

Abstract
Introduction
A previous phase 2b study supported the use of the 5‐HT6 receptor antagonist intepirdine as adjunctive therapy to donepezil for Alzheimer's disease (AD) dementia. A phase 3 study, MINDSET, was performed to test this hypothesis.
Methods
MINDSET was a global, double‐blind, randomized, placebo‐controlled trial in 1315 mild‐to‐moderate AD dementia patients on stable donepezil. Patients received 35 mg/day intepirdine or placebo for 24 weeks. The co‐primary endpoints were change from baseline to week 24 on the Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) and Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL).
Results
There were no statistically significant differences between intepirdine and placebo groups (adjusted mean [95% confidence interval]) on the co‐primary endpoints ADAS‐Cog (−0.36 [−0.95, 0.22], P = 0.2249) and ADCS‐ADL (−0.09 [−0.90, 0.72], P = 0.8260). Intepirdine demonstrated a favorable safety profile similar to placebo.
Discussion
Intepirdine as adjunctive therapy to donepezil did not produce statistical improvement over placebo on cognition or activities of daily living in mild‐to‐moderate AD dementia patients.
Keywords: 5‐HT6, Alzheimer's disease, clinical trial, intepirdine, phase 3
1. INTRODUCTION
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by deterioration of memory, cognition, and the ability to perform daily tasks. AD is the most common cause of dementia in adults and causes a massive global health burden that is expected to worsen substantially in the coming decades. The four currently available AD medications produce only modest cognitive improvement, highlighting the urgent need for novel treatment approaches. 1
Deficiencies in neurotransmitters, particularly acetylcholine, are a key feature in the pathophysiology of AD dementia. Acetylcholinesterase inhibitors (AChEIs: donepezil, galantamine, and rivastigmine) can improve AD symptoms by preventing synaptic acetylcholine breakdown in the brain. 2 However, AChEI benefits are limited, possibly because dosing is restricted by peripheral cholinergic stimulation, which induces several side effects. 3
Targeting the serotonin (5‐hydroxytryptamine) subtype 6 (5‐HT6) receptor, which is located primarily in brain regions critical to memory and learning, has garnered significant interest as a therapeutic mechanism in AD. Inhibition of the 5‐HT6 receptor has been shown to increase neuronal release of acetylcholine, as well as other neurotransmitters that are dysregulated in AD, leading to cognitive improvement in preclinical models. 3 , 4 Because the 5‐HT6 receptor is located almost exclusively in the central nervous system, 5‐HT6 antagonism has the potential to increase central acetylcholine release, while minimizing peripheral side effects. 3 , 4 This mechanism is distinct and complementary to that of the AChEIs, supporting adjunctive use to enhance therapeutic benefit.
Intepirdine (also called RVT‐101 or SB‐742457), a potent 5‐HT6 receptor antagonist, is an orally administered small molecule that has been studied in four phase 2 clinical trials in patients with mild‐to‐moderate AD dementia. Mixed or negative results were seen when intepirdine was administered as monotherapy in three of the trials. 5 , 6 , 7 In contrast, the most encouraging results were observed in the fourth trial, which was a large phase 2b study (N = 684) administering intepirdine as an adjunct to donepezil (Study 866). 7 Study 866 demonstrated statistically significant superiority of 35 mg/day intepirdine over placebo at week 24 on the co‐primary endpoint 11‐item Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog; 8 P = 0.012), but not on the co‐primary endpoint Clinical Dementia Rating Scale‐Sum of Boxes (CDR‐SB; 9 P = 0.462). The 35 mg/day group achieved significance over placebo on the CDR‐SB at week 12, but not at later timepoints, although numerical advantage was maintained at weeks 24, 36, and 48. Treatment with 35 mg/day intepirdine also demonstrated a significant benefit versus placebo at week 24 on the secondary endpoint 23‐item Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL; 10 P = 0.033). In Study 866 and all other phase 2 AD trials, 35 mg/day intepirdine was well tolerated. 3 , 7
The beneficial effects observed in Study 866 provided sufficient rationale for intepirdine's continued development. At the same time, another 5‐HT6 receptor antagonist, idalopirdine, had demonstrated statistically significant benefit as an adjunct to donepezil in a separate phase 2 AD dementia trial, providing additional support for intepirdine's mechanism of action. 11 Here, we report the results of a phase 3 clinical trial of intepirdine (MINDSET). The primary study objectives were to determine the effects of 35 mg/day intepirdine versus placebo on cognition and activities of daily living in mild‐to‐moderate AD dementia patients on background donepezil.
2. METHODS
2.1. Trial design
MINDSET was a multicenter, double‐blind, randomized, placebo‐controlled, parallel‐group study in patients with mild‐to‐moderate AD dementia who were on stable donepezil. The efficacy and safety of 35 mg intepirdine versus placebo administered orally once daily was evaluated over a 24‐week treatment period. A total of 207 clinical sites were initiated across 19 countries (Argentina, Australia, Bulgaria, Canada, Chile, Croatia, Czech Republic, France, Germany, Italy, Poland, Serbia, Singapore, Slovakia, Spain, Republic of Korea, Taiwan, United Kingdom, United States). The Protocol, the Statistical Analysis Plan, and an Addendum to the Statistical Analysis Plan written in response to European Medicines Agency comments are provided in Supporting Information. The study was reviewed and approved by a national, regional, or investigational independent ethics committee or institutional review board for each clinical site. An independent safety monitoring committee reviewed safety data on an ongoing basis. A list of site investigators and safety monitoring committee members is provided in Supporting Information. The study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. This trial is registered with ClinicalTrials.gov, NCT02585934.
