

Summary
Mechanical forces are important in (patho)physiological processes, including how host epithelial cells interact with intracellular bacterial pathogens. As these pathogens disseminate within host epithelial monolayers, large mounds of infected cells are formed due to the forceful action of surrounding uninfected cells, limiting bacterial spread across the basal cell monolayer. Here, we present a protocol for mound volume measurement and biophysical characterization of mound formation. Modifications to this protocol may be necessary for studying different host cell types or pathogenic organisms.
For complete details on the use and execution of this protocol, please refer to Bastounis et al. (2021).
Subject areas: Biophysics, Cell Biology, Cell culture, Immunology, Microbiology, Microscopy
Graphical abstract
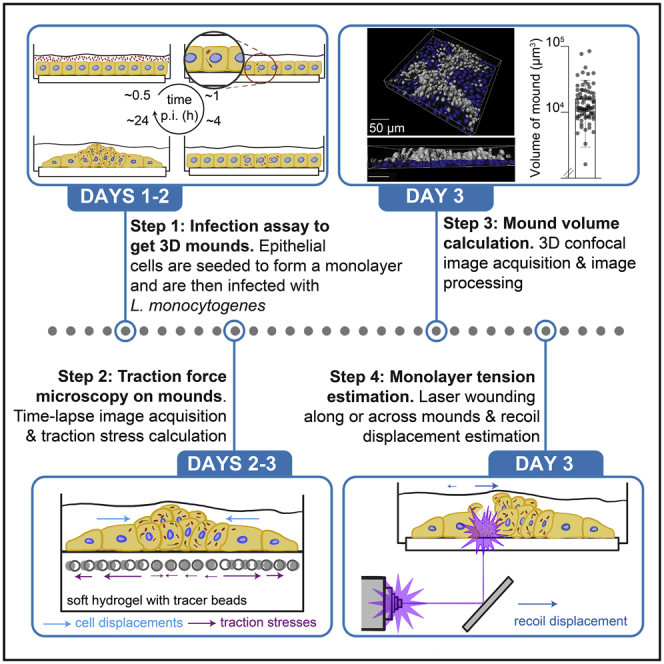
Highlights
-
•
Protocol allows for formation of mounds of extruded infected cells in cell monolayers
-
•
Confocal microscopy and image processing to calculate volume of extruded domains
-
•
Laser wounding protocol for tension estimation built around mounds
-
•
TFM incorporated to measure traction stresses of infected mounders and surrounders
Mechanical forces are important in (patho)physiological processes, including how host epithelial cells interact with intracellular bacterial pathogens. As these pathogens disseminate within host epithelial monolayers, large mounds of infected cells are formed due to the forceful action of surrounding uninfected cells, limiting bacterial spread across the basal cell monolayer. Here, we present a protocol for mound volume measurement and biophysical characterization of mound formation. Modifications to this protocol may be necessary for studying different host cell types or pathogenic organisms.
Before you begin
The protocol below describes specific steps for infecting host epithelial MDCK cells with a low multiplicity of infection (MOI) of Listeria monocytogenes (L.m.) to produce distinct infection mounds at 24 h post-infection (hpi). We empirically determined that a MOI∼200 bacteria/cell generates distinct infection foci, which are well-spaced (>300 μm away from neighboring foci) and represent ∼20% of all cells/well at 24 hpi. For different host cell types or other intracellular pathogens, the infection procedure might have to be altered to observe distinct infection foci and resulting mounds at late stages of infection.
Streaking of bacterial cells on plates for single colonies
Timing: 2–3 h
For all our studies we have been using the 1043S L.m. strain, which is a clinical isolate used commonly in laboratories studying L.m. infection and for which we have a variety of fluorescently-tagged or mutant strains.
-
1.
Preparation of stock antibiotic solutions.
Note: All water used in the preparation of reagents was type 1 ultrapure water.
Dissolve 0.5 g streptomycin sulfate into 10 mL water to prepare a 50 mg/mL stock solution. Dissolve 75 mg of chloramphenicol into 10 mL of 100% ethanol to prepare a 7.5 mg/mL stock solution. Dissolve 0.2 g of gentamicin sulfate into 10 mL water to prepare a 20 mg/mL stock solution. Use a 0.2-μm syringe filter to sterilize and store solutions at −20oC.
-
2.Preparation of brain heart infusion (BHI) medium, BHI agar plates and streaking of bacteria
-
a.BHI medium preparation
-
i.Add a magnetic stir bar to a 1 L glass flask.
-
ii.Add 37 g of BHI to the flask, fill it with water up to 1 L, and mix the solution by placing the flask on a magnetic stir plate until the powder is dissolved.
-
iii.Cover the flask loosely and autoclave using the liquid setting of your autoclave (or according to the autoclave’s specifications).Note: The sterilized BHI medium can be used as needed for up to a month.
-
i.
-
b.BHI agar plate preparation
-
i.Add a magnetic stir bar to a 1 L glass flask.
-
ii.Mix together 37 g of BHI powder, 15 g of granulated agar and water up to 1 L until the solid components are dissolved.Note: Leave the stir bar inside so it can be used to aid in mixing in the antibiotics.
-
iii.Loosely cover the flask and autoclave using the liquid setting of your autoclave. After removing the flask from the autoclave, let the solution inside cool down to around 55°C.
-
iv.Add 7.5 μg/mL chloramphenicol and 200 μg/mL streptomycin (or other appropriate antibiotic depending on the bacterial strain) and mix by placing the flask on a magnetic stir plate.Note: The antibiotics mentioned above are specific to our L.m. strains which are resistant to streptomycin. They are also conjugated with a plasmid that contains the chloramphenicol acetyltransferase open reading frame, and are thus resistant to chloramphenicol (for details refer (Bastounis et al., 2021)). Any other necessary antibiotics can be used depending on the strain used for infection.
-
v.Pour approximately 20 mL of the mixture into a 10 cm culture plate and repeat until the BHI agar plate is depleted.Note: Eradicate any bubbles by flaming the upper surface of the plates briefly.
-
vi.Let the plates cool down for 8 h at 25°C and then store in a bag at 4oC for up to a month.
-
i.
-
c.Streaking of bacteria into BHI agar plates
-
i.Up to 2 weeks before the day of infection, streak out the L.m. strain to be used from a glycerol stock (stored at −80oC) onto a BHI-agar plate.
-
ii.Incubate the plates at 37°C for 48 h until you can observe discrete colonies.
-
iii.Use parafilm to seal the edges of the plates so that they do not dry out over time and store the plates for up to 2 weeks at 4°C.
-
i.
-
a.
Culturing and passaging of the host epithelial MDCK cells
We used MDCK II cells because they form polarized monolayers in culture, because we have various fluorescently-tagged and otherwise mutated cell lines that are isogenic (e.g., MDCK II E-cadherin-RFP, αΕ-catenin knockout MDCK II), and because their mechanobiology has been extensively studied in recent years. They have also been extensively used to study L.m. infection in vitro (Ortega et al., 2019; Pentecost et al., 2006; Robbins et al., 1999).
