

Abstract
Background
Genetic diversity and parasite relatedness are essential parameters for assessing impact of interventions and understanding transmission dynamics of malaria parasites, however data on its status in Plasmodium falciparum populations in Uganda is limited. Microsatellite markers and DNA sequencing were used to determine diversity and molecular characterization of P. falciparum parasite populations in Uganda.
Methods
A total of 147 P. falciparum genomic DNA samples collected from cross-sectional surveys in symptomatic individuals of 2–10 years were characterized by genotyping of seven highly polymorphic neutral microsatellite markers (n = 85) and genetic sequencing of the Histidine Rich Protein 2 (pfhrp2) gene (n = 62). ArcGIS was used to map the geographical distribution of isolates while statistical testing was done using Student's t-test or Wilcoxon's rank-sum test and Fisher’s exact test as appropriate at P ≤ 0.05.
Results
Overall, 75.5% (95% CI 61.1–85.8) and 24.5% (95% CI14.2–38.9) of parasites examined were of multiclonal (mixed genotype) and single clone infections, respectively. Multiclonal infections occurred more frequently in the Eastern region 73.7% (95% CI 48.8–89.1), P < 0.05. Overall, multiplicity of infection (MOI) was 1.9 (95% CI 1.7–2.1), P = 0.01 that was similar between age groups (1.8 vs 1.9), P = 0.60 and regions (1.9 vs 1.8), P = 0.43 for the < 5 and ≥ 5 years and Eastern and Western regions, respectively. Genomic sequencing of the pfhrp2 exon2 revealed a high level of genetic diversity reflected in 96.8% (60/62) unique sequence types. Repeat type AHHAAAHHATD and HRP2 sequence Type C were more frequent in RDT−/PCR + samples (1.9% vs 1.5%) and (13% vs 8%), P < 0.05 respectively. Genetic relatedness analysis revealed small clusters of gene deleted parasites in Uganda, but no clustering with Eritrean parasites.
Conclusion
High level of genetic diversity of P. falciparum parasites reflected in the frequency of multiclonal infections, multiplicity of infection and variability of the pfhrp2 gene observed in this study is consistent with the high malaria transmission intensity in these settings. Parasite genetic analysis suggested spontaneous emergence and clonal expansion of pfhrp2 deleted parasites that require close monitoring to inform national malaria diagnosis and case management policies.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12936-021-03763-6.
Keywords: Malaria, Rapid diagnostic tests, Genetic diversity, Multiplicity of infection, Multiclonal infections, Parasite relatedness, Pfhrp2 deletion, Microsatellite markers
Background
In 2019, the World Health Organization (WHO) estimated that there were 229 million cases of and 409,000 deaths due to malaria globally. The WHO African region accounts for a disproportionately high share of the global burden (94% of malaria cases in 2019 alone) [1, 2]. Nearly all malaria cases in the WHO African region are caused by Plasmodium falciparum. Uganda is ranked among the top six countries with the highest malaria burden [1–3]. Despite the improved coverage of interventions, malaria remains a major public health problem in Uganda accounting for 30% of outpatient visits to health facilities (HF), 14–20% of hospital admissions and 8–10% of inpatient deaths [4–6]. Although the epidemiology of malaria is heterogenous and varies in space and time, it is endemic throughout the whole country, and transmission occurs year-round. Plasmodium falciparum accounts for > 95% of malaria infections in Uganda [6–11]. In the past 10 years, the Uganda national malaria programme registered great milestones in reducing malaria transmission as shown by the last three national population-based malaria indictor surveys [5, 11, 12]. There is evidence of declining parasite prevalence from 44% in 2009 to 19% in 2014 and 9.1% in 2019 [5, 11, 12]. Based on this evidence, the country developed an ambitious malaria control and elimination strategic plan 2021–2025 that aims to further reduce parasite prevalence to less than 2% by 2025 including transformation of targeted districts from control to elimination phase [13, 14]. This requires enhanced monitoring the impact of the malaria control interventions.
Individuals living in high malaria transmission settings often carry several genetically distinct clones of the same parasite species (multiclonal infection) that can be the result of independent bites of infected mosquitoes (also called superinfection), or a single mosquito bite transmitting a genetically diverse sporozoite inoculum. As seen elsewhere, the number of co-infections (multiclonal) within a host might be an important indicator of transmission intensity and annual incidence rates (API) and, therefore, a measure of impact of interventions [15] and may have negative effect on treatment and drug efficacy trials, diagnosis and surveillance.
The high degree of genetic diversity within the P. falciparum HRP2 protein antigen epitopes can potentially affect the functionality of HRP-based rapid diagnostic tests (RDTs) that currently account for > 85% total malaria testing in Uganda [16–20]. A number of studies to date have documented extensive length, size and amino acid structural variations in the exon 2 of pfhrp2 gene between parasite strains [16, 20]. The pfhrp2 exon2 protein is composed of varying numbers of different types of amino acid repeat motifs (sequences) with each motif having variable copies, which give rise to large variation in the size of PCR fragments that may pose parasite detection challenges with the current HRP2 RDTs [16–18, 20]. Therefore, understanding genetic diversity, genetic relatedness between parasite population structures and transmission dynamics in P. falciparum populations could provide important information to inform current and future malaria control intervention efforts in Uganda.
Accurate measurement and estimation of genetic diversity and parasite relatedness require identification of all clones in multiclonal infections. However identification of the within-host parasite component in multiclonal infection is often difficult due to lack of appropriate molecular diagnostic tools [15]. Previous investigations of genetic diversity and parasite relatedness in Uganda have mostly relied on size-polymorphic antigenic markers such as MSP1, MSP2 and GLURP that can be amplified by PCR and the size of the amplicon determined by either gel or capillary electrophoresis. However, these molecular markers are under strong immune selection which affects the distribution of alleles in the population. Selection pressure could bias diversity measurements with these markers, especially in endemic areas where individuals could harbour several parasite clones [15]. Also the limited discrimination of alleles of similar sizes on agarose gel electrophoresis makes it difficult to discriminate allele size difference of less than 20 bp resulting in underestimation of diversity and multiplicity based on polymorphic genes [15, 16, 20]. Amplicon ultra-deep sequencing and whole genome sequencing techniques have high sensitivity and specificity particularly when detecting minority clones in cases of multiclonal infections. However, the costs and time factors attached to the use of these techniques are relatively high thereby limiting their application especially in resource limited settings.
Microsatellite markers are highly polymorphic with tandem repeats of 2–6 bp, abundant and widely distributed throughout the Plasmodium parasite genome [15, 16, 21]. Microsatellites are expected to be selection neutral with no phenotypic consequences, tend to be more reliably variable in multiple populations than other markers and are readily amplifiable using relatively cheaper methods, such as PCR [15–17, 20–22]. In this study, highly polymorphic microsatellite markers were deployed to investigate the genetic diversity, multiplicity of infections and parasite relatedness in a set of P. falciparum isolates collected from the different geographical settings in Uganda that have been confirmed with presence of mutant parasites carrying pfhrp2/3 gene deletions.
Methods
Study design and setting
Plasmodium falciparum genomic DNA samples that were available from a previous study that characterized the status of pfhrp2 and pfhrp3 parasite genes were analysed [23]. Details of the study population, data collection and DBS sample collection have been published elsewhere [24–26]. Details of genomic DNA extraction method was also published elsewhere [23].
Origin and selection of genomic DNA samples
The available DBS samples consisted of 160 (microscopy + /RDT−) and 140 (microscopy + /RDT +) samples whose genomic DNA had been extracted for pfhrp2/3 gene deletion study [23]. From these DNA samples, 85 were randomly selected from different pfhrp2/3 status groups and used for microsatellite genotyping. An additional 62 samples were randomly selected from the pfhrp2 positive sample group to undergo Sanger sequencing. The geographical distribution of the 85 microsatellite and 62 sequencing samples is shown in Fig. 1.
Fig. 1.

