

Abstract
Piper was used to cure certain human afflictions. It has various biological processes in literary works. Our work aims to identify and to explain the molecular base in silico in phytoderived anti-viral compounds in Piper nigrum against the major protease enzyme COVID-19. The thesis includes docking and molecular dynamic modelling review of 8 phenolic compounds from Piper extracted from the PubMed database. Docking analysis with Autodock programme was conducted. Our analysis reveals that the two Piper phytochemicals are very susceptible to the COVID-19 major protease enzyme. These phyto-compounds from piper may use contemporary techniques to create a stable drug or help the detection of lead. In order to assess their efficacy against COVID-19, identified hit compounds can be taken further in vitro and in vivo tests.
Keywords: Autodock, Dynamics simulation, Molecular docking, Phenolic compounds, Piper nigrum, SARS-CoV-2
1. Introduction
SARS-CoV-2 (Severe Acute Respiratory Syndrome-Coronavirus-2), a highly contagious and pathogenic pandemic agent, has quickly spread to the global community, representing a health hazard of an unparalleled scale. In Wuhan, Hubei province, the novel SARS-CoV-2 virus was found for the first time in December 2019 in the live wildlife sector, which triggered local people mystical respiratory diseases of the pneumonic kind [1]. According to the ‘WHO’ (World Health Organization), COVID-19 condition update, on 18 March 2021, more than 120.383.919 people worldwide were affected by the virus, including 2,664,386 fatalities, at the cumulative death rate of greater than 3,2% [2]. No effective antiviral or vaccine against COVID-19 is available at present despite the urgent and massive development efforts of the world science community. Therefore, a possible therapy to monitor and control the pandemic of SARS-CoV-2 is an immediate requirement to be found.
Repurposing drugs are useful techniques in the field of medicinal chemistry to provide quick and better solutions to unsatisfied patient needs [3], [4]. In view of COVID-19′s accelerated dissemination across the globe, recycling medications from natural products are considered a vital treatment option [5], [6]. Vast varieties of complementary and Natural medicines are offered in Piper nigrum and other herbal plants to help overcome certain mysteries that accompany many viral diseases [7]. We have selected Piper Nigrum, a powerful source of antivirals, in our present investigation.
P. nigrum is often used as a remedy in India, and is considered the “king of spices.” It is classified as “Black Pepper.” The plant has anti-apoptotic, anti-depressant, antibacterial, anti-colonial, antimicrobial, anti-inflammatory, anti-diarrhoeal, anti-pasmodic, anti-metastatic, antitumor, anti-microbiotic, anti-thyroid, anti-myrospiratory, anti-hypothyroid, hepatic, gastric and insecticidal functioning [8]. SARS-CoV-2 has yet to be examined in core protease inhibitor activities of phenolic compounds. Our analysis therefore aimed at testing the behaviour of P. nigrum phenolic compounds against the principal protease of SARS-CoV-2 in Silico.
2. Materials and methods
2.1. Data source
In order to achieve the three-dimensional configuration of the phenolic substances, a ‘PubChem database’ (https://pubchem.nlm.nih.gov) was used. A database of PDB (https://www.rksb.org/) was used to achieve the whole configuration of the enzyme protease. The PDB database has obtained the access number 6lu7 structure [9].
2.2. Molecular docking studies
2.2.1. Preparation of protein
The data database RCSB PDB (Protein Data bank) has been used to acquire the Primary Protease Domain Inhibitor N3 X-ray crystal structures (PDB ID: 6LU7). The simulations of the docking were prepared, carried out and analysed using the 'Auto-Dock Tools' interactive user interface programme. Since the AutoDock stores the potential energy produced by the docking, pre-calculated grid maps are needed, one per atom shape and are placed on the ligand. The grid must surround the active site of the macromolecule (region of interest). Fig. 1 shows the molecular target’s 3D crystal, primary protease COVID-19 [10].
Fig. 1.

3-D crystal structure of the COVID-19 Main protease (6LU7).
2.2.2. Preparation of ligands
The following active Piper nigrum compound molecules were taken from the database of PubChem (https://pubchem.ncbi.nlm.nih.gov). The selected compounds are: Kadsurenin L (CID: 3083437); Kavain (CID: 5281565); Methysticin (CID: 4281567); Dihydrocavine (CID: 10220256), Dihydromethysticine (CID: 88308). (CID: 13962928). To translate the Structure Date File (SDF) to protein data bank (PDB) co-orders of the molecules, the Open Babel (http://openbabel.orge) converter has been used.
