

Abstract
Background: [11C]-Erlotinib is a radiolabeled analogue of a tyrosine kinase inhibitor used to treat non-small cell lung cancer (NSCLC) which expresses specific kinase domain mutations of the epidermal growth factor receptor (EGFR). In this study, 10 subjects with NSCLC and assorted EGFR mutation status underwent a dynamic, multi-bed positron emission tomography (PET) scan using [11C]-erlotinib. Data were analyzed using a variety of quantitative techniques common in PET (graphical methods, kinetic models, and uptake value-based endpoints). Our primary goal was to determine the most reliable imaging endpoint given the need for maintaining minimal patient burden and recognizing the advantage of simple calculations in future trials. Results: Standard uptake values (a semi-quantitative endpoint) were well correlated with both binding potential and volume of distribution (fully quantitative endpoints). Normalized tracer uptake was found to stabilize approximately 60 minutes post tracer injection. Conclusions: The kinetic properties of [11C]-erlotinib varied greatly across subjects. Our novel scanning protocol produced an important dataset which highlights the great heterogeneity of NSCLC and its apparent impact on [11C]-erlotinib kinetics. A lack of correlation between EGFR mutational status and quantitative endpoints appears to be due to disease heterogeneity and low tracer uptake. The most reliable fits of the dynamic data were based on the one-tissue compartmental model which were well correlated with mean SUV. Due to this correlation and good stability at late-time, SUV seems sufficiently well-suited to quantitative imaging of NSCLC lesions in the whole body with [11C]-erlotinib.
Keywords: Erlotinib, TKI, NSCLC, PET, quantitative imaging
Introduction
Erlotinib is a tyrosine kinase inhibitor (TKI) used for treating non-small cell lung cancer (NSCLC). It has demonstrated profound treatment benefits compared to chemotherapy in response rate (58-83% versus 10-13%) and progression-free survival (9.7-13 months versus 4.6-5.2 months) [1,2]. However, erlotinib and other TKIs are most effective in individuals bearing mutations of the epidermal growth factor receptor (EGFR) which confer greater affinity for the drug [3,4]. These mutations are present in 10-15% of Western populations and approximately 50% of Asian populations [5,6].
[11C]-Erlotinib is the radiolabeled analogue of erlotinib that has been used in a limited number of positron emission tomography (PET) studies. Initial experiments used human cancer cell lines xenografted onto rodents [7,8]. More recent investigations have analyzed tumors in NSCLC patients [9-11]. These studies have applied various standard analyses used in neuroimaging, including normalized tracer uptake and compartmental modeling.
Interpreting quantitative imaging data in NSCLC patients relies on knowledge of a number of unique biological factors. (I) Acquired resistance to TKIs can alter the affinity of TKIs for the EGFR binding site, the kinase domain [12]. (II) Consequently, changes in imaging endpoints which are affected by ligand-receptor affinity (e.g. binding potential) cannot be attributed solely to changes in receptor availability (a common assumption in neuroimaging). (III) Necrosis and other variations in a tumor’s microenvironment limit receptor availability and affinity. Metastases are often located outside of the lung, limiting the utility of a single chest scan [13,14]. In order to gain a more wholistic snapshot of subject health, multiple scans or multi-bed imaging are required. (IV) Treatment of cancer has become increasingly focused on cumulative tumor burden compared to changes in isolated regions. For the health and comfort of NSCLC subjects, arterial lines should be avoided when possible, limiting the calculation of “gold standard” imaging endpoints. Quantitative neuroimaging practices and analyses may need to be adapted to fit the unique challenges of oncology imaging and cancer patients.
The objectives of this study were to evaluate various image analysis methods taking into account observed biological heterogeneity in NSCLC and to calculate fully quantitative endpoints non-invasively in dynamic, multi-bed [11C]-erlotinib imaging. Prior studies focused on quantitative imaging techniques on isolated lesions collected from a single bed position. These studies employed traditional neuroimaging quantification without addressing biological concerns unique to cancer. We employed a semi-automated region selection technique in order to compare multiple lesions across the body. To our knowledge, this is the first study to collect dynamic TKI PET data at multiple bed positions in order to image multiple sites simultaneously. We examined the results of different model methods as applied to NSCLC subjects of differing EGFR mutation, cancer site, and metastatic status.
Materials and methods
Subjects
Subjects with stage IV NSCLC bearing measurable disease of at least 1 cm (confirmed with computed tomography (CT) imaging as per the RECIST 1.1 criteria [15]) were invited to participate. Mutational status of subjects’ primary tumor was confirmed with biopsy prior to inclusion in the study. Subjects were required to be TKI naïve at the time of scanning but were not excluded based on prior TKI treatment or other therapy methods such as chemotherapy or tumor resection. Subjects were excluded if participation would have caused them to exceed the FDA’s guideline for annual experimental radiation exposure, 5 rem [16]. Informed consent was obtained prior to the initiation of study procedures.
Subjects 1-7 possessed EGFR mutations suitable for treatment with TKIs. Specifically, subjects 1-5 expressed an exon 19 in-frame deletion and subject 6 expressed an exon 21 substitution (L858R), and subject 7 expressed a rare compound mutation (G719S+S768I). Exon 19 in-frame deletions and L858R are the most common mutations that sensitize NSCLC to TKIs [17]. Subject 8 expressed L861Q, an uncommon exon 21 mutation that sensitizes NSCLC to TKIs. This subject had previously undergone TKI treatment until the onset of resistance. Subjects 9-10 had tumors that were negative for EGFR mutations. A summary of subject characteristics is listed in Table 1.