2.2. Patients
MINDSET included patients who (1) met the criteria for a clinical diagnosis of AD dementia in accordance with the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups; 12 (2) had a magnetic resonance imaging (MRI) or computed tomography (CT) scan consistent with the exclusionary diagnosis of AD and performed during screening or within 12 months before screening; (3) were between 50 and 85 years of age, inclusive; (4) had a Mini‐Mental State Examination (MMSE) score of 12 to 24, inclusive, at screening and a score of 10 to 26, inclusive, at baseline (range 0 to 30, higher score indicates lower impairment); 13 (5) had a modified Hachinski Ischemic score ≤4 at screening (range 0 to 12, higher score indicates greater degree of ischemia); 14 and (6) had a documented history of at least 4 months of ongoing donepezil therapy, with at least 2 months of stable dosing (5 or 10 mg/day) and with no intent to change during the study. Patients were excluded if they (1) showed evidence of dementia not related to AD; (2) had a history of significant neurological or psychiatric illness; (3) had a history of negative amyloid positron emission tomography (PET) scan (n.b. amyloid scan was not required); (4) were taking memantine, AChEIs other than donepezil, or other agents to treat cognitive impairment; or (5) exhibited unacceptable laboratory values. Before study participation, informed consent or assent was obtained from all patients and caregivers.
2.3. Randomization and blinding
Patients were randomized in a 1:1 ratio to receive 35 mg/day intepirdine or placebo, with all patients maintaining their pre‐study stable donepezil dose. Randomization was performed using a validated interactive voice/web response system, with block sizes of four. Randomization was stratified by patients’ baseline MMSE scores (20%–30%: MMSE 10–15; 40%–60%: MMSE 16–20; 20%–30%: MMSE 21–26). This stratification methodology replicated that of Study 866 and was chosen because baseline AD severity has been correlated with extent of clinical decline and the investigational drug could have differential effects based on patient severity. The randomization sequence was generated by an independent statistician who had no further involvement with the study. Study staff, patients, and caregivers were blinded to treatment group assignment. Intepirdine and placebo were identical in tablet appearance and packaging to ensure adequate blinding.
RESEARCH IN CONTEXT
Systematic Review: We searched PubMed with terms “5‐HT6 receptor antagonist” AND “Alzheimer's disease.” We identified two 5‐HT6 receptor antagonists (intepirdine and idalopirdine) that had demonstrated statistically significant improvements on the Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) when administered as an adjunct to donepezil in phase 2 trials. MINDSET was the first phase 3 study of intepirdine and sought to corroborate the previous phase 2 results.
Interpretation: 35 mg/day intepirdine as adjunctive therapy to donepezil failed to demonstrate statistically significant improvement versus placebo on the co‐primary endpoints ADAS‐Cog and Alzheimer's Disease Cooperative Study‐Activities of Daily Living over 24 weeks. A statistical benefit over placebo was observed on the key secondary endpoint Clinician's Interview‐Based Impression of Change plus caregiver interview. Intepirdine was well tolerated with a safety profile comparable to placebo.
Future Directions: New research into Alzheimer's disease heterogeneity as well as the functionality of the 5‐HT6 receptor and its ligands may help elucidate the underlying reasons behind the conflicting data observed to date between the phase 2 and 3 trials.
2.4. Procedures
The study consisted of a 4‐week screening period, a 3‐week single‐blind placebo run‐in period, and a 24‐week randomized double‐blind treatment period. Patients who completed the last on‐treatment visit of MINDSET were eligible to enroll in an open‐label extension study (NCT02586909). Patients who did not enter the extension study had a follow‐up safety visit at week 26. During the run‐in period, patients received single‐blinded placebo and their pre‐study donepezil dose to evaluate the variability of their baseline status. During the double‐blind treatment period, patients received 35 mg/day intepirdine or placebo in addition to their background donepezil. Pill count was monitored to assess compliance.
Scheduled visits were at weeks 3, 6, 12, 18, and 24 during the double‐blind treatment period. The co‐primary endpoint assessments, ADAS‐Cog and ADCS‐ADL, were performed at each visit, except screening. The first key secondary assessment Clinician's Interview‐Based Impression of Change plus caregiver interview (CIBIC+) 15 was performed at weeks 12, 18, and 24 and was rated relative to a Clinician's Interview‐Based Impression of Severity (CIBIS) administered at baseline. The CIBIC+ and CIBIS were assessed by a rater independent of other efficacy outcomes. The second key secondary assessment Dependence Scale (DS) 16 was performed at baseline and at week 24. All raters passed a rigorous qualification and certification process. Variability and rating errors were monitored in a blinded manner.