-
3.
Culture Type II Madin-Darby Canine Kidney (MDCK) cells in high glucose DMEM medium containing 4.5 g/L glucose and supplemented with 10% fetal bovine serum (FBS) in T-75 flasks in a suitable incubator at 37°C and in the presence of 5% CO2.
-
4.Whenever cells become confluent, passage them by splitting 1:6 to 1:12.
-
a.For passaging, remove and discard the culture medium and rinse the cells once with PBS.
-
b.Add 2 mL of 0.25% trypsin-EDTA in the T-75 flask and place the flask in the incubator for 10 min or until the cells are detached from the surface of the flask.
 CRITICAL: To avoid clumping of cells do not shake/hit the flask while waiting for the cells to detach but rather let them sit in the incubator for the appropriate amount of time until they are released from the substrate. Mechanical agitation will lift the cells, but it is possible that they might detach in clumps. This part is particularly important when seeding cells in coverslips where one wants to ensure homogeneous cell spatial distribution in the monolayer.
CRITICAL: To avoid clumping of cells do not shake/hit the flask while waiting for the cells to detach but rather let them sit in the incubator for the appropriate amount of time until they are released from the substrate. Mechanical agitation will lift the cells, but it is possible that they might detach in clumps. This part is particularly important when seeding cells in coverslips where one wants to ensure homogeneous cell spatial distribution in the monolayer. -
c.When cells have detached, add culture medium upto a final volume of 12 mL and mix cells by gently pipetting.
-
d.Add appropriate volumes of the cell suspension to new flasks (so that the cell density as compared to the confluent flask is diluted between 1:6 and 1:12), and supplement with appropriate volumes of full culture medium.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 40% Acrylamide solution | Sigma | Cat#A4058 |
| 2% Bis-acrylamide solution | Thermo Fisher | Cat#BP1404-250 |
| Sulfo-SANPAH | Thermo Fisher | Cat#22589 |
| FluoSpheres™ Carboxylate-Modified Microspheres, 0.2 μm, yellow-green fluorescent (505/515), 2% solids | Invitrogen | Cat#F8811 |
| Rat tail collagen type I | Sigma | Cat#C3867 |
| HEPES, free acid | J.T. Baker | Cat#4018-04 |
| (3-Aminopropyl)triethoxysilane | Sigma | Cat#3648 |
| Ammonium persulfate | Fisher | Cat#BP17925 |
| TEMED | Sigma | Cat#T9281 |
| 25% Glutaraldehyde | Sigma | Cat#G6257 |
| Leibovitz's L-15 Medium, no phenol red | Thermo Fisher | Cat#21083027 |
| High-glucose DMEM medium | Thermo Fisher | Cat#11965092 |
| Foundation Fetal Bovine Serum, lot: A37C48A | Gemini Bio-Prod | Cat#900108 |
| Brain heart infusion (BHI) | BD | Cat#211059 |
| Phosphate buffered saline (PBS), no calcium, no magnesium | Fisher | Cat#SH30028FS |
| Gentamicin sulfate | MP Biomedicals | Cat#194530 |
| Chloramphenicol | Sigma | Cat#C0378-5G |
| Streptomycin sulfate | Thermo Fisher | Cat#3810-74-0 |
| DifcoTM agar, granulated | BD | Cat#214530 |
| 0.25% Trypsin-EDTA, phenol red | Thermo Fisher | Cat#25200056 |
| Hoechst 33342, trihydrochloride, trihydrate - 10 mg/μL | Invitrogen | Cat#H3570 |
| Formaldehyde 16% EM grade | Electron Microscopy Sciences | Cat#15710-S |
| Experimental models: Cell lines | ||
| MDCK II | Bakardjiev Lab (Faralla et al., 2018) | N/A |
| MDCK II E-cadherin-RFP | Nelson Lab (Perez et al., 2008) | N/A |
| Experimental models: Organisms/strains | ||
| L. monocytogenes: L.m.-ActAp::mTagRFP | Theriot Lab(Ortega et al., 2017) | N/A |
| L. monocytogenes: L.m.-GFP | Theriot Lab (Ortega et al., 2017) | N/A |
| Software and algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Micro-Manager | N/A | https://www.micro-manager.org/ |
| MATLAB | MathWorks | http://www.mathworks.com/products/matlab/?requestedDomain=www.mathworks.com |
| Mound volume measurement code | This paper | https://gitlab.com/theriot_lab/theriot-toolbox/infection-mound-volume |
| Laser wounding recoil displacement code | This paper | https://gitlab.com/theriot_lab/infection_mound_laser_wounding |
| GraphPad Prism v6 | GraphPad | http://www.graphpad.com/scientific-software/prism/ |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| Other | ||
| 24-Well plates with glass bottom | MatTek | Cat#P24G-1.5-13-F |
| T-75 Flasks | Falcon | Cat#353118 |
| Plastic cuvettes | Sarstedt | Cat#67.746 |
| 50 mL Conical tubes | Falcon | Cat#352070 |
| 15 mL Conical tubes | Falcon | Cat#352196 |
| Disposable serological pipettes (1 mL, 2 mL, 5 mL, 10 mL) | Falcon | various |
| Bacterial cell spreader | Sigma | Cat#Z376779 |
| Pipette tips 1-200 μL | Denville | Cat#P1122 |
| Pipette tips 101–1000 μL | Denville | Cat#P1126 |
| Powder-free examination gloves | Microflex | Cat#XC-310 |
| 0.2 μm Vacuum bottle top filters | Thermo Fisher | Cat#566-0020 |
| 20 mL Syringes | BD | Cat#302830 |
| 0.2 μm Syringe filters | Thermo Fisher | Cat#723-2520 |
| 10 cm Culture plates | Falcon | Cat#351029 |
| Biosafety cabinet | Baker | Cat#SG504 |
| Hemocytometer | Sigma | Cat#Z359629 |
| Bacterial incubator | Thermo Scientific | Cat#IGS180 |
| CO2 culture incubator | NuAire | Cat#NU-8700 |
| Vacuum chamber/degasser | Bel-Art | Cat#42025 |
| Cage incubator (for the microscope) | Haison | Custom |
| Pipettor | Drummond | Cat#4-000-110 |
| Pipet tips (10 μL, 200 μL, 1000 μL) | Gilson | Cat#F144802, F123601, F123602 |
| pH meter | Mettler Toledo | Cat#30019028 |
| 1 L Flask | Fisherbrand | Cat#FB5011000 |
| Autoclave | Amsco | Cat#3021 |
| Stir magnet plate | Bellco | Cat#7760-06000 |
| Magnet stirring bars | Bellco | Cat#1975-00100 |
| Spectrophotometer | Beckman | Cat#DU 640 |
| Firefly system | Rapp OptoElectronic | Cat#UGA-42 Firefly |
Note: We used an inverted Nikon Eclipse Ti2 with an EMCCD camera (Andor Technologies) for the traction force microscopy experiments. For confocal imaging of the mounds and laser wounding experiments, we used a Yokogawa W1 Spinning Disk Confocal with Borealis upgrade on a Leica DMi6 inverted microscope with a 50 μm disk pattern. Any inverted epifluorescence or confocal microscope should work for the purposes of the protocol presented below.
