATAD2 [16]. Due to the specificity of ATAD2 structure, Magnuson et al. briefly reviewed ATAD2 inhibitors in the summarization of Bromodomain inhibitors [17]. Steinman et al. discussed the cellular functions and conformational dynamics of AAA ATPase (including p97, dynein, and midasin) inhibitors [18]. Additionally, Shimizu summarized a few dynein inhibitors [19]. In summary, all these reviews mainly focus on small-molecule inhibitors against p97, ATAD2, or dynein. In this review, we discussed and analyzed the three-dimension structures of the well-studied p97, RuvBL1/2, ATAD2, and spastin as well as small molecules inhibitors because structural information for inhibitor binding pocket are available. We also picked two additional AAA ATPases, midasin and dynein, because they are potential drug targets. Due to the complicated structural arrangement of six not identical AAA domains in a single polypeptide for midasin and dynein, the inhibitor developments are mainly based on cell-based screenings and future works are needed to identify binding sites of inhibitors of midasin and dynein. We proposed the binding mode for some representative inhibitors by docking studies in this review. Because the p97 inhibitors have been summarized by several reviews, herein we only include some p97 inhibitors.
2. Selected AAA ATPases
2.1. p97/VCP/Cdc48/TER ATPase
p97, also known as the Valosin-containing Protein (VCP), is a homolog of the cell division cycle protein 48 (Cdc48) in Saccharomyces cerevisiae, and of translational ER ATPase in Drosophila. In 1982, Moir and co-authors identified Cdc48 as one of the 18 cold-sensitive cell-division-cycle (cdc) mutants in Saccharomyces cerevisiae [20]. Five years later, Koller and co-authors identified a supposed valosin-containing protein and concluded that valosin is an artifact during purification [21]. Since then, the name VCP has be used widely. In 1990, Peters and co-authors discovered a 97 kDa protein from Xenopuslaevis oocytes and mapped its amino acids to VCP [22]. In 1996, Ghislain and co-authors identified that Cdc48 is required for degrading the ubiquitin-fusion-degradation substrates, which is the first reported link of Cdc48 to the protein degradation pathway [23].
p97/VCP is highly conserved and involved in many biological processes, including endoplasmic reticulum-associated degradation (ERAD), Golgi reassembly, transcription regulation, and cell cycle control [24]. p97 is mainly localized in the cytosol and also present on organelle membranes including the endoplasmic reticulum (ER), Golgi, mitochondria, and endosomes [25–29]. p97 is composed of a substrate and cofactor binding N domain followed by two AAA ATPase domains termed D1 and D2, C-terminal and could form a hexameric doublering structure [30, 31] (Figure 1). D2 has major ATPase activity at physiological temperatures and ATP concentration, whereas D1 is involved in the regulation of heat-induced ATPase activity [32] and hexamerization [33, 34]. The D1 domain binds to ATP/ADP tighter than the D2 domain and has 10-fold higher catalytic efficiency via reduction in Km [35]. There are conserved arginine finger, walker A (GPPGCGKT), and walker B (VLFFDELD) in each domain 1 and 2. The structures of p97 with truncated or full-length sequences have been resolved using x-ray diffraction, electron microscopy, and solution NMR (Table 1).
Figure 1.
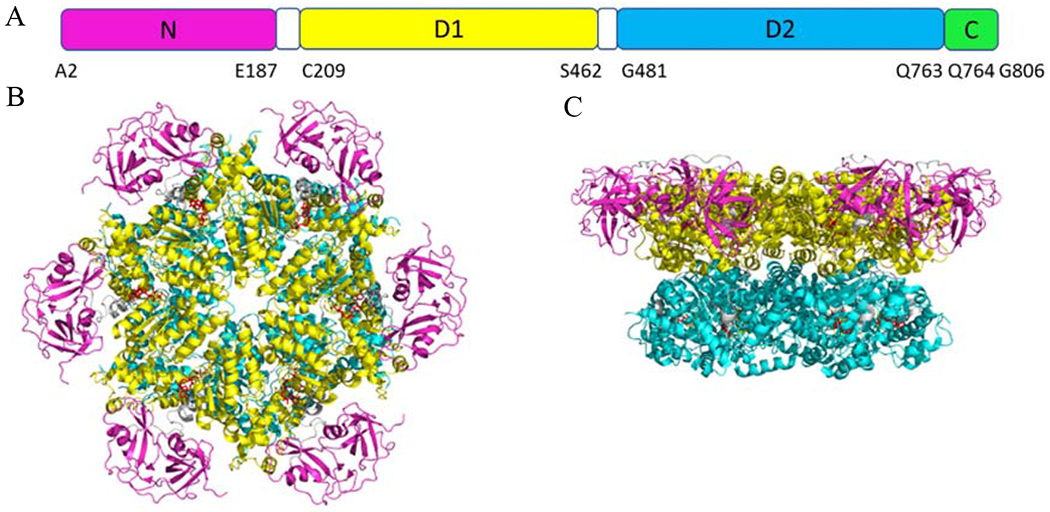
(A) Schematic diagram of the p97 domain organization. (B) Top view of the crystal structure of p97 (PDB code 5FTK). (C) Side view of p97. N domain, D1, D2, and C-terminal are presented in magenta, yellow, cyan, and green, respectively.
Table 1.
Crystal Structures of p97
| PDB Code | Methods (Å) | P97 Domains | Ligands | Organisms | References |
|---|---|---|---|---|---|
| 1E32 | X-ray (2.9) | N-D1:N21-Q458 (sequence:M1-G481) | ADP in D1 | mmu | [36] |
| 1R7R | X-ray (3.6) | N-D1-D2-C: L17-K336, A339-D609, N616-S664,D669-S702,P727-H735 (sequence: M1-G806+AALEHHHHHH) | ADP in D1 | mmu | [37] |
| 1S3S | X-ray (2.9) | N-D1:K18-Q458 (sequence: M1-Q458) | - | mmu | [38] |
| 3CF0 | X-ray (3.0) | D2#:A463-I707, V728-Q763(sequence: A463-Q763) | ADP | mmu | [39] |
| 3CF1 | X-ray (4.4) | N-D1-D2-C#: N21-I707, V728-Q763 (sequence: M1-G806) | ADP in D1 and D2 AF3 in D2 | mmu | [39] |
| 3CF2 | X-ray (3.5) | N-D1-D2-C#: N21-I582, A597-A622, N624-I645, K651-S664, A667-D669, D686-E704, V728-E730, H735-M740, E756-A759 (sequence: M1-G806) | ADP in D1, ANP in D2 | mmu | [39] |
| 3CF3 | X-ray (4.3) | N-D1-D2-C#: N21-I707, V728-Q763(sequence: M1-G806) | ADP | mmu | [39] |
| 4KOD | X-ray (3.0) | R155H N-D1*: K18-S462 (sequence: M1-G481+RSHHHHHH) | ADP in D1 | hsa | [40] |
| 5C1B | X-ray (3.1) | N-D1-D2-C: N21-G587, G594-R708, P729-S770 (sequence: A2-R708; P729-G806) | AGS in D1 and D2 | hsa | [41] |
| 5C18 | X-ray (3.3) | N-D1-D2-C: K20-G588, A596-R708, P729-G769 (sequence: A2-R708; P729-G806) | AGS in D1 and D2 | hsa | [42] |
| 5DYI | X-ray (3.7) | N-D1*: N21-P461 (sequence: M1-G481+RSHHHHHH) | ADP in D1 | hsa | [43] |
| 5FTJ | EM (2.3) | N-D1-D2-C: N21-I707, V728-Q763 (sequence: M1-G806) | ADP in D1 and D2, UPCDC30245 in the junction of D1 and D2 | hsa | [44] |
| 5FTK | EM (2.4) | N-D1-D2-C: N21, V728-Q763 (sequence: M1-G806) | ADP in D1 and D2 | hsa | [44] |
| 5FTL | EM (3.3) | N-D1-D2-C: N21-I707, V728-Q763 (sequence: M1-G806) | ADP in D1 and D2 | hsa | [44] |
| 5FTM | EM (3.2) | N-D1-D2-C: N21-I707, V728-F768 (sequence: M1-G806) | ADP in D1, AGS in D2 | hsa | [44] |
| 5FTN | EM (3.3) | N-D1-D2-C: L12-I707, V728-F768 (sequence: M1-G806) | AGS in D1 and D2 | hsa | [44] |
| 6CHS | EM (4.3) | P97-Np14 complex N-D1-D2 N215-E725, P745-A783 (sequence: M1-T819) | AGS in D1 and D2 | cthr | [45] |
| 6G2V | X-ray (1.9) | D2: V468-T549, S555-K584, A596-E712, V728-Q764 (sequence: GPG+S462-T549; S555-A585; A597-E712; V728-Q764) | ADP | hsa | [46] |
| 6G2W | X-ray (2.7) | D2: V468-T549, S555-K584, A596-E712, P727-Q763 (sequence: GPG+S462-Q764) | ADP | hsa | [46] |
| 6G2X | X-ray (2.1) | D2: V468-T549, S555-K584, A596-E712, P727-Q764 (sequence: GPG+S462-Q764) | ADP | hsa | [46] |
| 6G2Y | X-ray (2.2) | D2: V468-T549, S555-K584, A596-E712, P727-Q763 (sequence: S462-Q764) | ADP | hsa | [46] |
| 6G2Z | X-ray (1.9) | D2: V468-T549, S555-K584, A596-E712, P727-Q763 (sequence: GPG+S462-Q764) | ADP | hsa | [46] |
| 6G30 | X-ray (2.4) | D2: V468-T549, S555-K584, A596-E712, P727-Q763 (sequence: GPG+S462-Q764) | ADP | hsa | [46] |
| 6MCK | X-ray (3.8) | D1-D2*: R210-R424, V447-R586, A596-T715, E730-Q763 (sequence: M+R210-G806+RSHHHHHH) | CB5083 in D2 | hsa | [47] |
Dodecamer;
heptamer;
- no ligands.
Hsa, cthr, and mmu stand for Homo sapiens, Chaetomium thermophilum, and Mus musculus, respectively.
Human and mouse p97 amino acid sequences are identical.
PDB accessed on April 30th, 2020.
It is known that p97 was upregulated in many cancer types and associated with poor prognosis in some cancers [48, 49] and involved in neurodegenerative diseases [50]. The downregulation of p97 with siRNAs could cause endoplasmic reticulum (ER) stress and activate apoptosis through the unfolded protein response (UPR), a pathway that acts both to resolve unfolded protein stress and to trigger cell death when the buildup of such unfolded proteins becomes irresolvable [51, 52]. In addition, the downregulation of p97 could also dramatically suppress the genesis and progression of tumors in vivo [53]. Therefore, p97 has been pursued as a cancer drug target. Currently, there are many p97 inhibitors reported, including ATP competitive, uncompetitive, and non-competitive inhibitors, some of which are reversible inhibitors and others are irreversible inhibitors. Here we describe the main inhibitors of p97 (Figure 2).
Figure 2.

Chemical structure of p97 inhibitors 1-8.
Dibenzylquinazolines
N2, N4-dibenzylquinazoline-2,4-diamine (DBeQ, 1) is a reversible, ATP-competitive p97 inhibitor with IC50 of <10 μM in the ATPase activity assay [54, 55]. DBeQ also showed 50-fold selectivity over other unrelated ATPase activities, including the N-ethylmaleimide-sensitive factor (NSF) and the ATP-dependent chymotryptic activity of the 26S proteasome. DBeQ upregulated C/EBP-homologous protein (CHOP) resulted in cell death, suggesting that DBeQ-dependent inhibition of p97 resulted in a lethal ER stress response. DBeQ also enhanced lipidation of the microtubule-associated protein light chain (LC3 or LC3-B), which indicated that DBeQ induced autophagy [54, 56], Inspired by DBeQ, the structure-activity relationship (SAR) studies that were performed on the dibenzylquinazoline analogs led to the discovery of two compounds, ML240 (2) and ML241 (3), with different substitutions at the N2 position on quinazoline scaffolds. Both ML240 and ML241 were ATP-competitive p97 inhibitors and howed similar in vitro ATPase activity. However, they showed mechanistically different modes of action [56, 57]. ML240 induced the accumulation of LC3-B and was more potent than ML241 in the antiproliferative effects assessed in the NCI-60 cancer cell line panel.
CB-5083 (4), the first p97 inhibitor enter clinical trials in the US is analog of the dibenzylquinazoline, was developed as a D2 domain selective, first-in-class p97 inhibitor with an IC50 of 11 nM [58, 59]. The crystal structure CB-5083 showed it competes with ATP to bind at the binding site in Domain 2 [53] (PDB Code 6MCK, Figure 3A), which was consistent with DBeQ, ML240, and ML241. All the dibenzylquinazolines share the same binding site. CB-5083 affected the expression of mediators of the UPR [9] and significantly altered genes, including CHOP, DR5, HSPA5, HERPUD1, SEL1L, SYVN1 and EDEM1. CB-5083 showed promising antitumor responses in colorectal cancer, lung cancer, plasmacytoma tumor xenografts, and patient-derived xenograft models of colorectal cancer [9]. Despite its promising anti-cancer activity, CB-5083 failed its Phase I clinical trials against relapsed/refractory multiple myeloma and advanced solid tumors, due to an inhibition of PDE6 that caused blindness. This side effect was investigated in studies on gene expression and cell viability [9]. In 2019, LC-1028 (5) was developed by introducing a Michael acceptor to replace the amido group in CB-5083. LC-1028 showed promising potency, with Kiapp = 33.2 ± 4.0 nM and EC50 = 436 nM in antiproliferative assays (MIA PaCa-2 cells) [60]. The docking study indicated that the Cys522 thiol in the active site of p97 is indeed poised for a nucleophilic attack onto the electrophilicynamide moiety of LC-1028. MS/MS studies supported covalent modification of Cys522. Interestingly, LC-MS/MS analysis of p97 incubated with LC-1028 has also identified Cys105 as an off-target labelling site, which was further evidenced by slow adduct formation (t1/2 = 16.4 h) with intracellular GSH [60].