Table 1.
Subject characteristics table. The “EGFR Mutation” column indicates the presence of any mutations found in biopsied tissue. The location of this biopsy is labeled in the “Area of Primary Tumor” column, as identified by a clinical oncologist
| Subject | Gender | EGFR Status | Biopsy Site | Additional Disease Area(s) |
|---|---|---|---|---|
| 1 | F | Exon 19 deletion | Adrenal gland | n/a |
| 2 | M | Exon 19 deletion | Lung | n/a |
| 3 | F | Exon 19 deletion | Liver | Lung |
| 4 | M | Exon 19 deletion | Lung | Liver |
| 5 | F | Exon 19 deletion | Iliac bone | Lung |
| 6 | F | L858R | Lung | n/a |
| 7 | F | G719S+S768I | Pleural wall | n/a |
| 8 | F | L861Q | Lung | n/a |
| 9 | M | Wild-type | Lung | n/a |
| 10 | M | Wild-type | Lung | n/a |
[11C]-erlotinib synthesis
[11C]-Erlotinib was produced as described in the literature by O-methylation of the precursor 6-O-desmethyl ERLO with [11C]methyl iodide in dimethylformamide in the presence of sodium hydride [7,8]. After purification by HPLC followed by solid-phase extraction, the radiolabeled product was formulated in 10 mL saline containing 1 mL ethanol and 0.04 mL of 4.2% sodium bicarbonate (all reagents were USP grade). Finally, the PET drug product solution was passed through a 0.22 µm membrane filter and collected in a sterile dose vial affording a sterile solution ready for dispensing and administration. Radiochemical purity and molar activity (Am) was determined by analytical HPLC (Phenomenex Luna C18 (2)), 5 µm, 4.6 × 250 mm; mobile phase: 39% acetonitrile and 61% of 0.1 M aqueous ammonium acetate (v/v) with 0.5% acetic acid (pH = 4.2); flow rate: 2 mL/min; UV detector wavelength: 246 nm). The radiochemical purity of the final product was >99%, and the Am was 20.5 ± 13.7 Ci/µmol (n = 21, range 2.7-49.7) at the end of synthesis (EOS). The average synthesis time was 47 ± 4 min from end of target bombardment (EOB).
[11C]-erlotinib imaging
A physical assessment of subjects (blood pressure, heart rate, etc.) was performed prior to imaging. An intravenous line with a catheter was placed in the arm in order to facilitate radiotracer delivery. During scanning, vital signs were monitored continuously to ensure subject wellbeing. Approved medical personnel were present throughout all procedures in the instance of an adverse event.
PET scans were performed with a Biograph mCT (axial FWHM 4.4 mm, Siemens/CTI, Knoxville, TN [18]). Subjects were placed in the center of the field of view. A whole-body CT transmission scan (X-ray source) was performed for attenuation correction. Subjects were injected with [11C]-erlotinib at a mean activity of 9.14 ± 1.46 mCi and mean mass of 1.57 ± 1.74 μg. Scans were performed from the top of the head to the lowest known site of cancer in the body. Emission data were collected dynamically in multiple bed positions (between 3 to 7 bed positions requiring 10 to 14 passes) for approximately 90 min.
PET emission data were reconstructed using ordered subset expectation maximization (OSEM) with 2 iterations and 21 subsets at a voxel size of 2.04 × 2.04 × 2.00 mm3. Standard corrections for attenuation, scatter, dead time, detector sensitivity, randoms, radionuclide decay, and frame-wise motion were applied.
Image analysis
Semi-automated region of interest drawing
A PET reference image was created for each subject by averaging late-time (40 min and later) emission images and correcting them for subject weight and injected activity (standard uptake value, SUV). Each of the reference images were blurred with a 2.5 × 2.5 × 2.5 mm3 gaussian filter and overlaid on the respective CT image. Regions of interest (ROIs) were manually drawn around disease areas in ITK Snap [19] using the CT or reference PET information for guidance. Thresholding was performed by removing voxels within the ROI whose SUV values were less than 40% of the maximum. Each distinct, non-contiguous cluster of ROI voxels was then analyzed independently. ROI clusters containing less than 25 voxels (~0.2 cm3) were discarded. At the end of the refinement process, ROIs were assigned a type based on structural information: primary tumor, non-primary tumor, tumor sub-region (ROI on a tumor which does not comprise the entire tumor), or cancerous area (diseased tissue with borders not well defined by CT). This ROI refinement process is depicted for all subjects in Figure 1. A separate, spherical ROI (8.2 cm3) was placed on healthy shoulder muscle for use in reference ROI methods. Time activity curves (TACs) were created by superimposing ROIs on the dynamic PET emission data and averaging the voxel intensities within the region for each time frame using in-house software.
Figure 1.