2.3. Protein and ligand docking
As a docking software, Auto Dock was used. Auto Dock Tools 1.5.6 was used to repair the grid box at the active site of the protein [10]. The final ligand and protein were prepared for docking, and the remaining software parameters were left at normal, taking into account the movable ligand and rigid protein. Chimera 1.12 and LigPlot were used to visualise the docking effect [11] .
2.4. Molecular dynamics simulation
In MD simulations the conformational dynamics of protease-binding phenolic compounds were better understood. A GROMACS 4.6.5 package and gromos53e6 force field have been used to calculate the best score of two phenolic (cadsurenine and methysticine) complexes [12]. For the generation of the topology of the docked complex the pdb2gmx modules were used by GROMACS [13]. Finally, a 30 ns ‘molecular dynamics simulation’ was executed on the systems in order to see how stable each complex of protein lines.
2.5. MD simulations analysis
The subsequent trajectories for study have been sampled using GROMACS [14]. For protein dynamics and ligand interactions, both trajectories have been observed. The RMSD (root mean square variance) of the C-alpha backbone protein atoms has been determined for each system. The RMSF values of protein atoms remained also determined to distinguish fluctuating residues during the simulation. The gyration radius (Rg) was used to establish the protein structure's compactness after binding the ligand.
3. Results and discussion
3.1. In silico molecular docking analysis
This study examined the binding mode of the major protease using computer docking analyses. All eight compounds have been docked and evaluated according to the target enzyme of COVID-19. In silico study, all the phenolic compounds interacted well in the key protease's active site showing that they may inhibit COVID-19. For COVID-19 restraint, components with dock values of 7.0 or lower are considered to be a stronger representation. Table 1 shoes, the highest docked values for SARS-CoV2, i.e. 8.43 kilo calories per mole, −8.20 kilo calories per mole, −7.44 kilo calories per mole and −7,70 kcal-mol have been achieved for Kadsurenin, Methysticin, Kavin and Dihydrokavain (6LU7).
Table 1.
Binding energy and Hydrogen bonded interaction between phenolic compounds of P. negrum and COVID-19 main protease enzyme.
| S.No | Compound name | 3D-Chemical structure of the compound | Binding Energy (Kcal/mol) | H bond interacting residues | Distance (Ao) |
|---|---|---|---|---|---|
| 1 | Kadsurenin L |  |
−8.43 | Gly143 Cys145 His163 |
2.19 2.98 2.38 |
| 2 | Methysticin |  |
−8.20 | Gly143 Cys145 His163 |
2.75 3.86 2.99 |
| 3 | Kavain |  |
−7.44 | Gly:143 Ser:144 Cys:145 His:163 |
2.77 2.30 3.20 3.00 |
| 4 | Dihydrokavain |  |
−6.97 | Gly143 Cys145 His163 |
2.12 1.71 1.71 |
| 5 | Dihydromethysticin |  |
−7.70 | His:163 Ser:144 Gly:143 Cys:145 |
2.82 2.96 2.93 3.11 |
| 6 | Pulmatin |  |
−6.12 | Ser144 Cys145 Lue141 |
2.30 2.91 1.75 |
| 7 | Sinapaldehydeglucoside |  |
−5.15 | Glu166 Gln189 |
2.11 1.91 |
| 8 | 1-O-feruloyl-beta-D-glucose |  |
−4.45 | Gly143 His163 Gln192 Thr190 Leu141 Ser144 |
1.95 2.39 2.26 2.07 2.44 2.22 |
The lowest docking scores for all four phenolic compounds are kadsurenin and methysticin, meaning that these two compounds are more affinity to the main protease of the other compounds. The primal protease is well associated with binding energies of 6.97, −6.12, −5.15, and −4.45 kcal/mol respectively, Dihydrocavain, Pulmatine, Sinapaldehydeglucoside and 1-O-feruloyl-beta-D-glucoside.
Fig. 2 shows the molecular associations with the Covid-19 main protease for the four (4) top phenolic compounds. In the synthesis of hydrophobic interactions with cadsureninin, the main protéase was the formation of 3 hydrogen bonds, namely Gly143 (2,19 Ao), Cys:145 (2,05 Ao), His:164 (2,38 Ao) and 3 amino acids, Phe140 (3,75 Ao) (3.66 Ao). The primary protease of Methysticin formed three hydrogen bonds with the Gly 143 (2.75 Ao), Cys 145 (3.86 Ao) and His 163 amino acids (2.99 Ao). After examination of all docked compounds, four are interacted on the central protease-binding site of COVID-19 through hydrogen bonding with Gly143, Cys145 and His163 based on observations. The simulation was conducted for 30 ns to examine the compositional stability of the complexes.