Figure 3.
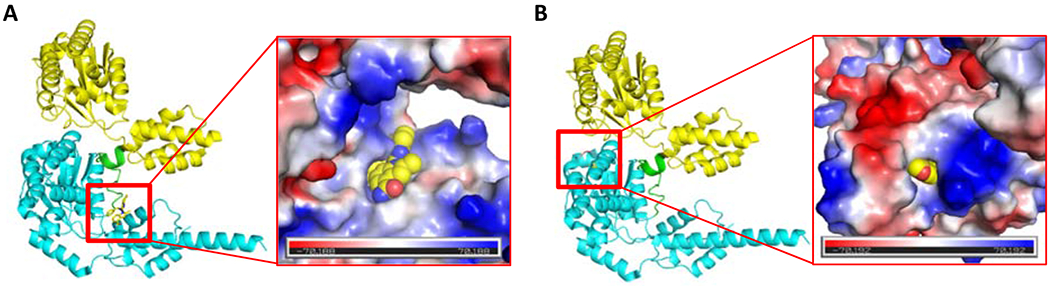
CB-5083 (PDB: 6MCK, A) and NMS-873 (proposed binding mode, PDB: 6MCK, B) in binding sites of p97 with a vacuum electrostatic surface. The D1, D1D2 linker, and D2 are presented as cartoons in yellow, green and cyan, respectively.
Alkylsulfanyl-1,2,3-triazoles
NMS-873 (6), is an allosteric p97 inhibitor, which induces the accumulation of polyubiquitinated proteins, leading to ER stress and inhibition of autophagosome maturation [61, 62]. NMS-873 is thought to bind to the linker domain between the D1 and D2 ATPase domains of p97, which stabilizes D2-ADP bound p97 [63]. We performed docking study to investigate the binding mode of NMS-873, which was proposed in Figure 3B [64]. Bastola et al. validated the binding site of NMS-873 by the mutations at nearby residues, which abrogated or significantly reduced the activity of NMS-873 [65]. NMS-873 showed potent inhibition against cell proliferation on HCT116 cells with IC50 of 400 nM [66]. Cellular assays indicated a dose-dependent accumulation of ubiquitinated proteins, activation of the unfolded protein response, as well as stabilization of key proteins (such as Mcl-1, cyclin E) at concentrations with demonstrated antiproliferative activity. Compared to the approved proteasome inhibitor bortezomib, NMS-873 showed a distinct profile with broader effects in tumor cell line panels. We have shown that allosteric p97 inhibitors can inhibit CB-5083 resistant p97 both with purified mutants and cell lines [67].
Despite the notable in vitro activity, NMS-873 showed poor bioavailability (around 16%), modest solubility (7 μM), and high clearance (based on in vitro microsomal data) [66]. Additionally, NMS-873 exhibited antiviral properties by inhibiting the production of the infectious virus in primary human fibroblast cells (targeting p97), with IC50 of 0.13 μM (the clinically used ganciclovir IC50 = 1.3 μM) [68]. NMS-873 was also reported to be effective against influenza and prevented viral replication in H1N1 strains, as well as in drug-resistant strains, with EC50 and selectivity indices ranging from 30-55 nM and 50-100 (CC50/EC50), respectively [69]. The related analogs bearing an imidazole core (7, IC50=8 nM) or aryl isostere (8, IC50=7 nM) demonstrate significant biochemical potencies [70, 71], Both the ATP-competitive inhibitors (DBeQ, ML240 and ML241) as well as the allosteric inhibitor NMS-873 have differential effects on the D1 and D2 ATPase activity of p97. DBeQ and NMS-873 inhibit ATPase activity from both the D1 and D2 domains, while ML240 and ML241 seem to inhibit selectively the D2 domain of p97 [35].
Tetrahydrocarbazoles
MSC1094308 (9, Figure 4A) was identified as an allosteric p97 inhibitor and exhibited moderate biochemical potency (IC50 = 7.2 μM) in the high-throughput screening campaign in 2018 [72], Kinetic analysis of the interaction between p97 and MSC1094308 showed non-competitive inhibition. Structure-activity relationship analysis showed the changes to the carbazole system, and the diphenyl-methane moiety didn’t improve the potency significantly. Additionally, MSC1094308 is also a VPS4B inhibitor with an IC50 value of 0.71 μM. Although VPS4B affinity over p97 could be increased up to 40-fold in this series, no p97-selective compounds were discovered. The binding pocket for MSC1094308 series was further validated by exposing p97 to a MSC1094308-based photoaffinity probe which cross-linked residues at the same site occupied by UPCDC30245 and NMS-873 (Figure 4B). The reduced potency of MSC1094308 against the N616F p97 mutant showed a common allosteric binding site for these three different scaffolds, which is considered a sensor for the NMS-873 allosteric binding site. Despite its low biochemical potency, MSC 1094308 was effective in cellular assays when dosed at 10 μM and caused the accumulation of ubiquitinated substrates, as measured by Western blot.
Figure 4.
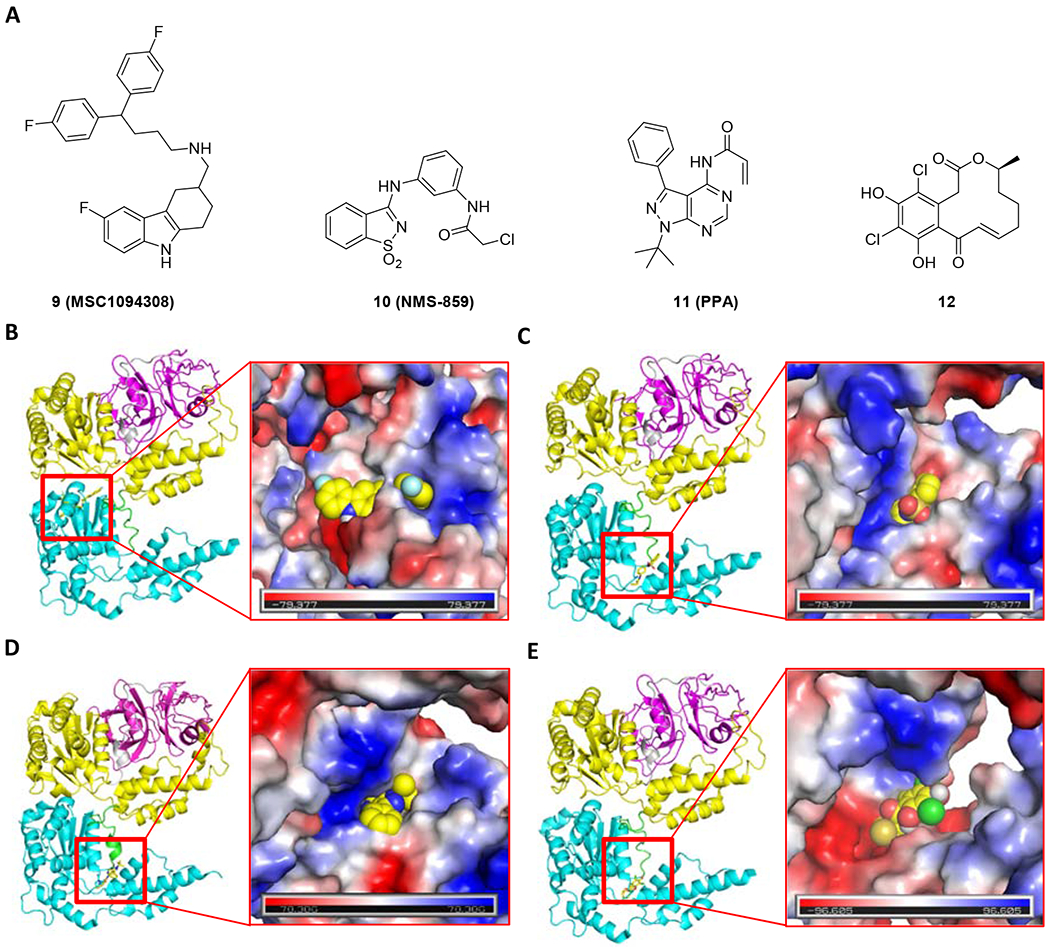
Chemical structure (A) and the proposed binding mode of MSC1094308 (9, PDB code 5FTJ, B), NMS-859 (10, PDB code 5FTJ, C), PPA (11, PDB code 5FTK, D) and curvularin (12, PDB code 5FTJ, E) with p97. The N, ND1 linker, D1, D1D2 linker, and D2 are presented as cartoons in magenta, gray, yellow, green, and cyan, respectively.
NMS-859
NMS-859 (10, Figure 4A), sharing an α-chloroacetamide, is a highly potent p97 inhibitor. The enzymatic inhibition by NMS-859 is time-dependent and covalent, which is validated by the mass spectrum of p97 incubated with NMS-859 [61], Compared to the nonspecific agents, such as N-ethylmaleimide targeting 6-8 cysteine residues of p97, NMS-859 bonded covalently at Cys522, which is proximal to the key residue Lys524 at D2 domain catalytic site. In order to investigate the binding mode, we performed a covalent docking study for NMS-859 to p97 (5FTJ) (Figure 4C). In addition to the covalent bond with C522, some significant interactions were proposed, such as the hydrophobic interaction from L526, the hydrogen interaction from T688, and the carbonyl-π interaction from N660. NMS-859 induced protein dysregulation and wild type Hela cell death with an IC50 of 3 μM. The p97-C522T mutant showed resistance to NMS-859-mediated inhibition. This effect was also recapitulated at the cellular level where NMS-859 showed a 3-fold increase in IC50 and no downstream effects on Hela cells expressing the mutant protein. As expected, NMS-859 was inactive against a panel of other AAA ATPases. The binding pocket of NMS-859 was defined using the key residues Leu526, Asn660, and Thr688 identified by computational studies [73]. Due to the selectivity, NMS-859 could be a promising starting point to design the covalent p97 inhibitors.
Pyrazolo [3,4-d] pyrimidines
Recently, a series of pyrazolo[3,4-d]pyrimidine analogs were reported to show p97 inhibition activity [74]. A covalent inhibitor, PPA (11, Figure 4D) from this series, exhibited at least 450-fold and 150-fold selectivity against wild type p97 over the yeast Cdc48 and the N-ethylmaleimide sensitive factor, respectively. The covalent action mode of PPA was confirmed by tandem mass spectrometry, verifying that PPA covalently bound with Cys522. PPA could effectively inhibit the proliferation of HCT116, HeLa, and RPMI8226 with IC50 values of 2.7 μM, 6.1 μM, and 3.4 μM. Additionally, PPA showed inhibition on CB-5083-resistant and NMS-873-resistant HCT116 cell lines. Therefore, PPA could serve as a lead compound to further structural optimization.
Curvularins
The dehydrocurvularin (DHC) and its chlorinated variants were identified as covalent p97 inhibitors by interfering with its ATPase activity, which are members of the macrocyclic lactone family of curvularins produced by several fungal species [75]. The dehydrocurvularin inhibited p97 by covalently bonding to Cys522 (12, Figure 4E) in a dose-dependent manner with an IC50 of 15.3 ± 9.9 μM, which caused the p97-dependent accumulation of polyubiqutinated substrates in cellular assay. The chlorinated variant 4,6-chlorinated derivative was more selective against p97-dependent ERAD than general proteasomal effects.
2.2. RUVBLs
RUVBL1 (TIP49, TIP49a, Rvbl, Pontin) and RUVBL2 (TIP48, TIP49b, Rvb2, Reptin) are highly conserved AAA ATPases which are involved in various cellular processes. Originally, RUVBL proteins were isolated from Escherichia coli and were designated as RuvA and RuvB [3]. Later, the homologues of RUVBL1 and RUVBL2 in humans, rats, and yeast (TIP49/TIP49a and TIP49b/TIP48) were characterized to interact with TATA-box binding protein (TBP) [76–79] They share the conserved motifs Walker A and Walker B, which have been characterized to bind and hydrolyze ATP [95]. RUVBL1 and RUVBL2 share 43% sequence identity and 65% sequence similarity and work together as part of chromatin remodeling complexes [96–100]. Although RUVBL1 and RUVBL2 are present in the cytoplasm and associated with the cell membranes, they are mainly localized in the nucleus and associated with the nuclear matrix or in the nuclear cytosol.
Both RUVBL1 and RUVBL2 contain N-terminal, Domain 1, Domain 2, Domain 3, and C-terminal. Together with the internal region of Domain 2, the Domains 1 (the typical Rossman-like αβα fold) and 3 (the canonical all-α subdomain) provide typical oligomeric rings of an AAA core, which is formed by a conserved Walker A motif (required for nucleotide binding), a conserved Walker B motif (required for ATP hydrolysis) (Figure 5). Domain 2 of RUVBLs contains two sub-domains, including an oligonucleotide-binding fold (external D2 or D2 ext or α/β Tip) and a base region closer to the AAA ring (internal D2). The D2 domain was involved in several protein–protein interactions as observed in the INO80 complex [101], the yeast SWR1 complex [102], the PAQosome [103], and the R2TP complex [104]. The conserved motif HSH in RUVBL2 is responsible for the nucleotide binding but not hydrolysis [81,83,87,89,91]. Although RUVBL1 and RUVBL2 share homology with the bacterial RuvB helicase [3, 105, 106], their functions as helicases are not yet established and remain controversial. It is observed that their oligomeric states were shown as homo-hexamers, hetero-hexamers, and even hetero-dodecamers [80,81,107,108]. Although the heptamer of the RUVBL1/2 complex could be observed in a preliminary negative staining EM analysis [90], no available heptameric structure was reported. The different oligomeric states were dependent on different functional contexts based on their cellular activities [99, 109].
