

Abstract
Diabetic retinopathy (DR) is a common consequence in type 2 diabetes (T2D) and a leading cause of blindness in working-age adults. Yet, its genetic predisposition is largely unknown. Here, we examined the polygenic architecture underlying DR by deriving and assessing a genome-wide polygenic risk score (PRS) for DR. We evaluated the PRS in 6079 individuals with T2D of European, Hispanic, African and other ancestries from a large-scale multi-ethnic biobank. Main outcomes were PRS association with DR diagnosis, symptoms and complications, and time to diagnosis, and transferability to non-European ancestries. We observed that PRS was significantly associated with DR. A standard deviation increase in PRS was accompanied by an adjusted odds ratio (OR) of 1.12 [95% confidence interval (CI) 1.04–1.20; P = 0.001] for DR diagnosis. When stratified by ancestry, PRS was associated with the highest OR in European ancestry (OR = 1.22, 95% CI 1.02–1.41; P = 0.049), followed by African (OR = 1.15, 95% CI 1.03–1.28; P = 0.028) and Hispanic ancestries (OR = 1.10, 95% CI 1.00–1.10; P = 0.050). Individuals in the top PRS decile had a 1.8-fold elevated risk for DR versus the bottom decile (P = 0.002). Among individuals without DR diagnosis, the top PRS decile had more DR symptoms than the bottom decile (P = 0.008). The PRS was associated with retinal hemorrhage (OR = 1.44, 95% CI 1.03–2.02; P = 0.03) and earlier DR presentation (10% probability of DR by 4 years in the top PRS decile versus 8 years in the bottom decile). These results establish the significant polygenic underpinnings of DR and indicate the need for more diverse ancestries in biobanks to develop multi-ancestral PRS.
Introduction
Type 2 diabetes (T2D) is a globally prevalent disease with high mortality, morbidity and healthcare costs (1,2). Insulin resistance causes hyperglycemia in T2D (3), resulting in retinal pericyte dysfunction, basement membrane thickening and ultimately diabetic retinopathy (DR) (4,5), the foremost cause of vision loss in working-age adults worldwide (6). In the United States, the number of persons with DR is predicted to rise 2-fold to 16 million by 2050 (7), with annual healthcare expenditures totaling $4 billion (8). Anxiety, depression and financial hardships are all common in DR (9). Earlier risk stratification and screening for DR can improve outcomes; however, an approximately 7-year lag between onset of T2D and development of any DR (10) burdens the healthcare system with maintaining adequate retinal surveillance in patients with T2D. Earlier stages of DR occur asymptomatically, while visual disturbances typically arise during later severe stages (11). Thus, there is heightened interest to enhance screening for the silent phase of this disease. The American Diabetes Association (ADA) recommends those with T2D undergo retinal screening annually and more frequently if retinopathy is detected (12). Though this strategy is validated for the population as a whole, individual risk for DR varies substantially due in part to each person’s unique genetic composition (13–17).
A surge in linked genetic and clinical data has enabled integrated genetic and clinical risk profiles that personalize an individual’s healthcare (18). Polygenic risk scores (PRS) have been developed to assess disease risk by aggregating genetic variants with small effect sizes into a single risk score. PRS has been implemented for many cardiovascular, neurological, psychiatric and ophthalmological diseases (19–21) and is strongly associated with disease at the tails of the PRS distribution. While elevated hemoglobin A1c (HbA1c), systolic blood pressure (SBP) and serum lipids [e.g. low-density lipoprotein cholesterol (LDL-C)] are correlated with increased risk of DR (22), a PRS may help identify high-risk patients who need more frequent screenings. PRS are overwhelmingly derived from genome-wide association studies (GWAS) of European individuals, and most PRS studies characterize disease risk solely in Europeans (23). A recent study showed that PRS derived from a European population for complex traits have lesser performance in non-European populations (23). As DR disproportionately affects minority populations (24), it is important to determine whether a DR PRS has generalizability across diverse ancestries.
In the present study, we performed a GWAS of DR in 17 567 individuals with T2D in the UK Biobank (UKB). The results were used to construct a genome-wide PRS, which was evaluated for association with DR in 6079 individuals with T2D in the BioMe Biobank (BioMe). We then examined transferability of the PRS to diverse ancestries. Lastly, we assessed the clinical validity and utility of PRS by testing its association with DR symptoms, conditions and time to diagnosis.
Results
GWAS of DR in UKB
A flowchart of the study design is provided (Supplementary Material, Fig. S1). The derivation of PRS required summary statistics of variant association with DR. Thus, we performed a GWAS of DR in 17 567 individuals with T2D (866 cases with DR; 16 701 controls without DR) in the UKB (Supplementary Material, Table S1). No significant difference in genetically defined type 1 diabetes (T1D) between DR cases and controls was detected with a previously published T1D genetic risk score (GRS) (25) (Supplementary Material, Fig. S2). In the GWAS, no loci reached genome-wide significance (all P > 5.0 × 10−8) and 28 loci were of suggestive significance (all P < 1.0 × 10−5) (Supplementary Material, Table S2; Supplementary Material, Fig. S3). There was minimal evidence of population stratification with a genomic control inflation factor (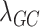 ) of 1.007.
) of 1.007.
Study population from an electronic health record-linked Biobank (BioMe)
We included 6079 participants with T2D (963 cases with DR; 5116 controls without DR) in BioMe who had genotype information and passed quality control (QC). There was no significant difference in T1D GRS between DR cases and controls (Supplementary Material, Fig. S2). DR cases had a greater proportion of hypertension (95 versus 83%), with higher median glucose (143 mg/dl versus 116 mg/dl) and HbA1c concentrations [8.0% (64 mmol/mol) versus 7.3% (56 mmol/mol)] than controls (Table 1). The proportion of DR cases varied by self-reported ancestry. Cases had a greater ratio of Hispanic Americans (53 versus 43%), but smaller ratios of European Americans (9 versus 17%) and other ancestries (6 versus 9%) compared to controls (Table 1); the ratio of African Americans was not statistically different (33 versus 31%).