2.5. Outcomes
The co‐primary endpoints were (1) ADAS‐Cog score change from baseline to week 24 (range 0 to 70, higher score indicates worse function) and (2) ADCS‐ADL score change from baseline to week 24 (range 0 to 78, higher score indicates better function). Pre‐specified key secondary endpoints were (1) CIBIC+ score at week 24 (range 0 to 7, higher score indicates worsened function from baseline) and (2) DS Total Score change from baseline to week 24 (range 0 to 15, higher score indicates higher dependency and worse function). Other efficacy endpoints included change from baseline to week 24 on the Neuropsychiatric Inventory (NPI) Total Score 17 and ADAS‐Cog plus delayed word recall and total digit cancellation score (ADAS‐Cog‐13). 18
Safety endpoints included treatment‐emergent adverse events (TEAEs); physical examinations; suicidality assessments; and measurements of vital signs, electrocardiograms, and laboratory tests. TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA).
2.6. Statistical analysis
Sample size estimates were based on the results of Study 866, which demonstrated an ADAS‐Cog difference between treatment groups of 1.5 points (standard deviation [SD] 6) and an ADCS‐ADL difference of 1.9 points (SD 9). 7 At a two‐sided 0.05 significance level, we calculated that 435 patients per arm would allow a difference of 1.6 points (SD 6.5) to be detected on the ADAS‐Cog with 95% power and a difference of 2 points (SD 9) to be detected on the ADCS‐ADL with 90% power. Assuming a drop‐out/missing data rate of ≈25%, 1150 patients were planned to be randomized.
The safety population consisted of all patients who received at least one dose of double‐blind study medication. The primary population for efficacy analysis consisted of all randomized patients who took at least one treatment dose and had at least one baseline and one post‐baseline primary efficacy assessment (modified intent‐to‐treat [mITT] population).
Treatment comparisons of the co‐primary endpoints were based on the Observed Case dataset (no imputation for missing values) using a mixed model for repeated measures (MMRM) with restricted maximum likelihood estimation and an unstructured covariance matrix. Countries were grouped into five regions for analysis. The statistical model was fitted with terms for treatment group, visit, treatment by visit interaction, baseline score, baseline MMSE, baseline score by visit interaction, and region. The interaction term of baseline MMSE by visit was evaluated at the 10% level of significance. If the interaction term was found to be significant, it was included in the MMRM model. Primary inferences were drawn from treatment differences for the changes from baseline derived from the MMRM models at week 24. Adjusted means and 95% confidence intervals (CI) are presented.
All hypothesis tests were two‐sided at a significance level of 0.05. Efficacy would be concluded only if both primary endpoints showed a significant drug–placebo difference. Nominal P‐values are reported, given the negative results on the co‐primary endpoints. All analyses were performed using SAS software, version 9.4. The Statistical Analysis Plan and Addendum were finalized prior to unblinding the treatment allocation codes.
3. RESULTS
MINDSET randomized patients between October 21, 2015 and March 8, 2017. A total of 2173 patients were screened, and 1315 patients were randomized (Figure 1). The primary reason for unsuccessful screening (observed in 296 patients) was a failure to meet the MMSE score inclusion criteria. A total of 654 patients received placebo, and 661 patients received 35 mg/day intepirdine, with 581 placebo‐treated and 592 intepirdine‐treated patients completing the study through week 24. Patient withdrawal rates were similar between placebo (11.2%) and intepirdine (10.4%) groups. The safety population (n = 1307) comprised 651 placebo‐treated patients and 656 intepirdine‐treated patients. The mITT population (n = 1276) comprised 633 placebo‐treated patients and 643 intepirdine‐treated patients.
FIGURE 1.
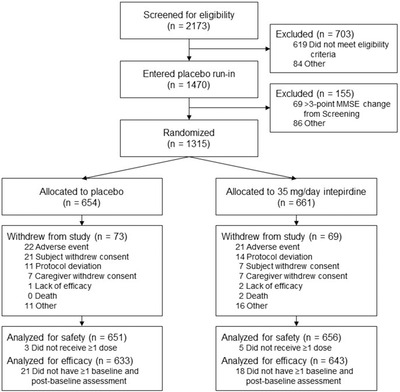
Patient flow through the MINDSET study. A total of 1315 patients were randomized across 19 countries. A total of 1307 patients were included in the safety population, and 1276 patients were included in the modified intent‐to‐treat (mITT; efficacy) population
Demographics, baseline characteristics, regional distributions, and donepezil dose distributions were similar between treatment groups (Table 1). Average age at diagnosis of probable AD dementia was 70.2 years, and patients were diagnosed a median of 2.0 years prior to screening. The average baseline MMSE score of 18.5 points for each treatment group reflected the stratified design of the study and a population with mild‐to‐moderate AD dementia. Baseline efficacy endpoint values were similar between treatment groups. Almost all patients were considered compliant with study drug and donepezil.
TABLE 1.