Figure 5.
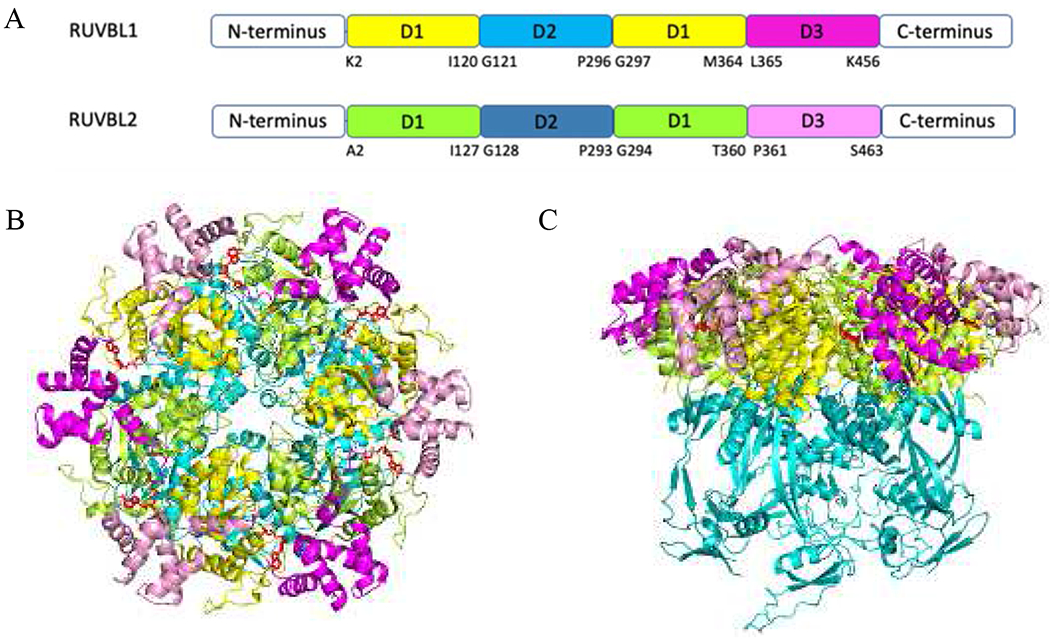
(A) Schematic diagram of the RUVBL1 and RUVBL2 domain organization. (B) Top view of the crystal structure of RUVBL1/2 complex (PDB code 5OAF). (C) Side view of RUVBL1/2 complex. For RUVBL1, D1, D2 and D3 are presented as a cartoon in yellow, cyan, and magenta; For RUVBL2, D1, D2 and D3 are presented as cartoons in limon, teal, and pink. ADP are presented as red sticks.
There is an increasing number of structures of RUVBL proteins reported, which were resolved by x-ray diffraction, electron microscopy, or solution NMR (Table 2). In order to better understand the structure of the RUVBL1/2 complex, we employed molecular modeling to overlay the RUVBL1/2 complex bound with ADP (5OAF) and without ligands (6IGM) (RMSD=1.315). The external Domain 2 of the RUVBL1/2 complex without ADP showed significant disordered compared to the structure of RUVBL1/2 complex with ADP. Due to the RMSD between 5OAF and 6IGM, we used ADP in 5OAF as the reference during the structural comparison. Some residues in the binding site showed significant shift, such as His20 in the HSH motif, the Arg finger 353, and Arg378 (Figure 6 and Table 3), which greatly decreased the interaction with ADP. Some mutations, such as RUVBL1 E303Q and RUVBL2 E300Q, led to the loss of activity. The complex formed by W-T RUVBL1/RUVBL2 E300Q or RUVBL1 E303Q/W-T RUVBL2 showed lower ATPase activity by 2.5-fold and 6.4-fold decrease compared to W-T RUVBL1/W-T RUVBL2, which indicated that both RUVBL1 and RUVBL2 contribute to the ATPase activity [10]. The RUVBL1 and RUVBL2 are involved in many cellular activities— including transcriptional regulation, chromatin remodeling, DNA damage signaling and repair, assembly of macromolecular complexes, regulating cell cycle/mitotic progression, and cellular motility— all of which contribute to their central roles in promoting cell proliferation and survival [99, 110–114]. RUVBL1 and RUVBL2 were shown to be essential for tumor cell growth of many cancers [79, 115] and were found to be overexpressed in many cancer types, which makes them ideal protein targets for developing the anti-cancer agents.
Table 2.
Crystal Structures of RUVBL1, RUVBL2 and RUVBL1/2 complex
| PDB Code | Methods (Å) | Macromolecules | Ligands | Organisms | References |
|---|---|---|---|---|---|
| 2C9O | X-ray (2.2) | RUVBL1: K9-C141, H156-N247, G277-D449 (Sequence: M1-K456) | ADP | hsa | [80] |
| 2CQA | NMR (NA) | RUVBL2 C-terminal: GSSGSSG+K132-R213+SGPSSG (Sequence: GSSGSSG+K132-R213+SGPSSG) | - | hsa | - |
| 2XSZ | X-ray (3.0) | RUVBL1 ΔDII: E5-R123, I235-R249, T269-K453 (Sequence: MVHHHHHHLLVPRGS+K2-E126+GPPG+I234-K456)/RUVBL2 ΔDII: I22-I131, H240-T254, E267-K456 (Sequence: MDYKDDDDKENLYFQG+A2-E133+GPPG+V238-S463) | ATP | hsa | [81] |
| 3UK6* | X-ray (3.0) | RUVBL2 ΔDII: T8-E12, D15-I131, T241-F261, E267-K444 (Sequence: M1-K132+AGA+V239-S463+LEHHHHHH) | ADP | hsa | [82] |
| 4WVY* | X-ray (3.6) | RUVBL1: I4-E142, T157-P251, I257-T449 (Sequence: M1-S462)/RUVBL2: H26-I145, Q157-A209,F223-D457 (Sequence: MGS SHHHHHHHHSSGLEVLFQGPGS+M1-S488) | ATP | cthr | [83] |
| 4WW4* | X-ray (2.9) | RUVBL1: Q3-V136, S156-K169, L173-D174, Q182-C201, R206-P251, D256-T451, G455-H458 (Sequence: M1-S462)/RUVBL2: T7-R149, G153-R212, F223-G229, Q232-V456 (Sequence: MGSSHHHHHHHHSSGLEVLFQGPGS+M1-S488) | ADP | cthr | [83] |
| 5FM6* | X-ray (3.0) | RUVBL1: Q3-T140, T157-T198, G205-S450 (Sequence: GA+M1-S462)/RUVBL2: S8-E14, H26-R149, G158-R210, T221-V455 (Sequence: GA+M1-S488) | ADP | cthr | [84] |
| 5FM7* | X-ray (2.9) | RUVBL1: Q3-E138, I160-T198, G205-P251, Q255-S450 (Sequence: GA+M1-S462)/RUVBL2: S8-L15, I21-R149, K159-A209, K222-V455 (Sequence: GA+M1-S488) | ADP | cthr | [84] |
| 5OAF | EM (4.1) | INO80 complex RUVBL1: K11-D452 (Sequence: M1-K456)/RUVBL2: V16-R211, P228-A449 (Sequence: M1-S463) | ADP | hsa | [85] |
| 5OUN | NMR (NA) | RUVBL2 DII: GPHM+E132-T234 (Sequence: E132-T234) | - | sce | [86] |
| 6FHS | EM (3.8) | INO80 complex RUVBL1: I4-S462 (Sequence: M1-S462)/RUVBL2: L20-F458 (Sequence: M1-S488) | ADP | cthr | [87] |
| 6FML | EM (4.3) | INO80 complex RUVBL1: I4-S462 (Sequence: M1-S462)/RUVBL2: L20-F458 (Sequence: M1-S488) | ADP | cthr | [87] |
| 6FO1 | EM (3.6) | R2TP Subcomplex RUVBL1: E5-I124, I235-R249, T269-K456 (Sequence: M1-K456)/RUVBL2: I22-I131, H240-R253, E267-N453 (Sequence: M1-S463) | ADP | hsa | [88] |
| 6GEJ | EM (3.6) | Nucleosome complex RUVBL1: V21-L463 (Sequence: M1-L463)/RUVBL2: L14-K460 (Sequence: M1-E471) | ADP | sce | [89] |
| 6GEN | EM (3.6) | Nucleosome complex RUVBL1: V21-L463 (Sequence: M1-L463)/RUVBL2: L14-K460 (Sequence: M1-E471) | ADP | sce | [89] |
| 6H7X | X-ray (2.9) | RUVBL2: S43-I147, K160-K184, Q188-I202, P228-S252, G256-K456 (Sequence: M1-S463) | - | hsa | [90] |
| 6HTS | EM (4.8) | INO80 Complex RUVBL1: T12-Y454 (Sequence: M1-K456)/RUVBL2: D15-P150, S156-N453 (Sequence: M1-S463) | ADP | hsa | [91] |
| 6IGM | EM (4.0) | SRCAP Complex RUVBL1: Q13-E142, I154-P250, T269-I446 (Sequence: M1-K456)/RUVBL2: V16-P150, K157-T183, D191-I202, Q226-A449 (Sequence: M1-S463) | - | hsa | [92] |
| 6K0R* | X-ray (2.5) | RUVBL1 ΔDII: E5-I124, I235-R249, T269-Q451 (Sequence: GPS+K2-E126+GPPG+ I234-K456)/RUVBL2 ΔDII: H25-I131, H240-N251, E267-F450 (Sequence: GGS+M1-E133+GPPG+V238-S463) | ADP, 3AT, CUU, CU0 | hsa | [93] |
| 6QI8 | EM (3.8) | RUVBL1: K2-I120, D237-R249, T272-K456 (Sequence: M1-K456)/RUVBL2: T8-I131, S243-N453 (Sequence: M1-S463) | ADP | hsa | [94] |
| 6QI9 | EM (4.6) | RUVBL1: K2-I120, D237-R249, T272-K456 (Sequence: M1-K456)/RUVBL2: T8-I131, S243-N453 (Sequence: M1-S463) | ADP# | hsa | [94] |
Hsa, cthr, and sce stand for Homo sapiens, Chaetomium thermophilum, and Saccharomyces cerevisiae, respectively.
The complex was assembled as Dodecamer
The hexameric RUVBL1/2 complex only binds with 5 ADP molecules.
NA: not applicable.
PDB accessed on May 13th, 2020
Figure 6.

Overlay of RUVBL1/2 complex with (5OAF) or without (6IGM) ADP. (A) Overlay of full-length RUVBL1/2 complex, RUVBL1/2 complex with ADP (5OAF) is in green (ADP in red), RUVBL1/2 complex without ADP (6IGM) is in cyan. (B) Overlay of the binding site of ADP. ADP is in yellow. The interacted residues are presented as lines. The conserved HSH motif, walker A, walker B from RUVBL1 are presented as a cartoon.
Table 3.
Distance between ADP and the residues at the binding site of 5OAF (with ADP) and 6IGM (without ADP)
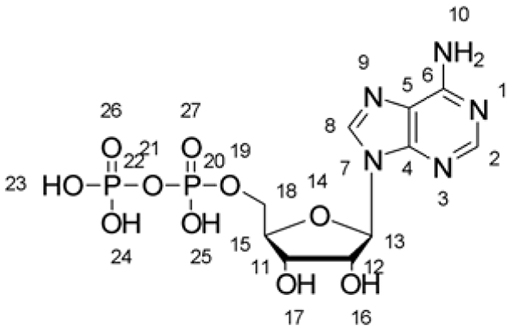 | ||||
|---|---|---|---|---|
| Residues | Groups | Labels (ADP) | Distance (Å) | |
| 5OAF | 6IGM | |||
| His20-A | NH | 16 | 2.5 | 5.0 |
| Val40-A | NH | 1 | 2.8 | 3.1 |
| NH | 10 | 3.0 | 3.3 | |
| C=O | 10 | 3.0 | 3.2 | |
| Gly73-A | NH | 26 | 3.0 | 2.7 |
| Thr77-A | NH | 27 | 2.9 | 3.1 |
| Arg378-A | NH2 | 16 | 2.6 | 4.2 |
| NH2 | 17 | 2.5 | 3.9 | |
| Arg404-A | NH | 19 | 2.5 | 2.8 |
| Glu317-B | C=O | 25 | 2.9 | 2.5 |
| Arg353-B | NH | 23 | 2.8 | 2.6 |
| NH2 | 23 | 2.9 | 4.9 | |
Elkaim et al. identified the first small-molecule inhibitors of RUVBL1 by employing virtual screening and enzymatic assay [116]. Three compounds 13, 14, and 15 with IC50 values in HuH7 and Hep3B cells of 0.57 and 0.25 μM for Rottlerin, 4.0 and 3.5 μM for Akt1/2 inhibitor, and 63.7 and 34.3 μM for Pranlukast tested reduced cell growth at concentrations close to those inhibiting the ATPase activity of RUVBL1 in vitro. However, their anti-proliferative action would require further study. Encouraged by the promising results of the first small-molecule inhibitors of RUVBL1, Elkaim et al. performed another virtual screening to hunt for RUVBL1 inhibitors, which led to the identification of two original scaffolds [117]. Two compounds 16 and 17 showed RUVBL1 inhibition in ATP-dependent mode with an IC50 at 9 and 18 μM, respectively. Compound 16 demonstrated the antiproliferative activity with the IC50s for KB and HL60 cells of 9 and 15 μM, which induced apoptotic cell death together with necrosis.