Table 1.
Overview of baseline demographic and clinical traits in DR cases and controls in BioMe
| Trait | Case (n = 963) | Control (n = 5116) |
|---|---|---|
| Male, n (%) | 399 (41%) | 2281 (45%) |
| Age in years, mean (SD) | 68 (12) | 67 (13) |
| European American, n (%) | 90 (9%) | 888 (17%) |
| African American, n (%) | 317 (33%) | 1608 (31%) |
| Hispanic American, n (%) | 507 (53%) | 2182 (43%) |
| Other, n (%) | 49 (6%) | 438 (9%) |
| BMI in kg/m2, mean (SD) | 32 (7) | 31 (7) |
| Glucose in mg/dl, mean (SD) | 151 (57) | 128 (50) |
| Hemoglobin A1c in % [mmol/mol], mean (SD) | 8.0 (2.0) [64 (22)] | 7.3 (1.9) [56 (20)] |
| Hypertension, n (%) | 917 (95%) | 4247 (83%) |
Overview of baseline demographic and clinical traits in DR cases and controls in BioMe. Other, other ancestry (includes Asian American, Native American and miscellaneous ancestries); BMI, body mass index; n, number.
Association of PRS with DR
We assessed the PRS for association with DR diagnosis in BioMe. In a meta-analysis, we observed robust association of PRS with DR: every standard deviation (SD) increase in PRS was accompanied by an increased odds ratio (OR) of 1.12 [95% confidence interval (CI) 1.04–1.20; P = 0.001] (Fig. 1) for DR diagnosis. We then evaluated transferability of the PRS across diverse ancestries by comparing its effect size and heterogeneity between ancestries. The PRS was associated with DR in European, African and Hispanic ancestries, but effect sizes varied (Fig. 1). European ancestry had the highest OR of DR diagnosis (OR = 1.22, 95% CI 1.02–1.41; P = 0.049) per SD increase in PRS, with lower performance measured by lower effect size in African (OR = 1.15, 95% CI 1.03–1.28; P = 0.028) and Hispanic ancestries (OR = 1.10, 95% CI 1.00–1.10; P = 0.050). In addition, there was increasing heterogeneity of PRS in European ancestry compared with African ancestry (heterogeneity test Q = 0.21; P = 0.69), Hispanic ancestry (Q = 0.80; P = 0.37) and other ancestry (Q = 2.46, P = 0.12), though none were statistically significant due in part to the relatively small sample sizes. Overall heterogeneity in the meta-analysis was Q = 2.77 (P = 0.43). The European ancestry was expected to have the highest OR as the PRS was derived from a European GWAS. The reduced trans-ancestral transferability of the DR PRS was consistent with previous reports for other diseases (23,26) and can be attributed to differences in linkage disequilibrium (LD), allele frequency and/or lifestyle factors between populations. Among all participants, the top PRS decile had an OR of 1.80 (95% CI 1.28–2.55; P = 0.002) for DR compared with the bottom decile (Table 2). The OR of DR increased monotonically across ascending PRS strata (Supplementary Material, Fig. S4).
Figure 1.
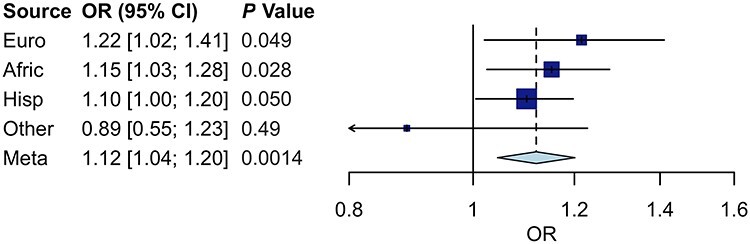
Forest plot of PRS on OR of DR diagnosis in different ancestries and meta-analysis in BioMe. Four box plots depict the middle 50% estimate (blue boxes) and corresponding 95% CI (horizontal lines) of PRS on OR of DR among individuals in four ancestries, with the size of each box proportional to the sample size of its representative ancestry. One diamond plot depicts the point estimate (dashed vertical line) and corresponding 95% CI (horizontal points) of PRS on OR of DR in a fixed-effects meta-analysis of all four ancestries. Euro, European ancestry; Afric, African ancestry; Hisp, Hispanic ancestry; Other, other ancestry (includes Asian, Native American and miscellaneous ancestries); Meta, meta-analysis of all ancestries.
Table 2.
OR of DR status in BioMe by PRS
| PRS % comparison | OR | 95% CI | P value |
|---|---|---|---|
| T 10 | B 10 | 1.80 | (1.28, 2.55) | 0.0017 |
| T 10 | M 50 | 1.70 | (1.22, 2.49) | 0.039 |
| T 10 | B 90 | 1.26 | (1.01, 1.59) | 0.048 |
| T 25 | B 75 | 1.15 | (0.98, 1.36) | 0.11 |
OR of DR status in BioMe in different PRS percentiles after adjusting for age, sex, BMI, 20 genetic principal components, history of hypertension and glucose levels. PRS % comparison, PRS percentile comparison; T 10 | B 10, top decile of PRS compared with bottom decile; T 10 | M 50, top decile of PRS compared with middle 50%; T 10 | B 90, top decile of PRS compared with bottom 90%; T 25 | B 75, top quartile of PRS compared with bottom 75%.