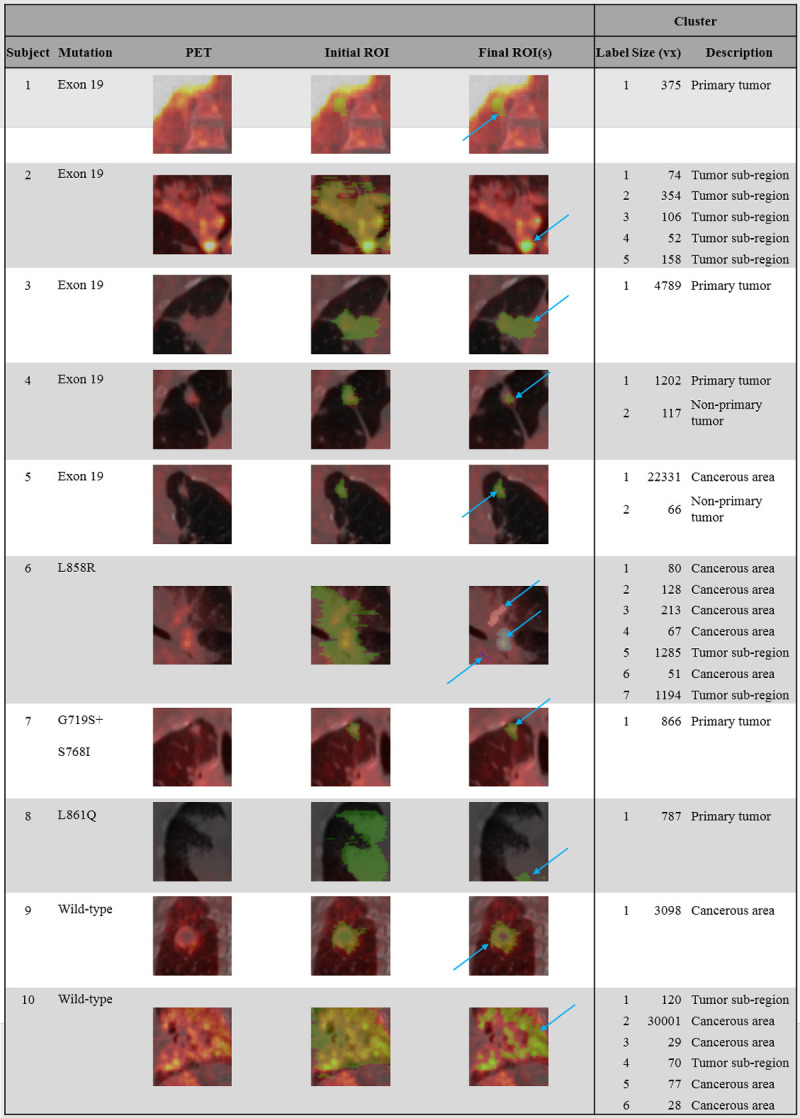
Subject tumor biology (Mutation column) compared with late-time PET scans (PET column). PET images are overlaid on attenuation correction CT images. All PET images are windowed at 0-3 SUV (hot color scale). The Initial ROI and Final ROI (s) columns display the ROIs manually placed over the functional images as well as their final volumes after the automated refinement was applied, respectively. ROIs are shown in green (if multiple clusters are segmented, they are depicted in alternative colors). Blue arrows highlight the final ROI (s). The Cluster columns give quantitative information on the number, size, and location of labels after refinement.
Image-derived input functions
Image-derived input functions (IDIFs) were created by manually delineating the left ventricle based on the CT information. This region was eroded in 3D by 1 mm (using ITK Snap) to a volume of approximately 50 mL to minimize spill in/spill out effects to/from the myocardium and to standardize sampling of the blood pool between subjects. A TAC was extracted from the eroded ventricle region in the same manner as described for tumor ROIs. The resultant TAC was corrected for radioactive metabolites using an average metabolite fraction versus time curve (extrapolated from arterial plasma data in erlotinib-naïve subjects collected in Bahce et al. [10]). The processed IDIF was used as an approximation of the concentration of parent tracer in the plasma, CP(t).
Calculated endpoints
SUV-based measures were calculated using late-time emission images: mean, standard deviation, and maximum. Skew, kurtosis, and AUC-CSH (area under the curve of the cumulative SUV histogram) are texture analysis endpoints calculated from histograms of late-time SUV images [20].
Graphical analysis using the Logan plot [21] was performed with the IDIF serving as CP(t) order to estimate volume of distribution (VT Logan). Non-displaceable binding potential (BPLogan) was calculated by normalizing the VT Logan of a target ROI by the VT Logan of the respective subject’s healthy muscle. BPLoganReference was estimated directly using the Logan graphical method modified for a reference region (muscle) input [22].
Non-displaceable binding potential was estimated by fitting the simplified reference tissue model (SRTM [23]) to the target TACs (BPSRTM) using the muscle TAC as the reference curve (implemented with non-linear regression via in-house software). Using the IDIF as input, the reversible one-tissue (1T-3k) and reversible two-tissue (2T-5k) models were fit to TACs in order to estimate VT (in-house software). In both of these compartmental models, blood volume fraction was an estimated parameter in order to account for differences in tumor vascularization.