Compound 18 was initially identified as the RUVBL1/2 complex inhibitor by screening and structural optimization of the hits from a custom library [118] that has a half maximal inhibitory concentration of 59 nM against RUVBL1/2. By inhibition of RUVBL1/2 in cells, compound 18 depleted PIKK-family members DNA-PKcs (PRKDC), ATM, and ATP, all of which require RUVBL1/2 ATPase activity for stability [119]. In mice bearing subcutaneous NSCLC xenografts, compound 18 inhibited RUVBL1/2 and reduced the levels of ATM, ATR, and DNA-PKcs [103]. Compound 18 could arrest NSCLC cells in S phase, which eventually culminated in apoptosis indicated by the sub-G1 population in DNA content analysis. However, compound 18 in NSCLC in vivo exhibited a modest therapeutic window.
CB-6644 (19 in Figure 7), an allosteric small-molecule inhibitor of the RUVBL1/2 complex, showed potent inhibition against p97 with an IC50 of 15 nM in an ATP-dependent manner, which interacts specifically with RUVBL1/2 in cancer cells, leading to cell death [10]. In a panel of 123 cancer cell lines, CB-6644 potently killed cells with an EC50 range of 41 to 785 nM. Despite the maximal inhibition of around 50% in the biochemical ATPase assay, more than 95% of cells from all the cell lines were killed at the concentration range of 81 nM to 6.59 μM. CB-6644 demonstrated potent inhibition against tumor growth without obvious toxicity in xenograft models of Burkitt lymphoma and multiple myeloma. Although CB-6644 showed promising activity as RUVBL1/2 inhibitor, the binding site on RUVBL1/2 remains to be clarified.
Figure 7.

Chemical structure of 13-19
2.3. ATAD2
ATPase family AAA domain-containing 2 (ATAD2) is a chromatin regulator harboring an AAA ATPase domain and a bromodomain and has been identified as a critical modulator involved in cell proliferation and invasion. ATAD2 is co-expressed with genes involved in DNA replication in various cancer types and predominantly expressed in S phase cells where it localized on nascent chromatin (replication sites). Overexpression of ATAD2 has been linked to poor prognosis in prostate, lung, and triple-negative breast cancers, as well as in hepatocellular carcinoma [11, 120–122]. Downregulation of ATAD2 via siRNA shows an increase in apoptotic activity [11]. ATAD2 is recruited to replication sites through a direct interaction with di-acetylated histone H4 at K5 and K12, indicative of newly synthesized histones during replication-coupled chromatin reassembly [123]. Drug discovery efforts for oncology so far have focused on the ATAD2 bromodomain rather than its ATPase domain due to the tractability of most screening approaches [16]. Herein we summarized all the available crystal structures of ATAD2 (Table 4), which mainly focus on ATAD2 bromodomain.
Table 4.
The structures of ATAD2
| PDB Code | Methods (Å) | Macromolecules | Ligands | Organisms | References |
|---|---|---|---|---|---|
| 3DAI | X-ray (2.0) | SM+Q981-R1108 | - | hsa | [128] |
| 4QSP | X-ray (1.6) | SM+Q981-R1108 | N(6)-ACETYLLYSINE | hsa | [129] |
| 4QSQ | X-ray (1.8) | SM+Q981-R1108 | - | hsa | [129] |
| 4QSR | X-ray (2.0) | SM+Q981-R1108 | - | hsa | [129] |
| 4QSS | X-ray (2.0) | SM+Q981-R1108 | - | hsa | [129] |
| 4QST | X-ray (2.1) | SM+Q981-R1108 | 1-METHYLQUINOLIN-2(1H)-ONE | hsa | [129] |
| 4QSU | X-ray (1.9) | SM+Q981-R1108 | THYMINE | hsa | [129] |
| 4QSV | X-ray (1.9) | SM+Q981-R1108 | THYMIDINE | hsa | [129] |
| 4QSW | X-ray (1.8) | SM+Q981-R1108 | 5-methyluridine | hsa | [129] |
| 4QSX | X-ray (1.9) | SM+Q981-R1108 | 1-[(2R,5S)-5-(hydroxymethyl)tetrahydrofuran-2-yl]-5-methylpyrimidine-2,4(1H,3H)-dione | hsa | [129] |
| 4QUT | X-ray (1.7) | SM+Q981-R1108 | - | hsa | [130] |
| 4QUU | X-ray (1.8) | SM+Q981-R1108 | - | hsa | [130] |
| 4TT2 | X-ray (2.5) | SM+Q981-R1108 | - | hsa | [131] |
| 4TT4 | X-ray (2.7) | SM+Q981-R1108 | - | hsa | [131] |
| 4TT6 | X-ray (2.0) | SM+Q981-R1108 | - | hsa | [131] |
| 4TTE | X-ray (1.8) | SM+Q981-R1108 | methyl 3-amino-5-(3,5-dimethyl-1,2-oxazol-4-yl)benzoate | hsa | [131] |
| 4TU4 | X-ray (1.7) | SM+Q981-R1108 | 3-(3,5-dimethyl-1,2-oxazol-4-yl)-5-[(phenylsulfonyl)amino]benzoic acid | hsa | [131] |
| 4TU6 | X-ray (2.3) | SM+Q981-R1108 | - | hsa | [131] |
| 4TYL | X-ray (1.9) | SM+Q981-R1108 | 5-amino-1,3,6-trimethyl-1,3-dihydro-2H-benzimidazol-2-one | hsa | [124] |
| 4TZ2 | X-ray (1.7) | SM+Q981-R1108 | 3-(5-phenyl-4H-1,2,4-triazol-3-yl)aniline | hsa | [124] |
| 4TZ8 | X-ray (2.2) | SM+Q981-R1108 | 2-amino-7,7-dimethyl-5,6,7,8-tetrahydro-4H-[1,3]thiazolo[5,4-c]azepin-4-one | hsa | [124] |
| 5A5N | X-ray (2.0) | SM+Q981-R1108 | (2S)-2,6-diacetamido-N-methyl-hexanamide | hsa | [125] |
| 5A5O | X-ray (2.0) | SM+Q981-R1108 | 3-METHYL-1,2-DIHYDROQUIN OLIN-2-ONE | hsa | [125] |
| 5A5P | X-ray (2.0) | SM+Q981-R1108 | 8-[2-(dimethylamino)ethylamino]-3-methyl-1,2-dihydroquinolin-2-one | hsa | [125] |
| 5A5Q | X-ray (2.0) | SM+Q981-R1108 | 3-methyl-8-[(piperidin-4-yl)amino]-1,2-dihydro-1,7-naphthyridin-2-one | hsa | [125] |
| 5A5R | X-ray (2.0) | SM+Q981-R1108 | 5-(5-methoxypyridin-3-yl)-3-methyl-8-[(piperidin-4-yl)amino]-1,2-dihydro-1,7-naphthyridin-2-one | hsa | [125] |
| 5A81 | X-ray (2.0) | SM+Q981-R1108 | (3R,4R)-3-(cyclohexylmethoxy)piperidin-4-yl]amino}-3-methyl-1,2-dihydro-1,7-naphthyridin-2-one | hsa | [126] |
| 5A82 | X-ray (1.9) | SM+Q981-R1108 | 8-[[(3R,4R)-3-[[1,1-bis(oxidanylidene)thian-4-yl]methoxy]piperidin-4-yl]amino]-3-methyl-1H-1,7-naphthyridin-2-one | hsa | [126] |
| 5A83 | X-ray (2.1) | SM+Q981-R1108 | 8-[[(3R,4R)-3-[[1,1-bis(oxidanylidene)thian-4-yl]methoxy]piperidin-4-yl]amino]-3-methyl-5-(5-methylpyridin-3-yl)-1H-quinolin-2-one | hsa | [126] |
| 5LJ0 | X-ray (1.8) | SM+Q981-R1108 | 8-(((3R,4R,5S)-3-((4,4-difluorocyclohexyl)methoxy)-5-methoxypiperidin-4-yl)amino)-3-methyl-5-(5-methylpyridin-3-yl)-1,7-naphthyridin-2(1H)-one | hsa | [127] |
| 6CPS | X-ray (1.9) | GPL+Q981-R1108 | 4-(2-HYDROXYETHYL)-1-PIPERAZINE ETHANESULFONIC ACID | hsa | [132] |
| 6HDN | X-ray (1.9) | SM+Q981-R1108 | 3-methyl-8-((8-methyl-8-azabicyclooctan-3-yl)amino)-1,7-naphthyridin-2(1H)-one | hsa | [133] |
| 6HDO | X-ray (2.6) | SM+Q981-R1108 | 8-(((1R,2R,3R,5S)-2-(2-(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)ethyl)-8-azabicyclo[3.2.1]octan-3-yl)amino)-3-methyl-5-(5-methylpyridin-3-yl)quinolin-2(1H)-one | hsa | [133] |
| 6S55 | X-ray (2.1) | SM+Q981-R1108 | N-(4-bromo-3-((3-methylpiperidin-1-yl)sulfonyl)phenyl)-2-(2,5-dioxoimidazolidin-1-yl)acetamide | hsa | [134] |
| 6S56 | X-ray (2.0) | SM+Q981-R1108 | N-(4-chloro-3-(N,N-dimethylsulfamoyl)phenyl)-2-(2,5-dioxo-3′,4′-dihydro-2′H-spiro[imidazolidine-4,1′-naphthalen]-1-yl)acetamide | hsa | [134] |
| 6S57 | X-ray (1.8) | SM+Q981-R1108 | N-(3-(azepan-1-ylsulfonyl)-4-methylphenyl)-2-(4,4-dimethyl-2,5-dioxoimidazolidin-1-yl)acetamide | hsa | [134] |
| 6YB4 | X-ray (1.9) | SM+Q981-R1108 | N-(4-bromo-3-(3-methylpyrrolidin-1-yl)sulfonyl)phenyl)-2-(-4-cyclopropyl-4-methyl-2,5-dioxoimidazolidin-1-yl)acetamide | hsa | [135] |
hsa: Homo sapiens; -: no ligands. PDB Accessed on June 3, 2020.
Several studies were performed on the discovery of ATAD2 inhibitors. Harner et al. identified some hits against ATAD2 using NMR spectroscopy by a fragment-based approach [124]. These hits bound the bromodomain of ATAD2 with Kd values of 600, 350 and 500 μM. The co-crystals of ATAD2 suggested that these three hits occupied the same general space with the Kac binding pocket. There is still ample opportunity to optimize these structures and improve drug-like properties.
Demont et al. identified two ATAD2 bromodomain inhibitors by fragment-based discovery, with the same pIC50 of 5.9 against ATAD2 [125]. Due to the selectivity, compound 20 was used as the lead compound to further optimize the structure and improve the potency as well as selectivity. The introduction of the thiopyran ring on 3’-position led to compound 21, which showed pIC50 of 6.9 and >100-fold selectivity over the BET bromodomains [126]. However, some disadvantages, such as greater hydrophilicity and the passive permeability, limited further development. The methylated 21 on the piperidyl ring, compound 22, mitigated these disadvantages with some off-target activity against other bromodomains. In order to improve cell permeability and selectivity over the BET bromodomains, the structure of naphthyridones was optimized by replacing SO2 with the polar hydrophobic isostere CF2 [127].
Compound 23 (GSK8814, Figure 8) retained the favorable ATAD2 interactions of the sulfone and selectivity over the BETs yet resulted in a dramatic improvement in logD and passive permeability. Inspired by the different conformation of piperidine in ATAD2 and BRD4 BD1, further optimization by an orthogonal conformational restriction strategy showed potent and selective tropane inhibitors (such as compound 24 with 60-fold selectivity) [133]. The tropane derivatives were, however, less selective than their disubstituted piperidine analogues (compound 23 with 500-fold selectivity) and much less selective than the corresponding compounds with SO2 group (1,300-fold selectivity). Thereafter, the phenylsulfonamides were identified as ATAD2 inhibitors by high-throughput screening [134]. Compound 25 has the most potent inhibition with single-digit micromolar IC50 (pIC50 5.0-6.0) in the ATAD2 TR-FRET assay. Optimization of compound 25 led to compound 26, which showed sub-micromolar ATAD2 inhibition and at least 10-fold selectivity over the BET bromodomains, such as CECR2, TAF1 and BRPF3. Using compound 26 as the starting point, further optimization toward ATAD2 and CECR2 (cat eye syndrome chromosome region, candidate 2) gave GSK232 (27), which showed high selectivity, cellular penetration, and excellent physicochemical properties [135]. The phenyl sulfonamides exhibited a novel mode of binding to non-bromodomain and non-BET bromodomains through displacement of a normally conserved network of four water molecules (Figure 9).
Figure 8.

Chemical structure of 20-27
Figure 9.

Overlay of the binding site of phenyl sulfonamide (cyan, PDB code 6S55) and KAc (yellow, PDB code 5A5N). The four conserved water molecules (PDB code 5A5N) are presented as red spheres.
2.4. Spastin
Like other AAA+ ATPases, there is an AAA domain in spastin, which is composed of a nucleotide-binding domain (NBD) and a four-helix bundle domain (HBD) [136, 137] (Figure 10). Walker A (P-loop) motif and walker B (Switch II) motif at NBD are involved in ATP hydrolysis. The hydrophobic region (HR) at the other side of MIT possibly forms an intramembrane hairpin loop. The microtubule interacting and trafficking (MIT) domain at N-terminal is connected to the AAA domain by a flexible linker containing a putative microtubule binding domain (MTBD) that binds to tubulin in ATP-independent manner [136]. Spastin is identified as a novel target of the multi-functional homeodomain interacting protein kinase 2 (HIPK2) [138]. Knockdown of spastin in zebrafish leads to shorter axons in both branchiomotor and spinal motor neurons [139]. Overexpression of spastin decreases the density of cellular microtubules [140]. The defining feature of the AAA proteins is a structurally conserved AAA domain which assembles into an oligomer. Chemical cross-linking and gel filtration chromatography showed that spastin could oligomerize into a hexamer [141].
Figure 10.