PRS and risk factors independently associate with DR
In a multivariable logistic regression, the PRS was associated with DR even after adjusting for clinical risk factors (OR = 1.14, 95% CI 1.05–1.23; P = 0.001; Table 3). T2D duration, hypertension, hyperglycemia, elevated HbA1c, hypercholesterolemia and hyperlipidemia have been reported as risk factors for DR (6,22) and were associated with DR here (all P < 0.001). Other studies have suggested a role for insomnia and sleep apnea in DR (27,28), both of which were significant in our analysis (P = 0.002 and P = 0.02, respectively). A multivariable model with continuous clinical measurements corresponding to these clinical risk factors showed similar association of the PRS with DR (Supplementary Material, Table S3). We then ascertained phenotypes of hypertension, hyperlipidemia and insomnia via medication usage and tested their association with DR to further confirm their risk factor status (Supplementary Material, Table S4). Three out of 17 hyperlipidemia medications were associated with DR (three statins; all P < 0.0001), 10 out of 33 hypertension medications were associated with DR (calcium channel blockers, loop diuretics, thiazides, beta blockers, angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers; all P < 0.002) and 2 out of 7 sleep medications were associated with DR (melatonin and ramelteon; both P ≤ 0.0001).
Table 3.
Multivariable model of standardized PRS and clinical risk factors on DR in BioMe
| Variable | OR | 95% CI | P value |
|---|---|---|---|
| PRS | 1.14 | (1.05, 1.23) | 1.3 × 10−3 |
| T2D duration | 1.48 | (1.35, 1.61) | 2.0 × 10−16 |
| T2D medication | 1.54 | (1.31, 1.86) | 9.2 × 10−7 |
| Hyperglycemia | 1.26 | (1.17, 1.36) | 5.0 × 10−9 |
| Elevated HbA1c | 1.39 | (1.30, 1.48) | 2.0 × 10−16 |
| Hypertension | 1.45 | (1.28, 1.65) | 6.7 × 10−9 |
| Hypercholesterolemia | 1.14 | (1.06, 1.23) | 3.7 × 10−4 |
| Hyperlipidemia | 1.27 | (1.17, 1.38) | 4.0 × 10−8 |
| Insomnia | 1.09 | (1.02, 1.17) | 1.5 × 10−3 |
| Sleep apnea | 1.10 | (1.02, 1.19) | 1.5 × 10−2 |
| Age | 1.04 | (0.95, 1.13) | 4.3 × 10−1 |
| Male | 0.99 | (0.91, 1.07) | 8.0 × 10−1 |
| BMI | 0.97 | (0.89, 1.06) | 5.0 × 10−1 |
Multivariable model of standardized PRS and clinical risk factors on DR in BioMe, adjusted for 20 genetic principal components (not shown). All variables were centered by subtracting by the mean and standardized by dividing by the SD. Diabetes medication was indicated if the patient was taking one or more T2D medications. T2D duration was calculated as time (in days) since first ICD-10 diagnosis code of T2D in the EHR. Clinical risk factors of hyperglycemia, hypertension, hypercholesterolemia, hyperlipidemia, insomnia and sleep apnea were defined by ICD-10 diagnosis codes. The SD was 3.0 years for T2D duration, 13 years for age and 7.2 kg/m2 for BMI. Elevated HbA1c, hemoglobin A1c >9% (75 mmol/mol); age, age in years; BMI, body mass index in kg/m2.
Association of PRS with ocular symptoms and conditions of DR
To examine clinical manifestations of the DR PRS in BioMe, we first quantified the difference in DR-related visual symptoms experienced by participants according to their PRS percentile. Individuals in the top (n = 502) and bottom (n = 524) PRS deciles lacking a DR diagnosis were assessed for visual symptoms indicative of DR. The proportion of participants with DR symptoms in the top PRS decile was significantly greater than in the bottom PRS decile (5.8 and 2.4%, respectively; P = 0.008). We also observed that the PRS was significantly associated with a greater risk of retinal hemorrhage (OR = 1.44, 95% CI 1.03–2.02; P = 0.03) and diplopia (OR = 1.31, 95% CI 1.02–1.70; P = 0.03). Bleeding from fragile retinal vessels and diplopia due to cranial nerve palsy, typically of the abducens or oculomotor nerves, are two common yet serious conditions in DR (29,30).
Temporal differences in DR presentation explained by polygenic risk
To evaluate if PRS may account for temporal differences in clinical presentation of DR, we assessed the association of PRS with time to diagnosis of DR. On average, the time between the dates of T2D and DR diagnoses was ~4.1 years (Supplementary Material, Fig. S5). We applied Cox proportional hazards tests and observed that PRS was associated with an elevated risk of DR over time (hazard ratio = 1.13 for DR diagnosis per SD increase in PRS, 95% CI 1.05–1.21; P = 9.4 × 10−4). Kaplan–Meier curves stratified by the top PRS decile, middle 50% PRS and bottom PRS decile revealed substantial differences in DR probability over time (Fig. 2). There was a 10% chance of DR by 4.4 years in the top PRS decile, by 5.7 years in the middle 50% PRS and by 7.7 years in the bottom PRS decile.
Figure 2.
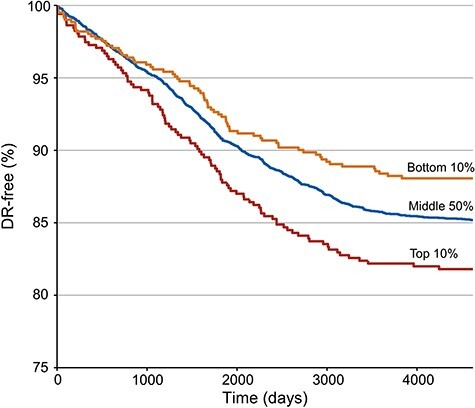
Cox proportional hazards model of time to DR diagnosis stratified by PRS in BioMe. The model adjusted for age, sex, body mass index, 20 genetic principal components, history of hypertension and glucose levels. PRS was associated with increased probability of DR over time (hazard ratio = 1.13 for DR diagnosis per SD increase in PRS, 95% CI 1.05–1.21; P = 9.4 × 10−4). Three Kaplan–Meier curves depict the probability of being DR-free over time in the bottom decile (orange), middle 50% (blue) and top decile (red) groups of PRS. Time to DR diagnosis was calculated as the difference in days between the date of diagnosis of T2D and the date of diagnosis of DR.