Step-by-step method details
Seeding of host cells and preparation of bacterial solutions – day 1
Host MDCK cells are seeded onto glass coverslips (or hydrogels) and allowed to form a monolayer for 24 h. Bacteria are picked from the BHI agar plate and grown in liquid broth for approximately 16 h in preparation for the next day’s infection assay.
-
1.Seeding of ΜDCK cells on collagen I-coated glass coverslips (Figure 1A)
-
a.Mix 1:100 collagen I solution (stock concentration is 3.1 mg/mL) in PBS and add 500 μL of the solution in each well of the multi-well plate. Incubate for 30 min at 37oC.
-
b.Detach the MDCK cells from their flasks using 0.25% trypsin/EDTA by aspirating out the media from the flask, washing the cell surface with 10 mL PBS once and adding 2 mL of 0.25% trypsin/EDTA to the cells. Place the flask in the incubator at 37oC for 10 min or until all cells detach.
-
c.Add 8 mL of complete DMEM media to the cells, count the number of cells per mL using a hemocytometer to prepare a solution of 2×105 cells/mL.Note: Concentrate the cells by centrifugation or dilute them as appropriate to obtain the correct concentration.
-
d.Remove the multi-well plate from the incubator, aspirate the collagen solution, wash the wells with PBS once and add 1 mL of the cell solution in each well.CRITICAL: You can vary the concentration of the host cells as needed. However, due to the impact of cell confluency on intracellular bacterial spread and mound formation, consistency is important when repeating experiments.
-
e.Let MDCK cells attach and proliferate in the incubator to form a monolayer for 24 h prior to infection. On the day of infection, the concentration of cells per well is estimated to be double what was seeded (i.e., 4×105 cells/well) and by that point, the cells should have formed a confluent monolayer.
-
a.
-
2.Preparation of bacterial liquid cultures
-
a.Add 2 mL of liquid BHI and the appropriate antibiotic in a 15 mL falcon tube.
-
b.Using a sterile pipette tip, select a single colony from the BHI agar plate and drop the tip into the liquid.
-
c.Loosely screw on the cover of the tube and place the tubes in dark without agitation.
-
d.Let the culture grow for 16–18 h at 25°C .
-
e.Prior to the infection, the concentration of the bacteria should be measured using a spectrophotometer. To do so, dilute the bacterial solution 1:2 in BHI and place it in a disposable cuvette.
-
f.Measure the optical density (OD) at λ= 600 nm which should be ∼ 0.8 after 16 h of growth in BHI solution. If OD600 is lower than 0.8 let the bacteria grow for longer time. If it is slightly more that 0.8 prior to infection dilute appropriately.
-
a.
Figure 1.

Schematic of infection assay for formation of mounds late in infection
Sketch depicting the main steps (A–F) for seeding and infection of host epithelial MDCK cells with low MOI of L.m. so that single infection mounds are formed between 16–24 hpi.
Infection of cells in a monolayer with low MOI of L.m.– day 2
MDCK cells in a monolayer are infected with low MOI of L.m. to allow for the observation of distinct infection foci and mounds between 16 to 24 hpi (Figure 1).
-
3.Preparation of the bacterial infection mix
-
a.Concentrate 2 mL of 0.8 OD bacterial suspension by centrifugation for 4 min at 2000 × g.
-
b.Remove the supernatant and resuspend the bacterial pellet in 2 mL of tissue culture PBS in a biosafety cabinet.
-
c.Wash the bacteria by centrifugation twice more as above and resuspend in 2 mL PBS.CRITICAL: Washing the bacteria is necessary to remove soluble pyrogens from the bacterial culturing process.
-
d.Prepare the infection mix by mixing the bacterial–PBS suspension with complete DMEM media at a ratio of 1:16 (1 mL bacterial suspension and 15 mL of DMEM media).
-
a.
Note: Note you might need to use slightly more or less bacteria in the suspension to ensure you get nicely separated infection foci overtime (see Troubleshooting 1).
-
4.Infection of host epithelial cells
-
a.Remove the culture media from the wells, wash cells with 1 mL media and then add 1 mL of bacterial mix to each well (Figure 1B).Note: Keep the rest of the infection mix for determining the multiplicity of infection, MOI (see step 5).
-
b.Cover the plate with its lid and place the plates in the incubator for 30 min to allow for bacterial invasion of host cells.
-
c.Wash the samples 4 times with media to remove most extracellular bacteria and move the plate back into the tissue culture incubator.
-
d.After an additional 30 min of incubation, replace the media in the wells with fresh DMEM media supplemented with 20 μg/mL gentamicin, to ensure that any remaining extracellular bacteria are killed (Figure 1C).
-
a.
-
5.Determination of MOI
-
a.To determine the MOI, plate 100 μL of the bacterial solution used for infection at different dilutions (1:102, 1: 103, 1: 104, 1: 105) on BHI agar plates with appropriate antibiotics.
-
b.Use a bacterial spreader or beads to ensure that bacteria are homogeneously spread on the plates and place the plates in an incubator at 37oC.
-
c.After 48 h, count the number of colonies formed in the higher dilution plates (i.e., 1: 104, 1: 105) and average out to calculate the resulting MOI.
-
a.
Note: MOI (number of bacteria/ cell) is calculated as: , where : number of colonies grown in the plate under consideration, : dilution factor, : volume of bacterial solution added on agar plates, : volume of bacterial solution added on host cells, : number of host cells in the infected well.
Sample fixation and mound volume measurement – day 3
Since host cell samples were infected with a low MOI of L.m., single infection foci can be easily observed by epifluorescence microscopy starting 4–5 hpi (Figures 1D and 1E). These foci are surrounded by large domains (>300 μm) of uninfected cells. Under these conditions, infection mounds comprised of hundreds of squeezed and/or extruded infected cells start forming at about 16 hpi (Figures 1F and 2A). Bacteria at these late times of infection continue to grow exponentially suggestive of gentamicin not being harmful in any way to the host cells. Here, we describe a method based on acquisition of 3D confocal microscopy images and subsequent image processing to characterize the resulting volume of infection mounds at 24 hpi.
Note: We have opted to use a fluorescently-tagged L.m. strain to avoid additional steps required for immunostaining the bacteria after sample fixation.
-
6.MDCK cell nuclei staining and sample fixation at 24 hpi
-
a.Prior to fixation, incubate the samples for 10 min at 37oC with 1 μg/mL Hoechst in culture media to stain the host cells’ nuclei.