Geographical distribution of DNA samples used for genotyping. The red and green dots indicate the location where the sequenced and microsatellite typed samples were collected respectively. Black lines indicate district boundaries
Laboratory analysis
Microsatellites genotyping of P. falciparum
Genotyping of the same 7 neutral microsatellite loci/markers previously analysed to reveal genetic relatedness of parasites with and without gene deletions in South America [27] and Africa [28] was conducted for each of the 85 samples as previously described [27]. Locations of these markers have been described previously [27, 29, 30]. The microsatellite markers TA1, PolyA, PfPK2, TA109 and 2490 were semi-nested PCRs while 313 and 383 were single round PCR reactions. Briefly, the primary reaction for each marker was carried out in a solution of total volume 15 µL containing 4.7 µL Nuclease Free Water, 1.8 µL MgCl2 (25 mM), 1.5 µL 10 × Reaction Buffer II, 0.6 µL dNTPs (1.25 mM), 0.6 µL Forward Primer(10 μM), 0.6 µL Reverse Primer(10 μM), 0.25 µL AmpliTaq Gold (5U/μL), and 5 µL DNA Template. Round 1 PCR conditions were 94 °C for 5 min, (94 °C for 30 s, 42 °C for 30 s, 40 °C for 65 s) 25 cycles, 65 °C for 5 min. The secondary reaction contained the same reagents as the primary reaction for each marker with the exception of the 0.6 µL each of the labelled primer pair. TA1-F, TA109-F, 2490-R and 313-F primers were labelled with -6-FAM. PolyA-R primer was labelled with –PET, PK2-R primer with -NED and 383-F primer with –VIC. One µL of the primary reaction product was used in a final volume of 15 µL in the semi-nested PCR reactions. Round 2 PCR conditions were 94 °C for 5 min, (94 °C for 30 s, 45 °C for 30 s, 40 °C for 65 s) 25 cycles, 65 °C for 5 min.
PCR conditions for the 313 and 383 PCRs were as follows: 94 °C for 5 min, (94 °C for 30 s, 50 °C for 30 s, 60 °C for 30 s) for 5 cycles, (94 °C for 30 s, 45 °C for 30 s, 60 °C for 30 s) for 40 cycles, then 60 °C for 2 min. A 5µL sample of the PCR product was then run on a 2% agarose gel to determine the factor prior to assay on the sequencer. Fluorescently labelled PCR products were assayed for size on an ABI 3100 Genetic Analyzer sequencer (Applied Biosystems, Foster City, CA, USA). The fragments/alleles were scored manually using Peak Scanner Software version 1.0 (Applied Biosystems), using a height of 300 relative fluorescence units as the minimal peak threshold (bands smaller than 300 relative fluorescence units (rfu) were defined as background). For samples producing more than one peak, peaks with height of > 300 rfu and also > 30% of the highest peak are classified as positive peaks [28]. Samples for which no amplification was obtained in some markers were repeated and re-analysed or excluded if they failed to amplify more than 3 of the 7 markers the second time [27, 28]. A laboratory P. falciparum line of 3D7 was included in each run as a control and sizes of seven microsatellite markers were calibrated against those of 3D7 before conducting genetic relatedness analysis.
Polyclonality
Samples were classified as either single clone infections (containing only one allele per marker at all 7 markers) or multiclonal/polyclonal infections (containing two or more alleles at any one or more of the 7 markers. The proportion of polyclonal infections were calculated as: number of samples with polyclonal infections/number of samples typed × 100%.
Multiplicity of infection (MOI)
MOI for each of the 7 microsatellite markers was calculated as: the total number of alleles revealed /total number of samples typed for that marker. The overall MOI was calculated as: the sum of the highest number of alleles at any of the 7 microsatellite markers typed for all samples/the total number of samples typed.
Haplotype construction
Seven-microsatellite marker haplotypes were constructed for samples with single clone infections or multiple clone infections with multiple alleles only detected on one of the 7 markers. For a proportion of samples where microsatellite data were obtained only for five or six of the seven markers typed, five- or six-microsatellite markers haplotypes were constructed.
Genetic relatedness of parasites
Five to seven microsatellite haplotypes were used to determine the relatedness among Ugandan parasite isolates and between parasites from Uganda and Eritrea by using the PHYLOViZ software (www.phyloviz.net, version 1.1) [28]. Sample data including individual microsatellite size, pfhrp2/3 status and geographical origin of the samples were imported into the online software for analysis. Plots were produced using Phyloviz software at cutoff value of 3 minimum differences for 4 loci).
Pfhrp2 DNA and putative amino acid sequence analyses
Exon 2 of the pfhrp2 gene was amplified and sequenced as previously described Baker et al. [20]. DNA sequences from samples with single clone infections or from multiple clone infections but producing clear sequences for a dominant clone (one that masks or overrides other variants) were used for sequence diversity analysis. Putative amino acid repeat types and HRP2 sequence types were determined following Baker et al. [16]. DNA sequences were imported into MEGA4.0.2 software [28] and were used to generate phylogeny bootstrap consensus neighbor-join tree using the software. Exon1 of the pfhrp2 gene was not sequenced as exon1 encodes a conserved signal peptide.
Data management and statistical analysis
Demographics and variables linked to the genomic DNA samples were extracted from the previous survey database. All data were entered and managed in Excel before it was exported to STATA for analysis. ArcGIS software version 10.8, Environmental Systems Research Institute (Esri), California U.S) was used to map the geographical distribution, locations and sites where the genomic DNA samples were collected and where the different pfhrp2 amino acid sequences/types actually occurred. Data analysis was done with STATA Ver 14, college Station, Tx: StataCorp LP), GraphPad Prism 7.00 for Windows (GraphPad Software, La Jolla, CA, USA). Descriptive analysis was done for baseline characteristics and presented as proportions. Proportions of the different sequence types were assessed for statistical significance using the Student's t-test or Wilcoxon's rank-sum test as appropriate while MOIs and multi-clones were analysed with Fisher’s exact test at level of significance p ≤ 0.05.
Ethical approval
The study was approved by the Makerere University School of Medicine Research and Ethics committee (#REC REF 2017-111), the Uganda National Council of science and technology (Ref No: HS271ES), and the Australian Department of Defence and Veterans’ Affairs Human Research Ethics Committee (DDVA HREC 096–18). In the primary surveys from where samples were collected, participants were enrolled after providing written informed consent for future use of biological samples for molecular analysis.
Results
Characterization and profile of samples
A total of 147 PCR confirmed parasite genomic DNA samples were available for molecular characterization in this study. Seven-microsatellite-marker typing was performed for 85 samples, however only 49 samples produced positive allele peaks at > 5 microsatellite markers and were considered for analysis. Thirty-six samples (n = 36) that produced allele peaks at 1–4 microsatellite markers only were excluded from further analyses on MOIs and parasite genetic relatedness. Clean DNA sequences of the pfhrp2 exon 2 was obtained from 62 samples. The entire sample of 147 genomic DNA and their demographics came from a study that previously characterized the pfhpr2 and pfhrp3 genes [23].
Baseline characteristics samples
The majority of these samples was from males (54.4%), aged ≥ 5 years (65.9%) and from the eastern region of Uganda (53.7%). Most had a parasite density ≥ 1000/μl (65.3%). All the genomic DNA were from PCR confirmed P. falciparum infected samples (Table 1).
Table 1.
