

Significance
Autosomal dominant mutations in one of the most ubiquitous isoforms of the inhibitory subunit (R1α) of protein kinase A (PKA) are linked to a generalized tumor predisposition, referred to as Carney complex (CNC). The molecular phenotype of CNC mutants is PKA overactivation. However, explaining PKA overactivation remains an unmet challenge for some of the most harmful CNC mutants, which are expressed and exhibit significantly reduced affinity for the cAMP activator but not for the PKA catalytic subunit (C). Here, we address this challenge by proposing a mechanism of PKA activation, distinct from the classical cAMP-dependent dissociation of the R2:C2 complex and based on oligomerization of R1α into assemblies, which are incompetent to inhibit PKA C.
Keywords: cAMP, PKA, Carney, aggregation, oligomerization
Abstract
Familial mutations of the protein kinase A (PKA) R1α regulatory subunit lead to a generalized predisposition for a wide range of tumors, from pituitary adenomas to pancreatic and liver cancers, commonly referred to as Carney complex (CNC). CNC mutations are known to cause overactivation of PKA, but the molecular mechanisms underlying such kinase overactivity are not fully understood in the context of the canonical cAMP-dependent activation of PKA. Here, we show that oligomerization-induced sequestration of R1α from the catalytic subunit of PKA (C) is a viable mechanism of PKA activation that can explain the CNC phenotype. Our investigations focus on comparative analyses at the level of structure, unfolding, aggregation, and kinase inhibition profiles of wild-type (wt) PKA R1α, the A211D and G287W CNC mutants, as well as the cognate acrodysostosis type 1 (ACRDYS1) mutations A211T and G287E. The latter exhibit a phenotype opposite to CNC with suboptimal PKA activation compared with wt. Overall, our results show that CNC mutations not only perturb the classical cAMP-dependent allosteric activation pathway of PKA, but also amplify significantly more than the cognate ACRDYS1 mutations nonclassical and previously unappreciated activation pathways, such as oligomerization-induced losses of the PKA R1α inhibitory function.
Protein kinase A (PKA) plays a central role in how cells respond to multiple G protein-coupled receptor (GPCR)-binding hormones, such as thyrotropin or the parathyroid hormone (1, 2). The physiological responses controlled by PKA range from metabolic regulation to cellular differentiation and proliferation (3, 4). PKA includes catalytic and regulatory subunits denoted here as C and R, respectively (4). Under resting conditions, C is inhibited by the R2 dimer through the formation of a stable holoenzyme complex (R2C2). Upon cellular stimulation and activation of adenylyl cyclase by GPCRs, cAMP levels increase sufficiently for cAMP to bind the tandem cAMP-binding domains of R (CBD-A and B; Fig. 1) and release C, which in turn phosphorylates downstream substrates that control a wide array of signaling pathways (4–8).
Fig. 1.

The PKA R1α system and disease-related mutations. (A) Domain organization of PKA R1α with non-haploinsufficient CNC and ACRDYS1 mutants within the tandem cAMP-binding domains (CBDs) indicated in green and red, respectively. The blue star in CBD-A denotes Ala-211, which is mutated to Asp in CNC and to Thr in ACRDYS1, and in CBD-B denotes Gly-287, which is mutated to Trp in CNC and to Glu in ACRDYS1. Further details are available in SI Appendix, Fig. S1A. (B) Structure of R1α CBDs in the cAMP-bound state [dark colors; PDB: 1RGS (26)] and C-subunit-bound state [light colors; PDB: 2QCS (5)]. CNC and ACRDYS1 mutation sites are highlighted with green and red spheres, while cAMP is shown with black sticks. The N3A motif (αN-helix, 310-loop, and αA helix) and phosphate binding cassette (PBC) are highlighted in red and yellow, respectively, while the rest of CBDs is in cyan. The Insets show details of the cAMP-binding sites adjacent to the A211 and G287 mutations, with selected hydrogen bonds and capping interactions highlighted by dashed lines and surfaces.
Autosomal dominant inherited mutations in the 1α isoform of PKA R have been associated with type 1 acrodysostosis (ACRDYS1) or the Carney complex (CNC) (1, 9). ACRDYS1 is a severe skeletal dysplasia leading to defects in the development of facial, finger, and toe bones as well as dwarfism (1, 2). CNC refers to a multiple endocrine neoplasia often linked to a generalized predisposition for tumors, including pituitary and breast ductal adenoma, heart and breast myxomas, adrenocortical tumors, pancreatic and liver cancers, as well as skin tumors characterized by spotty pigmentation (9–12). ACRDYS1 arises from hormonal resistance caused by an impaired PKA R1α response to cAMP (1, 2). In fact, most ACRDYS1 PKA R1α mutations are found in the vicinity of the cAMP-binding site of CBD-B. Since CBD-B functions as a “gatekeeper” for binding of cAMP also to CBD-A, these ACRDYS1 mutants exhibit elevated activation constants, which explain the decrease sensitivity to cAMP and lower degree of C activation in ACRDYS1 vs. wild-type (wt) PKA (1, 13, 14).
Unlike ACRDYS1, the CNC pathology reflects increased PKA activity arising from losses of the tumor-suppressor function of PKA R1α (11). Most CNC PKA R1α mutations incur nonsense-mediated mRNA decay (NMD), resulting in PKA R1α haploinsufficiency and overactivation of the PKA C kinase (15, 16). However, the most harmful CNC PKA R1α mutants often escape NMD and result in the expression of defective PKA R1α (16, 17). The majority of CNC PKA R1α mutations that are not subject to NMD cluster in CBD-A (Fig. 1A and SI Appendix, Fig. S1A) (15), which is essential for binding and inhibiting PKA C. Some of these CNC mutants, such as R144S and S145G, are located at the protomer–protomer interface of the R1α dimer and have been shown to perturb the allosteric response to cAMP. These mutants result in lowered Hill coefficients and activation constants, thus explaining the hypersensitivity to cAMP and the consequent PKA overactivation in these CNC mutations (18). However, for other CNC mutations, such as A211D and G287W (Fig. 1 and SI Appendix, Fig. S1A), the opposite effect is observed (13). The cAMP activation constants of A211D and G287W are significantly higher than those of wt PKA R, while no significant losses are observed in the ability to inhibit PKA C (13). The A211 and G287 CNC mutant sites are of particular interest because of their unique ability to switch from the CNC to the opposite ACRDYS1 phenotype simply by changing the side chains of these residues. Specifically, A211T and G287E are classified as ACRDYS1 mutations, suggesting that the 211 and 287 sites are critical for understanding PKA-linked pathologies. Taken together, these observations indicate that the molecular mechanisms underlying CNC PKA R1α mutations are currently not fully understood, especially in the context of the classical model of cAMP-dependent PKA activation.