-
b.Following incubation, wash the samples once gently with 1 mL PBS.CRITICAL: Harsh or repetitive washing can detach the extruded infected cells from the mound (see Troubleshooting 2).Alternative: You can opt for first fixing the samples, permeabilizing them with 0.2% Triton X-100 in PBS and then staining with 1 μg/mL DAPI for 30 min.
-
c.Fix the samples for 10 min at 25°C in pre-warmed (37°C) freshly-made 4% paraformaldehyde in PBS.
-
d.Wash the samples with PBS twice and proceed to image the mounds.
-
a.
Pause point: You can store the samples at 4°C for future imaging. You can also immunostain for different cellular compartments or cytoskeletal elements (for detailed protocol regarding immunostaining refer to (Bastounis et al., 2021)).
-
7.Acquisition of images of infection mounds using 3D confocal microscopy
-
a.Place the multi-well plate on the appropriate stage insert on the stage of your confocal microscope.Note: We chose to use rapid spinning disk confocal microscopy (rather than slower laser-scanning confocal microscopy) due to the large number of images that need to be acquired to completely capture the shape of an infection mound. Given the size of our mounds we used a 60× 1.4NA Plan Apo oil objective.
-
b.Find the focal plane where the cell monolayer is located and use the bacterial fluorescence to identify a single infection focus.
-
c.Acquire confocal images of the Hoechst-stained host cell nuclei and of the fluorescent bacteria using a z spacing of 0.2 μm.
-
a.
Note: The field of view is selected so that the infection mound is approximately at the center. We typically used 0.2 μm z spacing and imaged just one field of view for mounds formed by MDCK cells. However human A431D cells form much larger mounds, so we used a z spacing of 0.7 μm, acquired 9 fields of view and stitched them together to accurately capture the whole infection mound. For both MDCK and A431D cells mounds are ∼50 μm tall but the resulting total volume of the mounds in A431D is 1 order of magnitude larger (∼1×106 μm3) than in MDCK cells (Bastounis et al., 2021).
-
8.Image processing for extraction of mound volume using custom-built scripts in MATLAB (MathWorks) (Figures 2B–2D)Note: All scripts used for subsequent analysis can be downloaded from our gitlab repository (see code availability) and are extensively commented.
-
a.To quantify the volume of the infected cells extruded from the monolayer, first crop out the lower z slices that refer to cells still attached on their substratum.Note: To infer what the height of the monolayer is you must also acquire confocal slices for uninfected cells. Once you know that, you can crop out the lower z slices that refer to cells still attaching on their substratum, you use the rest of slices to define domains of extruded infected cells based on their nuclei position. We use the host cell nuclei channel instead of the bacterial fluorescence for defining those domains as it enables us more accurately to determine which infected cells are in contact to the substratum versus which are extruded.
-
b.Segment the channel containing the host cell nuclei to form binary images of each mound by first using a background image of a dummy coverslip lacking cells that is treated identically to the experimental coverslips that you previously obtained and subtract this background image from the image of the host cell nuclei.
-
c.Correct for uneven illumination if the nuclei at the center of your image appear brighter than nuclei around the periphery, by dividing the nucleus channel by a flatfield image acquired by using a concentrated dye slide.
-
i.Add 5 μL of 50 mg/mL coumarin dye to a Micro90-cleaned slide, add a coverslip onto the slide and seal using nail polish.
-
ii.After finding your focal plane using your confocal microscope, obtain a z stack of images at multiple positions that extend well above and below the focal plane. Choose laser intensity and exposure settings that avoid saturation at any plane.
-
iii.Divert light to the eyepiece and capture 100 images in a time series using the same intensity and exposure settings chosen for the flatfield image. Then, perform a median projection to convert the stack into one dark image.
-
iv.Apply a median filter to every slice of the flatfield image and make a maximum intensity projection.
-
v.Subtract the dark image from the flatfield image at each position.
-
vi.Perform a median projection across positions to obtain one flatfield image.Note: If applying a flatfield correction produces artifacts (such as in z-slices where there aren’t many nuclei at the periphery, typical at higher values of z), divide by a uniform matrix of similar intensity to the flatfield image. As the flatfield image is acquired using a concentrated dye slide, it is much brighter than a nucleus image. Correct for the difference in intensity between the flatfield and nucleus images (see our code repository for more details).
-
i.
-
d.Convert the grayscale into a binary image by applying an appropriate threshold. As the nuclei become dimmer at increasing mound heights, check the binary mask against the grayscale nucleus image at intervals of 10 z-slices and adjust the value of the threshold if necessary.Note: Perform adjustments, such as opening and closing steps, to perfect the segmentation. An opening step (performed using the imopen command in MATLAB) removes objects in the binary image that are below a specified size, while a closing step (performed using the imclose command in MATLAB) connects neighboring objects below a specified size into one larger object.
-
e.Draw a boundary around the segmented nuclei and only consider the fluorescence that falls within that region while binarizing the next z-slice.Note: This approach of nested segmentation allows capturing dim nuclei at higher z-slices and avoiding fluorescence artifacts in regions of the image that contained cells at lower values of z but are empty for the z-slice under consideration.
-
f.An alpha shape is a non-convex boundary drawn around a set of points, where the tightness of the shape around the points is determined by the alpha radius that is specified. Input the co-ordinates of the nuclei within the binarized image into the alphaShape function in MATLAB along with the alpha radius of 50 pixels. Then, feed the shape generated into the area function to determine the area enclosed within the alpha shape for each z slice.Note: The optimal choice of the alpha parameter needs to be determined experimentally for different host cell types (Figure 2E).
-
g.Add up the area of each z slice after converting the area into μm2 and multiply by the increment between each z slice to obtain the volume of the mound in μm3.
-
a.
Figure 2.

Visualization of infection mounds at 24 hpi with L.m. and calculation of mound volume
(A) Orthogonal views of uninfected (left) or L.m.-infected host MDCK cells at 24 hpi (right). Host nuclei are shown in yellow and L.m. in black. In the case of infection a single mound is shown. Barplot on the right shows the calculated volume of the extruded infected cell domains (N=77, mean+/-SD).
(B–D) Methodology followed for extracting the volume of extruded infected cell domains. From the confocal image of the region enclosing the mound (B, top), cells within the monolayer were cropped out, allowing us to calculate the volume of only extruded cells (silver cells in bottom images). Each confocal slice was converted into grayscale (representative z slices in C, left), binarized (middle), and contained within an alpha (α) shape (right, α=50). Note that z= 0 μm refers to the first confocal slice corresponding to extruded cells. (D) 3D reconstructions of the full mound volume generated by summing alpha (α) shapes determined for each confocal slice. Mound volume in μm3 is estimated by multiplying the area determined for the alpha (α) shape in each slide by the increment between z slices and summing over the total. Note that varying the alpha (α) parameter influences the shapes of the reconstructed mounds; compared to a typical choice of α=50 (left), decreasing α to 40 results in a smaller volume estimate (middle), and increasing α to 60 results in a larger volume estimate (right).