Baseline characteristics of samples
| Variable | Frequency | Proportion (%) | Sequenced (%) | Microsatellite typing (%) |
|---|---|---|---|---|
| Total (n = 147) | (n = 62) | (n = 85) | ||
| Age | ||||
| < 5 | 50 | 34.01 | 40.3 | 29.4 |
| ≥ 5 | 97 | 65.99 | 59.6 | 70.6 |
| Gender | ||||
| Male | 80 | 54.42 | 54.8 | 54.1 |
| Female | 67 | 45.58 | 45.2 | 35.9 |
| Region | ||||
| Western | 68 | 46.3 | 37.1 | 51.7 |
| Eastern | 79 | 53.7 | 62.9 | 48.2 |
| Density | ||||
| < 1000 | 51 | 34.69 | 38.7 | 45.8 |
| ≥ 1000 | 96 | 65.31 | 61.3 | 54.1 |
| Endemicity | ||||
| Low transmission | 87 | 59.18 | 45.2 | 69.4 |
| Moderate | 60 | 40.82 | 54.8 | 30.9 |
< 5 and ≥ 5 means less than 5 and equal or above years of age respectively. < 1,000 and ≥ 1,000 means parasite densities of less than 1,000 and equal or greater than 1,000 parasites/µL, respectively
Polyclonality of infections
Microsatellite typing revealed a high proportion samples examined had more than one allele on at least one of the seven markers indicative of multi-clone infections. Overall, 75.51 (95%, CI61.1–85.8) of samples examined were classified as multiple-clone (mixed genotype) infections, significantly higher compared to single clone 24.5% (95% CI 14.2–38.9), P < 0.05. This high level of polyclonality in samples indicates frequent infections consistent with high malaria transmission intensity in the study areas. Although not statistically significant, proportions of multiclonal infections were 71.8% (91% CI 53.4–85.1) and 82.4% (95% CI 55.5–94.6), P > 0.05 in > 5 and ≥ 5 years respectively. Multiclonal infections significantly occurred in isolates collected from Eastern 73.7% (95% CI 48.8–89.1) compared to Western region 26.3% (95% CI 10.9–51.2), P < 0.05.
Multiplicity of infection
The number of alleles at any of the 7 markers varied between markers. The highest number of alleles at any marker was 4 (Table 2). MOI at individual marker varied between 1 and 1.51. The overall MOI in this set of samples was 1.92 (95% CI1.72–2.12), consistent with high transmission intensity. Though not statistically significant the MOI was higher in children < 5 than ≥ 5 years old (1.9 and 1.8), respectively. No geographical differences in MOIs were seen in parasites collected from Eastern and Western Uganda (1.9 vs 1.8), P > 0.05, respectively (Table 2). Importantly, 5/7 samples with gene deletions were multi-clone infections (pfhrp2-/pfhrp3 + = 2, pfhrp2-/pfhrp3- n = 3, pfhrp2 + /pfhrp3- n = 1).
Table 2.
Microsatellite markers, Typing and overall MOI
| Microsatellite marker | No. of samples typed | No. of alleles revealed | Range in no. of alleles | MOI | 95%CI |
|---|---|---|---|---|---|
| 2409 | 49 | 65 | 1–3 | 1.33 | NA |
| TA1 | 48 | 56 | 1–2 | 1.17 | NA |
| 383 | 30 | 38 | 1–2 | 1.27 | NA |
| TA109 | 47 | 69 | 1–4 | 1.47 | NA |
| 313 | 19 | 19 | 1–1 | 1 | NA |
| PolyA | 49 | 74 | 1–3 | 1.51 | NA |
| PK2 | 42 | 50 | 1–4 | 1.19 | NA |
| Overall MOI* | 49 | 94 | 1–4 | 1.92 | 1.72–2.12 |
| Overall MOI* (RDT+/PCR +) | 33 | 65 | 1–3 | 1.97 | NA |
| Overall MOI* (RDT−/PCR +) | 16 | 29 | 1–4 | 1.81 | NA |
| Overall MOI* Age | |||||
| < 5 years | 26 | 48 | 1–4 | 1.88 | 1.52–2.24 |
| ≥ 5 years | 23 | 27 | 1–2 | 1.94 | 1.68–2.20 |
| Overall MOI* Region | |||||
| Western | 19 | 36 | 1–2 | 1.89 | 1.54–2.25 |
| Eastern | 31 | 51 | 1–4 | 1.93 | 1.68–2.19 |
*Calculated by selecting the highest number of alleles at any of the markers typed. NA = not applicable
Number of 5 to 7-microsatellite marker haplotypes
Five to 7 marker haplotypes were obtained from 29 samples: 12 single clone infected samples and 17 multi-clone infected samples where multiple alleles were observed in only one marker. This resulted in a total of 42 unique haplotypes from 29 samples. The remaining samples had multiple alleles at more than one marker resulting in haplotypes unassigned due to the complexity in number of possible haplotype combinations. The high level of diversity and multiplicity in haplotypes detected in this set of samples indicates large parasite population size consistent with high transmission intensity in the areas.
Only two of the 42 haplotypes (H29 and H31) were shared between 2 (D196 and D204) and between 3 (D130, D176 and D204) different samples. Interestingly, both these haplotypes H29 and H31 were identified from parasite samples that had both pfhrp2 and pfhrp3 deletions. Haplotypes H25, H37 and H38 were identified in samples determined to have deleted pfhrp2 only, while H26 and H27 were identified in a sample determined to have deleted pfhrp3 only (Table 3). This observation suggests that parasites having pfhrp2 or pfhrp3 gene deletions had a lower genetic diversity compared to parasites without gene deletions indicating clonal expansion of gene deleted parasites. Furthermore, parasites with 5 different haplotypes (H29, H31, H25, H37 and H38) and two different haplotypes (H26 and H27) were observed to have deleted the pfhrp2 gene and pfhrp3 gene respectively, suggesting that pfhrp2 and pfhrp3 gene deletions have occurred in parasites with different genetic backgrounds.
Table 3.
Haplotype determination in pfhrp2/3 deleted samples
| Haplotype | Identified in sample | pfhrp2/3 status |
|---|---|---|
| H29 | D196, D204 | pfhrp2−/pfhrp3− |
| H31 | D130, D176, D196 | pfhrp2−/pfhrp3− |
| H25 | D154 | pfhrp2−/pfhrp3 + |
| H37 | D169 | pfhrp2−/pfhrp3 + |
| H38 | D169 | pfhrp2−/pfhrp3 + |
| H26 | D263 | pfhrp2 + /pfhrp3− |
| H27 | D263 | pfhrp2 + /pfhrp3− |
pfhrp2/3 detected: + ; undetected:−
Genetic relatedness of parasites
In order to investigate the possible origin of pfhrp2/3 gene deleted parasites, genetic relatedness analysis was conducted among 42 Ugandan parasite haplotypes as well as between Ugandan and Eritrean parasites. Among parasites from Uganda, there was a major cluster of parasites having deleted both pfhrp2 and pfhrp3 (H29, H30 and H31), and small clusters of pfhrp2-/pfhrp3 + (H37 and H38), pfhrp2 + /pfhrp3- (H26 and H27) suggesting that these parasites are genetically closely related (Fig. 2a). There were several small clusters of closely related pfhrp2 + /hrp3 + parasites. The biggest of clustering consisted of parasites with dual pfhrp2/3 deletions indicating possible clonal expansion of these RDT undetectable parasites possibly selected for by RDT usage.
Fig. 2.

Genetic relatedness of P. falciparum populations. a and b Legend: Genetic relatedness of P. falciparum populations from Uganda differing in pfhrp2/3 genetic gene status (a) and comparison of parasite populations from Uganda and Eritrea (b). The relatedness among parasites was constructed using seven neutral microsatellite markers/loci indicated in Additional file 1: Table S1. Plots were produced using Phyloviz software at cutoff value of 3 minimum differences for 4 loci). Numbered circles indicate specific haplotypes. Circle sizes are proportional to the number of samples with a particular haplotype. The number of samples are indicated inside each circle. Pfhrp2/pfhrp3 status is indicated as: Positive/Positive (wild type), Negative/Negative (dual pfhrp2/3 deletion), Positive/Negative (pfhrp3 deletion) and Negative/Positive (Pfhrp2 deletion)
Haplotypes of Ugandan parasites (gene deleted and not deleted) did not cluster with parasite haplotypes from Eritrea (E1–E14), suggesting parasites with gene deletions in Uganda did not share common genetic origins with Eritrean parasites (different genetic lineages) and hence, were not likely to have resulted from spread from Eritrea. There is a trend indicating that parasites have formed small clusters within the Western and Eastern regions. However, there were two clusters that included parasites from both regions (Fig. 2b).