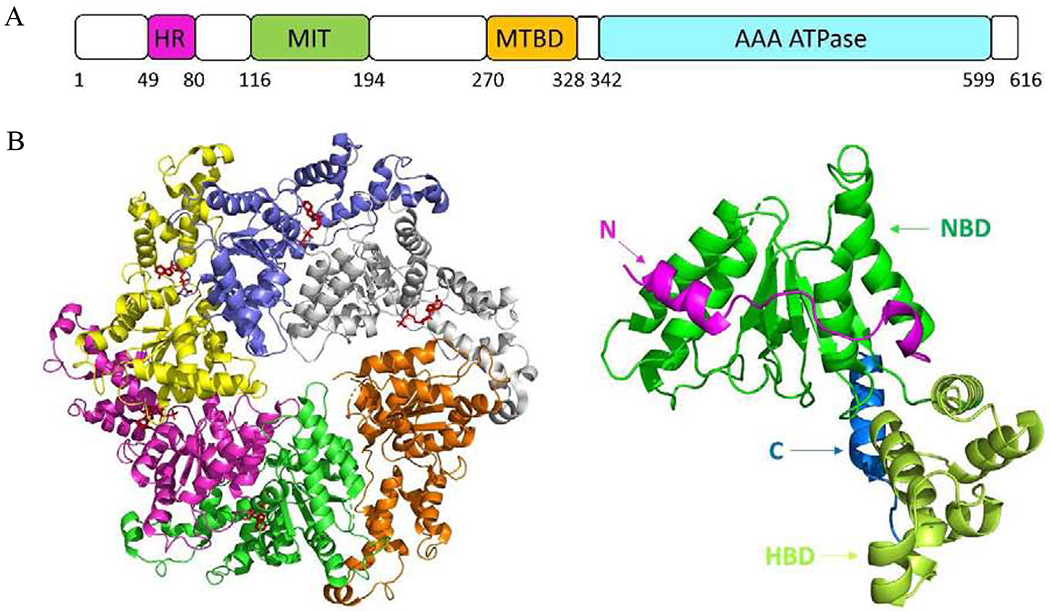
(A) Schematic diagram of the spastin domain organization. (B) The top view of the crystal structure of hexameric spastin (PDB code 6PEN). (C) The structure of the monomeric spastin.
Spastazoline (28 in Figure 12A) is an ATP-competitive chemical inhibitor of human spastin with IC50 of 99 nM [142]. Its parent compound, compound 29, has an IC50 of 4.4 ± 2.1 μM against Hs-spastin. It only inhibited one of the 65 kinases tested (NTRK1) by 50%. It did not appreciably inhibit any of the four related AAA proteins including Xl-katanin, Hs-FIGL1, Mm-VCP, or Hs-PCH2 at 10 μM. Spastazoline inhibits intercellular bridge disassembly by interfering with spastin activity, as the inhibitor-dependent phenotype was observed in wild-type cells but not in cells expressing the inhibitor resistance-conferring allele of spastin.
Figure 12.
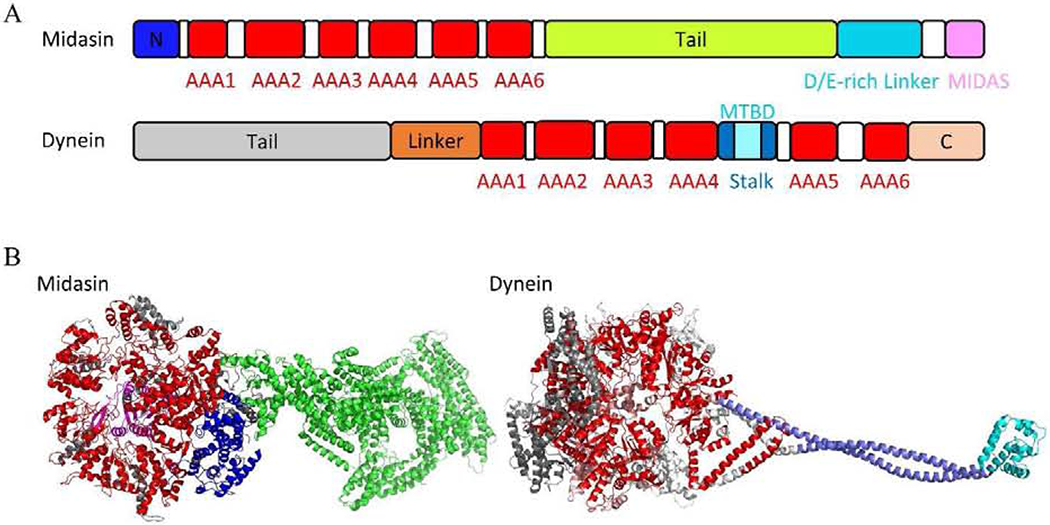
(A) Schematic diagram of the Midasin and dynein. (B) Crystal structure of Midasin (PDB code 6YLH) and dynein (PDB code 4RH7). Midasin: N domain, AAA domains, Tail, and MIDAS are presented in blue, red, green, and pink, respectively. Dynein: Tail, Linker, AAA domains, Stalk, C domain, and MTBD are presented in gray, orange, red, teal, cyan, and violet, respectively.
The mutant N527C did not disturb spastin’s ATPase activity. Based on the co-crystal structure of spastin-AAA-WT with compound 29 (Figure 11B), the authors designed compound 30 for targeting spasin N527C. Compound 30 showed 20-fold less potency for WT spastin (IC50 = 4.84 ± 1.46 μM) and 3-fold more potency for spastin N527C (IC50 = 0.23 ± 0.11 μM). The docking study of compound 30 with WT spastin showed compounds 29 and 30 shared the similar conformations at the binding site. The electron-withdrawing property and the unfavored angle (~112°) between F⋯C=O may decrease the binding affinity of compound 30 to WT spastin (Figure 11C). However, the N527A, -S, and -T mutant constructs are inhibited ~15-100 fold less potently than N527C, which is likely due to favorable stereoelectronic interactions of the fluorine atom with the Cys527 residue [143]. Furthermore, two similar inhibitors (compound 31 and 32, Figure 11A) were identified by the RADD (resistance analysis during design) involved in engineering point mutations in the target to generate active alleles. The superimposition of compounds 31 and 32 from crystal structures showed the core, phenyl(1H1,2,4-triazol-1-yl)methanone, occupied the same site and the substituents on 1,2,4-triazol ring were facing the opposite direction (Figure 11D). [144].
Figure 11.
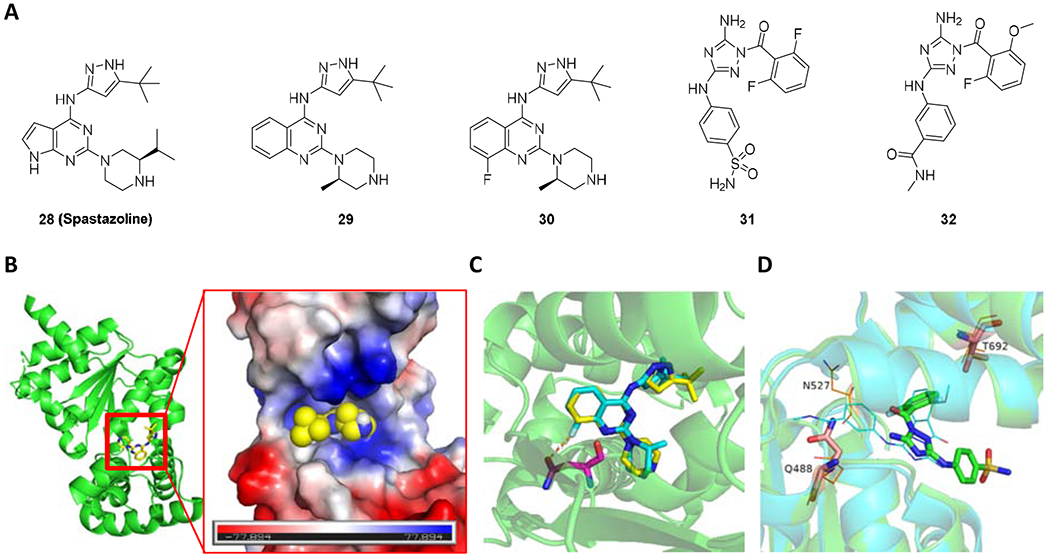
(A) Chemical structure of 28-32. (B) The binding site of compound 29 on spastin with a vacuum electrostatic surface (PDB code 6NYV). (C) The superimposition of compounds 29 and 30 in the binding site (PDB code 6NYV). Compounds 29 and 30 are presented as yellow and cyan sticks, respectively. (D)The superimposition of compound 31 (PDB code 6P10) and compound 32 (PDB code 6P12) at the binding site of spastin. Compound 31 and 32 are presented as green sticks and cyan lines, respectively. The key residues are presented as sticks or lines in salmon.
2.5. Midasin and Dynein
Midasin (Mdn1, REA1) is an essential AAA (ATPase Associated with various Activities) protein that removes assembly factors from distinct precursors of the ribosomal 60S subunit. Mdn1 was involved in 28S rRNA maturation [145, 146]. Knockdown of Mdn1 by small interfering RNA (siRNA) resulted in a severe growth defect in HeLa cells. Mdn1 is assembled by six non-equivalent AAA domains linked in a single polypeptide, which are similar to those of the microtubule-based motor protein dynein [147–149] (Figure 12). The ATPase activity of AAA2, AAA3, AAA4, and AAA5 and ATP-binding ability of AAA6 are likely essential for Mdn1’s cellular functions [150]. However, the AAA domains in Mdn1 share <15% identity with those in dynein.
Ribozinoindoles (or Rbins) were identified as potent, reversible, and specific inhibitors of Midasin [150]. Ribozinoindole-1 (Rbin-1, 33 in Figure 13) was discovered from a 10,353-member library of diverse chemicals aiming for a chemical synthetic lethal screen, which was more toxic to the MDR-sup cells that contained a mutation in cut2, compared to those with a wild-type cut2+ or a mutation in cut1. The structural optimization of Rbin-1 (GI50 = 136 ± 7 nM) led to Rbin-2 with a GI50 of 14 ± 1 nM. Rbin-2 (34 in Figure 13) does not strongly suppress the ATPase activity of the Mdn1-F1093L and inhibits less of the strain expressing the resistance-conferring mutation (mdn1-F1093L), indicating the physiological target of Rbins is Mdn1. The inhibition of Rbin-1 against Mdn1 led to the accumulation of pre-60S particles in Fission yeast cells.
Figure 13.

Chemical structure of 33 - 37
Dynein is a family of cytoskeletal motor proteins that regulates ciliary trafficking [151], mitotic spindle formation [152], and organelle transport [153]. Although dynein was discovered 50 years ago, the research progress in understanding dynein has been slow due to its huge size and complicated structure. The motor domain is located at the dynein heavy chain and composed of an N-terminal mechanical linker, a central ring of six AAA+ modules, and a long stalk extending from the AAA+ ring with a microtubule-binding domain (MTBD) at the tip (Figure 12). The tail is associated with accessory proteins and binds various cargoes. Although there are six AAA+ modules, only four AAA+ modules containing P-loops have nucleotide-binding sites [154]. The first site, AAA1, is the major site for ATP hydrolysis [155], whereas the other three P-loop containing AAA+ domains (AAA2-4) are thought to play a regulatory role. AAA3 can also hydrolyze ATP at a speed that is an order of magnitude slower than AAA1 [156]. Dynein transports various cellular cargos, provides forces and displacements important in mitosis, and drives the beat of eukaryotic cilia and flagella. Two of dynein’s ATPase sites contribute to the overall ATP hydrolysis and directional transport activity [157].
Ciliobrevins are the first specific small-molecule antagonists of cytoplasmic dynein. Starting from a high-throughput screen for inhibitors of the Hedgehog (Hh) pathway [158], a key mediator of embryonic development and oncogenesis [159], HPI-4, namely Ciliobrevin A, was discovered to block Hh pathway activation in cells lacking the negative regulator Suppressor of Fused (Sufu) [158]. Both ciliobrevins A and D (Figure 13, 35 and 36) induced ciliary Gli2 levels comparable to that in Shh-N-stimulated cells. Ciliobrevin D reversibly disrupts bipolar spindles, chromosomes, and the pre-formed spindles of metaphase-arrested cells and reduces overall microtubule levels [160]. Ciliobrevins are specific, reversible inhibitors of disparate cytoplasmic dynein-dependent processes.
Ciliobrevins do not perturb cellular mechanisms that are independent of dynein function, including actin cytoskeleton organization and signaling pathways of the mitogen-activated protein kinase as well as phosphoinositol-3-kinase. Ciliobrevins, therefore, will be useful reagents for studying cellular processes that require this microtubule motor and may guide the development of additional AAA ATPase superfamily inhibitors. The studies indicate that ciliobrevins can inhibit both cytoplasmic dynein 1 and 2, and, accordingly, the compounds will be broadly applicable probes of dynein-dependent processes [160]. Further development of ciliobrevin-like molecules could lead to isoform-selective inhibitors of this minus-end-directed microtubule motor and perhaps specific antagonists of other AAA1 ATPase superfamily members.
Dynarrestin (37 in Figure 13) is a novel inhibitor of cytoplasmic dyneins 1 and 2 [161]. Dynarrestin inhibits Hh-dependent signaling and proliferation of primate and human tumor cells. It also inhibits cytoplasmic dynein 1-dependent processes at 25 μM, including endosome movement, mitosis, and microtubule gliding without inducing mitochondrial toxicity or having off-target effects on protein kinases. Dynarrestin reversibly inhibits dynein 1 without affecting ATP hydrolysis. Dynarrestin could serve as a promising lead compound for drug development of anticancer therapeutics.