(E) Plot showing the number of infected cells within an infection focus versus the area of the focus (in a single confocal slice) calculated based on α shapes for three different values of α considered. The resulting R2 is also provided and suggests that α=50 represents an optimum value of α that maximizes R2.
Parts of the figure reprinted with permission from (Bastounis et al., 2021).
Monolayer tension estimation through laser wounding – day 3
Laser wounding provides a fast perturbation to intercellular tension and can be used to determine the direction and relative magnitude of pre-existing stresses in epithelial cell sheets (Bastounis et al., 2021) (Figure 3). Here, we present the protocol we used to determine the relative tension between infected cells contained in the mounds and surrounding uninfected cells by measuring the rapid recoil of the cells away from the wound over the first few minutes after wounding, and the slower wound closure response at later times (Figure 4).
-
9.Perform the laser wounding on a segment of E-cadherin-RFP expressing MDCK cells infected with L.m.-GFP a day before.Note: The host cells can express any other fluorescent protein of interest, however the resulting fluorescent images need to have some texture and minimal spaces devoid of fluorescence to facilitate subsequent image analysis.
-
a.Supplement Leibovitz’s L-15 with 10% FBS and 20 μg/mL gentamicin and warm to 37oC. Replace the DMEM-based existing medium in which host cells are immersed with 1 mL of the live-microscopy medium at ∼ 24 hpi.Alternative: If you have a CO2 control chamber in your microscope it is not necessary to use L-15 medium. However, note that phenol red will interfere with the bacterial or host cell fluorescence and must be avoided.
-
b.When mounds are readily visible at 24 hpi, transfer your multi-well plate on the stage of a confocal microscope equilibrated to 37oC.
-
c.Locate an infection focus and decide what type of wound you will create. You can wound the focus vertically (line that crosses along the middle of the mound), horizontally (tangent to the mound) or around the mound (Figure 4B).Note: The choice of the cut you perform depends on the question you want to answer. For more details refer to (Bastounis et al., 2021). The size of the ablated region should be proportional to the focus size, which is dependent on the specific host cell type and bacterial strain used (Figure 3B) (see Troubleshooting 3).
-
d.Start acquiring confocal images of the E-cadherin-RFP and L.m. fluorescence using a 60× 1.4NA Plan Apo oil objective and a z spacing 0.7 μm. The E-cadherin-RFP is imaged with a 561 nm laser at full power with 300 ms exposure, the L.m.-GFP fluorescence with a 488 nm laser at full power with 20 ms exposure and the brightfield image with a 50 ms exposure. 1 image pre-wounding is sufficient.
-
e.To achieve a laser wound, with a FRAP type system with a 405 nm laser, scan a line segment (or ROI) 270 times over a time period of 1 min. Acquire images every 1 min for up to 1 h post-wounding (or longer if you are interested in the wound healing process post-wounding).
-
a.
Note: These settings need to be determined empirically based on your specific setup (which includes the entire optical path, laser type, excitation power, the specific fluorophore used etc.). For instance, using full power in a different microscope to image E-cadherin-RFP could result in damaging the cells. If this scenario holds could be checked post-wounding by assessing whether the host cells are intact and thus able to mount a migratory wound-healing response or not, in which case it would be possible that they might be damaged.
-
10.Analysis of the displacements of cells upon laser wounding (recoil or relaxation) and their corresponding wound healing response using custom-built scripts in MATLAB (MathWorks).Note: All code and example data can be found in our gitlab repository (see code availability) and are extensively commented.
-
a.Calculate the maximum intensity projections of the E-cadherin-RFP images of the cells overtime.
-
b.Perform correlations comparing subsequent frames using the maximum intensity projections. We used a particle image velocimetry like-technique and employed windows of 36×36 pixels with a 50% overlap (Figure 4A) (Gui and Wereley, 2002).
-
i.For each experiment run the run_PIV.m script which calls the urapiv.m function to calculate the displacement vectors and store them as .txt files.
-
ii.After generating and storing the .txt files you can inspect the vector displacements by running the script readu_laser_ablation.m (line 138 in the script).
-
i.
-
c.Using the component of the velocity that is perpendicular to the ablated segment, you can calculate the mean displacement along the axis of the laser wound and construct kymographs that show the mean displacement across time and position away from the wound (Figure 4B).
-
i.When running the readu_laser_ablation.m, you are storing the mean displacement perpendicular to the wound in the variable “kymographm”.
-
ii.After having calculated the mean displacement for all frames you plot and save the kymograph in lines 173 to 178 of the readu_laser_ablation.m script.
-
iii.This representation allows for a detailed quantitative analysis of the recoil behavior of cells upon laser wounding and calculation of the magnitude of recoil, spatial location and temporal evolution.
-
i.
-
d.To quantitatively characterize recoil displacement based on the behavior of cells in response to laser wounding, calculate the mean uy along the x-axis but also along the y-axis for each experiment considering just the first 100 μm away from the ablated region (Figure 4C), as cell behavior will depend on distance from the wound margin.Note: The mean recoil displacement is calculated in lines 182 to 184 of the readu_laser_ablation.m script.
-
a.
Figure 3.

Schematic of laser wounding assay for monolayer tension estimation and examples of different wound configurations
(A) Sketch depicting the principles of the laser wounding assay for an uninfected epithelial cell monolayer. Local damage induced by the focused laser normally results in cells of the monolayer recoiling away from the wound margin, due to pre-existing stress in the monolayer.
(B) Representative examples of different wound configurations (columns) for L.m.-infected MDCK cells at 24 hpi when infection mounds emerge. First row shows the brightfield image of host cells with L.m. fluorescence superimposed (green) immediately after laser wounding and second row shows the corresponding fluorescence images of E-cadherin.
Figure 4.

Analysis of laser wounding experiments
(A) Laser wounding in monolayers of MDCK cells expressing E-cadherin-RFP for uninfected (top) or L. m.-infected wells (bottom) 24 hpi. Columns show: the brightfield image superimposed with L. m. fluorescence (green) prior to wounding (t=0 min); the corresponding E-cadherin localization prior to wounding (t=0 min); E-cadherin localization immediately after 1 min of laser exposure (t=1 min); the cell displacement vectors u (50-fold larger) calculated by performing PIV; the average uy at t=1 min (positive/negative values point towards/away from wound); and the average uy over the time period 30–40 min.
(B) Kymographs of the average with respect to the wound displacement vectors, uy as a function of time, t (min, x-axis) and vertical position, y (μm, y-axis) for the same examples as shown in panel A (see single and double crosses).
(C) Barplots of the mean uy along a distance of 100 μm away from the wound and calculated immediately after wounding, for cells originating from uninfected or from infected wells (N= 3 experiments, mean+/-SD, unpaired Student’s T-Test: ∗ p<0.01).
Parts of the figure reprinted with permission from (Bastounis et al., 2021).