Genetic diversity in the pfhrp2 gene
To investigate genetic diversity of the pfhrp2 positive parasite population, Exon 2 of the pfhrp2 gene was sequenced from 62 pfhrp2 positive samples to reveal genetic diversity in the gene. Sequencing analysis revealed 60 unique sequence types in 62 samples sequenced, i.e. almost every sample had a different pfhrp2 sequence, demonstrating a high level of genetic diversity in pfhrp2 of the sequenced parasite population. Only two sequence types were shared between two different samples: the unique sequence types 39 (shared by D102 and D103) and 49 (shared by C041 and D165). Details are indicated in Additional file 2: Table S2.
Sequence length of pfhrp2 exon2 which encodes the HRP2 protein
Exon2 length of the 62 sequences ranged between 537 and 801 bp. Generally, sequence length in the two sample sub-sets was comparable between the RDT−/LM + and the RDT+/LM + samples (p = 0.28) (Fig. 3). Detailed data on pfhrp2 exon length is included in Additional file 2: Table S2.
Fig. 3.
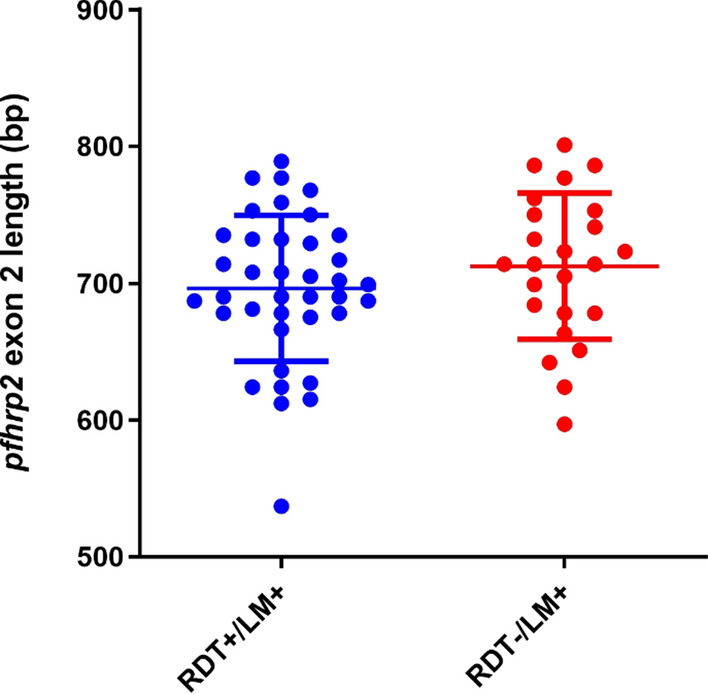
Sequence length of pfhrp2 exon2. LM means light microscopy
Phylogeny and sequence clustering
A phylogeny bootstrap consensus neighbor-join tree based on 62 pfhrp2 exon2 sequences was constructed. No major clustering of parasites was observed in relation to RDT positivity, sequence length or sequence types. Interestingly, the three pfhrp2 + /hrp3- parasites (D126, D146 and C139) did not appear to be clustered as they are on 3 separate branches (Fig. 4). This suggests that parasites with pfhrp3 deletions have emerged independently from different genetic backgrounds.
Fig. 4.

Phylogeny bootstrap consensus neighbour-join tree. D = discordant samples (RDT−/LM +), C = concordant samples (RDT+/LM +)
Putative amino acid repeat types in HRP2
Among 62 sequences, 15 types of amino acid repeats were identified. Their numbers and ranges are shown in Table 4. There was no significant correlation between the number of any repeats and geographical origin or RDT positivity Table 4. The geographical distribution of the different HRP2 sequences is shown in Fig. 5.
Table 4.
pfhrp2 Amino Acid Repeat types, translated sequences and frequencies (n = 62)
| PfHRP2 | Total (N = 62) | Region | RDT and LM | p-value | |||
|---|---|---|---|---|---|---|---|
| East | West | RDT−/LM + | RDT+/LM + | ||||
| Code | Repeat sequences | (N = 40) | (N = 22) | (N = 24) | (N = 38) | ||
| 1 | AHHAHHVAD | 2.7 ± 1.4 | 1–7 | 0–6 | 2.9 ± 1.5 | 2.6 ± 1.3 | 0.504 |
| 2 | AHHAHHAAD | 11.8 ± 2.2 | 10–17 | 8–13 | 12.1 ± 1.9 | 11.7 ± 2.4 | 0.427 |
| 3 | AHHAHHAAY | 1.2 ± 0.7 | 0–2 | 0–3 | 1.1 ± 0.7 | 1.3 ± 0.7 | 0.457 |
| 4 | AHH | 0.5 ± 0.8 | 0–3 | 0–1 | 0.3 ± 0.6 | 0.6 ± 0.9 | 0.146 |
| 5 | AHHAHHASD | 0.7 ± 0.5 | 0–2 | 0–1 | 0.8 ± 0.5 | 0.6 ± 0.5 | 0.221 |
| 6 | AHHATD | 3.0 ± 1.4 | 1–6 | 2–5 | 3.0 ± 1.3 | 3.0 ± 1.4 | 1 |
| 7 | AHHAAD | 6.5 ± 2.4 | 4–11 | 4–8 | 6.2 ± 2.1 | 6.8 ± 2.5 | 0.317 |
| 8 | AHHAAY | 1.1 ± 0.6 | 0–2 | 1–2 | 1.2 ± 0.8 | 1.1 ± 0.5 | 0.85 |
| 9 | AAY | 0.0 ± 0.0 | 0.0 | 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NA |
| 10 | AHHAAAHHATD | 1.6 ± 0.7 | 0–3 | 0–2 | 1.9 ± 0.7 | 1.5 ± 0.6 | 0.018 |
| 11 | AHN | 0.0 ± 0.0 | 0.0 | 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NA |
| 12 | AHHAAAHHEAATH | 0.0 ± 0.0 | 0 | 0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NA |
| 13 | AHHASD | 0.0 ± 0.0 | 0 | 0 | 0.0 ± 0.0 | 0.0 ± 0.0 | NA |
| 14 | AHHAHHATD | 0.3 ± 0.4 | 0–1 | 0–1 | 0.4 ± 0.5 | 0.2 ± 0.4 | 0.097 |
| *New | 0.0 ± 0.3 | 0–2 | 0 | 0.0 ± 0.0 | 0.1 ± 0.3 | 0.324 | |
*New = are sequences observed in Ugandan isolates that had not been reported before. East and West are the Eastern and Western regions of Uganda. Data was given by mean + -SD. P-value was calculated by Student’s t-test or Wilcoxon’s Rank Sum test as appropriate. LM = microscopy
HRP2 Amino acid sequence grouping
The amino acid sequences were grouped following the previously published formula (Type 2 × type 7) [16]. Proportions of samples with Type A sequence (≥ 100), Type B (44–99) and Type C (≤ 43) were: 12.9% (8/62), 77.4% (48/62) and 9.7% (6/62), respectively. Similar to sequences reported from other countries [20] the majority of parasites in the Ugandan samples had a Type B pfhrp2 sequence. Interesting to note is that 10% of HRP2 sequences are of type C that was initially reported to be associated with reduced performance of some HRP2-based RDTs [16] but later found to have no association when a larger sample size was analysed [20]. The proportion of Type C sequence accounted for 13% in the RDT−/LM + compared to 8% in RDT+/LM + samples (Table 5).
Table 5.