Here, we hypothesize that a viable mechanism to explain the CNC phenotype of the A211D and G287W PKA R1α mutations is the activation of PKA C through noncovalent oligomerization-induced losses of the PKA R inhibitory function. Upon partial denaturation, wt PKA R1α tends to aggregate (19). If a CNC mutant is more prone to unfold than wt, either because of reduced cAMP affinity or intrinsic destabilization of R1α (20), it is more likely to oligomerize into open-ended assemblies than wt (21). Bioinformatics analyses indicate that PKA R1α aggregates sequester and shield key loci of the inhibitory R:C interface (19), suggesting that PKA R1α aggregation reduces the inhibitory competency of PKA R, thus explaining the PKA C overactivation typical of CNC mutants. In order to test this hypothesis, here we comparatively analyze the structure, unfolding, aggregation, and inhibition profiles of wt PKA R1α and the CNC mutants A211D and G287W. We also extend our comparative analyses to the cognate ACRDYS1 mutations A211T and G287E, which, unlike the corresponding CNC mutants, result in PKA overinhibition. The ability to compare CNC vs. ACRDYS1 mutations of the same residue eliminates possible positional biases from the comparisons.
Structural perturbations were evaluated by combining NMR and X-ray crystallography. Unfolding was investigated at progressive degrees of resolution (i.e., full-length, domain and residue-resolution) by combining urea unfolding monitored by intrinsic fluorescence, bioinformatics, and hydrogen/deuterium exchange (HDX) monitored by NMR. Aggregation was probed by size-exclusion chromatography (SEC), extrinsic fluorescence using the ThT and ANS fluorophores, NMR, dynamic light scattering (DLS), and transmission electron microscopy (TEM), while PKA C activation was gauged through luminescence-based kinase assays. Our results show that the A211D and G287W CNC mutations unfold the tandem CBDs of PKA R1α to a larger extent than wt and the cognate ACRDYS1 mutants, leading to correspondingly higher propensities to oligomerize into open-ended assemblies, which are incompetent to inhibit PKA C. Taken together, our data support the hypothesis that PKA R oligomerization sequesters R from C, thus defining a noncanonical, yet viable, mechanism for PKA C activation that was not previously appreciated but is relevant for CNC mutations.
Results
The CNC Mutation A211D Destabilizes the PKA R1α Fold Significantly More than Does the Cognate ACRDYS1 Mutation A211T.
As a first step toward assessing the impact of the A211 mutations on the stability of PKA R1α, we monitored the urea unfolding of wt, A211D, and A211T PKA R1α through intrinsic Trp fluorescence (22, 23) (Fig. 2A). Fig. 2A shows that both mutants unfold at lower urea concentrations than wt. While the Cm of wt PKA R1α is ∼6.4 M under our conditions, the Cm values of the A211T and A211D mutants decrease to ∼5.6 and ∼3.2 M, respectively (Fig. 2A). The urea unfolding data in Fig. 2A are in agreement with the changes in mutant vs. wt stability (ΔΔG) computed through the structure-based Eris software (see SI Appendix, Table S1) (24). Positive or negative ΔΔG values indicate that the mutant destabilizes or stabilizes the structure, respectively (24). As shown in SI Appendix, Table S1, the ΔΔG values computed for the A211D mutant are consistently higher than those calculated for the A211T mutant. This pattern applies irrespective of whether PKA R1α adopts a cAMP- or a C-binding competent structure and regardless of the specific PKA R1α construct utilized as input (SI Appendix, Table S1). Overall, our unfolding data and computations consistently show that the CNC A211 mutation causes a loss of folding stability in PKA R1α significantly larger than the cognate ACRDYS1 A211 mutant. However, Fig. 2A alone is insufficient to establish whether the major destabilization induced by the A211D mutation arises from losses of cAMP binding affinity and/or from an intrinsic destabilization of the PKA R1α structure. In order to better understand the cause of the A211D instability, we further investigated this mutation in the context of the one domain CBD-A construct PKA R1α (96–244), which preserves a fold similar to that observed for the tandem CBD constructs and for which apo vs. holo unfolding changes more directly reflect 1:1 cAMP binding affinities to the mutated domain (25–28). SI Appendix, Fig. S1B shows the urea unfolding profiles of A211D PKA R1α (96–244) in the presence and absence of cAMP. As a control, SI Appendix, Fig. S1B also includes the urea unfolding profile for the wt PKA R1α (96–244) construct.
Fig. 2.

PKA RIα destabilization caused by the CNC and ACRDYS1 A211 mutations as revealed by urea unfolding and HDX NMR. (A) Urea unfolding profiles monitored through intrinsic fluorescence for 5 μM of full-length wt, A211T, and A211D PKA R1α (1–379) in the presence of 100-fold excess cAMP. Color codes are shown in the figure. The ΔUFGoH2O unfolding free energies decrease in the order wt > A211T > A211D (i.e., the free energy of unfolding extrapolated to pure aqueous buffer for wt, A211T, and A211D PKA R1α are ΔUFGoH2O = 10.7 ± 1.0, 8.5 ± 0.8, and 5.4 ± 1.1 kcal/mol, respectively). N is 3 and error is SD of plateau region. (B–E) Residue-specific solvent exposure as gauged based on H/D exchange monitored by HSQC spectra for the PKA R1α (119–379) construct in the presence of 0.7 mM excess cAMP. Residues are categorized in three groups: fully exposed, intermediate, and fully protected (SI Appendix, Fig. S2). (B) Frequency of occurrence for each exchange class based on the assigned residues. The vertical arrows show that the fraction of fully exposed residues increases in A211D compared to wt at the expense of losses in the other two classes. The cAMP-bound structure (26) is used for all HDX maps of (C) wt; (D) A211D mutant; and (E) A211T mutant. Color codes are shown in the figure. In D and E, residues that are more solvent exposed than wt are marked by black arrows, while residues that are more protected than wt are highlighted by orange arrows. The black box in D means that CBD-A is fully solved exposed. Surfaces in C–E highlight aggregation-prone regions identified by Aggrescan (SI Appendix, Fig. S3A).
In stark contrast to the wt construct, no appreciable changes in folding stability are observed for the A211D mutant upon addition of 500 μM cAMP (SI Appendix, Fig. S1B). This observation indicates that the A211D mutation reduces the cAMP affinity for the isolated CBD-A to greater than approximately submillimolar levels, i.e., approximately five orders of magnitude weaker than the corresponding wt construct. In addition, the A211D mutation significantly destabilizes also apo PKA R1α (96-244) (SI Appendix, Fig. S1B). Hence, the decreased stability of the A211D mutant relative to wt arises from dramatic losses of cAMP affinity at CBD-A as well as from an intrinsic destabilization of the CBD-A fold. In order to gain further insight on the nature of this dual loss of stability, we mapped at residue-resolution the effect of the A211 mutations on the solvent accessibility and structure of the PKA R1α CBDs through NMR monitored HDX (Fig. 2 B–E) and NMR chemical shifts (Fig. 3 A–F), respectively.