Traction force microscopy (TFM) during mound formation – day 3–4
This protocol is based on the assumption that 3 kPa 2-layered polyacrylamide hydrogels have been prepared on multi-well plates as described elsewhere (Bastounis et al., 2018). The upper hydrogel layer is embedded with fluorescent tracer beads and coated with collagen I (see Troubleshooting 4). MDCK cells are seeded on the hydrogels and infected with low dosage of fluorescently-tagged L.m. At 4–5 hpi the plate is placed on the stage of an epifluorescence microscope. Time-lapse acquisitions of the phase contrast image of cells, of the beads’ fluorescence and of L.m. fluorescence are acquired and analyzed to calculate the 2D traction stresses that infected and uninfected cells exert during the formation of infection mounds.
Note: As the hydrogels that we used for TFM are embedded with yellow fluorescent beads to avoid spectral overlap we infected host cells with L.m.-ActAp::mTagRFP. Any fluorescently-tagged strain or any color of fluorescent beads could be used providing there is no spectral overlap between the two.
-
11.Prepare the sample and start the TFM acquisition centering the field of view on a single infection focus (Figure 5A)
-
a.Replace the DMEM-based existing medium submerging host cells with 1 mL of the live-microscopy medium described above.
-
b.Cover the plate with its lid and place it in the microscope’s environmental chamber equilibrated to 37oC.Alternative: If you have a CO2 control chamber in your microscope it is not necessary to use L-15 medium. However, avoid phenol red.
-
c.Set the microscope up for phase contrast microscopy with a 20× or 40× phase-contrast objective.
-
d.Locate a single infection focus using the appropriate fluorescence filters, ensuring that host cells around this focus (in a radius 200–300 μm away) are not infected and that the center of the focus in the middle of the field of view.
-
e.Locate the fluorescent beads using the appropriate fluorescence filters and ensure that the distribution of beads in this location is even (Figure 5B).CRITICAL: Adjust the focus slightly to the plane where the intensity of the beads is maximal.
-
f.Start acquiring multi-channel time-lapse images every 10 min for 24 h (or more) while making sure the focus is maintained to avoid z drift.
-
a.
-
12.At the end of the recording acquire a reference image of the beads.
-
a.After an appropriate amount of time stop your recording.
-
b.Without moving the plate, remove the lid and add 100 μL of 10% SDS to cause host cells to detach from their substrate.Note: You can ensure that this occurs by observing them through phase contrast (see also Troubleshooting 6).
-
c.Acquire an image of the fluorescent beads upon removal of the host cells to get your reference image (Figure 5B).
-
a.
Note: After addition of the SDS the tracer beads go back to their initial configuration since the gel is elastic and the cells that have been exerting forces on it are now removed.
-
13.Analyze your images using TFM using custom-built scripts in MATLAB (MathWorks) (Figures 6A and 6B).Note: For more details please refer to our published protocol (Bastounis et al., 2018).
-
a.Determine the 2D deformation of the substrate at each point using a technique similar to particle image velocimetry (PIV) (Gui and Wereley, 2002). The local deformation vector is computed by performing a correlation between each experimental image and the unstretched reference image, using an interrogation window of 32×32 with a 8 pixel spacing (for more details see below).Note: You can run the urapiv.m function as in step 10.b.i., with the exception that in this case you are comparing the image of the tracer beads at each time interval with the reference image of the undeformed beads obtained at the end of your experiment when cells have been removed from the substrate.
-
b.Calculate the 2D traction stresses that cells exert on the gel as described elsewhere (del Álamo et al., 2007; Hur et al., 2012). Ensure that you provide as inputs the Young’s Modulus (stiffness) of your hydrogel (in our case 3 kPa), the finite thickness of the hydrogel measured after manufacturing the hydrogels (in our case 40 μm) (see Troubleshooting 7).
-
a.
Note: Codes and detailed protocols for TFM have been published (Teo et al., 2020) and can be downloaded from publicly available repositories written in Matlab (Saraswathibhatla et al., 2020) or as plugins working in ImageJ (Martiel et al., 2015).
Figure 5.

Representative images of TFM recording during infection
(A) Columns show representative phase contrast images of host MDCK cells in monolayer, L.m. fluorescence and fluorescence of the tracer beads embedded on the upper surface of a 3 kPa polyacrylamide hydrogel on which host MDCK cells reside. Rows show representative two time points at early (4 hpi) and late (24 hpi) infection. Note that brightness has been enhanced for the upper image (4 hpi) so that the bacterial fluorescence signal is visible.
(B) Reference image of the tracer beads for the representative recording shown in panel A (top-left) and examples of bad tracer beads’ distribution because: of the beads’ layer being too thick (top-right); of cracks on the upper hydrogel layer (bottom-left); or of the hydrogel not being flat so the beads in the bottom area in the field of view are out of focus (bottom-right).
Figure 6.

TFM during L.m. infection mound formation in MDCK cells in monolayer
Columns show: representative phase contrast images of MDCK cells in monolayer residing on a 3 kPa hydrogel and infected with low dosage of L.m. (same as example shown in Figure 5), corresponding L.m. fluorescence, cell-matrix deformation maps (color indicates deformation magnitude in μm), traction stresses exerted by cells on their matrix (color indicates stress magnitude in Pa) and radial cell-matrix deformation, ur maps (positive deformations values in μm indicate deformations pointing away from the center of the focus, outwards). Parts of the figure reprinted with permission from (Bastounis et al., 2021).
Expected outcomes
With the current protocol, users will be able infect host epithelial MDCK cells with a low multiplicity of infection of the intracellular bacterial pathogen Listeria monocytogenes (L.m.) and monitor infection focus growth and mound formation. Moreover, users will learn how the host cell traction stresses (Figure 5 and 6) and intercellular tension built within the infected cell monolayer (Figures 3 and 4) are dynamically modulated. In addition, users will be able to quantitatively characterize the volume of the extruded infected cell domain that emerges at late infection (Figure 2).
Quantification and statistical analysis
All image processing was performed using custom-built script in MATLAB (MathWorks). We applied image processing and segmentation based on alpha shapes to quantify the volume of extruded infected cell domains (mounds) as described above. The codes to measure mound volume are available at our gitlab repository together with instructions on how to run them (https://gitlab.com/theriot_lab/theriot-toolbox/infection-mound-volume). Sample data are also provided to allow users to repeat our approach. We used a particle image velocimetry like-technique to quantify cell displacements post-wounding and a kymographic representation to inspect cellular displacements perpendicular to the wound as a function of distance away from the wound and time post-wounding. The codes for this analysis are uploaded in out gitlab repository together with instructions on how to run them (https://gitlab.com/theriot_lab/infection_mound_laser_wounding). Sample data are also provided to allow users to repeat our approach. We performed TFM on cellular monolayers during infection to quantify the traction stresses that infected cells at the base of mounds or surrounding uninfected cells exert. To that end we used our particle image velocimetry like-technique to determine cell-matrix displacements and TFM to calculate cellular traction stresses, using codes published elsewhere (Lamason et al., 2016; Saraswathibhatla et al., 2020; Teo et al., 2020). Statistical parameters and significance are reported in the corresponding Figures and the Figure Legends. Boxplots represent mean+/-SD. In Figure 4C data are determined to be statistically significant when p < 0.05 by an unpaired Student’s T-Test. Statistical analysis was performed in GraphPad PRISM 8.