Unique pfhrp2 sequences, sequence types and their proportions (n = 62)
| Genomic DNA sub-sets | No. sequenced | Unique Sequence types identified | Sequence type A | Sequence type B | Sequence type C | ||||
|---|---|---|---|---|---|---|---|---|---|
| Total | N | % | N | % | N | % | N | % | |
| RDT+/LM + | 38 | 38 | 100% | 5 | 13% | 30 | 79% | 3 | 8% |
| RDT−/LM + | 24 | 23 | 96% | 3 | 13% | 18 | 75% | 3 | 13% |
| Overall | 62 | 61 | 98% | 8 | 13% | 48 | 77% | 6 | 10% |
RDT+/LM + and RDT−/LM + are genomic DNA samples that were initially (HRP2 RDT negative and microscopy positive) and those that were both HRP2 RDT and microscopy positive respectively
Discussion
Under the national malaria control and elimination strategic plan 2021- 2025, the Uganda National Malaria Programme aims to achieve malaria elimination in targeted districts by 2025. However incomplete surveillance data hampers efforts to assess parasite transmission dynamics and the impact of ongoing interventions. As P. falciparum population studies have revealed increasing genetic diversity and proportion of polyclonal infections correlating with increasing transmission intensity [29], molecular epidemiological surveillance of parasite diversity and relatedness has the potential to provide useful information for malaria transmission intensity, particularly in the elimination context where detecting ongoing local transmission and identifying the origin of infections may be relevant. The current study presents molecular characterization of the parasite population using microsatellite markers to determine genetic diversity and parasite relatedness with a special interest in the emergence of parasites with pfhrp2/3 deletions, and use of genetic sequencing to investigate variability of the pfhrp2 gene in P. falciparum populations from a wide geographical coverage and epidemiological settings in Uganda. The study is a follow up investigation that used genomic P. falciparum DNA from a previous survey that investigated and reported the presence of pfhrp2/3 gene deletions in parasite isolates [23].
Microsatellites are abundant in P. falciparum; an average of one microsatellite locus is found every 2–3 kb of genome sequence [31, 32]. A set of 7 microsatellite markers that had been used to compare genetic relatedness between parasites with and without pfhrp2/3 deletions in different areas of the world including South America and Eritrea were used, providing important information on whether parasites with gene deletions were of de novo emergence. For comparison purposes, exactly the same set of microsatellite markers were used in this study. The 3D7 parasite DNA was included in every microsatellite run as a common control for calibrating data. Using 7 Microsatellite loci scattered throughout the genome of P. falciparum, markedly high population-level parasite genetic diversity was found in the study samples that is consistent with malaria transmission intensities in the survey areas (Fig. 5).
Fig. 5.

GIS mapping for distribution of sequences across sites. The different colored dots represent the different HRP2 sequence types
High proportion of polyclonal infections
In this study, most of the samples, 75.5% (95% CI 61.1–85.8), were of multiclonal (mixed genotype) P. falciparum infections. The proportion of samples with polyclonal infections observed in this study is higher than 60.2% (50/83) reported for Entebbe area in Uganda in the late 1990s also using microsatellite makers [29]. Using 12 microsatellite markers, Anderson et al. reported a strong positive correlation between proportion of polyclonal infections and transmission intensity: < 20% polyclonal infections were detected in low transmission settings of South America, intermediate proportions in Thailand, while > 45% polyclonal infections were detected in Africa and Papua New Guinea where malaria prevalence is high. Hence, the proportion of polyclonal infection observed in the current study indicates malaria transmission intensity remains high in Uganda. This conclusion is consistent with high malaria transmission levels reported in the study areas [5].
Previous studies in similar high transmission settings in Africa have reported similar high levels of multiclonal and genetically diverse P. falciparum infections [15, 33–37]. In contrast, other studies in Africa [38, 39], found a lower frequency of patients with multiclonal infections. This variation likely reflects heterogeneity in malaria transmission intensity resulted from geographical variability, seasonality and population variations. The frequency of multiclonal infections has also been reported to increase with age until late childhood before declining [35]. In this study, the isolates were collected from children between 2 and 10 years of age however no significant differences in multiclonal infections were observed between the < 5 and ≥ 5 years. This can be explained partly by the fewer samples tested in the < 5 age group (29%) vs 71% for > 5 age. Interestingly, multiclonal infections occurred more frequently in the Eastern compared to the Western region 73.7% (95% CI 48.8–89.1), P < 0.05). This observation is consistent with the relatively high level of malaria transmission in the Eastern region [5, 11, 14].
High multiplicity of infection
MOI is defined as the number of genetically distinct parasite strains co-infecting a single host, which is an important indicator of malaria epidemiology [15, 40]. The overall MOI in the samples was 1.92 (95% CI 1.7–2.1). MOI did not differ between age groups < 5 and ≥ 5 groups (P = 0.60). This observation is in line with previous findings in Uganda that showed that parasitaemia is concentrated in children between 2 and 15 years of age which falls within the age groups (2–10 years) where the study samples were collected [41]. Although differences in MOIs were not significant between regions, the frequency of occurrence was higher in the Eastern compared to Western region (1.9 vs 1.8), consistent with the high transmission setting in the Eastern region [5, 11, 14]. Previous studies elsewhere in Africa have reported comparably high MOIs in parasite populations in similar transmission settings [21, 33, 40]. In Uganda, similarly high MOIs were reported in two localized studies at two hospitals in Kampala (MOI = 3.0) and Arua (MOI = 2.2) conducted 13 and 8 years ago, respectively [42, 43], when malaria prevalence in the country was much higher than recent years [5, 11, 12]. It is important to note however both studies used the MSP1 and MSP2 genes that are under immune selection pressure [15].
The number of co-infections within a host might be an important indicator of transmission intensity and annual incidence rates (API) and, therefore, a measure of impact of interventions [15]. In addition to determination of malaria transmission intensity, multiplicity of infections in P. falciparum are essential parasite indices that can impact on the effectiveness of malaria intervention due to their effect on treatment, diagnostics, surveillance and parasite transmission dynamics [15, 21]. There is indication that MOI can also help to monitor vaccine efficacy since vaccine-induced immune responses show strain-transcending specificity depending on the polymorphic alleles of the vaccine candidate antigens as reported previously by Zhong et al. [15]. The study reported greater RTS, S vaccine activity against malaria parasites with matched circumsporozoite protein allele than against mismatched parasite strains. A similar significance for studying MOIs is seen in therapeutic efficacy studies where detection of all clones is important, as only one of them might be drug resistant and will result in recrudescence. Non- detection of all clones at baseline might lead to false identification of these clones as new infections even when they existed at baseline, and thus an underestimate of treatment failure [15]. Several studies in Uganda have reported correlations between clinical symptoms and higher MOIs [42, 43]. Understanding MOIs in parasite populations in wider surveys could provide important information on transmission dynamics and inform current and future malaria control intervention efforts in Uganda.
High HRP2 genetic diversity
Previous studies have reported the presence of pfhrp2/3 gene deletions in Ugandan parasites, however genetic variation in the pfhrp2 gene that may affect the functionality of HRP2-based RDTs has not been described on a wider scale [23, 44, 45]. Certain amino acid repeats within the pfhrp2 gene have been shown to contain epitopes targeted by the monoclonal antibodies used to detect the HRP2 protein [16, 20, 46]. Variation in the pattern and sequence of histidine repeat tandems, number and frequency of repeat tandems and composition of amino acids within the HRP2 may affect the accuracy and functionality of HRP2-based RDTs [16, 20]. In the current study, the pfhrp2 exon 2 was sequenced and examined to determine genetic variability of the pfhrp2 gene in the samples. A high level of pfhrp2 genetic diversity was found as revealed in 60 unique sequences from 62 samples sequenced 96.8% (60/62). This provides further evidence indicating high transmission intensities in the study areas.