Correlation of DR PRS with genetic and clinical risk factors
We examined the correlation between the DR PRS and traditional clinical risk factors, as well as its correlation with other GRS, to better understand the characteristics of the DR PRS. None of the eight clinical risk factors tested (age, body mass index, T2D duration, glucose, HbA1c, LDL-C, triglycerides and SBP) were significantly correlated with the DR PRS (all P > 0.05). However, there was significant correlation between the DR PRS and four out of six previously published GRS that were tested (Supplementary Material, Fig. S6). The DR PRS was positively correlated with GRS for T2D [correlation coefficient (r) = 0.14; P = 1.6 × 10−29], glucose (r = 0.027; P = 3.7 × 10−2), LDL-C (r = 0.054; P = 2.4 × 10−5) and SBP (r = 0.047; P = 2.7 × 10−4). These indicate partial overlap between the genetic predisposition for DR and the genetic risk for diabetogenic and cardiometabolic traits.
Discussion
Identifying individuals at elevated genetic risk for DR raises the possibility of targeting those who may benefit from more frequent retinal screenings. Yet numerous GWAS of DR have failed to reliably identify risk variants (13–17). Here, we employed an approach where variants that fail to reach genome-wide significance may be incorporated into a PRS. Our PRS associates with DR, establishing the significant polygenic architecture of DR. While previous studies used PRS to explore polygenicity of T2D (31), there has not been a genome-wide PRS study to date that assesses DR, the ophthalmological sequela of T2D.
Genetic biobanks such as UKB and BioMe have enabled interrogation of the polygenic nature of complex diseases. These include coronary artery disease, atrial fibrillation, major depressive disorder, alcohol dependence and glaucoma (19–21). In this study, we leveraged linked genetic and clinical data to test a PRS for DR. We show that the additive impact of an individual’s genome-wide variants associates with DR. We evaluated the generalizability of the PRS in different ancestries and confirm that the PRS, which is derived from a European GWAS, has imperfect transferability to non-European ancestries. This is concordant with a previous report (23) and highlights the need for development of multi-ethnic PRS. We further demonstrate that those in the topmost PRS decile are at 1.8 times increased risk of DR relative to the lowest PRS decile and present with DR earlier than those in lower PRS strata, underscoring a significant difference in polygenic risk. This addresses the ADA’s 2020 Consensus Report, which calls for assessing genetic variation alongside electronic health record (EHR) data to lay the foundations of personalized diabetes care (32). Further study in a randomized controlled trial will be needed to definitively assess the clinical utility of PRS for DR.
DR is often asymptomatic during early stages, sometimes even with extensive retinal neovascularization. It has therefore been subject to intense scrutiny for physicians to screen for DR before irreparable harm occurs. An integrative approach combining genetic and clinical risk profiles can inform screening strategies and risk assessment (33). Studies have identified clinical risk factors for DR, such as duration of T2D (34), hypertension, hyperglycemia, hyperlipidemia (6) and sleep disorders (27,28), all of which were correlated with DR in BioMe. Notably, we observed that the PRS is associated with DR independent of these risk factors and that a SD increase in PRS yields an OR equivalent to or greater than that of several established risk factors such as sleep disorders and hypercholesterolemia. Receipt of medications indicated for several of these risk factors was associated with DR, an approach supported by a recent GWAS of medication use in UKB (35), though further study of medication usage as a proxy for phenotypes is warranted.
To characterize the clinical presentation of our PRS, we examined ocular symptoms and conditions common in DR. Using physician notes in the EHR, we observed a preponderance of DR symptoms among those lacking a DR diagnosis in the top PRS decile versus the bottom decile. We also noted that retinal hemorrhage and diplopia were associated with PRS. Thus, genomes of individuals with higher PRS manifest clinically as common symptoms and complications of DR (29,30). We also observed that the PRS was associated with increased risk of earlier DR presentation. Together, these results highlight a PRS that is associated with DR diagnosis, earlier DR and a higher burden of DR-related ocular consequences, yet is not currently considered clinically when diagnosing DR. The increasing affordability and standardization of genome-wide genotyping panels provide an opportunity for PRS to be translated to the clinic and achieve precision medicine (18,33).
There were several limitations in the study. First, International Classification of Diseases-Clinical Modification 10 (ICD-10) codes and self-reported diagnoses were used to define DR diagnosis. Population-based studies of T2D (36) and ocular diseases (37) often use ICD codes, yet some misclassification may occur. It has been reported that about 5% of T2D cases are genetically defined as T1D in the UKB; however, we showed that there was no significant difference in T1D GRS between DR cases and controls in the present study. The prevalence of DR among those with T2D was 19% in BioMe and 4.9% in UKB, a difference due in part to BioMe’s patient-dominated composition while UKB consists primarily of healthy volunteers. Second, phenotypic information was sourced from EHRs. Despite biases inherent in EHR data, we controlled for important potential confounders—clinical risk factors and population stratification—in our application of PRS. Third, varying ORs of the PRS association with DR were seen in different ancestry groups. This is likely explained by the PRS derived from a European GWAS yet validated in a multi-ethnic sample in BioMe. Non-European ancestries are underrepresented in biobanks and GWAS (23), and addition of more diverse populations would enable the standardization of multi-ethnic PRS. Increasing heterogeneity, though not significant due to relatively small sample sizes, was observed between European and other ancestries and is consistent with previous PRS reports for other diseases (23,26). Fourth, a diagnosis code for any type of DR was used to define cases, in part due to absence of more granular diagnosis codes for DR subtypes in UKB. Although this maximized the number of cases and power in the GWAS and PRS, it prevented delineation of different DR stages. Further study with datasets rich in DR subtypes will be needed to address PRS in proliferative versus non-proliferative DR. Fifth, we employed PRS based on a GWAS that did not reveal genome-wide significant loci. Other GWAS of DR have not been able to identify consistent or reproducible loci (13–17), and while PRS incorporates subgenome-wide significant loci, larger studies will be needed to increase the power of GWAS discovery in DR. Finally, our study is retrospective, characterizing the association of PRS with the prevalence, not incidence, of DR. Future studies should therefore assess the PRS longitudinally and prospectively.