Demographics and baseline characteristics of the mITT population (N = 1276)
| Placebo (N = 633) | Intepirdine (N = 643) | |
|---|---|---|
| Age (years), mean (SD) | 72.5 (7.55) | 72.7 (7.68) |
| Age (years) at diagnosis, mean (SD) | 70.0 (7.70) | 70.4 (7.75) |
| Time (years) since diagnosis, mean (SD) | 2.62 (2.05) | 2.55 (2.25) |
| Female sex, n (%) | 394 (62.2%) | 386 (60.0%) |
| White race, n (%) | 592 (94.1%) | 595 (93.1%) |
| Non‐Hispanic ethnicity, n (%) | 519 (82.8%) | 518 (81.2%) |
| Region, n (%) | ||
| US | 169 (26.7%) | 171 (26.6%) |
| Non‐US English | 132 (20.8%) | 133 (20.7%) |
| West Europe | 115 (18.2%) | 110 (17.1%) |
| East Europe | 117 (18.5%) | 116 (18.0%) |
| Rest of World | 100 (15.8%) | 113 (17.6%) |
| MMSE score | ||
| Mean (SD) | 18.5 (3.63) | 18.5 (3.70) |
| Stratification, n (%) | ||
| 10–15 | 151 (24.0%) | 148 (23.0%) |
| 16–20 | 284 (44.9%) | 283 (44.0%) |
| 21–26 | 197 (31.1%) | 212 (33.0%) |
| Donepezil dosing, n (%) | ||
| 5 mg/day | 151 (23.9%) | 142 (22.1%) |
| 10 mg/day | 482 (76.1%) | 501 (77.9%) |
| ADAS‐Cog, mean (SD) | 24.8 (8.80) | 24.3 (8.87) |
| ADCS‐ADL, mean (SD) | 57.7 (12.6) | 58.1 (12.7) |
| CIBIS, mean (SD) | 3.8 (0.76) | 3.7 (0.74) |
| DS Total Score, mean (SD) | 5.20 (2.34) | 4.90 (2.34) |
| NPI Total Score, mean (SD) | 7.60 (9.59) | 7.70 (9.88) |
| ADAS‐Cog‐13, mean (SD) | 37.0 (10.3) | 36.5 (10.4) |
Notes: The treatment groups were comparable with respect to the distributions of baseline characteristics. Regions were pre‐specified for subgroup analyses per the stratifications in the table. US = United States; Non‐US English = United Kingdom, Canada, Australia; West Europe = Germany, Spain, Italy, France; East Europe = Bulgaria, Croatia, Czech Republic, Poland, Serbia, Slovakia; Rest of World = Argentina, Chile, Singapore, Republic of Korea, Taiwan
Abbreviations: ADAS‐Cog, Alzheimer's Disease Assessment Scale‐Cognitive Subscale; ADCS‐ADL, Alzheimer's Disease Cooperative Study‐Activities of Daily Living; CIBIS, Clinician's Interview‐Based Impression of Severity; DS, Dependence Scale; NPI, Neuropsychiatric Inventory; mITT, modified intent‐to‐treat; SD, standard deviation.
The results of the co‐primary endpoint analyses are shown in Figure 2A,B and Table 2. Intepirdine 35 mg/day as adjunctive therapy to donepezil failed to demonstrate statistically significant differences from placebo on the ADAS‐Cog (adjusted mean difference = −0.36 versus placebo, 95% CI [−0.95, 0.22], P = 0.2249) and ADCS‐ADL (adjusted mean difference = −0.09 versus placebo, 95% CI [−0.90, 0.72], P = 0.8260).
FIGURE 2.
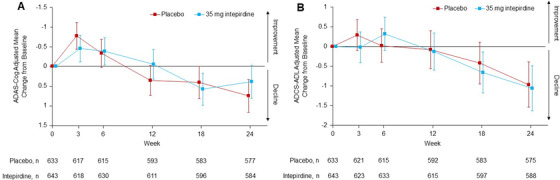
Adjusted mean changes on the co‐primary endpoints, (A) Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) and (B) Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL) scores, over the 24‐week treatment period (modified intent‐to‐treat [mITT] population). Treatment comparisons were based on a mixed model for repeated measures. Error bars are 95% confidence intervals.
TABLE 2.
Primary and secondary efficacy outcomes at end of double‐blind treatment period, week 24 (mITT population)
| Adjusted mean through week 24 | Pairwise comparison | ||||||
|---|---|---|---|---|---|---|---|
| n | Placebo | n | Intepirdine | Difference | 95% CI | P‐value | |
| Primary endpoints | |||||||
| ADAS‐Cog change from baseline | 577 | 0.75 (0.21) | 584 | 0.39 (0.21) | −0.36 (0.30) | −0.95, 0.22 | 0.2249 |
| ADCS‐ADL change from baseline | 575 | −0.97 (0.29) | 588 | −1.06 (0.29) | −0.09 (0.41) | −0.90, 0.72 | 0.8260 |
| Secondary endpoints | |||||||
| CIBIC+ | 568 | 4.30 (0.037) | 577 | 4.18 (0.037) | −0.12 (0.051) | −0.22, −0.02 | 0.0234 |
| DS Total Score change from baseline | 568 | 0.17 (0.071) | 580 | 0.30 (0.070) | 0.12 (0.10) | ‐0‐.07, 0.32 | 0.2096 |
| NPI Total Score change from baseline | 570 | 0.06 (0.34) | 583 | −0.08 (0.34) | −0.14 (0.48) | −1.09, 0.80 | 0.7650 |
| ADAS‐Cog‐13 change from baseline | 576 | 0.64 (0.24) | 583 | 0.26 (0.23) | −0.38 (0.33) | −1.03, 0.27 | 0.2472 |
A total of 633 placebo‐treated patients and 643 intepirdine‐treated patients made up the mITT population. The co‐primary endpoints, ADAS‐Cog and ADCS‐ADL, and the secondary endpoints, DS Total Score, NPI Total Score, and ADAS‐Cog‐13, were not statistically different between the intepirdine and placebo groups. The secondary endpoint CIBIC+ demonstrated a statistically significant improvement favoring intepirdine over placebo. Treatment comparisons were based on a mixed model for repeated measures. SE are in parentheses. n = number of patients with measurement of the indicated endpoint at week 24
Abbreviations: ADAS‐Cog, Alzheimer's Disease Assessment Scale‐Cognitive Subscale; ADCS‐ADL, Alzheimer's Disease Cooperative Study‐Activities of Daily Living; CIBIC+, Clinician's Interview‐Based Impression of Change plus caregiver interview; DS, Dependence Scale; NPI, Neuropsychiatric Inventory; mITT, modified intent‐to‐treat; SE, standard error.