Goodness of fit was assessed using the Akaike information criterion (AIC). The standard error of the endpoint (VT or BP) was calculated numerically from a sensitivity function using the weighted residual sum of squares. When the standard error of the estimated parameter exceeded 10, the result was excluded. See Table S1 for a complete list of calculated endpoints and their associated standard errors. Correlation between versions of calculated endpoints was calculated using the coefficient of determination. Linear fitting was performed between selected endpoints.
VT 1T-3k, VT 2T-5k, BPSRTM, and standard uptake value normalized by a reference (SUVR) were also re-calculated after truncating scan data at 45 min and 60 min. When calculating SUVR at shortened tmax values, at least 2 time frames were always used.
Results
ROI refinement
In 5 of 10 subjects, the refinement process segmented the initial, manually drawn ROI into 2 or more separate ROIs (subjects 2, 4, 5, 6, and 10) as shown in Figure 1. In the remaining 5 subjects, only 1 ROI was retained. The segmented ROI was equivalent to the initial ROI in subjects 1 and 7, but was reduced by SUV threshold in subjects 3, 8, and 9. In total, 27 distinct ROIs were produced from 10 subjects.
Correlating imaging endpoints
The estimated parameters from the graphical methods, modeling methods, and SUV-based methods for each ROI are listed in Table 2. Of the 27 ROIs, 21 were fitted successfully with 1T-3k, 9 with 2T-5k, and 22 with SRTM (VT and BP estimates with standard errors over 10 were excluded). Mutation status was not correlated with graphical, compartmental model, or SUV-based endpoints as shown in Figure 2.
Table 2.
Calculated endpoints for each ROI identified in the semi-automated process. Column groupings from left to right: ROI information, graphical endpoints, curve fitting endpoints, and SUV-based endpoints. Mean, standard deviation, maximum, and minimum values are listed in the bottom rows. Endpoints derived from non-linear fitting of TACs were excluded from this table if their standard error was estimated to be greater than 10. A complete list of endpoints with standard error estimates is provided in Table S1
| Subject | Cluster | VT Logan | BPLogan | BPReferenceLogan | VT 1T-3k | VT 2T-5k | BPSRTM | Mean | SD | Max | Skew | Kurtosis | AUIVH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1.23 | 1.59 | 1.37 | 0.75 | 1.39 | 1.41 | 0.32 | 1.84 | 0.76 | 0.28 | 0.59 | |
| 2 | 1 | 2.06 | 3.10 | 3.77 | 2.39 | 3.50 | 2.36 | 0.40 | 2.92 | 0.57 | -0.46 | 0.71 | |
| 2 | 2.50 | 3.98 | 4.71 | 3.08 | 4.31 | 2.71 | 1.00 | 4.10 | 1.32 | 1.50 | 0.41 | ||
| 3 | 1.98 | 2.94 | 3.88 | 2.74 | 3.45 | 2.58 | 0.70 | 3.54 | 1.41 | 1.94 | 0.49 | ||
| 4 | 1.50 | 1.97 | 3.08 | 2.93 | 2.32 | 0.30 | 2.72 | 0.16 | -0.57 | 0.76 | |||
| 5 | 2.61 | 4.18 | 4.09 | 3.15 | 3.93 | 2.40 | 0.47 | 3.03 | 0.58 | 0.13 | 0.63 | ||
| 3 | 1 | 0.97 | 1.05 | 1.07 | 0.96 | 0.76 | 0.52 | 0.15 | 0.73 | 1.18 | 1.87 | 0.41 | |