Fig. 3.

Chemical shift map of the perturbations caused by the CNC and ACRDYS1 A211 mutations on PKA R1α tandem CBDs and crystal structure of the A211D mutant. (A) Overlay of HSQCs spectra of A211D (blue) and wt PKA R1α (119–379) (green) in the presence of 2 mM cAMP. (B) As A, but for the A211T mutation. (C) Map of residues with A211D-induced CCS changes above the average plus 1 SD on the structure of the tandem PKA R1α CBDs bound to cAMP (26). Unassigned residues or broadened beyond detection are highlighted with a gray ribbon. (D) As C, but for the A211T mutation. (E) Correlation between the compounded chemical shifts (CCS) of A211D and wt PKA R1α (119–379) from the spectra in A. (F) As E, but for the A211T mutation. (G) The A211D PKA R1α (1–379) dimer structure with cAMP-bound to all four CBDs (SI Appendix, Table S2). (H) WT dimer structure (PDB: 4MX3). Domains are color coordinated as follows: Linker and N3A (red); rest of CBD-A (light cyan); CBD-B (dark cyan). A211D and wt exhibit a well-conserved structure (SI Appendix, Table S3), including the N3A motif dimer interface.
The A211 Mutations Cause Primarily Local Perturbations of the PKA R1α Ground Folded State, Confined to CBD-A and the CBD-A/B Interface Without Major Changes in the Global Fold.
We acquired 1H15N-heteronuclear single-quantum coherence (HSQC) spectra of the wt as well as A211D and A211T PKA R1α (119–379) construct, which spans both tandem CBDs (Fig. 3 A and B). Both mutations lead to intensity losses and chemical shift changes (Fig. 3 A and B). Although intensity losses are more pronounced for A211D than for A211T (Fig. 3 A and B), for both mutations the most significant chemical shift changes are local and confined to CBD-A, where A211 is situated, and part of the CBD-A/B interface (Fig. 3 C and D). However, the chemical shifts of the A211 mutants remain overall quite comparable to those of wt (Fig. 3 E and F), suggesting that the ground state conformation of the ensemble sampled by both A211 mutants preserves a global fold similar to wt PKA R1α.
The A211D vs. wt structural similarity is confirmed also at the level of full-length PKA R1α, as shown by the crystal structure of the full-length PKA RIα dimeric A211D mutant saturated with cAMP (Fig. 3 G and H). We solved the A211D PKA RIα dimer structure at 4.15-Å resolution, with R and Rfree values of 0.221 and 0.270, respectively (SI Appendix, Table S2). While the limited resolution of this structure likely reflects the lower stability of the A211D mutant relative to wt, the RMSD values compared to the wt (Fig. 3H; PDB: 4MX3) confirm the overall similarity between the A211D and wt structures (SI Appendix, Table S3) (18), in agreement with our Eris predictions (SI Appendix, Table S1) and chemical shift analyses (Fig. 3E). However, based on the urea unfolding data (Fig. 2A and SI Appendix, Fig. S1B) and the Eris stability predictions, we expect that the A211 mutations should make the unfolded and/or partially unfolded excited states more accessible compared to wt PKA R1α. We therefore anticipate the A211 mutants to display different solvent exposure relative to wt. To test this prediction, we examined the solvent accessibility of PKA R1α (119-379) through HDX monitored in real time at residue resolution by NMR (Fig. 2 B–E).
The A211D Mutation Increases the Solvent Exposure of Both CBDs More than Does the A211T Mutation Under Native Conditions.
Due to the limited signal-to-noise ratio exhibited by several peaks in the mutant A211 HSQC spectra (Fig. 3 A and B), we opted for a semiquantitative approach to the HDX analysis. We categorized residues in three groups: fully exposed, fully protected residues, and those with intermediate exposure, which fall between the extremes of the first two classes (SI Appendix, Fig. S2). Residues without detectable signal beyond the second HSQC acquired after the dead time of the HDX experiment are defined as fully exposed (SI Appendix, Fig. S2). Fully protected residues are those not subject to appreciable H/D exchange within the HDX experiment time frame (≤16 h; SI Appendix, Fig. S2). The remaining residues were assigned to the intermediate group (SI Appendix, Fig. S2). Based on this categorization (Fig. 2B), we mapped the results of our HDX experiments for wt, A211D, and A211T PKA R1α (119–379) on the structure of the PKA R1α CBDs (Fig. 2 C–E).
Fig. 2 B–E shows that both A211 mutants result in a loss of fully protected residues (Fig. 2B), pointing to an overall destabilization of the CBDs with respect to transient global unfolding. However, the changes elicited by the A211T and A211D mutations on the other two HDX classes are clearly distinct (Fig. 2B). For A211T, we observed an increase in the occurrence of residues with intermediate protection (Fig. 2B), without major changes for the fully exposed category. On the contrary, for A211D we detected a higher frequency of residues with full exposure and losses in the intermediate category (Fig. 2B). So overall, the solvent exposure appears to increase in the order wt < A211T < A211D, in agreement with the urea unfolding data (Fig. 2A and SI Appendix, Fig. S1B).
The CBD-A domain of the A211D mutant is the main region responsible for the reduction in full and intermediate protection and enhancement in full exposure (Fig. 2D). However, several CBD-B residues also become more solvent exposed in A211D relative to wt (Fig. 2D). Hence, the A211D mutation affects solvent exposure in both CBDs. Similarly, in the A211T mutant changes in solvent exposure are observed in both CBD-A and -B (Fig. 2E). However, for the A211T mutant we observed some residues becoming more solvent exposed than wt, and vice versa for other residues (Fig. 2E). These results further confirm the increased overall solvent exposure in A211D compared to both A211T and wt PKA R1α. Notably, A211D increases exposure through partial unfolding primarily in CBD-A, including the short αP helix in the phosphate binding cassette (PBC) and the β1 strand (Fig. 2D), which are aggregation prone motives (SI Appendix, Fig. S3 A and B) (19, 29, 30). Interestingly, the A211D mutation also causes intensity losses in these regions more than the cognate A211T mutation (SI Appendix, Fig. S3 C–E), consistent with A211D induced millisecond–microsecond dynamics due to conformational changes and/or self-association at these sites. Therefore, our HDX and NMR intensity changes data suggest that A211D is more likely to aggregate than wt and A211T. To test this hypothesis, we monitored the A211D PKA R1α self-association and amyloid formation through SEC, NMR intensity losses, fluorescence, TEM (Fig. 4 A–F), and DLS (SI Appendix, Fig. S4 A and B).
Fig. 4.