While sequence length of exon2 of the pfhrp2 that encodes the HRP2 protein antigen ranged between 537 and 801 bp in these samples, parasites with Type B sequences were the most common (48/62, 77.4%). Parasites with Type C sequences that usually contain fewer AHHAHHAAD and AHHAAD motifs which are possibly associated with reduced sensitivity of HRP2 RDTs [16, 18, 20, 47], were detected in low frequency (6/62, 10%) in the study samples. A slightly higher frequency of type C sequences was observed in RDT−/Microscopy + group compared to RDT+/microscopy + group (13% vs 8%) (Table 5). This may provide an additional explanation for the false negative RDT results not due to the quality of the RDTs or user related challenges reported by three previous studies in Uganda [23, 44, 45]. Further studies with larger sample size are required to validate this finding.
Parasite genetic relatedness analysis
Despite recent advances in genome sequencing of malaria parasites, microsatellite markers remain an important source of data to understand the genetic diversity and relatedness of parasite populations and understanding of transmission dynamics [15, 21, 22, 32, 33, 48–51]. In this study, the genetic relatedness among monoclonal Ugandan parasites and between monoclonal Ugandan and Eritrean parasites was investigated using 7-microsatellite-marker haplotypes. The interest was to assess if there was any clustering among Ugandan parasites and in particular any clustering of Ugandan parasites with Eritrean isolates because of the high levels of pfhrp2/3 gene deletions reported in Eritrea that led to the change of policy and replacement of HRP2 RDTs with non-HRP2-based RDTs [28] and because both parasite populations have been analysed by the same laboratory using the same set of microsatellite markers. It was assessed whether parasites with or without gene deletions in Uganda are genetically closely related to those reported in Eritrea. The analysis informs whether mutant parasites emerged locally or somehow spread from Eritrea. Analysis was performed using 42 unique haplotypes from 29 samples. A major cluster of parasites having deleted both pfhrp2 and pfhrp3 (H29, 30 and 31), and small clusters of pfhrp2-/pfhrp3 + (H37 and H38), pfhrp2 + /pfhrp3- (H26 and H27) were observed suggesting that these parasites are genetically closely related. Clear clustering observed for pfhrp2/pfhrp3 deleted parasites suggests clonal expansion of double deleted parasites due to selection. Pfhrp2/3 gene deleted parasites in Uganda did not cluster with those from Eritrea (accessed using the same microsatellite markers), suggesting local emergence of gene deleted parasites. This observation suggests that the gene deleted parasites observed in Ugandan samples emerged independently by spontaneous genetic event rather than introduced through Eritrea. Future genetic studies on parasite populations in neighbouring countries may shed further light on the origin of gene deleted parasites in Uganda.
Implications for detection of pfhrp2 deleted parasites
Plasmodium falciparum parasite with pfhrp2 and pfhrp3 gene deletions have been reported in Uganda [23, 44, 45]. It is critical to continuously assess the prevalence of gene deleted parasites to inform malaria control policies in the country. The high proportion of polyclonal infections and MOI > 1 detected in the parasite populations in study areas presents a major challenge to the gene deletion surveillance. Co-infection that involves a non-deleted strain can mask a gene deleted strain giving incorrect results when conventional PCR methods are used for detecting parasites with gene deletions [52, 53]. In the context of RDTs functionality, multiclonal infections involving wild type parasites and gene deleted parasites will give positive RDT results ensuring diagnosis and treatment of infected people, although this masks the presence of gene deleted parasites. This effect could occur even if an infection with a deleted parasite occurs subsequent to an infection with a wild type parasite since circulating HRP2 can persist for up to 3–4 months [53]. In addition, parasites having deleted pfhrp2 only can be detected by some RDTs due to cross-reactivity of antibodies to HRP3 although at higher parasitaemias [16]. This can similarly mask the detection of pfhrp2 deleted parasites but on the other hand help diagnosis of patients.
General implication of findings
The higher proportion of MOIs and multiclonal infections observed in the study implies that these samples were mainly collected from high transmission settings suggesting persistent high transmission in these areas. This may call for strengthening control and intervention efforts. Importantly data obtained from this study can serve as baseline for assessing the impact of current and future control intervention measures. The presence of high proportion of multiclonal infections in these samples calls for improvement in the detection capacity of malaria diagnostic tools to overcome challenges associated with the detection all parasite clones as genotyping might falsely identify them as new infections if missed at baseline in therapeutic efficacy studies.
Previous studies in Uganda reported higher MOIs in Kampala and Arua in West Nile respectively [42, 43]. In both studies children infected with multiple strains were more likely to have treatment failure and severe disease suggesting that these multiclonal infections have a role in disease progression and severity. Similarly higher MOIs of 3.39 were seen in P. falciparum samples in Western Kenya [33]. The relatively high MOI detected in the study areas have likely contributed to the high malaria disease burden reported in the areas. Currently there is no point of care test that detects multiclonal and multiplicity of infection to assist case management.
The study showed high genetic diversity in the pfhrp2 gene in Ugandan parasite population reflected in the presence of different types of repeats, as well as variations in sequence, repeat number, and arrangement of these repeats. The data provided further indication for high transmission intensity in the study areas. In addition, the observation of a slightly higher proportion of Type C HRP2 sequences that may be associated with reduced efficacy of HRP2 RDTS in the RDT−/microscopy + samples is of concern and may call for investigation of their occurrence in parasite isolates in other regions of Uganda.
It was observed in this study that parasites that harbored gene deletions formed clusters among Ugandan isolates suggesting close relatedness. However, these were genetically unrelated to the Eritrean parasites. This suggests that gene deleted parasites in Uganda have emerged independently by spontaneous genetic event and implies the potential for gene deletion to occur anywhere in Uganda under the selection of HRP2-based RDT use. This calls for periodic monitoring surveys.
Limitations of the study
The study was limited by the fact that the P. falciparum isolates analysed were obtained from only two regions of Uganda and from two selected groups: RDT−/microscopy + and RDT+/microscopy + , which leaves the status genetic diversity, MOIs and parasite relatedness in other regions unknown. It is recommended that future surveillance programmes should consider a more representative sample covering all regions of Uganda. Due to resource constraint, the pfhrp2 gene sequencing and microsatellite typing were performed on a limited number of genomic DNA samples. The study was unable to determine haplotypes for samples with polyclonal infections. Due to the high proportion of polyclonal infections and low parasite densities, haplotypes could only be determined for a relatively small number of samples.
Conclusion
High level of genetic diversity was observed in P. falciparum parasites reflected in the frequency of multiclonal infections, multiplicity of infection and variability of the pfhrp2 gene in these samples. These findings are consistent with the high malaria transmission intensity and endemicity in these settings despite the scaling up of malaria interventions. Genetic relatedness analysis suggested spontaneous de novo emergence and clonal expansion of pfhrp2 deleted parasites that requires continuous close monitoring to inform malaria diagnosis and case management policies. Future molecular epidemiological surveys on parasite genomics that are more representative with wider coverage are recommended.
Supplementary Information
Additional file 1: Table S1. Summary of samples successfully genotyped on 4–7 microsatellite markers (-s: single haplotype, -m2: 2 haplotypes, -m3: 3 haplotypes, -m4: 4 haplotypes)
Additional file 2: Table S2. Sequence length and unique sequence type of pfhrp2 exon 2.
Acknowledgements
We thank the primary study teams and the Makerere University malaria training grant staff for the administrative and technical support. We are grateful to the study participants who participated in the primary study and their families. We thank Mr. Peter Mutungi, Mr. Victor Asua and Mr. David Masanga for their support in processing the data and samples.