| 4 | 1 | 1.70 | 1.77 | 1.25 | 0.95 | 0.21 | 1.24 | 0.83 | 0.74 | 0.53 | |||
| 2 | 1.41 | 1.30 | 1.31 | 1.12 | 1.03 | 0.88 | 0.14 | 1.08 | 0.72 | -0.29 | 0.70 | ||
| 5 | 1 | 0.93 | 0.55 | 0.56 | 0.66 | 0.46 | 0.65 | 0.17 | 0.88 | 1.23 | 2.64 | 0.35 | |
| 2 | 0.86 | 0.44 | 0.41 | 0.54 | 0.95 | 0.35 | 0.63 | 0.08 | 0.69 | 0.67 | 0.33 | 0.73 | |
| 6 | 1 | 1.23 | 0.92 | 0.69 | 0.38 | 1.23 | 0.68 | 0.86 | 0.07 | 0.97 | 0.54 | -0.40 | 0.83 |
| 2 | 1.26 | 0.96 | 0.87 | 0.52 | 1.02 | 0.85 | 0.88 | 0.09 | 1.01 | 0.45 | -0.24 | 0.80 | |
| 3 | 1.17 | 0.81 | 0.72 | 0.58 | 1.33 | 0.66 | 0.85 | 0.08 | 0.95 | 0.56 | -0.04 | 0.76 | |
| 4 | 1.34 | 1.08 | 1.14 | 0.97 | 1.55 | 1.01 | 0.87 | 0.07 | 0.96 | 0.34 | -0.69 | 0.85 | |
| 5 | 1.37 | 1.13 | 1.04 | 0.64 | 1.04 | 0.99 | 0.97 | 0.24 | 1.31 | 1.52 | 2.51 | 0.46 | |
| 6 | 1.29 | 1.00 | 0.94 | 0.47 | 1.15 | 0.83 | 0.97 | 0.15 | 1.16 | 0.60 | -0.25 | 0.73 | |
| 7 | 1.45 | 1.25 | 1.07 | 0.80 | 1.32 | 1.02 | 1.05 | 0.36 | 1.54 | 1.64 | 2.62 | 0.42 | |
| 7 | 1 | 0.98 | 0.52 | 0.45 | 0.73 | 0.17 | 0.96 | 0.41 | -0.33 | 0.56 | |||
| 8 | 1 | 1.59 | 1.19 | 1.18 | 0.75 | 0.31 | 0.06 | 0.39 | 1.65 | 3.44 | 0.50 | ||
| 9 | 1 | 1.34 | 2.18 | 2.35 | 0.89 | 1.83 | 2.15 | 0.91 | 0.25 | 1.25 | 0.92 | 0.86 | 0.44 |
| 10 | 1 | 1.63 | 1.96 | 3.07 | 1.24 | 2.41 | 1.50 | 0.33 | 1.95 | 0.75 | 0.01 | 0.61 | |
| 2 | 1.81 | 2.28 | 3.14 | 1.13 | 2.49 | 1.45 | 0.31 | 1.86 | 1.17 | 2.24 | 0.44 | ||
| 3 | 1.45 | 1.63 | 2.49 | 1.48 | 0.12 | 1.62 | -0.43 | -0.69 | 0.89 | ||||
| 4 | 1.54 | 1.79 | 2.46 | 1.60 | 1.96 | 1.50 | 0.24 | 1.83 | 0.79 | 0.07 | 0.69 | ||
| 5 | 1.46 | 1.66 | 1.90 | 1.62 | 1.37 | 0.17 | 1.62 | 0.73 | -0.24 | 0.75 | |||
| 6 | 1.38 | 1.50 | 2.59 | 1.50 | 0.19 | 1.78 | 0.55 | -0.44 | 0.78 | ||||
| Mean | 1.48 | 1.66 | 1.91 | 1.21 | 1.27 | 1.76 | 1.28 | 0.25 | 1.63 | 0.80 | 0.61 | 0.62 | |
| SD | 0.42 | 0.96 | 1.26 | 0.87 | 0.28 | 1.21 | 0.66 | 0.21 | 0.91 | 0.47 | 1.24 | 0.16 | |
| Max | 2.61 | 4.18 | 4.71 | 3.15 | 1.83 | 4.31 | 2.71 | 1.00 | 4.10 | 1.65 | 3.44 | 0.89 | |
| Min | 0.86 | 0.44 | 0.41 | 0.38 | 0.95 | 0.35 | 0.31 | 0.06 | 0.39 | -0.43 | -0.69 | 0.35 |
Figure 2.
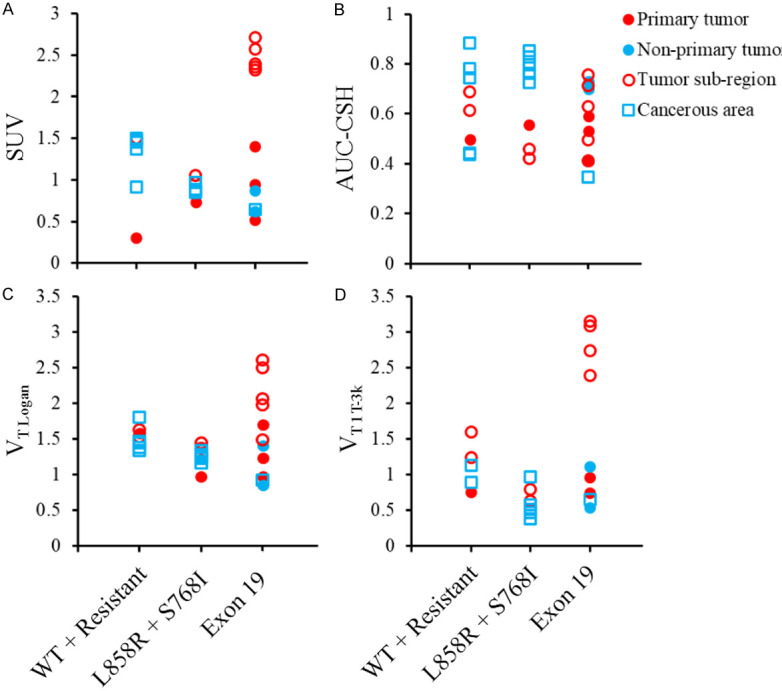
SUV (A), AUC-CSH (B), VT Logan (C), and VT 1T-3k (D) plotted by tumor biology and lesion type. Results were combined across all subjects. Values were similar within subjects but not across them. No associations were identified between endpoints and mutation status or lesion type.
VT Logan was more strongly correlated with VT 1T-3k (r2 = 0.81) compared to VT 2T-5k (r2 = 0.20). Graphical estimations of binding potential (BPLogan and BPReferenceLogan) were well correlated with one another (r2 = 0.88) and with BPSRTM (BPLogan r2 = 0.94, BPReferenceLogan r2 = 0.98). The greatest correlation between mean late-time SUV and a fully quantitative parameter was with BPSRTM (r2 = 0.90). Correlations between selected endpoints are depicted in Figure 3 and a complete matrix of correlation results can be found in Table 3.
Figure 3.