Effect of the CNC and ACRDYS1 A211 mutations on PKA R1α self-association, oligomerization, and inhibition of PKA kinase activity. (A) SEC profiles of full-length wt, A211D, and A211T PKA R1α prepared as for the kinase inhibition assays. The intensity of the dimer peaks are normalized to 1. Hence, the relative intensity of the >100-kDa oligomer peak reflects the extent of oligomerization of the PKA R1α dimer. (B) Aggregation probed through 1D 1H NMR methyl intensity losses upon mild heat treatment (i.e., 90-min incubation at 60 °C) of 8 μM PKA R1α in the presence of 10-fold excess cAMP. Mild heat treatment leads to a ∼70% intensity loss for A211D, but only ∼20% for wt and A211T. (C) Kinetics of cross-β-sheet formation as monitored by normalized ThT fluorescence while incubating 8 μM PKA R1α at 60 °C in the presence of 10-fold excess cAMP. The ThT fluorescence profile of Aβ (1–40) serves as a positive control for amyloid formation. Data acquired in triplicate and error is SD. (D) Difference of ANS fluorescence spectra of 8 μM R1α (1–379) with 10-fold cAMP excess before and after heat treatment. The enhanced ANS fluorescence intensity upon heat treatment points to increased exposure of hydrophobic residues. Data acquired in triplicate, and error is SD. (E and F) TEM images of WT, A211D, and A211T PKA R1α assemblies in the presence of 10-fold excess cAMP before, before (E) and after (F) heat treatment (i.e., 14-h incubation at 60 °C). PKA R1α oligomers with sizes of the order of 100 nm are already present prior to heat treatment (orange arrows), suggesting that the heat treatment accelerates oligomerization processes intrinsic to PKA R1α. (G) Nonoligomerized full-length PKA R1α inhibits the kinase activity of PKA C in a dose-dependent manner. This applies to wt, A211T, and A211D. Data acquired in triplicate and error is SD of the plateau region. (H) The PKA R1α oligomers are incompetent to inhibit the kinase activity of PKA C. Thick horizontal lines (shaded rectangles) denote the average (SDs) of all time points measured for each sample (A211D, or A211T, or WT) at each incubation temperature. The boxes with lighter (darker) shades are for incubation at 4 (37) °C. Other color codes are indicated in each panel. Filled gray dots (open circles) represent actual data points acquired with incubation at 4 (37) °C. The inhibitory potency at saturation of PKA C was measured for full-length PKA R1α incubated at 1 μM and 4 or 37 °C in the time range 2–8 h. Data acquired at least in triplicate and error is SD.
A211D Is Significantly More Aggregation Prone than Are A211T and wt PKA R1α.
We monitored the extent of PKA R1α aggregation through multiple techniques. First, the gel filtration elution profiles of wt, A211D, and A211T PKA R1α consistently show two distinct peaks at around 44 and 68 mL (Fig. 4A). The first peak corresponds to oligomers larger than 100 kDa, while the second peak corresponds to dimeric PKA R1α (Fig. 4A). Interestingly, the relative oligomer vs. dimer intensity increases approximately fourfold to fivefold in A211D vs. wt PKA R1α, but remains comparable to wt in A211T (Fig. 4A). Hence, the A211D mutant is markedly more prone to self-association than wt and A211T even in the absence of heating. To complement this result, we probed the aggregation of PKA R1α through heat-induced 1D 1H NMR methyl intensity losses (31–34) (Fig. 4B). For wt and A211T such losses are limited to ∼20%, but they increase to ∼70% for A211D, confirming the increased aggregation propensity of A211D relative to wt and A211T.
The Probability of Forming Cross-β-Sheet–Containing Oligomers with Exposed Hydrophobic Sites Is Higher in A211D than in A211T and wt PKA R1α.
To further explore the nature of the aggregates formed by wt, A211D, and A211T PKA R1α, we probed the PKA R1α oligomerization with the ThT and ANS fluorophores, which report on cross-β-sheets (31, 35) and hydrophobic exposure (36, 37), respectively (Fig. 4 C and D). ThT fluorescence for wt, A211D, and A211T PKA R1α was recorded during a 14-h incubation period at 60 °C (Fig. 4C), which results in the formation of PKA R1α oligomers with size 100 nm for both wt and the A211 mutants, as shown by DLS (SI Appendix, Fig. S4B). The dimension of these PKA R1α assemblies is comparable to that of oligomers formed under the same experimental conditions by the prototypical amyloidogenic peptide Aβ (1–40), which serves as positive control (SI Appendix, Fig. S4 A and B). In addition, TEM images show that for both wt and the A211 mutants the 14-h incubation period at 60 °C results in the growth of PKA R1α assemblies (Fig. 4 E and F). It is notable that oligomers are already present prior to heat treatment, as consistently shown by our TEM (Fig. 4E), DLS (SI Appendix, Fig. S4A), and SEC data (Fig. 4A). These observations indicate that the ability to oligomerize is an intrinsic property of PKA R1α, which is further accelerated through heating.
The comparative analysis of the ThT fluorescence time profiles for wt, A211D, and A211T PKA R1α reveals that A211D not only exhibits the highest cross-β-sheet content, but also the shortest lag time (Fig. 4C). Wt R1α also showed a rise in ThT signal over time (19), but, contrary to the A211 mutants, only to levels lower than the control peptide Aβ (1–40) (38, 39). Furthermore, wt exhibits the longest lag time among all samples tested (Fig. 4C). The ThT profile of A211T is intermediate between those of Aβ (1–40) and A211D (Fig. 4C). The enhanced ThT fluorescence for A211D vs. A211T and wt is also confirmed at 37 °C (SI Appendix, Fig. S4C). Overall, the order of cross-β formation propensity as gauged based on the lag time duration and the maximum ThT fluorescence is A211D > A211T > wt. A similar ranking order is observed also based on the heat-induced ANS fluorescence enhancements (Fig. 4D), indicating that A211D increases the exposure of hydrophobic surfaces more than A211T. Taken together, these results indicate that A211D exhibits a higher propensity than A211T to form cross-β-sheet aggregates with exposed hydrophobic sites.
Oligomerization of PKA R1α Shields Sites Mediating Inhibitory Interactions with PKA C.
Interestingly, some of the most aggregation-prone sites in PKA R1α, such as the αP helix in CBD-A (SI Appendix, Fig. S3 A and B) (19, 29, 30, 40, 41), are also critical sites in the PKA R:C binding interface (5). In addition, the PKA R1α agglomerates formed under native conditions, as supported by our SEC, TEM, DLS data (Fig. 4 A and E and SI Appendix, Figs. S4 A and B and S9A), may also shield key R1α inhibitor sites from the PKA C-subunit. While the intermolecular interactions underlying the formation of these R1α assemblies are currently only limitedly understood, the crystal structures of wt and A211D PKA R1α offer initial clues about how the PKA R1α dimers may assemble into open-ended superquaternary agglomerates.