3. Conclusions and future perspectives
AAA ATPases are essential for many cellular functions, and members of the AAA family are found in all organisms. To understand the role of each in cellular and molecular level, specific and selective small molecular inhibitors with distinct mode of action will be critical. Interestingly, the protein abundance for AAA ATPase is relatively high, for example p97/VCP is 1% of total cellular proteins. To knock out essential proteins is not feasible and to knock down abundant proteins is more challenging. Therefore, to develop small molecule compounds provide a valuable tool to study biology of these protein and the added values of potentially validating whether these proteins are good drug targets and eventually to develop a drug. For example, p97 is the most studied AAA ATPase, and due to the potent activity of p97 inhibition and the binding site, CB-5083 and NMS-873 could serve as tool compounds to further investigate p97 function and develop novel p97 inhibitors. CB-6644 as the RUVBL1/2 complex inhibitor showed promising activity and could serve as a tool compound for the further investigation on RUVBL1/2 complex function and signaling pathway. Due to the significance of AAA ATPases, the work done so far provides the foundation for identification and development of new inhibitors against AAA ATPases using the combination of chemical biology, structural biology, and proteomic approaches in the near future.
Highlights.
Comparison of the structures of p97, RUVBL1/2 complex, ATAD2, spastin, midasin, and dynein.
Comparison of the structures of hexameric and heptameric p97 Domain 2.
Comparison of the structures of RUVBL1 Domain 2 within the RUVBL1/2 complex with or without ADP bound.
Review of small-molecule inhibitors of p97, RUVBL1/2 complex, ATAD2, spastin, midasin, and dynein.
Acknowledgment
AAA ATPase related work in the Chou lab was funded in part by the National Institutes of Health, the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E, through the NExT Chemical Biology Consortium. National Institute of Neurological Disorders and Stroke, R01NS100815 and R01NS102279; National Institute of Child Health and Human Development R01 HD086596.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- [1].Ranson NA, White HE, Saibil HR, Chaperonins, Biochem. J, 333 (Pt 2) (1998) 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hirokawa N, Noda Y, Okada Y, Kinesin and dynein superfamily proteins in organelle transport and cell division, Curr. Opin. Cell Biol, 10 (1998) 60–73. [DOI] [PubMed] [Google Scholar]
- [3].Otsuji N, Iyehara H, Hideshima Y, Isolation and Characterization of an Escherichia coli ruv Mutant Which Forms Nonseptate Filaments After Low Doses of Ultraviolet Light Irradiation, J. Bacteriol, 117 (1974) 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee DG, Bell SP, ATPase switches controlling DNA replication initiation, Curr. Opin. Cell Biol, 12 (2000) 280–285. [DOI] [PubMed] [Google Scholar]
- [5].Yang W, Structure and function of mismatch repair proteins, Mutat. Res, 460 (2000) 245–256. [DOI] [PubMed] [Google Scholar]
- [6].Caruthers JM, McKay DB, Helicase structure and mechanism, Curr. Opin. Struct. Biol, 12 (2002) 123–133. [DOI] [PubMed] [Google Scholar]
- [7].Nishi T, Forgac M, The vacuolar (H+)-ATPases--nature’s most versatile proton pumps, Nat. Rev. Mol. Cell Biol, 3 (2002) 94–103. [DOI] [PubMed] [Google Scholar]
- [8].HGNC Database, HUGO Gene Nomenclature Committee (HGNC), European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, U.K. www.genenames.org. [Google Scholar]
- [9].Anderson DJ, Moigne RL, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M, Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis, Cancer Cell, 28 (2015) 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Assimon VA, Tang Y, Vargas JD, Lee GJ, Wu ZY, Lou K, Yao B, Menon MK, Pios A, Perez KC, Madriaga A, Buchowiecki PK, Rolfe M, Shawver L, Jiao X, Le Moigne R, Zhou HJ, Anderson DJ, CB-6644 Is a Selective Inhibitor of the RUVBL1/2 Complex with Anticancer Activity, ACS Chem. Biol, 14 (2019) 236–244. [DOI] [PubMed] [Google Scholar]
- [11].Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V, Barbry P, Debernardi A, Brambilla C, Brambilla E, Rousseaux S, Khochbin S, Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers, Oncogene, 29 (2010) 5171–5181. [DOI] [PubMed] [Google Scholar]
- [12].Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Durr A, Wincker P, Brottier P, Cattolico L, Barbe V, Burgunder JM, Prud’homme JF, Brice A, Fontaine B, Heilig B, Weissenbach J, Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia, Nat. Genet, 23 (1999) 296–303. [DOI] [PubMed] [Google Scholar]
- [13].Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM, Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin, EMBO J, 32 (2013) 2920–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huryn DM, Kornfilt DJP, Wipf P, p97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections, J. Med. Chem, 63 (2019) 1892–1907. [DOI] [PubMed] [Google Scholar]
- [15].Sui X, Pan M, Li Y-M, Insights into the Design of p97-targeting Small Molecules from Structural Studies on p97 Functional Mechanism, Curr. Med. Chem, 27 (2020) 298–316. [DOI] [PubMed] [Google Scholar]
- [16].Hussain M, Zhou Y, Song Y, Hameed HMA, Jiang H, Tu Y, Zhang J, ATAD2 in cancer: a pharmacologically challenging but tractable target, Expert Opin. Ther. Targets, 22 (2018) 85–96. [DOI] [PubMed] [Google Scholar]
- [17].Romero FA, Taylor AM, Crawford TD, Tsui V, Cote A, Magnuson S, Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors, J. Med. Chem, 59 (2016) 1271–1298. [DOI] [PubMed] [Google Scholar]
- [18].Steinman JB, Kapoor TM, Using chemical inhibitors to probe AAA protein conformational dynamics and cellular functions, Curr. Opin. Chem. Biol, 50 (2019) 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shimizu T, Inhibitors of the Dynein ATPase and Ciliary or Flagellar Motility, in: Methods Cell Biol., 1995, pp. 497–501. [DOI] [PubMed] [Google Scholar]
- [20].Moir D, Stewart SE, Osmond BC, Botstein D, Cold-Sensitive Cell-Division-Cycle Mutants of Yeast: Isolation, Properties, and Pseudoreversion Studies, Genetics, 100 (1982) 547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koller KJ, Brownstein MJ, Use of a cDNA clone to identify a supposed precursor protein containing valosin, Nature, 325 (1987) 542–545. [DOI] [PubMed] [Google Scholar]
- [22].Peters JM, Walsh MJ, Franke WW, An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF, EMBO J, 9 (1990) 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ghislain M, Dohmen RJ, Levy F, Varshavsky A, Cdc48p interacts with Ufd3p, a WD repeat protein required for ubiquitin-mediated proteolysis in Saccharomyces cerevisiae, EMBO J, 15 (1996) 4884–4899. [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Q, Song C, Li CC, Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions, J. Struct. Biol, 146 (2004) 44–57. [DOI] [PubMed] [Google Scholar]
- [25].Acharya U, Jacobs R, Peters J-M, Watson N, Farquhar MG, Malhotra V, The formation of golgi stacks from vesiculated golgi membranes requires two distinct fusion events, Cell, 82 (1995) 895–904. [DOI] [PubMed] [Google Scholar]
- [26].Latterich M, Fröhlich K-U, Schekman R, Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes, Cell, 82 (1995) 885–893. [DOI] [PubMed] [Google Scholar]
- [27].Rabouille C, Levine TP, Peters J-M, Warren G, An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic golgi fragments, Cell, 82 (1995) 905–914. [DOI] [PubMed] [Google Scholar]
- [28].Ramanathan HN, Ye Y, The p97 ATPase associates with EEA1 to regulate the size of early endosomes, Cell Res, 22 (2012) 346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu S, Peng G, Wang Y, Fang S, Karbowski M, The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover, Mol. Biol. Cell, 22 (2011) 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].DeLaBarre B, Brunger AT, Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains, Nat. Struct. Biol, 10 (2003) 856–863. [DOI] [PubMed] [Google Scholar]
- [31].Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM, Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle, Nat. Struct. Biol, 9 (2002) 950–957. [DOI] [PubMed] [Google Scholar]
- [32].Song C, Wang Q, Li CC, ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity, J. Biol. Chem, 278 (2003) 3648–3655. [DOI] [PubMed] [Google Scholar]
- [33].Wang Q, Song C, Li C-CH, Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities, Biochem. Biophys. Res. Commun, 300 (2003) 253–260. [DOI] [PubMed] [Google Scholar]
- [34].Wang Q, Song C, Yang X, Li CC, D1 ring is stable and nucleotide-independent, whereas D2 ring undergoes major conformational changes during the ATPase cycle of p97-VCP, J. Biol. Chem, 278 (2003) 32784–32793. [DOI] [PubMed] [Google Scholar]
- [35].Chou TF, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, Arkin MR, Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains, J. Mol. Biol, 426 (2014) 2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS, Structure of the AAA ATPase p97, Mol. Cell, 6 (2000) 1473–1484. [DOI] [PubMed] [Google Scholar]
- [37].Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, Ma J, Zhang X, Freemont PS, The crystal structure of murine p97/VCP at 3.6A, J. Struct. Biol, 144 (2003) 337–348. [DOI] [PubMed] [Google Scholar]
- [38].Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS, Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47, EMBO J, 23 (2004) 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Davies JM, Brunger AT, Weis WI, Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change, Structure, 16 (2008) 715–726. [DOI] [PubMed] [Google Scholar]
- [40].Tang WK, Xia D, Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants, J. Biol. Chem, 288 (2013) 36624–36635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hänzelmann P, Schindelin H, Characterization of an Additional Binding Surface on the p97 N-Terminal Domain Involved in Bipartite Cofactor Interactions, Structure, 24 (2016) 140–147. [DOI] [PubMed] [Google Scholar]
- [42].Hanzelmann P, Schindelin H, Structural Basis of ATP Hydrolysis and Intersubunit Signaling in the AAA+ ATPase p97, Structure, 24 (2016) 127–139. [DOI] [PubMed] [Google Scholar]
- [43].Tang WK, Xia D, Role of the D1-D2 Linker of Human VCP/p97 in the Asymmetry and ATPase Activity of the D1-domain, Sci. Rep, 6 (2016) 20037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, Falconieri V, Deshaies RJ, Milne JL, Huryn D, Arkin M, Subramaniam S, 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition, Science, 351 (2016) 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bodnar NO, Kim KH, Ji Z, Wales TE, Svetlov V, Nudler E, Engen JR, Walz T, Rapoport TA, Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4, Nat. Struct. Mol. Biol, 25 (2018) 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stach L, Morgan RM, Makhlouf L, Douangamath A, von Delft F, Zhang X, Freemont PS, Crystal structure of the catalytic D2 domain of the AAA+ ATPase p97 reveals a putative helical split-washer-type mechanism for substrate unfolding, FEBS Lett, 594 (2020) 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tang WK, Odzorig T, Jin W, Xia D, Structural Basis of p97 Inhibition by the Site-Selective Anticancer Compound CB-5083, Mol. Pharmacol, 95 (2019) 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yamamoto S, Tomita Y, Hoshida Y, Takiguchi S, Fujiwara Y, Yasuda T, Yano M, Nakamori S, Sakon M, Monden M, Aozasa K, Expression level of valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma, J. Clin. Oncol, 21 (2003) 2537–2544. [DOI] [PubMed] [Google Scholar]
- [49].Yamamoto S, Tomita Y, Nakamori S, Hoshida Y, Nagano H, Dono K, Umeshita K, Sakon M, Monden M, Aozasa K, Elevated expression of valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence, J. Clin. Oncol, 21 (2003) 447–452. [DOI] [PubMed] [Google Scholar]
- [50].Kakizuka A, Roles of VCP in human neurodegenerative disorders, Biochem. Soc. Trans, 36 (2008) 105–108. [DOI] [PubMed] [Google Scholar]
- [51].Wojcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN, Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells, Mol. Biol. Cell, 17 (2006) 4606–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wojcik C, Yano M, DeMartino GN, RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis, J. Cell Sci, 117 (2004) 281–292. [DOI] [PubMed] [Google Scholar]
- [53].Liu Y, Hei Y, Shu Q, Dong J, Gao Y, Fu H, Zheng X, Yang G, VCP/p97, down-regulated by microRNA-129-5p, could regulate the progression of hepatocellular carcinoma, PLoS One, 7 (2012) e35800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ, Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways, Proc. Natl. Acad. Sci. U.S.A, 108 (2011) 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Deshaies RJ, Chou T-F, Schoenen FJ, Li K, Frankowski KJ, Aube J, Gerritz SW, Zhou H-J, Methods and compositions for inhibition of the transitional endoplasmic reticulum ATPase, US8865708B2, 2014. [Google Scholar]