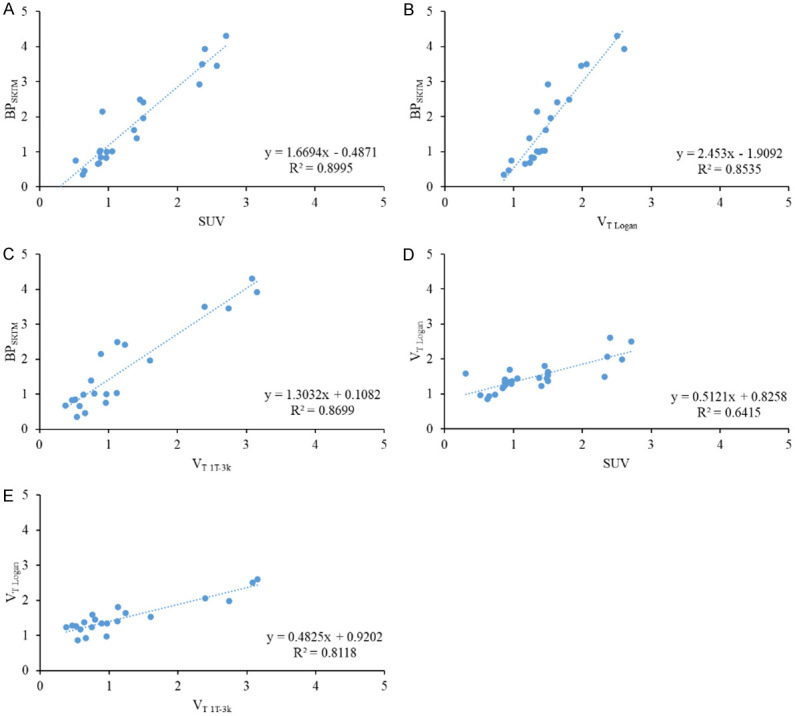
Linear correlation of BPSRTM with SUV (A), VT Logan (B), and VT 1T-3k (C). Coefficient of determination was greater than 0.85 in all instances, indicating that non-linear calculation of binding potential is well correlated with late-time uptake, graphical methods, and other non-linear endpoints. Correlation was not as strong between VT Logan and SUV (D) or VT 1T-3k (E).
Table 3.
Matrix of Pearson correlation coefficients between endpoints listed in the respective rows and columns. VT and BP from the 1T-3k model were well correlated (r2>0.80) when derived using either graphical or numeric methods. Mean late-time SUV was more strongly correlated with numeric modeling endpoints (except VT 2T-5k) compared to graphical endpoints
| Graphical | Modeling | SUV | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||||
| VT Logan | BPLogan | BPReferenceLogan | VT 1T-3k | VT 2T-5k | BPSRTM | Mean | SD | Max | Skew | Kurtosis | AUC-CSH | ||
| Graphical | VT Logan | 1.000 | 0.897 | 0.750 | 0.812 | 0.197 | 0.854 | 0.642 | 0.580 | 0.677 | 0.022 | 0.006 | 0.020 |
| BPLogan | 1.000 | 0.877 | 0.875 | 0.641 | 0.943 | 0.756 | 0.670 | 0.792 | 0.008 | 0.000 | 0.034 | ||
| BPReferenceLogan | 1.000 | 0.842 | 0.681 | 0.984 | 0.836 | 0.621 | 0.832 | 0.000 | 0.002 | 0.009 | |||
| Modeling | VT 1T-3k | 1.000 | 0.538 | 0.870 | 0.838 | 0.699 | 0.835 | 0.008 | 0.000 | 0.035 | |||
| VT 2T-5k | 1.000 | 0.653 | 0.069 | 0.055 | 0.080 | 0.001 | 0.004 | 0.061 | |||||
| BPSRTM | 1.000 | 0.900 | 0.705 | 0.899 | 0.003 | 0.000 | 0.039 | ||||||
| SUV | Mean | 1.000 | 0.663 | 0.965 | 0.010 | 0.031 | 0.001 | ||||||
| SD | 1.000 | 0.824 | 0.124 | 0.057 | 0.191 | ||||||||
| Max | 1.000 | 0.002 | 0.003 | 0.013 | |||||||||
| Skew | 1.000 | 0.811 | 0.685 | ||||||||||
| Kurtosis | 1.000 | 0.725 | |||||||||||
| AUC-CSH | 1.000 | ||||||||||||
Effect of scan length on quantitative endpoints
SUVR values calculated by truncating scan length at different tmax values (45 min, 60 min, and 90 min, full scan length) are shown in Figure 4. Between 45 and 60 min, the median percent change in SUVR values of all ROIs was approximately 10%. This trend was also true between 60 and 90 min.
Figure 4.
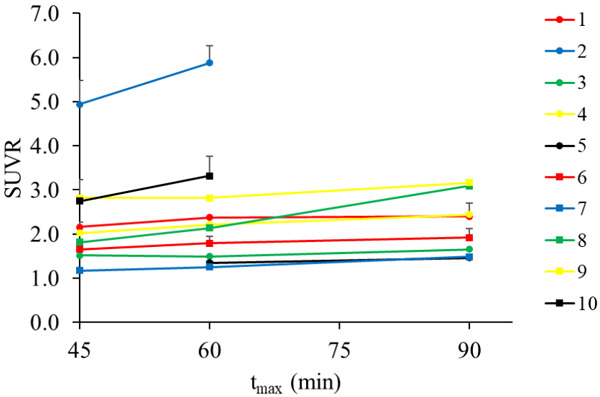
Average SUVR (muscle reference) for all ROIs in a subject utilizing the two closest frames to tmax. Data was truncated at tmax values of 45 min, 60 min, and 90 min (full scan length) in order to simulate scans of varying length. SUVR values stabilized between 60 and 90 minutes.