Abbreviations
- HRP2
Histidine Rich Protein 2
- pfhrp2
Plasmodium falciparum Histidine rich protein 2 gene
- RDTs
Rapid diagnostic tests
- MSP1
Merozoite surface antigen 1
- MSP2
Merozoite surface antigen 2
- PCR
Polymerase chain reaction
- WHO
World Health Organization
- LM
Light microscopy
- MOI
Multiplicity of infection
- DNA
Deoxyribonucleic acid
- rRNA
Ribosomal ribonucleic acid
- DBS
Dried blood spots
Authors' contributions
AB conceptualized the study, AB, MK, QC designed the study. AB, QC, KA, KG, CP, DS did the sample analysis, AB, JK, PM, JN, JNK supported the data analysis. AB drafted the manuscript, AB QC, KA, KG, CP, DS, AY, NS, JIN, JO, RN, SG, EA, PM, CSL, CK, JC, JKN all reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the U.S. Department of Defense Global Emerging Infections Surveillance (GEIS) under Award Number P0008-19-AM awarded to QC and the Fogarty International Center of the National Institutes of Health scholarship awarded to AB under grant number D43TW010526. The funders had no role in the study design, data collection or analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
Data for this study is available and the row dataset has been uploaded as additional information with the manuscript.
Declarations
Ethics approval and consent to participate
Ethical approval for the primary study was obtained from the Makerere University School of Medicine Research and Ethics Committee, the Uganda National Council of Science and Technology, the London School of Hygiene & Tropical Medicine Ethics Committee, and the University of California, San Francisco Committee on Human Research. Ethical approval to access and use participants’ samples from the primary study was obtained from the school of Medicine Research and Ethics Committee (SOMREC), the Uganda National Council of Science and Technology (UNCST) and the Australian Department of Defence and Veterans’ Affairs Human Research Ethics Committee (DDVA HREC 096–18).
Consent for publication
All authors read and approved the manuscript for publication.
Competing interests
All authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. World Malaria Report 2019. Geneva. World Health Organization. https://www.who.int/publications-detail/world-malaria-report-2019.
- 2.WHO. World Malaria Report 2020. Geneva. World Health Organization.
- 3.WHO. World Malaria Report 2018. Geneva. World Health Organization https://www.who.int/malaria/publications/world-malaria-report-2018/en/.
- 4.DHIS2. Routine District Health Information System, Uganda. 2020.
- 5.NMCP. Uganda Malaria Indicator Survey (MIS). Kampala, 2019. https://dhsprogram.com/pubs/pdf/ATR21/ATR21.pdf.
- 6.NMCP. Uganda National Malaria Control Reduction Strategy. Kampala, 2015–20. http://library.health.go.ug/publications/malaria/uganda-malaria-reduction-strategic-plan-2014-2020-0.
- 7.NMCP. Uganda Malaria strategic Plan Midterm Review. Kampala, 2017.
- 8.NMCP. Uganda National Malaria Indicator Survey. Kampala, 2014.
- 9.Uganda Ministry of Health. District Health Information System (DHIS2). Kampala, 2019.
- 10.NMCP. Uganda Malaria Control Policy. Kampala, 2011. https://www.severemalaria.org/sites/mmv-smo/files/content/attachments/2017-02-28/Uganda%20NATIONAL%20MALARIA%20CONTROL%20POLICY%20-%202011.pdf.
- 11.MIS. Uganda Malaria Indicator Survey. Kampala, 2009. https://dhsprogram.com/pubs/pdf/MIS21/MIS21.pdf.
- 12.MIS. Malaria Survey. Kampala, 2019. https://www.dhsprogram.com/pubs/pdf/MIS34/MIS34.pdf.
- 13.NMCP. National Malaria Control and Elimination strategic Plan. Kampala, 2020.
- 14.NMCP. Uganda Malaria Program Review (MPR) Report. Kampala, 2019.
- 15.Zhong D, Koepfli C, Cui L, Yan G. Molecular approaches to determine the multiplicity of Plasmodium infections. Malar J. 2018;17:172. doi: 10.1186/s12936-018-2322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker J, McCarthy J, Gatton M, Kyle DE, Belizario V, Luchavez J, et al. Genetic diversity of Plasmodium falciparum histidine-rich protein 2 (PfHRP2) and its effect on the performance of PfHRP2-based rapid diagnostic tests. J Infect Dis. 2005;192:870–877. doi: 10.1086/432010. [DOI] [PubMed] [Google Scholar]
- 17.Deme AB, Park DJ, Bei AK, Sarr O, Badiane AS, Gueye PEHO, et al. Analysis of pfhrp2 genetic diversity in Senegal and implications for use of rapid diagnostic tests. Malar J. 2014;13:34. doi: 10.1186/1475-2875-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar N, Singh JP, Pande V, Mishra N, Srivastava B, Kapoor R, et al. Genetic variation in histidine rich proteins among Indian Plasmodium falciparum population: possible cause of variable sensitivity of malaria rapid diagnostic tests. Malar J. 2012;11:298. doi: 10.1186/1475-2875-11-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rock EP, Marsh K, Saul AJ, Wellems TE, Taylor DW, Maloy WL, et al. Comparative analysis of the Plasmodium falciparum histidine-rich proteins HRP-I, HRP-II and HRP-III in malaria parasites of diverse origin. Parasitology. 1987;95:209–227. doi: 10.1017/S0031182000057681. [DOI] [PubMed] [Google Scholar]
- 20.Baker J, Ho MF, Pelecanos A, Gatton M, Chen N, Abdullah S, et al. Global sequence variation in the histidine-rich proteins 2 and 3 of Plasmodium falciparum: implications for the performance of malaria rapid diagnostic tests. Malar J. 2010;9:129. doi: 10.1186/1475-2875-9-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abukari Z, Okonu R, Nyarko SB, Lo AC, Dieng CC, Salifu SP, et al. The diversity, multiplicity of infection and population structure of P. falciparum parasites circulating in asymptomatic carriers living in high and low malaria transmission settings of Ghana. Genes (Basel). 2019;10:634. doi: 10.3390/genes10060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatei W, Gimnig JE, Hawley W, ter Kuile F, Odero C, Iriemenam NC, et al. Genetic diversity of Plasmodium falciparum parasite by microsatellite markers after scale-up of insecticide-treated bed nets in western Kenya. Malar J. 2015;14:495. doi: 10.1186/s12936-015-1003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosco AB, Anderson K, Gresty K, Prosser C, Smith D, Nankabirwa JI, et al. Molecular surveillance reveals the presence of pfhrp2 and pfhrp3 gene deletions in Plasmodium falciparum parasite populations in Uganda, 2017–2019. Malar J. 2020;19:300. doi: 10.1186/s12936-020-03362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonahasa S, Maiteki-Sebuguzi C, Rugnao S, Dorsey G, Opigo J, Yeka A, et al. LLIN evaluation in Uganda project (LLINEUP): factors associated with ownership and use of long-lasting insecticidal nets in Uganda: a cross-sectional survey of 48 districts. Malar J. 2018;17:421. doi: 10.1186/s12936-018-2571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rugnao S, Gonahasa S, Maiteki-Sebuguzi C, Opigo J, Yeka A, Katureebe A, et al. LLIN Evaluation in Uganda Project (LLINEUP): factors associated with childhood parasitaemia and anaemia 3 years after a national long-lasting insecticidal net distribution campaign: a cross-sectional survey. Malar J. 2019;18:207. doi: 10.1186/s12936-019-2838-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staedke SG, Kamya MR, Dorsey G, Maiteki-Sebuguzi C, Gonahasa S, Yeka A, et al. LLIN evaluation in Uganda project (LLINEUP) - Impact of long-lasting insecticidal nets with, and without, piperonyl butoxide on malaria indicators in Uganda: study protocol for a cluster-randomised trial. Trials. 2019;20:321. doi: 10.1186/s13063-019-3382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akinyi S, Hayden T, Gamboa D, Torres K, Bendezu J, Abdallah JF, et al. Multiple genetic origins of histidine-rich protein 2 gene deletion in Plasmodium falciparum parasites from Peru. Sci Rep. 2013;3:2797. doi: 10.1038/srep02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berhane A, Anderson K, Mihreteab S, Gresty K, Rogier E, Mohamed S, et al. Major threat to malaria control programs by Plasmodium falciparum lacking histidine-rich protein 2. Eritrea Emerg Infect Dis. 2018;24:462–470. doi: 10.3201/eid2403.171723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson TJC, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 30.Anderson T, Bockarie M, Lagog M, Day K. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–25. doi: 10.1017/S0031182099004552. [DOI] [PubMed] [Google Scholar]