For the CIBIC+, the intepirdine group was numerically superior to placebo at the 18‐ and 24‐week timepoints, with the difference at 24 weeks achieving statistical significance (adjusted mean difference = −0.12 versus placebo, 95% CI [−0.22, −0.02], P = 0.0234; Table 2, Figure 3). This result was largely consistent across subgroup and sensitivity analyses. There was no statistical benefit at week 24 over placebo for the DS Total Score (adjusted mean difference = 0.12 versus placebo, 95% CI [−0.07, 0.32], P = 0.2096). Other secondary endpoints were not statistically significant versus placebo (Table 2). Non‐CIBIC+ subgroup analyses and exploratory post hoc efficacy analyses generally showed no meaningful differences between treatment groups; however, there was evidence that patients who were taking donepezil the longest prior to study entry (i.e., greater than the mean) experienced statistical improvement on the ADAS‐Cog with interpirdine versus placebo.
FIGURE 3.
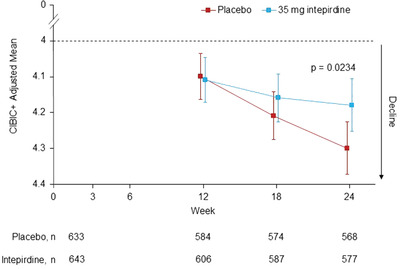
Adjusted means of key secondary endpoint Clinician's Interview‐Based Impression of Change plus caregiver interview (CIBIC+) over 24‐week treatment period (modified intent‐to‐treat [mITT] population). Scores between 0 and 4 indicate an improvement in global function from baseline, while scores between 4 and 7 indicate a decline from baseline. Treatment comparisons were based on a mixed model for repeated measures. Error bars are 95% confidence intervals
The incidence of TEAEs, treatment‐emergent serious adverse events (SAEs), and TEAEs leading to withdrawal were similar across treatment groups (Table 3). TEAEs were generally consistent with those expected for the trial's patient population. The most common TEAEs (≥2% in either treatment group) were fall, urinary tract infection, nasopharyngitis, headache, dizziness, diarrhea, nausea, back pain, bronchitis, and cough. Almost all TEAEs were reported at an incidence of <5%. Suicidal ideation and behavior were also low and comparable across groups. Increased incidence of elevated liver enzymes has been observed in the active group of other 5‐HT6 receptor antagonist studies; 11 , 19 this trend was not seen in MINDSET. No death was considered related to study treatment by the site investigator.
TABLE 3.
Adverse events seen in safety population (N = 1307)
| Placebo (N = 651) | Intepirdine 35 mg (N = 656) | |
|---|---|---|
| At least one TEAE, n (%) | 355 (54.5%) | 366 (55.8%) |
| Fall | 29 (4.5%) | 37 (5.6%) |
| Urinary tract infection | 26 (4.0%) | 25 (3.8%) |
| Nasopharyngitis | 19 (2.9%) | 23 (3.5%) |
| Headache | 18 (2.8%) | 17 (2.6%) |
| Dizziness | 12 (1.8%) | 19 (2.9%) |
| Diarrhea | 17 (2.6%) | 17 (2.6%) |
| Nausea | 13 (2.0%) | 17 (2.6%) |
| Back pain | 15 (2.3%) | 11 (1.7%) |
| Bronchitis | 10 (1.5%) | 14 (2.1%) |
| Cough | 7 (1.1%) | 15 (2.3%) |
| Elevated liver enzymes | 9 (1.5%) | 7 (1.1%) |
| Treatment‐emergent SAE, n (%) | 44 (6.8%) | 40 (6.1%) |
| TEAE leading to study withdrawal, n (%) | 21 (3.2%) | 21 (3.2%) |
| Death, n (%) | 2 (0.3%) | 3 (0.5%) |
Notes: All adverse events that occurred in at least 2% of patients in either treatment group are shown. An increase in the frequency of elevated liver enzymes has been observed in other 5‐HT6 receptor antagonist trials, but, in MINDSET, this TEAE was low and similar across treatment groups. No deaths were considered to be related to treatment by the study investigator.
Abbreviations: SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
4. DISCUSSION
The primary objectives of the phase 3 MINDSET study were not met: 35 mg/day intepirdine as adjunctive therapy to donepezil in mild‐to‐moderate AD dementia patients did not show statistically significant benefit over placebo on the co‐primary endpoints ADAS‐Cog and ADCS‐ADL over the 24‐week treatment period. The intepirdine group demonstrated a statistically significant benefit over placebo on the first key secondary endpoint CIBIC+ at 24 weeks (P = 0.0234). The second key secondary endpoint DS Total Score, as well as all other secondary endpoints, showed no significant difference between treatment groups. Intepirdine demonstrated a strong safety profile during the 24‐week treatment period, with no apparent safety differences compared to placebo.