Figure 5 displays endpoints derived from nonlinear model fitting In all models, endpoint values do not vary greatly between tmax = 60 min and tmax = 90 min. Overall, endpoint values increased from tmax = 45 min to tmax = 60 min. The VT derived from the 2T-5k model was the most sensitive to differences in tmax, however this model did not consistently converge to parameter estimates with acceptable standard errors.
Figure 5.
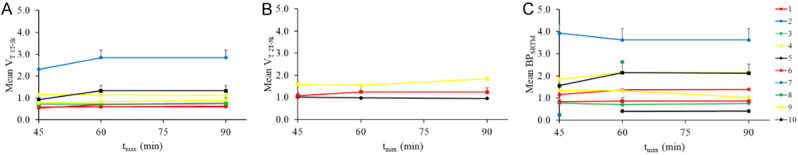
Average endpoint values for all ROIs in a subject using nonlinear fitting to the 1T-3k (A), 2T-5k (B), and SRTM (C) models. Across all models, endpoints were stable between 60 and 90 min.
Discussion
Experimental techniques
Whole-body cancer imaging is complex and nuanced. Applying and interpreting familiar techniques and endpoints commonly used in neuroimaging requires an added level of context and understanding.
No blood sampling was performed in this study in order to minimize patient discomfort. Without this quantitative information, direct comparison to a quantitative “gold standard” model (i.e. 2T-5k with arterial input) was not possible. The use of an image-derived input function partially compensated for the absence of blood sampling data.
This is the first study to use [11C]-erlotinib to image all disease areas, not just lesions of the lung. Because of time spent in different bed positions, continuous data corresponding to each ROI was not available for the entire scan duration. This impacted the IDIF: the first bed position during scanning was always the subject’s head and the second position captured the left ventricle. Therefore, the signal in the ventricle immediately following tracer injection was missed, causing an underestimation in the peak of the IDIF. Similarly, due to variability in the number of bed positions between subjects, the frequency and timing of data collection in the ROIs varied.
A second challenge for data analysis in this study was the absence of a fixed atlas for normalization. ROI definition was an important part of this study as subjects bore tumors in a wide variety of locations. The semi-automated technique outlined in the methods preserved areas of greater uptake, refining otherwise large and ambiguous ROIs. The segmentation approach that we employed focused on areas of greater tracer concentration which may have excluded portions of cancerous tissue without significant uptake.
Evaluating endpoints
Unlike in prior studies [9-11], no correlation was found between imaging endpoints and mutation status of subjects’ tumors. Here, cancerous tissue was analyzed from several parts of the body within and outside the lung. Lesions were largely heterogeneous within and across subjects, motivating the use of automated ROI refinement methods. The broader inclusion of cancerous tissue may account for the lack of direct correlation with known tumor biology. Even in studies demonstrating a connection between [11C]-erlotinib data and mutation, effect sizes are limited.
Because of the absence of arterial blood data and practical limitations in measuring an IDIF, fitting to the 2T-5k model was not consistently successful (9 of 27 ROIs fit by our error criterion). There was very limited correlation between VT 2T-5k and other endpoints (partially due to the smaller sample size). Fitting with SRTM was far more robust and BPSRTM correlated well with other outcomes. However, the reference tissue used (healthy muscle) does not strictly conform to the requirements of a true reference region. Tumor tissue differs from healthy tissue especially in vasculature, limiting the comparability of endpoints using reference regions. In general, tumor biology violates many of the assumptions required in common neuroimaging analysis techniques. Fitting ROIs to the 1T-3k model was the most robust despite limitations in the IDIF.
Graphical modeling techniques provide a fully quantitative endpoint with less stringent technical and experimental requirements of modeling. Importantly, the correlation between graphical and analytical endpoints was high (r2~0.8).
SUVR also serves as a desirable endpoint due to its relative simplicity to calculate and reasonable correlation with quantitative endpoints. Application of simplified texture analysis techniques (skew, kurtosis, and AUC-CSH) offers independent information from mean SUVR or modeling techniques. However, in our cohort, these endpoints were also not associated with tumor biology.
Analysis of endpoint stability
Truncating scan data at 45 and 60 minutes was analyzed to test the practical utility of shorter scans. For both semi-quantitative and fully-quantitative endpoints, a scan length of 60 min was sufficient to accurately represent 90 min scans. Although the 2T-5k model did produce results dependent on tmax, we believe its use in this experimental technique is sub-optimal due to the difficulties of consistently fitting dynamic data to the model. Subjects with a larger disease area tended to have more stable endpoints between 60 and 90 min.