- [56].Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G, Methods for assessing autophagy and autophagic cell death, Methods Mol. Biol, 445 (2008) 29–76. [DOI] [PubMed] [Google Scholar]
- [57].Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ, Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase, ChemMedChem, 8 (2013) 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon MK, Madriaga A, Kiss von Soly S, Kumar A, Parlati F, Yakes FM, Shawver L, Le Moigne R, Anderson DJ, Rolfe M, Wustrow D, Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083), J. Med. Chem, 58 (2015) 9480–9497. [DOI] [PubMed] [Google Scholar]
- [59].Zhou H, Parlati F, Wustrow D, Preparation of fused pyrimidines as inhibitors of p97 complex, WO2014015291, 1, 2014. [Google Scholar]
- [60].Ding R, Zhang T, Wilson DJ, Xie J, Williams J, Xu Y, Ye Y, Chen L, Discovery of Irreversible p97 Inhibitors, J. Med. Chem, 62 (2019) 2814–2829. [DOI] [PubMed] [Google Scholar]
- [61].Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A, Donati D, O’Brien T, Jackson PK, Isacchi A, Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death, Nat. Chem. Biol, 9 (2013) 548–556. [DOI] [PubMed] [Google Scholar]
- [62].Anderson D, Rolfe M, Letovsky S, Companion diagnostic for p97 inhibitor therapy and methods of use thereof, WO2016154110A1, 2016. [Google Scholar]
- [63].Segura-Cabrera A, Tripathi R, Zhang X, Gui L, Chou TF, Komurov K, A structure- and chemical genomics-based approach for repositioning of drugs against VCP/p97 ATPase, Sci. Rep, 7 (2017) 44912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pan M, Yu Y, Ai H, Zheng Q, Xie Y, Liu L, Zhao M, Mechanistic insight into substrate processing and allosteric inhibition of human p97, BioRxiv, (2021), doi: 10.1101/2021.02.08.430295. [DOI] [PubMed] [Google Scholar]
- [65].Bastola P, Wang F, Schaich MA, Gan T, Freudenthal BD, Chou TF, Chien J, Specific mutations in the D1-D2 linker region of VCP/p97 enhance ATPase activity and confer resistance to VCP inhibitors, Cell Death Discov, 3 (2017) 17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Polucci P, Magnaghi P, Angiolini M, Asa D, Avanzi N, Badari A, Bertrand J, Casale E, Cauteruccio S, Cirla A, Cozzi L, Galvani A, Jackson PK, Liu Y, Magnuson S, Malgesini B, Nuvoloni S, Orrenius C, Sirtori FR, Riceputi L, Rizzi S, Trucchi B, O’Brien T, Isacchi A, Donati D, D’Alessio R, Alkylsulfanyl-1,2,4-triazoles, a new class of allosteric valosine containing protein inhibitors. Synthesis and structure-activity relationships, J. Med. Chem, 56 (2013) 437–450. [DOI] [PubMed] [Google Scholar]
- [67].Wang F, Li S, Gan T, Stott GM, Flint A, Chou TF, Allosteric p97 inhibitors can overcome resistance to ATP-competitive p97 inhibitors for potential anticancer therapy, ChemMedChem, 15 (2020) 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lin YT, Prendergast J, Grey F, The host ubiquitin-dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication, PLoS Pathog, 13 (2017) e1006329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang J, Hu Y, Hau R, Musharrafieh R, Ma C, Zhou X, Chen Y, Wang J, Identification of NMS-873, an allosteric and specific p97 inhibitor, as a broad antiviral against both influenza A and B viruses, Eur. J. Pharm. Sci, 133 (2019) 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].HURYN DM, WIPF P, LAPORTE MG, Modulators of p97 AAA ATPase Activity, WO2017197080 A1, 2017. [Google Scholar]
- [71].HURYN DM, WIPF P, LAPORTE MG, Modulators of p97 AAA ATPase Activity, WO2018209083 A1, 2018. [Google Scholar]
- [72].Pohler R, Krahn JH, van den Boom J, Dobrynin G, Kaschani F, Eggenweiler HM, Zenke FT, Kaiser M, Meyer H, A Non-Competitive Inhibitor of VCP/p97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases, Angew. Chem. Int. Ed, 57 (2018) 1576–1580. [DOI] [PubMed] [Google Scholar]
- [73].Rendine S, Orrenius C, Dapiaggi F, Pieraccini S, Motto I, D’Alessio R, Magnaghi P, Isacchi A, Felder E, Sironi M, Molecular Dynamics Simulations of p97 Including Covalent, Allosteric and ATP-Competitive Inhibitors, Acta Chim. Slov, 66 (2019) 395–401. [PubMed] [Google Scholar]
- [74].Zhang G, Li S, Wang F, Jones AC, Goldberg AFG, Lin B, Virgil S, Stoltz BM, Deshaies RJ, Chou T-F, A Covalent p97/VCP ATPase Inhibitor can overcome resistance to CB-5083 and NMS-873 in colorectal cancer cells, Eur. J. Med. Chem, 213 (2021) 113148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tillotson J, Bashyal BP, Kang M, Shi T, De La Cruz F, Gunatilaka AA, Chapman E, Selective inhibition of p97 by chlorinated analogues of dehydrocurvularin, Org. Biomol. Chem, 14 (2016) 5918–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kanemaki M, Makino Y, Yoshida T, Kishimoto T, Koga A, Yamamoto K, Yamamoto M, Moncollin V, Egly JM, Muramatsu M, Tamura T, Molecular cloning of a rat 49-kDa TBP-interacting protein (TIP49) that is highly homologous to the bacterial RuvB, Biochem. Biophys. Res. Commun, 235 (1997) 64–68. [DOI] [PubMed] [Google Scholar]
- [77].Kanemaki M, Kurokawa Y, Matsu-ura T, Makino Y, Masani A, Okazaki K, Morishita T, Tamura TA, TIP49b, a new RuvB-like DNA helicase, is included in a complex together with another RuvB-like DNA helicase, TIP49a, J. Biol. Chem, 274 (1999) 22437–22444. [DOI] [PubMed] [Google Scholar]
- [78].Makino Y, Kanemaki M, Kurokawa Y, Koji T, Tamura T, A rat RuvB-like protein, TIP49a, is a germ cell-enriched novel DNA helicase, J. Biol. Chem, 274 (1999) 15329–15335. [DOI] [PubMed] [Google Scholar]
- [79].Wood MA, McMahon SB, Cole MD, An ATPase/Helicase Complex Is an Essential Cofactor for Oncogenic Transformation by c-Myc, Mol. Cell, 5 (2000) 321–330. [DOI] [PubMed] [Google Scholar]
- [80].Matias PM, Gorynia S, Donner P, Carrondo MA, Crystal structure of the human AAA+ protein RuvBL1, J. Biol. Chem, 281 (2006) 38918–38929. [DOI] [PubMed] [Google Scholar]
- [81].Gorynia S, Bandeiras TM, Pinho FG, McVey CE, Vonrhein C, Round A, Svergun DI, Donner P, Matias PM, Carrondo MA, Structural and functional insights into a dodecameric molecular machine - the RuvBL1/RuvBL2 complex, J. Struct. Biol, 176 (2011) 279–291. [DOI] [PubMed] [Google Scholar]
- [82].Petukhov M, Dagkessamanskaja A, Bommer M, Barrett T, Tsaneva I, Yakimov A, Queval R, Shvetsov A, Khodorkovskiy M, Kas E, Grigoriev M, Large-scale conformational flexibility determines the properties of AAA+ TIP49 ATPases, Structure, 20 (2012) 1321–1331. [DOI] [PubMed] [Google Scholar]
- [83].Lakomek K, Stoehr G, Tosi A, Schmailzl M, Hopfner KP, Structural basis for dodecameric assembly states and conformational plasticity of the full-length AAA+ ATPases Rvb1 . Rvb2, Structure, 23 (2015) 483–495. [DOI] [PubMed] [Google Scholar]
- [84].Silva-Martin N, Dauden MI, Glatt S, Hoffmann NA, Kastritis P, Bork P, Beck M, Muller CW, The Combination of X-Ray Crystallography and Cryo-Electron Microscopy Provides Insight into the Overall Architecture of the Dodecameric Rvb1/Rvb2 Complex, PLoS One, 11 (2016) e0146457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Aramayo RJ, Willhoft O, Ayala R, Bythell-Douglas R, Wigley DB, Zhang X, Cryo-EM structures of the human INO80 chromatin-remodeling complex, Nat. Struct. Mol. Biol, 25 (2018) 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bragantini B, Rouillon C, Charpentier B, Manival X, Quinternet M, NMR assignment and solution structure of the external DII domain of the yeast Rvb2 protein, Biomol. NMR Assign, 12 (2018) 243–247. [DOI] [PubMed] [Google Scholar]
- [87].Eustermann S, Schall K, Kostrewa D, Lakomek K, Strauss M, Moldt M, Hopfner KP, Structural basis for ATP-dependent chromatin remodelling by the INO80 complex, Nature, 556 (2018) 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Martino F, Pal M, Munoz-Hernandez H, Rodriguez CF, Nunez-Ramirez R, Gil-Carton D, Degliesposti G, Skehel JM, Roe SM, Prodromou C, Pearl LH, Llorca O, RPAP3 provides a flexible scaffold for coupling HSP90 to the human R2TP co-chaperone complex, Nat. Commun, 9 (2018) 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Willhoft O, Ghoneim M, Lin CL, Chua EYD, Wilkinson M, Chaban Y, Ayala R, McCormack EA, Ocloo L, Rueda DS, Wigley DB, Structure and dynamics of the yeast SWR1-nucleosome complex, Science, 362 (2018) eaat7716. [DOI] [PubMed] [Google Scholar]
- [90].Silva STN, Brito JA, Arranz R, Sorzano COS, Ebel C, Doutch J, Tully MD, Carazo JM, Carrascosa JL, Matias PM, Bandeiras TM, X-ray structure of full-length human RuvB-Like 2 - mechanistic insights into coupling between ATP binding and mechanical action, Sci. Rep, 8 (2018) 13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ayala R, Willhoft O, Aramayo RJ, Wilkinson M, McCormack EA, Ocloo L, Wigley DB, Zhang X, Structure and regulation of the human INO80-nucleosome complex, Nature, 556 (2018) 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Feng Y, Tian Y, Wu Z, Xu Y, Cryo-EM structure of human SRCAP complex, Cell Res, 28(2018)1121–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ju D, Zhang W, Yan J, Zhao H, Li W, Wang J, Liao M, Xu Z, Wang Z, Zhou G, Mei L, Hou N, Ying S, Cai T, Chen S, Xie X, Lai L, Tang C, Park N, Takahashi JS, Huang N, Qi X, Zhang EE, Chemical perturbations reveal that RUVBL2 regulates the circadian phase in mammals, Sci. Transl. Med, 12 (2020) eaba0769. [DOI] [PubMed] [Google Scholar]
- [94].Munoz-Hernandez H, Pal M, Rodriguez CF, Fernandez-Leiro R, Prodromou C, Pearl LH, Llorca O, Structural mechanism for regulation of the AAA-ATPases RUVBL1-RUVBL2 in the R2TP co-chaperone revealed by cryo-EM, Sci. Adv, 5 (2019) eaaw1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hishida T, Iwasaki H, Yagi T, Shinagawa H, Role of walker motif A of RuvB protein in promoting branch migration of holliday junctions. Walker motif a mutations affect Atp binding, Atp hydrolyzing, and DNA binding activities of Ruvb, J. Biol. Chem, 274 (1999) 25335–25342. [DOI] [PubMed] [Google Scholar]
- [96].Gerhold CB, Hauer MH, Gasser SM, INO80-C and SWR-C: guardians of the genome, J. Mol. Biol, 427 (2015) 637–651. [DOI] [PubMed] [Google Scholar]
- [97].Jha S, Gupta A, Dar A, Dutta A, RVBs are required for assembling a functional TIP60 complex, Mol. Cell Biol, 33 (2013) 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jonsson ZO, Dhar SK, Narlikar GJ, Auty R, Wagle N, Pellman D, Pratt RE, Kingston R, Dutta A, Rvb1p and Rvb2p are essential components of a chromatin remodeling complex that regulates transcription of over 5% of yeast genes, J. Biol. Chem, 276 (2001) 16279–16288. [DOI] [PubMed] [Google Scholar]
- [99].Nano N, Houry WA, Chaperone-like activity of the AAA+ proteins Rvb1 and Rvb2 in the assembly of various complexes, Philos. Trans. R. Soc. Lond., B, Biol. Sci, 368 (2013) 20110399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhou CY, Stoddard CI, Johnston JB, Trnka MJ, Echeverria I, Palovcak E, Sali A, Burlingame AL, Cheng Y, Narlikar GJ, Regulation of Rvb1/Rvb2 by a Domain within the INO80 Chromatin Remodeling Complex Implicates the Yeast Rvbs as Protein Assembly Chaperones, Cell Rep, 19 (2017) 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tosi A, Haas C, Herzog F, Gilmozzi A, Berninghausen O, Ungewickell C, Gerhold CB, Lakomek K, Aebersold R, Beckmann R, Hopfner KP, Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex, Cell, 154 (2013) 1207–1219. [DOI] [PubMed] [Google Scholar]
- [102].Nguyen VQ, Ranjan A, Stengel F, Wei D, Aebersold R, Wu C, Leschziner AE, Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1, Cell, 154 (2013) 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yenerall P, Das AK, Wang S, Kollipara RK, Li LS, Villalobos P, Flaming J, Lin YF, Huffman K, Timmons BC, Gilbreath C, Sonavane R, Kinch LN, Rodriguez-Canales J, Moran C, Behrens C, Hirasawa M, Takata T, Murakami R, Iwanaga K, Chen BPC, Grishin NV, Raj GV, Wistuba II, Minna JD, Kittler R, RUVBL1/RUVBL2 ATPase Activity Drives PAQosome Maturation, DNA Replication and Radioresistance in Lung Cancer, Cell Chem. Biol, 27 (2020) 105–121 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rivera-Calzada A, Pal M, Munoz-Hernandez H, Luque-Ortega JR, Gil-Carton D, Degliesposti G, Skehel JM, Prodromou C, Pearl LH, Llorca O, The Structure of the R2TP Complex Defines a Platform for Recruiting Diverse Client Proteins to the HSP90 Molecular Chaperone System, Structure, 25 (2017) 1145–1152 e1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kurokawa Y, Kanemaki M, Makino Y, Tamura TA, A notable example of an evolutionary conserved gene: studies on a putative DNA helicase TIP49, DNA Seq, 10 (1999) 37–42. [DOI] [PubMed] [Google Scholar]