The analysis of the wt PKA R1α dimer structure reveals not only an intradimer interface between the N3A motives of the two protomers belonging to the same dimer (Table 1, interface 1; Fig. 5 A, Left), as previously observed (18, 42), but also multiple interdimer interfaces (Fig. 5 A, Center and Right; Fig. 5 B and C and SI Appendix, Fig. S5 and Table 1). Interestingly, most residues mediating hydrogen bonds or salt bridges at these interdimer interfaces are also prone to serve as self-association sites (“hot spots”), as independently predicted by sequence-based algorithms (Table 1 and SI Appendix, Fig. S3 A and B). This colocalization with self-association loci is unique to the interdimer interfaces, as it is not observed for the intradimer interface (Table 1). Out of the multiple interdimer interfaces, interface 2a is particularly interesting (Fig. 5 A, Center and Fig. 5B and Table 1). Like the N3A dimer (18), this 2a interface is unlikely to reflect merely crystal-packing (ΔiG P value < 0.5; Table 1), and it is conserved in higher-resolution structures of shorter PKA R1α constructs [Table 1; PDB codes 1RL3 (43) and 1NE4 and 1NE6 (44)].
Table 1.
PISA analysis of intra- and interdimer interfaces in wt PKA R1α
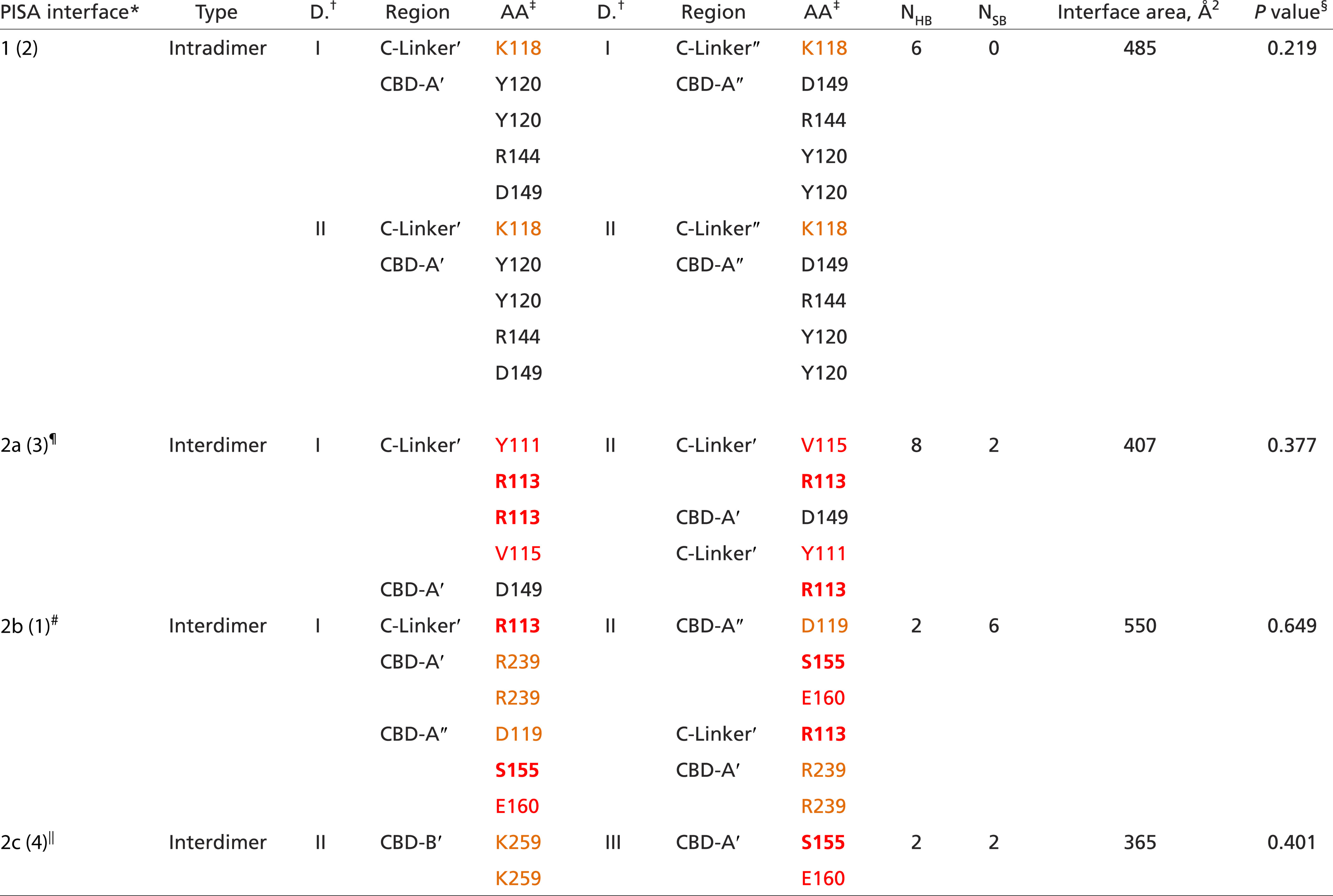 |
The 4MX3 structure was used. The numbers in parentheses indicate the interface numbering used in the PISA analysis of structure 4MX3 (68). Only interfaces including at least one hydrogen bond and/or salt bridge are included.
Dimer involved in the interface.
Residues involved in interfacial hydrogen bonds and/or salt bridges. Red means they are identified by sequence-based AGGRESCAN as belonging to an aggregation “hot spots.” Orange means they are within two residues in the primary sequence from an AGGRESCAN aggregation hot spots. Residues in bold are also predicted by AmylPred2 as consensus sites for amyloid propensity.
A value of P > 0.5 indicates that the interface is likely to reflect primarily crystal packing, while a value of P < 0.5 points to interfaces with hydrophobicity higher than average, suggesting an interaction-specific surface.
This interface includes all residues in the 108–117 segment as well as S145 and D149. A similar interface is found with P value of 0.292 in structure 1RL3 of PKA R1α 91–379 with cGMP bound to CBD-A and apo CBD-B solved at 2.70-Å resolution. Even lower values of P < 0.25 are associated with a similar interface in structures 1NE4 and 1NE6 of PKA R1α 91–379 with Rp- or Sp-cAMPS bound at both CBDs, respectively, solved at 2.40- and 2.30-Å resolution.
This interface includes also residues 108, 111, 137, 138, 140–142, 145, 146, 236, 240, 243, 244, 271, 273, 344 from C-linker′ (I) and CBD-A′ (I), and 117, 120, 121, 125, 154, 156–159, 177, 183, 214–216, 218, 220 from C-linker″ (II) and CBD-A″ (II) and the symmetrical counterparts.