In conclusion, we demonstrate that PRS associates with DR diagnosis in a multi-ethnic biobank, independent of risk factors. The PRS is associated with DR symptoms, complications and earlier presentation, yet is not currently used when screening or diagnosing DR. This underscores an important polygenic component of DR and warrants further investigation into PRS as a tool for risk stratification and diagnosis of DR.
Materials and Methods
Use of human subjects
De-identified EHR information from human subjects were used in this study. The Institutional Review Board/Ethics Committee at the Icahn School of Medicine at Mount Sinai approved the study. All participants provided informed consent. All research adhered to the tenets of the Declaration of Helsinki.
Identification of DR cases in UKB and BioMe
In the UKB, we obtained T2D and DR status using a combination of diagnosis codes. Participants were positive for T2D if they (1) had an ICD-10 from E11.0-E11.9, (2) were diagnosed with T2D by a physician or (3) self-reported T2D. Participants with self-reported T1D were excluded, leaving 17 567 individuals. DR cases (n = 866) were identified by an ICD-10 of E11.3 or self-report of DR. Dates for the combination of these T2D diagnoses were not readily available and duration of T2D was not included in the UKB analysis. In BioMe, participants with T2D (n = 6079) were obtained if they had an ICD-10 from E11.0-E11.9. Among these, 963 cases of DR were ascertained with an ICD-10 of E11.3. BioMe ICD-10 codes were searched between 2004 and 2019, including those converted from equivalent ICD-9 codes in 2016, and dates of diagnoses of DR and T2D were used to calculate duration of T2D. To assess whether DR case status overlaps with genetically defined T1D, we evaluated and compared a previously published T1D GRS derived from 29 variants (25) in DR cases versus controls for both UKB and BioMe.
GWAS of DR in UKB
The derivation of PRS required summary statistics for variant association with DR. Thus, we performed a GWAS of DR across autosomal chromosomes in 17 567 quality-controlled UKB individuals of British ancestry with T2D (Supplementary Material, Table S1; Supplementary Material, Fig. S3). UKB is a population-based cohort of ~500 000 participants predominantly of British ancestry between ages 40 and 69 years old enrolled across the UK between 2006 and 2010 (38). Genetic data in UKB are described elsewhere with QC (38). Briefly, genotyping of ~0.8 million markers was conducted using Affymetrix Axiom UK Biobank (n = 450 000) and UK BiLEVE arrays (n = 50 000). Imputation of variants was performed using IMPUTE2 (39) to a reference panel of UK10K haplotype and 1000 Genomes Phase 3 data (40) for a total of ~96 million variants. Samples were excluded if they lacked QC data (n = 14 705), were outliers in heterozygosity or missingness (n = 968), were not ancestrally British (n = 77 706) or were related to another sample up to the third degree (n = 69 272). Samples passing QC (n = 340 431) were used in analyses. We also applied QC on single-nucleotide polymorphism (SNP) data: SNPs were restricted to (1) minor allele frequency >0.1%, (2) Hardy–Weinberg equilibrium (HWE) P > 1 × 10−10 and (3) imputation info score ≥0.9 (1 is no uncertainty about genotype). After QC, we tested 10.8 million SNPs for association with DR in 17 567 individuals with T2D (866 cases with DR; 16 701 controls without DR) using SAIGE (41). This method implements a generalized mixed model association test with saddle-point approximation while controlling for imbalanced case–control ratios. The association test adjusted for sex, assessment center, age at recruitment, genotype measurement batch and 20 principal components (PCs). PCs were generated using Principal Component Analysis (PCA) on a subset of SNPs and samples that passed QC.
Derivation of PRS
PRS quantify the aggregate effect of common genetic variants across the genome on disease risk. Scores are calculated by summing the dosage of risk variants present multiplied by each variant’s effect size. Using GWAS summary statistics from UKB (n = 4 418 057 variants) and an LD reference panel from 1000 Genomes Europeans, we derived PRS models with LDpred (42). This method adjusts effect sizes of variants to account for LD and allows for tuning of a parameter rho (the assumed proportion of causal SNPs). Between rho = 1 (all SNPs assumed to be causal) and rho = 0.001, we adjusted effect sizes for several rho values and calculated polygenic scores: PRS = ∑jwj × Xj, where wj = adjusted effect size of a risk allele for the jth SNP and Xj = number of copies of the risk allele. We selected an optimal PRS with rho = 0.001 based on maximal association with DR (Supplementary Material, Table S5). This optimal PRS included 3 537 914 variants and was used for all analyses. Scores were standardized by centering at mean zero with a SD of one.
Calculation of PRS in BioMe
BioMe is a repository for biological data linked to the EHR for ~50 000 patients of African, Hispanic, European and Other (Asian, Native American and miscellaneous) ancestries at the Mount Sinai Hospital. Participants (n = 32 595) recruited between 2007 and 2019 were genotyped on the Illumina Global Screening Array (GSA) platform. Individuals with an ancestry-specific heterozygosity rate exceeding ±6 SDs of the mean or a call rate <95% were removed (n = 684). Individuals with discordant or missing sex (n = 88) or duplications (n = 102) were removed, leaving 31 911 individuals. Lastly, individuals with a relative up to the third degree (n = 2873) or lacking laboratory glucose data (n = 88) were excluded. The remaining individuals with T2D (n = 6079) were included for analysis. Of 635 623 variants in the GSA data, 19 253 variants with call rate <95% were excluded, along with 11 503 variants that significantly violated HWE when stratified by ancestry: P < 1 × 10−5 in African and European ancestries, and P < 1 × 10−13 in Hispanic ancestry. The 604 869 sites that passed QC were then imputed with IMPUTE2.3.2.2 (39) to 1000 Genomes Phase 3 data (40). PCA was then performed on the imputed genotype data using SMARTPCAv10210 software from EIGENSOFTv5.0.1 (43).