Fig. 2.

Molecular interactions of the phenolic compounds, (A) Kadsurenin, (B) Methysticin (C) Kavain and (D) Dihydromethysticin against Civid-19 main protease (PDB ID: 6LU7) from docking study. The interactions image was visualized using Chimera 1.12 and interacted amino acids are highlighted in Ligplot.
3.2. Molecular dynamics
In addition to the simulations of docking results, the two (2) top-docked ligand (binding energy 8 k cal/mol) complexes (Kadsurenin and methysticin) were conducted for 30 ns Molecular Dynamics. In order to verify receptor-ligand conformational possessions, including consistency and flexibility we have analysed RMSD (root-mean-square variance), RMSF (root-mean-square-fluctuation) and Rg (gyration radius).
3.3. RMSD analysis
Fig. 3 shows the graph from RMSD vs. time. The RMSD is an sign of the stability of receptor-ligand complex. Protease complexes of cadsurenine and methysticine were subjected to 30 ns of Molecular Dynamic simulations to provide dynamic information. Fig. 3A and B shows, the Kadsurenin and methysticin RMSD showed that 5 ns and 10 ns respectively achieved a simulation balance for the ligand–protein complexes of cadsurenin and methysticin.
Fig. 3.

Analyzed RMSD-plots of (A) Kadsurenin and (B) Methysticin during conformational stability investigation at a 30 ns time.
During the simulation, the typical RMSD backbone was 2,75 Ao and 2,8 Ao for both Kadsurenin and Methysticin. The binding of all phenolic compounds in accordance with the RMSD profiles significantly stabilised the COVID-19 core protease structure. Also calculated were individual RMSD compound values. Normal RMSD values were 1.1 Ao and 1.4 Ao for Kadsurenin and Methysticin. Fig. 4 shows, they were perfectly superimposed between 2.5 and 10 ns. The results showed that the dynamic balance stability of the complexes was correct and that the pathways could be used to collect snapshots for further analysis. In addition, the stability of the device demonstrated the pertinence of the docking results.
Fig. 4.
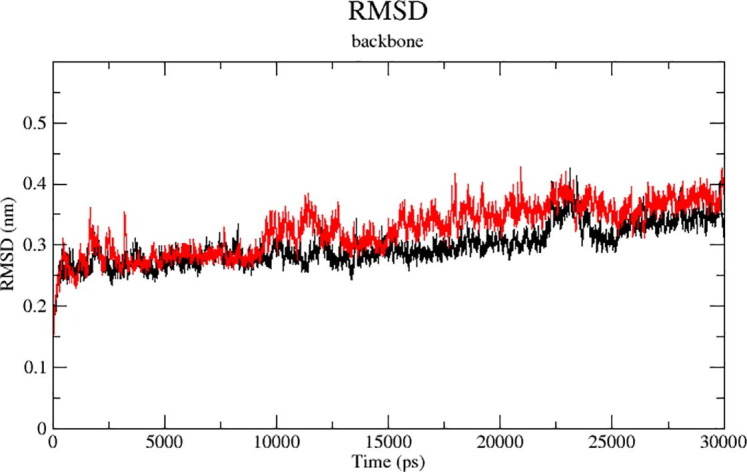
Analyzed RMSD-plots of Kadsurenin (Red) and Methysticin (Black) during conformational stability investigation at a 30 ns time. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. RMSF analysis
The RMSF has been designed to provide more accurate knowledge about protease movements during phenolic compound binding based on the residue amount of the last 30 ns trajectories. Fig. 5 shows, the RMSF values for Kadsurenine were strong, low and medium at 0.90, 0.14 and 0.52 nm, while 0.64, 0.12 and 0.38 nm for the Methysticin values. During the MD simulation, specific amino acid residues demonstrated stability in the complex protein state centred on both phenolic compounds’ low average RMSF values.
Fig. 5.