This interface could not be found in the A211D structure. Further details are available in SI Appendix, Fig. S5.
Fig. 5.

Structure of wt and A211D PKA R1α dimer agglomerates. (A) Interprotomer interfaces in WT PKA R1α (PDB code: 4MX3). Gray surfaces are shown for residues involved in interfacial hydrogen bonds and/or salt bridges. (Left) Intradimer interfaces (denoted as interface 1 in Table 1); (Center and Right) interdimer interfaces between adjoining dimers (i.e., 2a and 2b in Table 1). Dimer I is composed of two protomers. One protomer of dimer I includes CBD-A′ (I) and CBD-B′ (I), while the other protomer of dimer I includes CBD-A″ (I) and CBD-B″ (I). A similar notation applies to dimer II. N3A motives of CBD-A′ and CBD-A″ are shown in red for both dimers. (B and C) Close-up views of interdimer interfaces 2a and 2b. (D) Interdimer 2a and 2b interfaces (gray) in A211D PKA R1α illustrating the formation of an open-ended assembly of dimers through a chain of interdimer 2a interfaces (dashed rectangle; “2a spine”). The visible 2b interfaces are highlighted with circles (black for dimers I–II and III–IV, but red between dimers II and III).
Each dimer is comprised of two chains referred to as protomer′ and protomer″. The interdimer interface 2a is homotypic and is formed by the C-linker region preceding the N3A motif (residues 108-117) as well as N3A residues S145 and D149 of protomer′ from dimer II and protomer′ from dimer I (Fig. 5 A, Center and Fig. 5B and SI Appendix, Fig. S5 and Table 1). This interface formed by protomers from dimer I and II is further stabilized by an antiparallel β-sheet between dimers I and II, while the two other protomers (protomer″) remain available to generate similar interfaces with other R1α dimers (Fig. 5 A, Center and SI Appendix, Fig. S5A). Specifically, protomer″ in dimer II forms a homotypic 2a interface with protomer″ from dimer III. Hence, interface 2a (Table 1) may serve as a means to assemble open-ended agglomerates of R1α dimers. This is clearly evident in the structure of full-length A211D R1α, in which interdimer 2a interfaces bring together four dimers in the asymmetric unit, forming a “2a spine” that stabilizes the agglomerate of dimers (Fig. 5D).
The interdimer contacts defined by interface 2a are complemented by additional dimer-to-dimer interactions provided by another interface denoted as 2b, which is primarily mediated by two sets of hydrogen bonds and salt bridges in the A211D structure: 1) a set between R239 in the C-helix of protomer′ from dimer I and residues 155–160 in the β1–β2 loop C-terminal to the N3A motif of protomer″ of dimer II (Fig. 5C and Table 1); 2) a similar set of hydrogen bonds/salt bridges form between residues R239 and 155–160 of the other two protomers, but with a swapped arrangement, i.e., R239 is contributed by protomer′ of dimer II and residues 155–160 by protomer″ of dimer I (Fig. 5C and Table 1).
In the wt RIα structure, residues S155 and E160 are also involved in another interdimer interface with K259 (interface 2c, Table 1 and SI Appendix, Fig. S5B). In this case, the S155 and E160 in one protomer interact with R239 in an adjacent dimer, while in the other protomer residues S155 and E160 interact with K259 in the adjacent dimer. This is thus a heterotypic interaction in wt RIα and introduces asymmetry. In contrast, in the A211D structure discussed above, where there are four dimers in the asymmetric unit as opposed to a single dimer in the wt RIα structure, S155 and E160 in each protomer interact exclusively with R239 in an adjacent dimer (Table 1 and SI Appendix, Fig. S5A and Fig. 5C). The creation of this homotypic interface introduces symmetry and presumably stabilizes the oligomerized state. While the limited resolution of the A211D PKA R1α structure does not warrant further analyses into the atomic details of these interactions, taken together these results describing the importance of interfaces 1 and 2a–c (Table 1 and Fig. 5 and SI Appendix, Fig. S5) highlight the central role played by the N3A motif (i.e., the αN-helix, 310-loop, and αA helix), and the residues flanking it, in serving as a pivotal module for both intradimer and interdimer contacts leading to open-ended agglomerates.
Interestingly, the N3A motif is not only important for the R1α dimer but is also critical for one of the conformations of the holoenzyme (5, 38). For example, residues 137, 138, and 140, which are part of the 2b interface, are also close to the R1α:C interface (Table 1 and Fig. 5 A, Right and Fig. 5C), while residues 113–119 are anchored onto the extended B/C/N helix of the R-subunit by the inhibitor site that precedes the C-linker. Based on this analysis of the holoenzyme and the observation that selected aggregation-prone sites (e.g., PBC) are adjacent to residues that also contributed to the R:C interface, we hypothesize that PKA R oligomerization, which is amplified in the CNC mutant, will reduce the PKA C inhibition competency of PKA R. To test this hypothesis and gauge the kinase inhibition competency of the assemblies promoted by the CNC mutant, we reverted to kinase assays (Fig. 4 G and H).
The A211D Aggregation Causes a Loss of PKA C Inhibition.
PKA C kinase inhibition was probed for wt, A211D, and A211T full-length PKA R1α (Fig. 4 G and H). The wt PKA R1α served as our positive control. Prior to oligomerization, wt, A211D, and A211T PKA R1α inhibit the kinase activity of PKA C in a dose-dependent manner (Fig. 4G). Similar to wt and A211T, the A211D PKA R1α mutant remains competent to inhibit PKA C if incubated at 4 °C for up to 8 h (Fig. 4H). However, upon incubation at 37 °C, the inhibitory competency of A211D is nearly fully lost within 2 h (Fig. 4H), while such losses are not observed for wt or A211T (Fig. 4H). These results indicate that the heat-induced A211D assemblies cause a loss of inhibitory function in PKA R1α. Such loss is not simply caused by precipitation because UV absorbance at 280 nm indicates that the protein is still present in solution even after heat treatment. While other in vivo mechanisms cannot be ruled out (45) (e.g., limited proteolysis; see SI Appendix, Fig. S4D), our data show that oligomerization suppresses the inhibitor potency of A211D PKA R1α, suggesting a viable mechanism to explain the kinase overactivation typical of this CNC mutant. As a first step toward checking whether such mechanism of PKA C overactivation through mutation-induced PKA R1α oligomerization applies also to other CNC mutants, we extended a similar comparative analysis of unfolding, aggregation, and kinase inhibition to the CNC G287W mutant (SI Appendix, Figs. S6–S9 and Table S4). Unlike A211D, the G287W mutation is situated in CBD-B (Fig. 1 and SI Appendix, Fig. S1A), but similar to A211D it is paired to a cognate ACRDYS1 mutation at the same site (G287E), which serves as a benchmark term of comparison.