Statistical analysis
Differences in categorical and continuous variables were assessed with a chi-square test and t-test, respectively. Association analyses of PRS with DR diagnosis and symptoms were performed using a multivariable logistic regression, adjusted for clinical covariates and 20 PCs. Association analyses were stratified by ancestry with results combined in a fixed-effects meta-analysis using the inverse variance weighted method in the meta R package (44). Differences in PRS between ancestries were further assessed using tests of heterogeneity (Cochran’s Q test) in the meta R package (44). Medications indicated for hyperlipidemia, hypertension and insomnia were individually tested for association with DR in univariable analyses, and those achieving Bonferroni-corrected significance were included in a multivariable analysis of PRS and medications on DR. Association of PRS with time to DR diagnosis was evaluated with a Cox proportional hazards model, and Kaplan–Meier curves were generated using the survminer R package (45). Lastly, correlations between the PRS and clinical risk factors for DR, and between the DR PRS and previously published GRS for T2D (46), glucose (47), HbA1c (48), LDL-C (47), SBP (47) and c-reactive protein (47), were examined using Pearson correlation tests.
PRS association with DR independent of clinical risk factors
We investigated the association of PRS with DR, adjusted for clinical risk factors and 20 PCs. In a multivariable logistic regression of DR, we included PRS, 20 PCs, age, sex, body mass index, duration of T2D, glucose level, hypertension history and elevated HbA1c level [>9% (75 mmol/mol)] indicating ineffective glycemic control (49,50), as well as ICD-10-coded traits of hypercholesterolemia, hyperlipidemia, insomnia and sleep apnea. T2D medication (insulin, insulin analogs, pramlintide, glucagon-like peptide 1 agonists, metformin, sulfonylurea, dipeptidyl peptidase 4 inhibitors, glitazone, sodium-glucose transport protein 2 inhibitors or alpha-glucosidase inhibitors) was also included. The use of lipid-lowering medication (statins, PCSK9 inhibitors, bile acid sequestrants, omega-3 fatty acid derivatives, ezetimibe, fibrates and niacin), antihypertensive medication (angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, thiazides, calcium channel blockers, beta blockers, loop diuretics, aldosterone antagonists, renin inhibitors and alpha-1 blockers) and sleep-promoting medication (melatonin, ramelteon, zolpidem, zaleplon, eszopiclone, doxepin and suvorexant) were separately assessed for association with DR.
PRS association with ocular symptoms and conditions of DR
Though DR can be asymptomatic early on, we sought to examine clinical manifestations of DR by PRS. First, we mined physician notes in the EHR for visual complaints common in DR: blurred or decreased vision (e.g. inability to read), floaters (e.g. cobwebs), ocular nerve palsy (e.g. double vision) and eye pain were included. Then, the prevalence of DR-related symptoms in the top and bottom PRS deciles was compared. Lastly, we assessed the association of PRS with two ICD-10-coded conditions common in DR, retinal hemorrhage and diplopia (29,30).
Data Availability
No datasets were generated during the current study. UKB data may be accessed by registering at https://www.ukbiobank.ac.uk/register-apply/. More information about BioMe can be found at https://icahn.mssm.edu/research/ipm/programs/biome-biobank/researcher-faqs.
Supplementary Material
Acknowledgements
The BioMe healthcare delivery cohort at Mount Sinai was founded and maintained with a generous gift from the Andrea and Charles Bronfman Philanthropies. We thank the individuals who were involved in the quality control and/or file handling for the exome sequencing and genome-wide genotyping data, including Aayushee Jain, Arden Moscati, Kumardeep Chaudhary, Lisheng Zhou, Michael Preuss, Quingbin Song, Stephane Wenric and Steve Ellis. This research has been conducted using the UK Biobank Resource under Application Number ‘16218’.
Conflict of Interest statement: R.D. reported receiving grants from AstraZeneca, grants and non-financial support from Goldfinch Bio, being a scientific co-founder, consultant and equity holder for Pensieve Health and being a consultant for Variant Bio. G.N.N. reported being a scientific co-founder, consultant, advisory board member and equity owner of Renalytix AI; is a scientific co-founder, consultant and equity holder for Pensieve Health; being a consultant for Variant Bio and receiving grants from Goldfinch Bio and receiving personal fees from Renalytix AI, BioVie, Reata, AstraZeneca and GLG Consulting. L.R.P. is supported by National Eye Institute and is also a consultant for Bausch+Lomb, Eyenovia, Verily, Nicox and Emerald Bioscience.
Author Contributions
I.S.F., G.N.N. and R.D. designed the study. I.S.F., R.J.F.L., J.C., G.N.N. and R.D. obtained the data. I.S.F., K.C., I.P., L.R.P., H.M.T.V., C.M.-L., G.R., A.S., L.C. and T.V.V. analyzed and/or interpreted the data. I.S.F., L.R.P., G.N.N. and R.D. drafted and revised the manuscript. All authors read and provided critical comments on the manuscript.