Conclusions
This study is a thorough exploration of [11C]-erlotinib cancer imaging which includes a broad sampling of relevant tumor biology, advanced experimental techniques (dynamic, full body imaging), and a comparison of several quantitative and semi-quantitative analysis methods. Based on comparisons between endpoints, we believe that mean SUV, Logan graphical methods, and the 1T-3k analyses (with 60 min scan lengths) represent appropriate characterizations of [11C]-erlotinib in NSCLC subjects. The variability of disease location and type in this subject population is complex and more studies must be performed to better understand this tracer and its uptake by target receptors.
Acknowledgements
Financial support was provided by the Yale Clinical Center for Investigation Scholar Award (UL1RR024139 and KL2RR024138), the Kalimeris fund, Yale PET Center, Yale Cancer Center Radiation and Radiobiology Section, and R01 CA195493-01 (to EDM and JNC). JR Petrulli is supported by the National Science Foundation Graduate Research Fellowship under grant DGE-1122492. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. Special thanks to I Bahce et al. for providing plasma data.
Disclosure of conflict of interest
None.
Abbreviations
- 1T-3k
reversible one-tissue compartmental model (with blood volume fraction estimate)
- 2T-5k
reversible two-tissue compartmental model (with blood volume fraction estimate)
- AIC
Akaike information criterion
- AUC-CSH
area under the curve of the cumulative SUV histogram
- BP
binding potential
- CP(t)
concentration of parent tracer at time t
- CT
computed tomography
- EGFR
epidermal growth factor receptor
- IDIF
image derived input function
- NSCLC
non-small cell lung cancer
- OSEM
ordered subset expectation maximization
- PET
positron emission tomography
- ROI
region of interest
- SUV
standard uptake value
- SUVR
standard uptake value normalized by a reference region
- TAC
time activity curve
- TKI
tyrosine kinase inhibitor
- VT
total volume of distribution
Supporting Information
References
- 1.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, You C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Munoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, Kuhlmann GL, Han M, Goldberg JS, Settleman J, Iafrate AJ, Engelman JA, Haber DA, Johnson BE, Lynch TJ. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 6.Yang CH, Yu CJ, Shih JY, Chang YC, Hu FC, Tsai MC, Chen KY, Lin ZZ, Huang CJ, Shun CT, Huang CL, Bean J, Cheng AL, Pao W, Yang PC. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J. Clin. Oncol. 2008;26:2745–2753. doi: 10.1200/JCO.2007.15.6695. [DOI] [PubMed] [Google Scholar]
- 7.Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009;69:873–878. doi: 10.1158/0008-5472.CAN-08-3118. [DOI] [PubMed] [Google Scholar]
- 8.Petrulli JR, Sullivan JM, Zheng MQ, Bennett DC, Charest J, Huang Y, Morris ED, Contessa JN. Quantitative analysis of [11C]-erlotinib PET demonstrates specific binding for activating mutations of the EGFR kinase domain. Neoplasia. 2013;15:1347–1353. doi: 10.1593/neo.131666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahce I, Smit EF, Lubberink M. Development of [11C] erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin Cancer Res. 2013;19:183–193. doi: 10.1158/1078-0432.CCR-12-0289. [DOI] [PubMed] [Google Scholar]
- 10.Bahce I, Yaqub M, Errami H, Schuit RC, Schober P, Thunnissen E, Windhorst AD, Lammertsma AA, Smit EF, Hendrikse NH. Effects of erlotinib therapy on [(11)C]erlotinib uptake in EGFR mutated, advanced NSCLC. EJNMMI Res. 2016;6:10. doi: 10.1186/s13550-016-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yaqub M, Bahce I, Voorhoeve C, Schuit RC, Windhorst AD, Hoekstra OS, Boellaard R, Hendrikse HN, Smit EF, Lammertsma AA. Quantitative and simplified analysis of 11C-erlotinib studies. J Nucl Med. 2016;57:861–6. doi: 10.2967/jnumed.115.165225. [DOI] [PubMed] [Google Scholar]
- 12.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langer CJ, Mehta MP. Current management of brain metastases, with a focus on systemic options. J. Clin. Oncol. 2005;23:6207–6219. doi: 10.1200/JCO.2005.03.145. [DOI] [PubMed] [Google Scholar]
- 14.Schuette W. Treatment of brain metastases from lung cancer: chemotherapy. Lung Cancer. 2004;45(Suppl 2):S253–57. doi: 10.1016/j.lungcan.2004.07.967. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.United States Food and Drug Administration. Code of Federal Regulations. 2016;21:361.1. [Google Scholar]
- 17.Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010;277:301–308. doi: 10.1111/j.1742-4658.2009.07448.x. [DOI] [PubMed] [Google Scholar]
- 18.Jakoby BW, Bercier Y, Conti M, Casey ME, Bendriem B, Townsend DW. Physical and clinical performance of the mCT time-of-flight PET/CT scanner. Phys Med Biol. 2011;56:2375–2389. doi: 10.1088/0031-9155/56/8/004. [DOI] [PubMed] [Google Scholar]
- 19.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 20.van Velden FH, Cheebsumon P, Yaqub M, Smit EF, Hoekstra OS, Lammertsma AA, Boellaard R. Evaluation of a cumulative SUV-volume histogram method for parameterizing heterogeneous intratumoural FDG uptake in non-small cell lung cancer PET studies. Eur J Nucl Med Mol Imaging. 2011;38:1636–1647. doi: 10.1007/s00259-011-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 22.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.