Limitations
Although the first steps (1–5) of the protocol describe infection of a particular host cell type (MDCK) with a specific intracellular bacterial pathogen (L.m.), the rest of the steps could be used to investigate the infection in additional host cell types with other bacterial or viral pathogens that spread intracellularly and could give rise to similar mechanical battles between infected and uninfected cells (Abaitua et al., 2013; Beerli et al., 2019). However, one would have to properly adjust the infection protocol for the particular host cell type or bacterial pathogen to be used. For infection protocols tailored for the intracellular bacterial pathogen Rickettsia parkeri, please refer to our paper (Bastounis et al., 2021).
To indirectly estimate the tension built between cells within infection mounds and their uninfected neighbors, we used laser wounding. Although this method can assist in determining the direction and relative magnitude of pre-existing stresses in epithelial cell sheets, it does not provide the magnitude of absolute intercellular stresses. Also, laser wounding provides a sense of how cells are behaving collectively rather than the level of tension experienced by individual cells in or near the mound. Continuing work on this aspect including usage of FRET-based force sensors could potentially facilitate a more absolute quantitation of intercellular stresses in and near infection mounds (Eder et al., 2017; Sim et al., 2015).
Troubleshooting
Problem 1
I do not get single infection foci, and over the course of time two distinct infection foci merge. As a result, I cannot locate single infection mounds.
Potential solution
Lower the dosage of L.m. you add on your host cells. The first time you perform your experiment you can seed cells on multiple wells and infect them with decreasing MOIs. 4 hpi or later you can inspect your samples under a microscope and determine empirically which MOI gives you single infection foci that are nicely separated (at least 300 μm) from neighboring foci.
Problem 2
Although I can observe infection mounds while cells are alive, I cannot locate any mounds after fixation and thus cannot acquire images for mound volume calculation.
Potential solution
Be very gentle when you wash the samples prior to fixation as you can accidentally wash off the extruded cells. If that does not remove the issue, you can stain the host cell nuclei with Hoechst prior to infection and then at 24 hpi acquire 3D confocal microscopy images for mound volume measurements using live samples.
Problem 3
Mound sizes vary greatly and so the area that must be ablated to create equivalent wounds across multiple mounds will also vary.
Potential solution
Rather than ablating cells for a fixed length of time, keep the number of laser runs consistent across multiple mounds to ensure that they are damaged to an equal extent.
Problem 4
The hydrogel layer with beads is too thick, or there are spurious structures and thus the bead distribution is not even.
Potential solution
To ensure having a thin upper hydrogel layer after you deposit the second polyacrylamide solution and cover it with a circular glass coverslip, immediately press downwards using a pair of forceps to ensure the thickness is minimal. To avoid getting uneven distribution of beads or to not observe spurious structures emerging from bead aggregation there are a number of things you can attempt: (1) When pipetting the beads from their stock bottle to add them in the polyacrylamide solution, do not immerse the pipette tip close to the bottom of the bottle as aggregates tend to accumulate there; (2) Consider making hydrogels in multi-well plates so you can discard the ones that do not look appropriate for TFM and use the rest; (3) Usually stiffer hydrogels’ bead distribution (> 3 kPa) is much more even that of softer ones. Avoid making very soft hydrogels for TFM unless that is really needed to address your research questions.
Problem 5
During the TFM image acquisition, I don’t see any tracer beads’ displacements, or I see too large beads’ displacements and beads move out of the focal plane imaged.
Potential solution
To perform TFM, host cells must be able to displace the beads embedded on the substrate on which host cells reside and exert traction stresses. To our experience ∼70 kPa is the upper limit of ECM stiffness where displacements produced by MDCK imparting stresses can still be observed. If a different host cell type is used, then this upper limit would have to be determined empirically. In addition, placing the host cells on very soft matrices (<2 kPa) can lead to significant z-displacement of the beads due to the cells imparting large normal deformations (in the vertical direction). In that case 2D TFM cannot be applied since the fluorescent beads will move in and out of the focal plane imaged. If the question that one wants to answer is how infection alters host cell traction stresses (and not how ECM stiffness modulates the host cell response to infection) then we recommend building a hydrogel of intermediate stiffness, since very soft hydrogels can also be challenging to make as uneven distribution of beads and cracks in the gel are often observed (Figure 5B).
Problem 6
After I run PIV to calculate the displacements of the tracer beads in TFM, I notice significant x, y or z drift which makes the displacement fields look extremely noisy.
Potential solution
It is possible that by removing the lid of the multi-well pate to add SDS, you accidentally slightly displaced the plate. When you take your reference image you can compare the position that you are imaging with the image of the tracer beads during your recording. In case you notice a slight drift, you can adjust the focus and center the image appropriately. Usually there are some features in the texture of your tracer beads’ image than can facilitate this adjustment.
Problem 7
The magnitude of the traction stresses that host cells exert varies significantly from one experiment to another.
Potential solution
Because cell confluency might vary for multiple reasons, one way of ensuring that monolayers in different experiments have similar cell densities is by staining host cell nuclei with Hoechst and acquiring multiple microscopy images of different fields of view. You can segment the host cell nuclei and count them, thus determining whether that density is similar. In addition, make sure the upper hydrogel layer is thin and confined into a single plane. If that is the case, you should be more or less in focus irrespective of whether you are looking at the fluorescence of the tracer beads or the phase contrast image of the host cells. Note that sometimes the upper layer is thicker and you might be imaging deeper into the hydrogel (thus estimating lower forces because these beads will also get displaced but to a lesser extent than beads right at the surface) (Figure 5B).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Julie A. Theriot (jtheriot@uw.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The codes generated during this study to measure mound volume are available at https://gitlab.com/theriot_lab/theriot-toolbox/infection-mound-volume. The codes generated during this study to measure cellular displacements after laser wounding are available at https://gitlab.com/theriot_lab/infection_mound_laser_wounding. Codes to perform TFM in cellular monolayers have been published elsewhere (Lamason et al., 2016; Saraswathibhatla et al., 2020; Teo et al., 2020).
Acknowledgments
We are grateful to Matthew Footer for discussions, experimental support, and reviewing the protocols and to Ellie Labuz for helping out with the creation of our code repository. We also thank Andrew Kennard for his assistance on the use of the confocal microscope and on performing the background and flat-field corrections on the resulting images we acquired. We thank our co-authors/collaborators from (Bastounis et al., 2021) Francisco Serrano Alcalde, Patrik Engström, María J. Gómez Benito, Mackenzi S. Oswald, Jason G. Smith, Matthew D. Welch, and José M. García Aznar, for discussions and powering in part the development of some of the protocols and approaches described herein. This work was supported by NIH R37AI036929 (J.A.T.), HHMI (J.A.T.), and the American Heart Association, Award number: 18CDA34070047 (E.E.B.).