Comparative Analysis of the G287 PKA R1α Mutants Confirms that PKA C Activation by PKA R Oligomerization Is a Viable Mechanism for CNC Mutations.
The chemical shift maps for both G287 mutants show that the mutation-induced perturbations to the ground-state PKA R1α (119–379) structure are primarily local and confined to CBD-B and the A/B domain interface (SI Appendix, Fig. S6 A–D), without significant changes in the global fold (SI Appendix, Fig. S6 E and F). However, HDX NMR reveals that the G287 mutations cause changes in solvent exposure in both domains (SI Appendix, Fig. S7), albeit the effect in the nonmutated domain is not as dramatic as for A211D. The G287W mutation leads to a loss of residues with full or intermediate protection and a concurrent gain of fully exposed residues (SI Appendix, Fig. S7A), with G287W being overall less protected than wt and G287E (SI Appendix, Fig. S7). The pattern of G287W vs. G287E destabilization revealed by HDX NMR is in full agreement with the computations of mutant stability, which show that G287W is consistently more destabilizing than G287E (SI Appendix, Table S4), and with the increased exposure of hydrophobic sites upon heating in G287W vs. G287E as revealed by ANS fluorescence (SI Appendix, Fig. S8B). Most importantly, the G287W mutation decreases the protection of PKA R1α sites known to be aggregation prone, such as the αP helix of the PBC of the CBDs (SI Appendix, Figs. S3 A and B and S7B) (29, 30), suggesting that G287W may aggregate more easily than wt and G287E. Consistent with this hypothesis, we observed enhanced aggregation for this CNC mutant, as evidenced by the higher ThT fluorescence of G287W vs. wt and G287E (SI Appendix, Fig. S8A) arising from elongated aggregates (SI Appendix, Fig. S9). The G287W mutant aggregation leads to a nearly quantitative loss of PKA C inhibition competency (SI Appendix, Fig. S8C). Overall, our G287W vs. G287E and A211D vs. A211T comparisons consistently support the notion that PKA C activation by PKA R oligomerization is a viable mechanism to explain the PKA overactivation typical of CNC mutants.
Discussion
PKA C Activation by PKA R Oligomerization as a Viable Mechanism for CNC Mutations.
The comparative analysis of the unfolding, aggregation, and kinase inhibition profiles for wt PKA R1α and the two CNC/ACRDYS1 mutant pairs A211D/T and G287W/E reveals a previously unappreciated pathway for PKA C activation distinct from the classical allosteric cAMP-dependent dissociation of the R2:C2 complex, i.e., kinase activation through oligomerization-induced losses of PKA R inhibitory function, as schematically summarized in Fig. 6A. The CNC mutations investigated here promote solvent exposure by lowering the binding affinity for cAMP and/or the free energy of unfolding of apo R (Fig. 6A, blue vertical arrows), as consistently shown by CNC vs. wt enhancements in activation constants (13), urea unfolding (Fig. 2A and SI Appendix, Fig. S1B), computations (SI Appendix, Tables S1 and S4), and HDX NMR (Fig. 2 B–E and SI Appendix, Figs. S2 and S7).
Fig. 6.

Proposed mechanism for the CNC PKA R1α mutations A211D and G287W. (A) Simplified free energy landscape diagram for PKA C activation and inhibition by wt (black) and CNC A211D and G287W (blue) PKA R1α, denoted here simply as R. Abbreviations: A, CBD-A of R; B, CBD-B of R; F, folded native structure of R; U, ensemble of unfolded and partially unfolded states of R (a single free energy level is shown to simplify the scheme); R2 oligomer, oligomerized form of the R dimer; G, Gibbs free energy; ΔUFGapo, unfolding free energy of apo R; ΔBindingGcAMP or C, free energy of cAMP or C binding to R. cAMP is shown as a circle. In the case of the CNC mutants, the circle is deformed to indicate that cAMP occupancies may change compared to wt. The specific mutant vs. wt free energy differences for apo U and apo F are unknown. In the CNC mutants, the ΔUFGapo and/or ΔBindingGcAMP free energies decrease relative to wt, resulting in higher populations of unfolded and/or partially unfolded conformers with exposed hydrophobic surfaces and aggregation-prone sites. Dashed arrows indicate processes that enhance PKA C activation by R oligomerization, which shields loci necessary for C binding and inhibition. (B) Intra- and interdimer protein:protein interaction sites linked to the N3A motif. Zoomed-in view of the interdimer contacts of dimer II in the structure of A211D PKA R1α. (Center) N3A motif where the residues (tan spheres) mediating the dominant intradimer contacts of interface 1 are localized. Key residues of interface 2a that promote interdimer contacts and correlate with the propensity to oligomerize are localized to the linker region that precedes the N3A motif (blue spheres). Interface 2a is conserved in both the wt dimer and in the A211D mutant. An additional interface, denoted as 2b in Table 1, highlights the importance of the β1–β2 loop, specifically, residues 155 and 160 (green spheres), following the N3A motif. (Left) Key contacts between dimers II and III (semitransparent ribbon); (Right) key contacts between dimer II and both adjacent dimers I and III (semitransparent ribbons). Further details are available in Table 1 and SI Appendix, Fig. S5.
The HDX data show that unfolding is not confined to the CBD where the CNC mutation is located but extends also to the other CBD. The pervasive unfolding caused by the CNC mutations exposes to the solvent PKA R sites that are prone to aggregation, such as the αP helices in the two PBCs, thus promoting the formation of cross-β-sheet–rich aggregates of PKA R (Fig. 6A). The propensity to aggregate in the A211D and G287W CNC mutants is consistently supported by fluorescence, NMR intensity losses, DLS, and TEM (Fig. 4 and SI Appendix, Figs. S4 A and B, S8 A and B, and S9). Such aggregation of PKA R1α is distinct from degradation or haploinsufficiency altering the PKA R1α levels. The formation of PKA R1α oligomers does not necessarily remove PKA R-subunits, but it shields from PKA C critical R1α sites needed to form a stable inhibitory R:C interface. Hence, upon aggregation, the PKA C inhibition competency of PKA R1α is compromised, as confirmed by kinase assays (Fig. 4H and SI Appendix, Fig. S9C).
The proposed model (Fig. 6A) shows that there are at least two viable pathways to activate PKA, one classical and one nonclassical. The classical pathway is the well-known allosteric cAMP-dependent dissociation of the inhibitory R2:C2 complex (Fig. 6, gray shaded area). The nonclassical pathway proposed here pertains to the formation of inhibition-incompetent R oligomers (Fig. 6, blue shaded area). While R oligomers can be formed also by wt R (Fig. 4 A, B, and E and SI Appendix, Figs. S4 and S9A) (19), the classical activation pathway prevails in wt PKA. However, the nonclassical activation pathway becomes more relevant for the CNC R mutants, as a result of the unfolding caused by the CNC mutations. Aggregation upon mutation-induced misfolding is a well-known process and applicable to proteins in general (46). However, here we show that this mechanism not only serves as a PKA C activation pathway, but it is also significantly more pronounced in CNC mutants relative to both wt and cognate ACRDYS1 variants.