- 31.Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, et al. Current trends in microsatellite genotyping. Mol Ecol Resour. 2011;11:591–611. doi: 10.1111/j.1755-0998.2011.03014.x. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Tessema SK, Murphy M, Xu S, Schwartz A, Wang W, et al. Confirmation of the absence of local transmission and geographic assignment of imported falciparum malaria cases to China using microsatellite panel. Malar J. 2020;19:244. doi: 10.1186/s12936-020-03316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Touray AO, Mobegi VA, Wamunyokoli F, Herren JK. Diversity and Multiplicity of P. falciparum infections among asymptomatic school children in Mbita Western Kenya. Sci Rep. 2020;10:5924. doi: 10.1038/s41598-020-62819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durand R, Ariey F, Cojean S, Fontanet A, Ranaivo L, Ranarivelo LA, et al. Analysis of circulating populations of Plasmodium falciparum in mild and severe malaria in two different epidemiological patterns in Madagascar. Trop Med Int Health. 2008;13:1392–1399. doi: 10.1111/j.1365-3156.2008.02156.x. [DOI] [PubMed] [Google Scholar]
- 35.Mohammed H, Hassen K, Assefa A, Mekete K, Tadesse G, Taye G, et al. Genetic diversity of Plasmodium falciparum isolates from patients with uncomplicated and severe malaria based on msp-1 and msp-2 genes in Gublak North West Ethiopia. Malar J. 2019;18:413. doi: 10.1186/s12936-019-3039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roh ME, Tessema SK, Murphy M, Nhlabathi N, Mkhonta N, Vilakati S, et al. High genetic diversity of Plasmodium falciparum in the low-transmission setting of the Kingdom of Eswatini. J Infect Dis. 2019;220:1346–1354. doi: 10.1093/infdis/jiz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong D, Afrane Y, Githeko A, Yang Z, Cui L, Menge DM, et al. Plasmodium falciparum genetic diversity in western Kenya highlands. Am J Trop Med Hyg. 2007;77:1043–1050. doi: 10.4269/ajtmh.2007.77.1043. [DOI] [PubMed] [Google Scholar]
- 38.Pringle JC, Wesolowski A, Berube S, Kobayashi T, Gebhardt ME, Mulenga M, et al. High Plasmodium falciparum genetic diversity and temporal stability despite control efforts in high transmission settings along the international border between Zambia and the Democratic Republic of the Congo. Malar J. 2019;18:400. doi: 10.1186/s12936-019-3023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oyebola MK, Idowu ET, Olukosi YA, Iwalokun BA, Agomo CO, Ajibaye OO, et al. Genetic diversity and complexity of Plasmodium falciparum infections in Lagos Nigeria. Asian Pacific J Trop Biomed. 2014;4:S87–S91. doi: 10.12980/APJTB.4.2014C1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahdi Abdel Hamid M, Elamin AF, Albsheer MMA, Abdalla AAA, Mahgoub NS, Mustafa SO, et al. Multiplicity of infection and genetic diversity of Plasmodium falciparum isolates from patients with uncomplicated and severe malaria in Gezira State Sudan. Parasit Vectors. 2016;9:362. doi: 10.1186/s13071-016-1641-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nankabirwa JI, Arinaitwe E, Rek J, Kilama M, Kizza T, Staedke SG, et al. Malaria transmission, infection, and disease following sustained indoor residual spraying of insecticide in Tororo Uganda. Am J Trop Med Hyg. 2020;103:1525–1533. doi: 10.4269/ajtmh.20-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiwuwa MS, Ribacke U, Moll K, Byarugaba J, Lundblom K, Färnert A, et al. Genetic diversity of Plasmodium falciparum infections in mild and severe malaria of children from Kampala Uganda. Parasitol Res. 2013;112:1691–1700. doi: 10.1007/s00436-013-3325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyabayinze DJ, Karamagi C, Kiggundu M, Kamya MR, Wabwire-Mangen F, Kironde F, et al. Multiplicity of Plasmodium falciparum infection predicts antimalarial treatment outcome in Ugandan children. Afr Health Sci. 2008;8:200–205. [PMC free article] [PubMed] [Google Scholar]
- 44.Bosco AB, Nankabirwa JI, Yeka A, Nsobya S, Gresty K, Anderson K, et al. Limitations of rapid diagnostic tests in malaria surveys in areas with varied transmission intensity in Uganda 2017–2019: implications for selection and use of HRP2 RDTs. PLoS ONE. 2021;15:e0244457. doi: 10.1371/journal.pone.0244457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomson R, Beshir KB, Cunningham J, Baiden F, Bharmal J, Bruxvoort KJ, et al. pfhrp2 and pfhrp3 gene deletions that affect malaria rapid diagnostic tests for Plasmodium falciparum: analysis of archived blood samples from 3 African countries. J Infect Dis. 2019;220:1444–1452. doi: 10.1093/infdis/jiz335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee N, Gatton ML, Pelecanos A, Bubb M, Gonzalez I, Bell D, et al. Identification of optimal epitopes for Plasmodium falciparum rapid diagnostic tests that target histidine-rich proteins 2 and 3. J Clin Microbiol. 2012;50:1397–1405. doi: 10.1128/JCM.06533-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gendrot M, Fawaz R, Dormoi J, Madamet M, Pradines B. Genetic diversity and deletion of Plasmodium falciparum histidine-rich protein 2 and 3: a threat to diagnosis of P. falciparum malaria. Clin Microbiol Infect. 2019;25:580–5. doi: 10.1016/j.cmi.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Addai-Mensah O, Dinko B, Noagbe M, Ameke SL, Annani-Akollor ME, Owiredu E-W, et al. Plasmodium falciparum histidine-rich protein 2 diversity in Ghana. Malar J. 2020;19:256. doi: 10.1186/s12936-020-03328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li P, Xing H, Zhao Z, Yang Z, Cao Y, Li W, et al. Genetic diversity of Plasmodium falciparum histidine-rich protein 2 in the China-Myanmar border area. Acta Trop. 2015;152:26–31. doi: 10.1016/j.actatropica.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metoh TN, Chen J-H, Fon-Gah P, Zhou X, Moyou-Somo R, Zhou X-N. Genetic diversity of Plasmodium falciparum and genetic profile in children affected by uncomplicated malaria in Cameroon. Malar J. 2020;19:115. doi: 10.1186/s12936-020-03161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mussa A, Talib M, Mohamed Z, Hajissa K, et al. Genetic diversity of Plasmodium falciparum histidine-rich protein 2 (PfHRP2) and its effect on the performance of PfHRP2-based rapid diagnostic tests. BMC Res Notes. 2019;12:334. doi: 10.1186/s13104-019-4361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agaba BB, Yeka A, Nsobya S, Arinaitwe E, Nankabirwa J, Opigo J, et al. Systematic review of the status of pfhrp2 and pfhrp3 gene deletion, approaches and methods used for its estimation and reporting in Plasmodium falciparum populations in Africa: review of published studies 2010–2019. Malar J. 2019;18:355. doi: 10.1186/s12936-019-2987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng Q, Gatton ML, Barnwell J, Chiodini P, McCarthy J, Bell D, et al. Plasmodium falciparum parasites lacking histidine-rich protein 2 and 3: a review and recommendations for accurate reporting. Malar J. 2014;13:283. doi: 10.1186/1475-2875-13-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Summary of samples successfully genotyped on 4–7 microsatellite markers (-s: single haplotype, -m2: 2 haplotypes, -m3: 3 haplotypes, -m4: 4 haplotypes)
Additional file 2: Table S2. Sequence length and unique sequence type of pfhrp2 exon 2.
Data Availability Statement
Data for this study is available and the row dataset has been uploaded as additional information with the manuscript.