Funding
This work was supported by the National Institutes of Health [T32GM728042 to I.S.F., R35GM124836 to R.D., R01HL139865 to R.D.]. I.S.F. was supported by T32GM007280, the Medical Scientist Training Program Training Grant, from the National Institute of General Medical Sciences of the National Institutes of Health. R.D. is supported by R35GM124836 from the National Institute of General Medical Sciences of the National Institutes of Health, and R01HL139865 from the National Heart, Lung and Blood Institute of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Zheng, Y., Ley, S.H. and Hu, F.B. (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol., 14, 88–98. [DOI] [PubMed] [Google Scholar]
- 2. Unnikrishnan, R., Pradeepa, R., Joshi, S.R. and Mohan, V. (2017) Type 2 diabetes: demystifying the global epidemic. Diabetes, 66, 1432–1442. [DOI] [PubMed] [Google Scholar]
- 3. Stolar, M. (2010) Glycemic control and complications in type 2 diabetes mellitus. Am. J. Med., 123, S3–S11. [DOI] [PubMed] [Google Scholar]
- 4. Frank, R.N., Keirn, R.J., Kennedy, A. and Frank, K.W. (1983) Galactose-induced retinal capillary basement membrane thickening: prevention by Sorbinil. Investig. Opthalmology Vis. Sci., 24, 1519–1524. [PubMed] [Google Scholar]
- 5. Engerman, R.L. and Kern, T.S. (1984) Experimental galactosemia produces diabetic-like retinopathy. Diabetes, 33, 97–100. [DOI] [PubMed] [Google Scholar]
- 6. Yau, J.W.Y., Rogers, S.L., Kawasaki, R., Lamoureux, E.L., Kowalski, J.W., Bek, T., Chen, S.J., Dekker, J.M., Fletcher, A., Grauslund, J. et al. (2012) Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care, 35, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willis, J.R., Doan, Q.V., Gleeson, M., Haskova, Z., Ramulu, P., Morse, L. and Cantrell, R.A. (2017) Vision-related functional burden of diabetic retinopathy across severity levels in the United States. JAMA Ophthalmol., 135, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wittenborn, J.S., Zhang, X., Feagan, C.W., Crouse, W.L., Shrestha, S., Kemper, A.R., Hoerger, T.J. and Saaddine, J.B. (2013) The economic burden of vision loss and eye disorders among the United States population younger than 40 years. Ophthalmology, 120, 1728–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mazhar, K., Varma, R., Choudhury, F., McKean-Cowdin, R., Shtir, C.J. and Azen, S.P. (2011) Severity of diabetic retinopathy and health-related quality of life: the Los Angeles Latino eye study. Ophthalmology, 118, 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kato, S., Takemori, M., Kitano, S., Hori, S., Fukushima, H., Numaga, J. and Yamashita, H. (2002) Retinopathy in older patients with diabetes mellitus. Diabetes Res. Clin. Pract., 58, 187–192. [DOI] [PubMed] [Google Scholar]
- 11. Jenkins, A.J., Joglekar, M.V., Hardikar, A.A., Keech, A.C., O’Neal, D.N. and Januszewski, A.S. (2015) Biomarkers in diabetic retinopathy. Rev. Diabet. Stud., 12, 159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Solomon, S.D., Chew, E., Duh, E.J., Sobrin, L., Sun, J.K., VanderBeek, B.L., Wykoff, C.C. and Gardner, T.W. (2017) Diabetic retinopathy: a position statement by the American Diabetes Association. Diabetes Care, 40, 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho, H. and Sobrin, L. (2014) Genetics of diabetic retinopathy. Curr. Diab. Rep., 14, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han, J., Lando, L., Skowronska-Krawczyk, D. and Chao, D.L. (2019) Genetics of diabetic retinopathy. Curr. Diab. Rep., 19, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pollack, S., Igo, R.P., Jensen, R.A., Christiansen, M., Li, X., Cheng, C.Y., Ng, M.C.Y., Smith, A.V., Rossin, E.J., Segrè, A.V. et al. (2019) Multiethnic genome-wide association study of diabetic retinopathy using liability threshold modeling of duration of diabetes and glycemic control. Diabetes, 68, 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng, W., Shah, K.P., Pollack, S., Toppila, I., Hebert, H.L., McCarthy, M.I., Groop, L., Ahlqvist, E., Lyssenko, V., Agardh, E. et al. (2018) A genome-wide association study suggests new evidence for an association of the NADPH oxidase 4 ( NOX4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol., 96, e811–e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burdon, K.P., Fogarty, R.D., Shen, W., Abhary, S., Kaidonis, G., Appukuttan, B., Hewitt, A.W., Sharma, S., Daniell, M., Essex, R.W. et al. (2015) Genome-wide association study for sight-threatening diabetic retinopathy reveals association with genetic variation near the GRB2 gene. Diabetologia, 58, 2288–2297. [DOI] [PubMed] [Google Scholar]
- 18. Hudson, K., Lifton, R., Patrick-Lake, B. and Denny, J.M. (2015) The Precision Medicine Initiative Cohort Program – Building a Research Foundation for 21st Century Medicine. National Institutes of Health, Bethesda, MD.
- 19. Khera, A.V., Chaffin, M., Aragam, K.G., Haas, M.E., Roselli, C., Choi, S.H., Natarajan, P., Lander, E.S., Lubitz, S.A., Ellinor, P.T. et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet., 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andersen, A.M., Pietrzak, R.H., Kranzler, H.R., Ma, L., Zhou, H., Liu, X., Kramer, J., Kuperman, S., Edenberg, H.J., Nurnberger, J.I. et al. (2017) Polygenic scores for major depressive disorder and risk of alcohol dependence. JAMA Psychiatry, 74, 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Craig, J.E., Han, X., Qassim, A., Hassall, M., Cooke Bailey, J.N., Kinzy, T.G., Khawaja, A.P., An, J., Marshall, H., Gharahkhani, P. et al. (2020) Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat. Genet., 52, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith, J.J., Wright, D.M., Scanlon, P. and Lois, N. (2020) Risk factors associated with progression to referable retinopathy: a type 2 diabetes mellitus cohort study in the Republic of Ireland. Diabet. Med., 37, 1000–1007. [DOI] [PubMed] [Google Scholar]