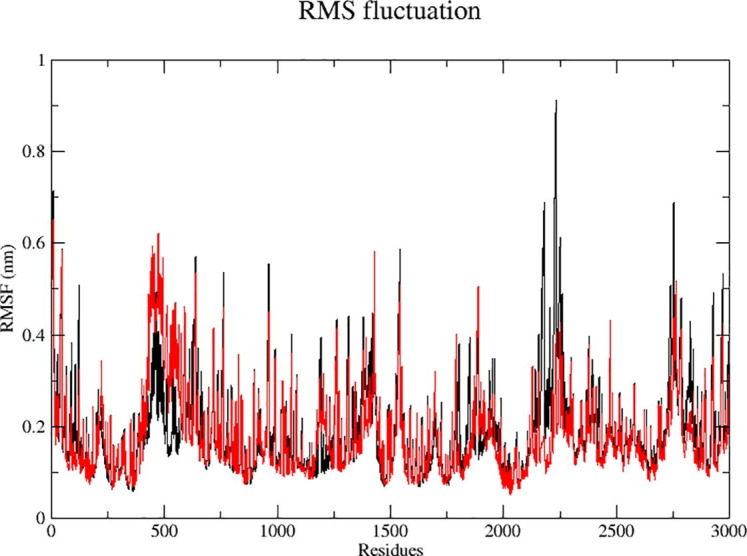
Analyzed RMSF-plots of Kadsurenin (Red) and Methysticin (Black) during conformational stability investigation at a 30 ns time. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. Radius of gyration (Rg) analysis
Figure 6 displays the Rg, defined by the protease-lining complex intrinsic dynamics. Rg tests are an excellent way to determine how lightweight the composition of a protein is. All two complexes have a stable mean Rg 2.2 nm throughout the 30 ns MD simulation. These results indicate that the major protease of Covid-19 was two ligands forming compact and stable complexes. Fig. 6(A) and (B) shows, a maximum of 10 ns of Rg of Kadsurenine for the Tyr101 trajectory, and a maximum of 11 ns for the Rg of Methysticin for the Gln110.
Fig. 6.

Radius of gyration (Rg) calculation of docked complexes (A) Kadsurenin and (B) Methysticin.
4. Conclusion
The current molecular docking studies indicate that the phenolic compounds Kadsurenin L and Methysticin of Piper nigrum are candidate ligands for inhibiting Covid-19 main protease and work by interactions with it. These piper phyto-compounds could be used to produce a safe drug or help in lead detection using modern methods. The identified hit compounds will then be tested in vitro and in vivo to see how successful they are against COVID-19.
CRediT authorship contribution statement
Rakesh Davella: Formal analysis, Data curation. Swapna Gurrapu: Investigation. Estari Mamidala: Conceptualization, Writing original draft.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We thank to the Department of Zoology, Kakatiya University for providing all the facilities related to this research.
References
- 1.Merkley E., Loewen P.J. Anti-intellectualism and the mass public’s response to the COVID-19 pandemic. Nat. Hum. Behav. 2021 doi: 10.1038/s41562-021-01112-w. [DOI] [PubMed] [Google Scholar]
- 2.WHO coronavirus disease (COVID-19) dashboard. https://covid19.who.int/. [Accessed 18 March 2021].
- 3.Khan R.J., Jha R.K., Amera G.M., Jain M., Singh E., Pathak A., Singh R.P., Muthukumaran J., Singh A.K. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021;39(8):2679–2692. doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta M.K., Vemula S., Donde R., Gouda G., Behera L., Vadde R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2021;39(7):2617–2627. doi: 10.1080/07391102.2020.1751300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enmozhi S.K., Raja K., Sebastine I., Joseph J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: an in silico approach. J. Biomol. Struct. Dyn. 2020;1–7 doi: 10.1080/07391102.2020.1760136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan T., Khan M.A., Mashwani Z.-u.-R., Ullah N., Nadhman A. Therapeutic potential of medicinal plants against COVID-19: the role of antiviral medicinal metabolites. Biocatal. Agric. Biotechnol. 2021;31:101890. doi: 10.1016/j.bcab.2020.101890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahfouz A. Assessment of antibacterial, antifungal, antioxidant and antiviral activities of black pepper aqueous seed extract. Al-Azhar Univ. J. Virus Res. Stud. 2020;2(1):1–18. doi: 10.21608/aujv.2020.108428. [DOI] [Google Scholar]
- 9.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 10.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seeliger D., De Groot B. tCONCOORD-GUI: visually supported conformational ampling of bioactive molecules. J. Comput. Chem. 2009;30(7):1160–1166. doi: 10.1002/jcc.21127. [DOI] [PubMed] [Google Scholar]
- 12.S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, E. Lindahl. ROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics 29(7) 2013 845–854. http://dx.doi.org/10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed]
- 13.Sinha S.K., Shakya A., Prasad S.K., Singh S., Gurav N.S., Prasad R.S., Gurav S.S. An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J. Biomol. Struct. Dyn. 2020;1–12 doi: 10.1080/07391102.2020.1762741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark, H.J.C. Berendsen. GROMACS: fast, flexible, and free, J. Comput. Chem. 26(16) 20051701–1718. http://dx.doi.org/10.1002/jcc.20291. [DOI] [PubMed]