- [106].Makino Y, Mimori T, Koike C, Kanemaki M, Kurokawa Y, Inoue S, Kishimoto T, Tamura T, TIP49, homologous to the bacterial DNA helicase RuvB, acts as an autoantigen in human, Biochem. Biophys. Res. Commun, 245 (1998) 819–823. [DOI] [PubMed] [Google Scholar]
- [107].Cheung KL, Huen J, Houry WA, Ortega J, Comparison of the multiple oligomeric structures observed for the Rvb1 and Rvb2 proteins, Biochem. Cell Biol, 88 (2010) 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Niewiarowski A, Bradley AS, Gor J, McKay AR, Perkins SJ, Tsaneva IR, Oligomeric assembly and interactions within the human RuvB-like RuvBL1 and RuvBL2 complexes, Biochem. J, 429 (2010) 113–125. [DOI] [PubMed] [Google Scholar]
- [109].Grigoletto A, Lestienne P, Rosenbaum J, The multifaceted proteins Reptin and Pontin as major players in cancer, Biochim. Biophys. Acta, 1815 (2011) 147–157. [DOI] [PubMed] [Google Scholar]
- [110].Boulon S, Bertrand E, Pradet-Balade B, HSP90 and the R2TP co-chaperone complex: building multi-protein machineries essential for cell growth and gene expression, RNA Biol, 9 (2012) 148–154. [DOI] [PubMed] [Google Scholar]
- [111].Gallant P, Control of transcription by Pontin and Reptin, Trends Cell Biol, 17 (2007) 187–192. [DOI] [PubMed] [Google Scholar]
- [112].Jha S, Dutta A, RVB1/RVB2: running rings around molecular biology, Mol. Cell, 34 (2009) 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kakihara Y, Saeki M, The R2TP chaperone complex: its involvement in snoRNP assembly and tumorigenesis, Biomol. Concepts, 5 (2014) 513–520. [DOI] [PubMed] [Google Scholar]
- [114].Rosenbaum J, Baek SH, Dutta A, Houry WA, Huber O, Hupp TR, Matias PM, The emergence of the conserved AAA+ ATPases Pontin and Reptin on the signaling landscape, Sci. Signal, 6 (2013) mr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Qiu XB, Lin YL, Thome KC, Pian P, Schlegel BP, Weremowicz S, Parvin JD, Dutta A, An eukaryotic RuvB-like protein (RUVBL1) essential for growth, J. Biol. Chem, 273 (1998) 27786–27793. [DOI] [PubMed] [Google Scholar]
- [116].Elkaim J, Castroviejo M, Bennani D, Taouji S, Allain N, Laguerre M, Rosenbaum J, Dessolin J, Lestienne P, First identification of small-molecule inhibitors of Pontin by combining virtual screening and enzymatic assay, Biochem. J, 443 (2012) 549–559. [DOI] [PubMed] [Google Scholar]
- [117].Elkaim J, Lamblin M, Laguerre M, Rosenbaum J, Lestienne P, Eloy L, Cresteil T, Felpin FX, Dessolin J, Design, synthesis and biological evaluation of Pontin ATPase inhibitors through a molecular docking approach, Bioorg. Med. Chem. Lett, 24 (2014) 2512–2516. [DOI] [PubMed] [Google Scholar]
- [118].Ebisawa M, Suzuki T, Haginoya N, Hamada T, Murata T, Uoto K, Murakami R, Takata T, Aminopyrazolone derivative, in, WO2015125786, 2015. [Google Scholar]
- [119].Izumi N, Yamashita A, Iwamatsu A, Kurata R, Nakamura H, Saari B, Hirano H, Anderson P, Ohno S, AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay, Sci. Signal, 3 (2010) ra27. [DOI] [PubMed] [Google Scholar]
- [120].Hsia EY, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen HW, Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer, Mol. Cancer Res, 8 (2010) 183–193. [DOI] [PubMed] [Google Scholar]
- [121].Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD, Chen HW, ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2, Cancer Res, 70 (2010) 9402–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW, ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification, Proc. Natl. Acad. Sci. U.S.A, 104 (2007) 18067–18072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Koo SJ, Fernandez-Montalvan AE, Badock V, Ott CJ, Holton SJ, von Ahsen O, Toedling J, Vittori S, Bradner JE, Gorjanacz M, ATAD2 is an epigenetic reader of newly synthesized histone marks during DNA replication, Oncotarget, 7 (2016) 70323–70335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Harner MJ, Chauder BA, Phan J, Fesik SW, Fragment-based screening of the bromodomain of ATAD2, J. Med. Chem, 57 (2014) 9687–9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Demont EH, Chung CW, Furze RC, Grandi P, Michon AM, Wellaway C, Barrett N, Bridges AM, Craggs PD, Diallo H, Dixon DP, Douault C, Emmons AJ, Jones EJ, Karamshi BV, Locke K, Mitchell DJ, Mouzon BH, Prinjha RK, Roberts AD, Sheppard RJ, Watson RJ, Bamborough P, Fragment-Based Discovery of Low-Micromolar ATAD2 Bromodomain Inhibitors, J. Med. Chem, 58 (2015) 5649–5673. [DOI] [PubMed] [Google Scholar]
- [126].Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Sheppard RJ, Barnett H, Diallo H, Dixon DP, Douault C, Jones EJ, Karamshi B, Mitchell DJ, Prinjha RK, Rau C, Watson RJ, Werner T, Demont EH, Structure-Based Optimization of Naphthyridones into Potent ATAD2 Bromodomain Inhibitors, J. Med. Chem, 58 (2015) 6151–6178. [DOI] [PubMed] [Google Scholar]
- [127].Bamborough P, Chung CW, Demont EH, Furze RC, Bannister AJ, Che KH, Diallo H, Douault C, Grandi P, Kouzarides T, Michon AM, Mitchell DJ, Prinjha RK, Rau C, Robson S, Sheppard RJ, Upton R, Watson RJ, A Chemical Probe for the ATAD2 Bromodomain, Angew. Chem. Int. Ed, 55 (2016) 11382–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S, Histone recognition and large-scale structural analysis of the human bromodomain family, Cell, 149 (2012) 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chaikuad A, Petros AM, Fedorov O, Xu J, Knapp S, Structure-based approaches towards identification of fragments for the low-druggability ATAD2 bromodomain, MedChemComm, 5 (2014) 1843–1848. [Google Scholar]
- [130].Morozumi Y, Boussouar F, Tan M, Chaikuad A, Jamshidikia M, Colak G, He H, Nie L, Petosa C, de Dieuleveult M, Curtet S, Vitte AL, Rabatel C, Debernardi A, Cosset FL, Verhoeyen E, Emadali A, Schweifer N, Gianni D, Gut M, Guardiola P, Rousseaux S, Gerard M, Knapp S, Zhao Y, Khochbin S, Atad2 is a generalist facilitator of chromatin dynamics in embryonic stem cells, J. Mol. Cell Biol, 8 (2016) 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Poncet-Montange G, Zhan Y, Bardenhagen JP, Petrocchi A, Leo E, Shi X, Lee G.R.t., Leonard PG, Geck Do MK, Cardozo MG, Andersen JN, Palmer WS, Jones P, Ladbury JE, Observed bromodomain flexibility reveals histone peptide-and small molecule ligand-compatible forms of ATAD2, Biochem. J, 466 (2015) 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Gay JC, Eckenroth BE, Evans CM, Langini C, Carlson S, Lloyd JT, Caflisch A, Glass KC, Disulfide bridge formation influences ligand recognition by the ATAD2 bromodomain, Proteins, 87 (2019) 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Watson RJ, Mitchell DJ, Barnett H, Prinjha RK, Rau C, Sheppard RJ, Werner T, Demont EH, Aiming to Miss a Moving Target: Bromo and Extra Terminal Domain (BET) Selectivity in Constrained ATAD2 Inhibitors, J. Med. Chem, 61 (2018) 8321–8336. [DOI] [PubMed] [Google Scholar]
- [134].Bamborough P, Chung CW, Demont EH, Bridges AM, Craggs PD, Dixon DP, Francis P, Furze RC, Grandi P, Jones EJ, Karamshi B, Locke K, Lucas SCC, Michon AM, Mitchell DJ, Pogany P, Prinjha RK, Rau C, Roa AM, Roberts AD, Sheppard RJ, Watson RJ, A Qualified Success: Discovery of a New Series of ATAD2 Bromodomain Inhibitors with a Novel Binding Mode Using High-Throughput Screening and Hit Qualification, J. Med. Chem, 62 (2019) 7506–7525. [DOI] [PubMed] [Google Scholar]
- [135].Lucas SCC, Atkinson SJ, Bamborough P, Barnett H, Chung CW, Gordon L, Mitchell DJ, Phillipou A, Prinjha RK, Sheppard RJ, Tomkinson NCO, Watson RJ, Demont EH, Optimization of Potent ATAD2 and CECR2 Bromodomain Inhibitors with an Atypical Binding Mode, J. Med. Chem, 63 (2020) 5212–5241. [DOI] [PubMed] [Google Scholar]
- [136].White SR, Evans KJ, Lary J, Cole JL, Lauring B, Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing, J. Cell Biol, 176 (2007) 995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Roll-Mecak A, Vale RD, Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin, Nature, 451 (2008) 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pisciottani A, Biancolillo L, Ferrara M, Valente D, Sardina F, Monteonofrio L, Camerini S, Crescenzi M, Soddu S, Rinaldo C, HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission, Cells, 8 (2019) 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wood JD, Landers JA, Bingley M, McDermott CJ, Thomas-McArthur V, Gleadall LJ, Shaw PJ, Cunliffe VT, The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo, Hum. Mol. Genet, 15 (2006) 2763–2771. [DOI] [PubMed] [Google Scholar]
- [140].Sherwood NT, Sun Q, Xue M, Zhang B, Zinn K, Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function, PLoS Biol, 2 (2004) e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pantakani DV, Swapna LS, Srinivasan N, Mannan AU, Spastin oligomerizes into a hexamer and the mutant spastin (E442Q) redistribute the wild-type spastin into filamentous microtubule, J. Neurochem, 106 (2008) 613–624. [DOI] [PubMed] [Google Scholar]
- [142].Cupido T, Pisa R, Kelley ME, Kapoor TM, Designing a chemical inhibitor for the AAA protein spastin using active site mutations, Nat. Chem. Biol, 15 (2019) 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Pisa R, Cupido T, Kapoor TM, Designing Allele-Specific Inhibitors of Spastin, a Microtubule-Severing AAA Protein, J. Am. Chem. Soc, 141 (2019) 5602–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Pisa R, Cupido T, Steinman JB, Jones NH, Kapoor TM, Analyzing Resistance to Design Selective Chemical Inhibitors for AAA Proteins, Cell Chem. Biol, 26 (2019) 1263–1273 e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Finkbeiner E, Haindl M, Muller S, The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex, EMBO J, 30 (2011) 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U, A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export, PLoS Biol, 8 (2010) e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Garbarino JE, Gibbons IR, Expression and genomic analysis of midasin, a novel and highly conserved AAA protein distantly related to dynein, BMC Genom, 3 (2002) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Schmidt H, Carter AP, Review: Structure and mechanism of the dynein motor ATPase, Biopolymers, 105 (2016) 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kater L, Mitterer V, Thoms M, Cheng J, Berninghausen O, Beckmann R, Hurt E, Construction of the Central Protuberance and L1 Stalk during 60S Subunit Biogenesis, Mol. Cell, 79 (2020) 615–628. [DOI] [PubMed] [Google Scholar]
- [150].Kawashima SA, Chen Z, Aoi Y, Patgiri A, Kobayashi Y, Nurse P, Kapoor TM, Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis, Cell, 167 (2016) 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Scholey JM, Intraflagellar transport, Annu. Rev. Cell Dev. Biol, 19 (2003) 423–443. [DOI] [PubMed] [Google Scholar]
- [152].Merdes A, Ramyar K, Vechio JD, Cleveland DW, A Complex of NuMA and Cytoplasmic Dynein Is Essential for Mitotic Spindle Assembly, Cell, 87 (1996) 447–458. [DOI] [PubMed] [Google Scholar]
- [153].Akhmanova A, Hammer JA 3rd, Linking molecular motors to membrane cargo, Curr. Opin. Cell Biol, 22 (2010) 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Mocz G, Gibbons IR, Phase partition analysis of nucleotide binding to axonemal dynein, Biochemistry, 35 (1996) 9204–9211. [DOI] [PubMed] [Google Scholar]
- [155].Gibbons BH, Gibbons IR, Vanadate-sensitized Cleavage of Dynein Heavy Chains by 365-nm Irradiation of Demembranated Sperm Flagella and Its Effect on the Flagellar Motility, J. Biol. Chem, 262 (1987) 8354–8359. [PubMed] [Google Scholar]
- [156].DeWitt MA, Cypranowska CA, Cleary FB, Belyy V, Yildiz A, The AAA3 domain of cytoplasmic dynein acts as a switch to facilitate microtubule release, Nat. Struct. Mol. Biol, 22 (2015) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Carter AP, Crystal clear insights into how the dynein motor moves, J. Cell Sci, 126 (2013) 705–713. [DOI] [PubMed] [Google Scholar]
- [158].Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, Solow-Cordero DE, Jiang J, Rowitch DH, Chen JK, Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade, Proc. Natl. Acad. Sci. U.S.A, 106 (2009) 14132–14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Jiang J, Hui CC, Hedgehog signaling in development and cancer, Dev. Cell, 15 (2008) 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, Chen JK, Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein, Nature, 484 (2012) 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Hoing S, Yeh TY, Baumann M, Martinez NE, Habenberger P, Kremer L, Drexler HCA, Kuchler P, Reinhardt P, Choidas A, Zischinsky ML, Zischinsky G, Nandini S, Ledray AP, Ketcham SA, Reinhardt L, Abo-Rady M, Glatza M, King SJ, Nussbaumer P, Ziegler S, Klebl B, Schroer TA, Scholer HR, Waldmann H, Sterneckert J, Dynarrestin, a Novel Inhibitor of Cytoplasmic Dynein, Cell Chem Biol, 25 (2018) 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]