- 23. Martin, A.R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B.M. and Daly, M.J. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet., 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barsegian, A., Kotlyar, B., Lee, J., Salifu, M. and McFarlane, S. (2017) Diabetic retinopathy: focus on minority populations. Int. J. Clin. Endocrinol. Metab., 3, 034–045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomas, N.J., Jones, S.E., Weedon, M.N., Shields, B.M., Oram, R.A. and Hattersley, A.T. (2018) Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross-sectional, genetically stratified survival analysis from UK Biobank. Lancet Diabetes Endocrinol., 6, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fahed, A.C., Aragam, K.G., Hindy, G., Chen, Y.-D.I., Chaudhary, K., Dobbyn, A., Krumholz, H.M., Sheu, W.H.H., Rich, S.S., Rotter, J.I. et al. (2020) Transethnic transferability of a genome-wide polygenic score for coronary artery disease. Circ. Genomic Precis. Med., 14, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leong, W.B., Jadhakhan, F., Taheri, S., Chen, Y.F., Adab, P. and Thomas, G.N. (2016) Effect of obstructive sleep apnoea on diabetic retinopathy and maculopathy: a systematic review and meta-analysis. Diabet. Med., 33, 158–168. [DOI] [PubMed] [Google Scholar]
- 28. Tan, N.Y.Q., Chew, M., Tham, Y.-C., Nguyen, Q.D., Yasuda, M., Cheng, C.-Y., Wong, T.Y. and Sabanayagam, C. (2018) Associations between sleep duration, sleep quality and diabetic retinopathy. PLoS One, 13, e0196399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamboj, A., Lause, M. and Kumar, P. (2017) Ophthalmic manifestations of endocrine disorders—endocrinology and the eye. Transl. Pediatr., 6, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trigler, L., Siatkowski, R.M., Oster, A.S., Feuer, W.J., Betts, C.L., Glaser, J.S., Schatz, N.J., Farris, B.K. and Flynn, H.W. (2003) Retinopathy in patients with diabetic ophthalmoplegia. Ophthalmology, 110, 1545–1550. [DOI] [PubMed] [Google Scholar]
- 31. Udler, M.S., McCarthy, M.I., Florez, J.C. and Mahajan, A. (2019) Genetic risk scores for diabetes diagnosis and precision medicine. Endocr. Rev., 40, 1500–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chung, W.K., Erion, K., Florez, J.C., Hattersley, A.T., Hivert, M.F., Lee, C.G., McCarthy, M.I., Nolan, J.J., Norris, J.M., Pearson, E.R. et al. (2020) Precision medicine in diabetes: a consensus report from the American Diabetes Association (ADA) and the European Association for the Study of diabetes (EASD). Diabetes Care, 43, 1617–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Torkamani, A., Wineinger, N.E. and Topol, E.J. (2018) The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet., 19, 581–590. [DOI] [PubMed] [Google Scholar]
- 34. Fong, D.S., Aiello, L., Gardner, T.W., King, G.L., Blankenship, G., Cavallerano, J.D., Ferris, F.L. and Klein, R. (2004) Retinopathy in diabetes. Diabetes Care, 27, s84–s87. [DOI] [PubMed] [Google Scholar]
- 35. Wu, Y., Byrne, E.M., Zheng, Z., Kemper, K.E., Yengo, L., Mallett, A.J., Yang, J., Visscher, P.M. and Wray, N.R. (2019) Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun., 10, 1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xue, A., Wu, Y.Y., Zhu, Z., Zhang, F., Kemper, K.E., Zheng, Z., Yengo, L., Lloyd-Jones, L.R., Sidorenko, J., Wu, Y.Y. et al. (2018) Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun., 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choquet, H., Paylakhi, S., Kneeland, S.C., Thai, K.K., Hoffmann, T.J., Yin, J., Kvale, M.N., Banda, Y., Tolman, N.G., Williams, P.A. et al. (2018) A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun., 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., Downey, P., Elliott, P., Green, J., Landray, M. et al. (2015) UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Howie, B.N., Donnelly, P. and Marchini, J. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Auton, A., Abecasis, G.R., Altshuler, D.M., Durbin, R.M., Bentley, D.R., Chakravarti, A., Clark, A.G., Donnelly, P., Eichler, E.E., Flicek, P. et al. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou, W., Nielsen, J.B., Fritsche, L.G., Dey, R., Gabrielsen, M.E., Wolford, B.N., LeFaive, J., VandeHaar, P., Gagliano, S.A., Gifford, A. et al. (2018) Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet., 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vilhjálmsson, B.J., Yang, J., Finucane, H.K., Gusev, A., Lindström, S., Ripke, S., Genovese, G., Loh, P.R., Bhatia, G., Do, R. et al. (2015) Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet., 97, 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Price, A.L., Patterson, N.J., Plenge, R.M., Weinblatt, M.E., Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet., 38, 904–909. [DOI] [PubMed] [Google Scholar]
- 44. Schwarzer, G., Carpenter, J.R. and Rücker, G. (2015) An Introduction to Meta-Analysis in R. In: Meta-Analysis with R. Springer, Cham, Switzerland, pp. 3–17.
- 45. R Core Team (2018) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. [Google Scholar]
- 46. Mahajan, A., Taliun, D., Thurner, M., Robertson, N.R., Torres, J.M., Rayner, N.W., Payne, A.J., Steinthorsdottir, V., Scott, R.A., Grarup, N. et al. (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet., 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie, T., Wang, B., Nolte, I.M., Van Der Most, P.J., Oldehinkel, A.J., Hartman, C.A. and Snieder, H. (2020) Genetic risk scores for complex disease traits in youth. Circ. Genomic Precis. Med., 13, 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wheeler, E., Leong, A., Liu, C.-T., Hivert, M.-F., Strawbridge, R.J., Podmore, C., Li, M., Yao, J., Sim, X., Hong, J. et al. (2017) Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med., 14, e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iqbal, N., Morgan, C., Maksoud, H. and Idris, I. (2008) Improving patients’ knowledge on the relationship between HbA1c and mean plasma glucose improves glycaemic control among persons with poorly controlled diabetes. Ann. Clin. Biochem., 45, 504–507. [DOI] [PubMed] [Google Scholar]
- 50. Centers for Medicare & Medicaid Services . (2020) Medicare Part B Claims Measure - Diabetes: Hemoglobin A1c Poor Control. Centers for Medicare & Medicaid Services, Woodlawn, Maryland.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated during the current study. UKB data may be accessed by registering at https://www.ukbiobank.ac.uk/register-apply/. More information about BioMe can be found at https://icahn.mssm.edu/research/ipm/programs/biome-biobank/researcher-faqs.