The CIBIC+, a measure of global functioning encompassing both cognition and activities of daily living, has been used as a co‐primary endpoint and measure of clinical meaningfulness in studies of all currently available AD medications. 20 , 21 , 22 , 23 In MINDSET, it was assessed by experienced clinicians independent of other efficacy variables to reduce bias. The treatment benefit of 0.12 points observed in this study is more modest than the 0.2‐ to 0.5‐point improvements seen in pivotal studies of approved AD medications 20 , 21 , 22 , 23 as well as the statistically significant 0.31‐point improvement observed in a previous monotherapy study of intepirdine. 5 In Study 866, a different measure of global functioning (CDR‐SB) was used, and the result for the 35 mg/day group was statistically significant at week 12 and non‐significantly different but numerically superior to placebo at weeks 24, 36, and 48. 7 Overall, the benefit of intepirdine on AD global function is unclear, and any effect size would likely be smaller than that of currently approved medications.
MINDSET's co‐primary endpoints, sample size, and intepirdine dose were selected based on the results of Study 866. Although MINDSET required a donepezil use history of only 4 months compared to 6 months in Study 866, the eligibility criteria of MINDSET generally matched that of Study 866. The sample size per arm of MINDSET was almost three times that of Study 866, so MINDSET was well powered to detect treatment effects on both co‐primary endpoints. Despite this increased power and the fact that the baseline characteristics of the two studies were largely comparable, MINDSET failed to corroborate the results of Study 866.
Study‐to‐study variability in AD trials is not uncommon. Idalopirdine, a selective 5‐HT6 receptor antagonist, and latrepirdine, an antihistamine capable of non‐selectively antagonizing the 5‐HT6 receptor, followed a similar development pattern to intepirdine. Idalopirdine demonstrated a statistical benefit on the ADAS‐Cog in a phase 2 study but then failed to meet the same endpoint in three separate phase 3 trials, although these studies used different dose regimens than the phase 2 study. 11 , 19 Latrepirdine showed positive results on multiple endpoints in phase 2 but failed on these endpoints in two larger phase 3 studies using an identical dose. 24 Interestingly, in both development programs, the phase 3 studies’ primary endpoint placebo declines from baseline were lower than the declines in the phase 2 studies. 11 , 19 , 24 Similarly, placebo declines in MINDSET were lower than Study 866 (ADAS‐Cog at week 24: 0.75‐point decline in MINDSET versus 1.2‐point decline in Study 866; ADCS‐ADL at week 24: 0.97‐point decline in MINDSET versus 3.2‐point decline in Study 866). 7 Because the placebo group did not act as predicted, separation between treatment groups was difficult to achieve.
The major difference between Study 866 and MINDSET was MINDSET's considerably larger geographical scope. Similar to other global AD trials, 25 , 26 MINDSET demonstrated striking heterogeneity across regions in placebo change from baseline to week 24 on the co‐primary endpoints, with one region (“Rest of World,” i.e., Argentina, Chile, Republic of Korea, Singapore, Taiwan) even experiencing mean improvements (ADAS‐Cog placebo change range: −1.19 to 1.85; ADCS‐ADL placebo change range: 1.11 to −3.37). Although all nine countries involved in Study 866 were included in MINDSET, the regional distribution within each study differed substantially. A total of 52% of dosed patients in Study 866 were recruited from West Europe (data unpublished), a region shown to produce relatively large placebo declines, 25 , 26 compared to only 17.7% in MINDSET. Thus, MINDSET's increased regional heterogeneity may have resulted in its lower‐than‐expected placebo decline, potentially contributing to the study's failure as the investigational drug could have affected non‐progressing and fast‐progressing patients differently.
Given their smaller size, phase 2 AD trials may be able to selectively recruit in regions with experienced sites and raters and a well‐characterized, progressive patient population. For phase 3 AD trials, sponsors often decide to recruit in new regions to increase the study's sample size/power and complete the study in a timely manner. However, this decision may introduce significant treatment, operational, and/or disease heterogeneity because new medical systems with different standards of care are added, executional standardization across sites and raters with varying linguistic and cultural backgrounds is harder to accomplish, and the underlying disease characteristics of the study's patient population may change. 25 , 26 , 27 Therefore, while initially well conceived, the decision to expand to new geographies may reduce probability of success and phase‐to‐phase translatability. Future large‐scale AD trials may benefit from restricting recruitment to well‐studied regions or periodically assessing placebo decline through a data monitoring committee.
In addition, although the patients in MINDSET met clinical criteria for AD, no biomarker confirmation was required for inclusion into the study. This approach may have contributed to the reduced placebo decline observed in the study, as has been documented in other trials lacking biomarker entry criteria. 28 Using biomarker‐based strategies may reduce diagnostic heterogeneity and increase the rate of placebo decline required for the demonstration of a drug–placebo difference.