Author contributions
E.E.B. designed and optimized the protocol for construction of polyacrylamide hydrogels on multi-well plates, the infection assays, and performed the TFM experiments. P.R. designed and performed the laser wounding experiments, and E.E.B. performed the analysis and calculation of recoil speed and subsequent migratory wound healing response. E.E.B. collected images of mounds, and P.R. designed the analysis pipeline for mound volume calculation. C.P. assisted E.E.B. on the design of the graphical abstract and of the sketches presented herein. E.E.B., P.R., and J.A.T. wrote and reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Effie E. Bastounis, Email: effie.bastounis@uni-tuebingen.de.
Julie A. Theriot, Email: jtheriot@uw.edu.
References
- Abaitua F., Zia F.R., Hollinshead M., O'Hare P. Polarized cell migration during cell-to-cell transmission of herpes simplex virus in human skin keratinocytes. J. Virol. 2013;87:7921–7932. doi: 10.1128/JVI.01172-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E.E., Ortega F.E., Serrano R., Theriot J.A. A multi-well format polyacrylamide-based assay for studying the effect of extracellular matrix stiffness on the bacterial infection of adherent cells. J. Vis. Exp. 2018;5:57361. doi: 10.3791/57361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E.E., Serrano-Alcalde F., Radhakrishnan P., Engström P., Gómez-Benito M.J., Oswald M.S., Yeh Y.-T., Smith J.G., Welch M.D., García-Aznar J.M. Mechanical competition triggered by innate immune signaling drives the collective extrusion of bacterially infected epithelial cells. Dev. Cell. 2021;56:443–460.e411. doi: 10.1016/j.devcel.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli C., Yakimovich A., Kilcher S., Reynoso G.V., Fläschner G., Müller D.J., Hickman H.D., Mercer J. Vaccinia virus hijacks EGFR signalling to enhance virus spread through rapid and directed infected cell motility. Nat. Microbiol. 2019;4:216–225. doi: 10.1038/s41564-018-0288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Álamo J.C., Meili R., Alonso-Latorre B., Rodríguez-Rodríguez J., Aliseda A., Firtel R.A., Lasheras J.C. Spatio-temporal analysis of eukaryotic cell motility by improved force cytometry. Proc. Natl. Acad. Sci. U S A. 2007;104:13343–13348. doi: 10.1073/pnas.0705815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder D., Basler K., Aegerter C.M. Challenging FRET-based E-Cadherin force measurements in Drosophila. Sci. Rep. 2017;7:13692. doi: 10.1038/s41598-017-14136-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faralla C., Bastounis E.E., Ortega F.E., Light S.H., Rizzuto G., Gao L., Marciano D.K., Nocadello S., Anderson W.F., Robbins J.R. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. PLoS Pathog. 2018;14:e1007094. doi: 10.1371/journal.ppat.1007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui L., Wereley S.T. A correlation-based continuous window-shift technique to reduce the peak-locking effect in digital PIV image evaluation. Exp. Fluids. 2002;32:506–517. [Google Scholar]
- Hur S.S., del Alamo J.C., Park J.S., Li Y.S., Nguyen H.A., Teng D., Wang K.C., Flores L., Alonso-Latorre B., Lasheras J.C. Roles of cell confluency and fluid shear in 3-dimensional intracellular forces in endothelial cells. Proc. Natl. Acad. Sci. U S A. 2012;109:11110–11115. doi: 10.1073/pnas.1207326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamason R.L., Bastounis E., Kafai N.M., Serrano R., Del Alamo J.C., Theriot J.A., Welch M.D. Rickettsia Sca4 reduces vinculin-mediated intercellular tension to promote spread. Cell. 2016;167:670–683.e610. doi: 10.1016/j.cell.2016.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiel J.-L., Leal A., Kurzawa L., Balland M., Wang I., Vignaud T., Tseng Q., Théry M. Chapter 15 - Measurement of cell traction forces with ImageJ. In: Paluch E.K., editor. Methods in Cell Biology. Academic Press; 2015. pp. 269–287. [DOI] [PubMed] [Google Scholar]
- Ortega F.E., Koslover E.F., Theriot J.A. Listeria monocytogenes cell-to-cell spread in epithelia is heterogeneous and dominated by rare pioneer bacteria. eLife. 2019;8:e40032. doi: 10.7554/eLife.40032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega F.E., Rengarajan M., Chavez N., Radhakrishnan P., Gloerich M., Bianchini J., Siemers K., Luckett W.S., Lauer P., Nelson W.J. Adhesion to the host cell surface is sufficient to mediate Listeria monocytogenes entry into epithelial cells. Mol. Biol. Cell. 2017;28:2945–2957. doi: 10.1091/mbc.E16-12-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentecost M., Otto G., Theriot J.A., Amieva M.R. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2006;2:e3. doi: 10.1371/journal.ppat.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez T.D., Tamada M., Sheetz M.P., Nelson W.J. Immediate-early signaling induced by E-cadherin engagement and adhesion. J. Biol. Chem. 2008;283:5014–5022. doi: 10.1074/jbc.M705209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J.R., Barth A.I., Marquis H., de Hostos E.L., Nelson W.J., Theriot J.A. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J. Cell Biol. 1999;146:1333–1350. doi: 10.1083/jcb.146.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswathibhatla A., Galles E.E., Notbohm J. Spatiotemporal force and motion in collective cell migration. Sci. Data. 2020;7:197. doi: 10.1038/s41597-020-0540-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C., Rasband W., Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim J.Y., Moeller J., Hart K.C., Ramallo D., Vogel V., Dunn A.R., Nelson W.J., Pruitt B.L. Spatial distribution of cell–cell and cell–ECM adhesions regulates force balance while maintaining E-cadherin molecular tension in cell pairs. Mol. Biol. Cell. 2015;26:2456–2465. doi: 10.1091/mbc.E14-12-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo J.L., Lim C.T., Yap A.S., Saw T.B. A biologist’s guide to traction force microscopy using polydimethylsiloxane substrate for two-dimensional cell cultures. STAR Protocols. 2020;1:100098. doi: 10.1016/j.xpro.2020.100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The codes generated during this study to measure mound volume are available at https://gitlab.com/theriot_lab/theriot-toolbox/infection-mound-volume. The codes generated during this study to measure cellular displacements after laser wounding are available at https://gitlab.com/theriot_lab/infection_mound_laser_wounding. Codes to perform TFM in cellular monolayers have been published elsewhere (Lamason et al., 2016; Saraswathibhatla et al., 2020; Teo et al., 2020).