Experience with 5‐HT6 receptor antagonists in the treatment of AD has been disappointing and hopes for this class of drugs have diminished. It is possible that underlying factors such as amyloid status and APOE genotype (neither of which were assessed in MINDSET) affect 5‐HT6 pharmacodynamics—an area that has not been fully explored so far. 29 Paradoxically, 5‐HT6 receptor agonists have also demonstrated pro‐cognitive effects, and a better understanding of the 5‐HT6 receptor and its ligands would help in clarifying the potential of 5‐HT6 receptor‐targeting agents in AD, either alone or in combination with an AChEI. 3
Although MINDSET's per‐site enrollment rate (≈0.4 patients per site per month) was not particularly rapid, the study's overall enrollment rate (≈80 patients per month) was unusually fast for a global AD trial. We made significant efforts to engage the patient community across the world and worked closely with study investigators to overcome any operational challenges. For example, while many previous AD studies have offered reimbursement for transportation to and from clinical sites, caregivers and study coordinators are burdened with the details of booking drivers and submitting reimbursement paperwork. We therefore leveraged a mobile ride‐share application, which provided convenient transportation that we easily reimbursed via electronic billing. We also conducted > 50 presentations at local senior centers and engaged grassroots media organizations to discuss the impact of AD in their community along with the MINDSET study. Innovative operational strategies may allow future studies to improve study participants’ satisfaction and reduce time to data readout.
Overall, the conflicting results of 5‐HT6 receptor antagonists in phase 2 and 3 studies highlight the importance of future research into underlying AD neurobiology and heterogeneity, with the goal of identifying patient populations that will respond better to certain therapeutic mechanisms. The worldwide burden of AD is expected to dramatically increase in the coming decades, and new treatment options are clearly needed.
CONFLICTS OF INTEREST
Frederick M. Lang is a paid consultant and former full‐time member of Axovant Sciences (Axovant) and is an employee and shareholder of Roivant Sciences (Roivant). Shari Coslett is a former employee and shareholder of Axovant. Sarah R. Friedhoff is a former employee of Axovant and Roivant. Yi Mo is a former employee of Axovant. Mark Harnett was a former paid consultant of Axovant and is a current paid consultant of Roivant.
Jeffrey L. Cummings has provided consultation to Acadia, Actinogen, Alkahest, Allergan, Alzheon, Annovis, Avanir, Axsome, BiOasis, Biogen, Bracket, Cassava, Cerecin, Cortexyme, Diadem, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, GemVax, Grifols, Hisun, Merck, Otsuka, Resverlogix, Roche, Samumed, Samus, Takeda, Third Rock, and United Neuroscience pharmaceutical and assessment companies. Jeffrey L. Cummings has stock options in Prana, Neurokos, ADAMAS, MedAvante, QR pharma, BiOasis. Jeffrey L. Cummings owns the copyright of the Neuropsychiatric Inventory. Jeffrey L. Cummings is supported by KMA; NIGMS grant P20GM109025; NINDS grant U01NS093334; and NIA grant R01AG053798.
Timo Grimmer certifies that there is no actual or potential conflict of interest in relation to this article. Outside the submitted work, Timo Grimmer reported having received consulting fees from Actelion, Biogen, Eli Lilly, Iqvia/Quintiles, MSD, Novartis, Quintiles, Roche Pharma; lecture fees from Biogen, Lilly, Parexel, Roche Pharma; and grants to his institution from Actelion and PreDemTech.
Bruno Dubois received fees for his participation in the scientific advisory board of the study.
The institutions of Paul Solomon, Merce Boada, Roy W. Jones, Giovanni B. Frisoni, Timo Grimmer, and Bruno Dubois received funding for the conduct of the study.
AUTHOR CONTRIBUTIONS
Frederick M. Lang led the effort to put together the manuscript and was the primary writer, drafting each section with input from all authors, assisting with data interpretation, and conducting the systematic review of the literature. Yi Mo supported data collection, cleaning, analysis, and interpretation. Jeffrey L. Cummings and Marwan Sabbagh contributed to protocol design, selection of endpoints, implementation and interpretation of outcomes. Paul Solomon, Merce Boada, Roy W. Jones, Giovanni B. Frisoni, Timo Grimmer, and Bruno Dubois were study investigators who participated in the design and/or enrollment of the trial, the discussion of the results, and the writing of the manuscript. Mark Harnett assisted with the statistical analysis plan and the analysis and interpretation of study outcomes. Shari Coslett was the head of clinical operations of the study. Sarah R. Friedhoff was responsible for strategy and implementation of the study's patient advocacy, patient engagement, and patient recruitment strategies. The corresponding author had access to all data in the study. All authors provided critical feedback and helped shape the research, analysis, and writing of the manuscript, in accordance with the criteria established by the International Committee of Medical Journal Editors (ICMJE) and Good Publication Practice Guidelines.
DATA SHARING STATEMENT
The funder will not share data from the study.
Supporting information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
ACKNOWLEDGMENTS
We thank the patients and caregivers for their participation in the MINDSET study. We thank all study investigators and staff at each clinical site for their support in conducting the study. We thank Ajith Karunakara (who led the statistical programming for the study), Benjamin Thorp (who led the data management of the study), Rizalyn Golnar (clinical operations), and David Jerchower (clinical supplies) for their roles in the study and for their help in compiling the data for the manuscript. MINDSET was funded and overseen by Axovant. During the time of the study, Roivant was the majority shareholder of Axovant. Roivant is the largest shareholder of Axovant at the time of manuscript submission.
Lang FM, Mo Y, Sabbagh M, et al. Intepirdine as adjunctive therapy to donepezil for mild‐to‐moderate Alzheimer's disease: A randomized, placebo‐controlled, phase 3 clinical trial (MINDSET). Alzheimer's Dement. 2021;7:e12136. 10.1002/trc2.12136
REFERENCES
- 1. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 2018;141:1917‐1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khoury R, Grysman N, Gold J, Patel K, Grossberg GT. The role of 5 HT6‐receptor antagonists in Alzheimer's disease: an update. Expert Opin Investig Drugs. 2018;27:523‐533. [DOI] [PubMed] [Google Scholar]
- 4. Ferrero H, Solas M, Francis PT, Ramirez MJ. Serotonin 5‐HT6 receptor antagonists in Alzheimer's disease: therapeutic rationale and current development status. CNS Drugs. 2017;31:19‐32. [DOI] [PubMed] [Google Scholar]
- 5. Maher‐Edwards G, Zvartau‐Hind M, Hunter AJ, et al. Double‐blind, controlled phase II study of a 5‐HT6 receptor antagonist, SB‐742457 in Alzheimer's disease. Curr Alzheimer's Research. 2010;7:374‐385. [DOI] [PubMed] [Google Scholar]
- 6. Maher‐Edwards G, Dixon R, Hunter J, et al. SB‐742457 and donepezil in Alzheimer disease: a randomized placebo‐controlled study. Int J Geriatr Psychiatry. 2011;26:536‐544. [DOI] [PubMed] [Google Scholar]
- 7. Maher‐Edwards G, Watson C, Ascher J, et al. Two randomized controlled trials of SB742457 in mild‐to‐moderate Alzheimer's disease. Alzheimers Dement (NY). 2015;1:23‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. AM J Psychiatry. 1984;141:1356‐1364. [DOI] [PubMed] [Google Scholar]
- 9. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 10. Galasko D, Bennet D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11(suppl 2):33‐39. [PubMed] [Google Scholar]
- 11. Wilkinson D, Windfeld K, Colding‐Jorgensen E. Safety and efficacy of idalopirdine, a 5‐HT6 receptor antagonist, in patients with moderate Alzheimer's disease (LADDER): a randomized, double‐blind, placebo‐controlled phase 2 trial. Lancet Neurol. 2014;13:1092‐1099. [DOI] [PubMed] [Google Scholar]
- 12. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute of Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Folstein MF, Folstein SE, McHugh PR. Mini‐Mental State”. a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 14. Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementia. Ann Neurol. 1980;7:486‐488. [DOI] [PubMed] [Google Scholar]
- 15. Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer's Disease Cooperative Study—clinical global impression of change. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11(suppl 2):22‐32. [DOI] [PubMed] [Google Scholar]
- 16. Stern Y, Albert SM, Sano M, et al. Assessing patient dependence in Alzheimer's disease. J Gerontol. 1994;49:M216‐222. [DOI] [PubMed] [Google Scholar]
- 17. Cummings JL, Mega M, Gray K, Rosenberg‐Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308‐2314. [DOI] [PubMed] [Google Scholar]
- 18. Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disorder. 1997;11(suppl 2):13‐21. [PubMed] [Google Scholar]
- 19. Atri A, Frolich L, Ballard C, et al. Effect of Idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA. 2018;319:130‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eisai . Aricept (donepezil hydrochloride) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020690s042,021720s014,022568s011lbl.pdf. Revised December 2018. Accessed February 5, 2020.
- 21. Forest Labs LLC. Namenda XR (memantine hydrochloride) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022525s015lbl.pdf. Revised November 2019. Accessed February 5, 2020.
- 22. Janssen Pharmaceuticals. Razadyne ER (galantamine hydrobromide) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021169Orig1s032,021224Orig1s030,021615Orig1s023lbl.pdf. Revised September 2016. Accessed February 5, 2020.
- 23. Novartis. Exelon Patch (rivastigmine transdermal system) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022083lbl.pdf. Accessed April 19, 2020.
- 24. Cano‐Cuenca N, Solis‐Garcia del Pozo JE, Jordan J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: a meta‐analysis. J Alzheimers Dis. 2014;38:155‐164. [DOI] [PubMed] [Google Scholar]
- 25. Cummings JL, Atri A, Ballard C, et al. Insights into globalization: comparison of patient characteristics and disease progression among geographic regions in a multinational Alzheimer's disease clinical program. Alzheimers Res Ther. 2018;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henley DB, Dowsett SA, Chen YF, et al. Alzheimer's disease progression by geographical region in a clinical trial setting. Alzheimers Res Ther. 2015;7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummings JL, Reynders R, Zhong K. Globalization of Alzheimer's disease clinical trials. Alzheimers Res Ther. 2011;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballard C, Atri A, Boneva N, et al. Enrichment factors for clinical trials in mild‐to‐moderate Alzheimer's disease. Alzheimers Dement (N Y). 2019;5:164‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roberts PD, Spiros A, Geerts H. Simulations of symptomatic treatments for Alzheimer's disease: computational analysis of pathology and mechanisms of drug action. Alzheimers Res Ther. 2019;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Supplementary information
Supplementary information