The C subunit stabilizes the R subunit with respect to unfolding (Fig. 6A), since no major losses in the inhibitory potency of R were previously observed for the A211D and G287W CNC mutations (13). In this respect, the C subunit acts similarly to a chaperone, as it prevents the oligomerization of the R1α subunits. However, in the cell the PKA R subunits are typically present at concentrations close to micromolar, an approximately one order of magnitude excess relative to PKA C (47), thus limiting the ability of PKA C to suppress the oligomerization of PKA R. Substrate competition is another factor limiting the stabilization of dimeric R by C. In general, while it is clear that the in vitro conditions do not fully recapitulate cellular complexity, including chaperone binding, ubiquitination, and degradation, the PKA C activation by PKA R oligomerization model proposed here (Fig. 6A) offers a simple yet viable explanation for the overactivation typically reported for the CNC PKA variants relative to both wt and ACRDYS1 mutants. In addition, mutation-induced aggregation of another tumor suppressor, p53, has been linked to losses of proapoptotic function and consequent carcinogenesis (48). The PKA R1α-oligomerization dependent pathway to activate PKA C is not necessarily exclusive, but it may be synergetic with the cAMP-dependent activation of PKA C.
Synergies between Classical and Nonclassical PKA C Activation Pathways.
The two viable pathways to activate the kinase function of PKA considered here, the canonical allosteric cAMP-dependent dissociation of the inhibitory complex and the noncanonical formation of R oligomers (Fig. 6A), are not necessarily mutually exclusive. For example, cAMP maintains sufficient affinity for the nonmutated domain of A211D (13) to preserve largely unaltered the cAMP-dependent allosteric response of CBD-B. While the allosteric conformational transition of CBD-B alone is not sufficient to fully activate PKA C (49), it does significantly prime the R:C interface for dissociation, thus limiting the ability of PKA C to suppress the oligomerization of PKA R (Fig. 6). Hence, synergies are anticipated between the canonical and noncanonical pathways of PKA C activation and such synergies may explain the enhanced suprabasal activation upon stimulation with low cAMP doses in cell extracts expressing the A211D PKA R1α mutant (13).
The nonclassical activation of PKA through formation of inhibition incompetent R oligomers may be synergetic also with respect to the recently proposed cAMP compartmentalization through liquid–liquid phase separation (LLPS) of PKA R1α in crowded physiological conditions (50). Such PKA R1α condensates not only act as dynamic “sponges” that compartmentalize cAMP, but they also increase the local PKA R1α concentration. The increased PKA R1α levels in these dense droplets may provide additional pathways for PKA R1α fibril formation, which are alternative but nonexclusive to those that rely on nucleation in dilute conditions (51).
Fibril formation causes losses of cAMP-sequestering R1α puncta, which in turn decreases cAMP buffering and compartmentalization. Loss of cAMP compartmentalization leads to spatial dysregulation of cAMP/PKA signaling, which is known to favor tumorigenesis (50), a phenotype also consistent with the Carney complex. Although the structure of such fibrils is not yet known, they may resemble amyloid fibrils in the case of partially unfolded CNC mutants, whereas they may be more closely represented by open-ended agglomerates (46) in the case of folded states of wt or mutant PKA R1α. While it is currently unknown where R1α oligomers fall between these two extreme scenarios, the analysis of the structures of wt and A211D PKA R1α provides unprecedented insight into the interfaces available to R1α dimers to assemble into open-ended agglomerates (Figs. 5 and 6B and SI Appendix, Fig. S5 and Table 1). A recurring and central element coordinating such interfaces is the highly conserved N3A motif of CBDs (52).
The N3A Motif Coordinates the Superquaternary Assembly of PKA R1α Dimers into Open-Ended Agglomerates.
The structures of full-length wt and A211D PKA R1α suggest that the assembly of PKA R1α dimers into open-ended agglomerates primarily relies on two homotypic interdimer interfaces, 2a and 2b (Figs. 5 A–C and 6B and SI Appendix, Fig. S5). Interface 2a is mediated by linker residues 108–117 of protomer′ from dimer I, which form an antiparallel β-sheet with residues 108–117 of protomer′ from dimer II (Fig. 5 A, Center; Fig. 5 B and D; Fig. 6B; and Table 1). Similarly, residues 108–117 from protomer″ from dimer II form an antiparallel β-sheet with residues 108–117 from protomer″ from dimer III (Figs. 5D and 6B), thus enabling the formation of open-ended agglomerates of PKA R1α dimers. This interface was seen in our previous structures of RIα (39, 40), but here we clearly show the role of these contacts in defining the “2a spine” that weaves together and stabilizes the PKA R1α dimer agglomerate (Fig. 5D). The pivotal role of residues 108–117 in agglomeration may also explain why the PKA R1α linker is necessary for the phase separation of PKA R1α into puncta (44).
Another set of interdimer contacts complementing interface 2a is defined by interface 2b between R239 from protomer′ from dimer II and residues 155–160 from protomer″ of dimer I and the corresponding twofold symmetric interactions between dimers I and II (Fig. 5 A, Right; Fig. 5C; Fig. 6B; and Table 1). Similar interactions occur at the interface of dimers II and III (Fig. 6B). Overall, our structures suggest that two adjacent dimers, for example dimers II and III, interact not only through the interface 2a primarily mediated by the 108–117 linker region, but also through the complementary 2b interfaces primarily mediated by residues 155–160 in one promoter and 239 in the other and vice versa (Fig. 6B). This symmetrical oligomerization is favored by the A211D mutant, whereas the 155–160 loop appears to toggle between two different states in the wt R1α.
Based on the analysis of interdimer interfaces in PKA R1α, the N3A motif emerges as a central coordinating unit to control the oligomerization of PKA R1α dimers. The N3A is a highly conserved structural element of CBDs and includes three N-terminal helices, i.e., αX:N, αA, and a short helical loop between them (SI Appendix, Fig. S1A). Multiple N3A residues contribute directly to both the intra- and interdimer interfaces (Figs. 5 and 6B and Table 1). In addition, N3A functions as a bridge between primary sites of interface 2a and 2b (Fig. 6B), i.e., residues 108–117 in the C-linker that precedes the N3A and residues 155–160 in the β1–β2 loop C-terminal to the N3A (SI Appendix, Fig. S1A and Fig. 6B). Therefore, the N3A motif, including the flanking residues, emerges as a core signaling unit not only for the formation of the inhibitory R:C complex and inter-CBD communication, as previously shown (5, 33, 41, 44), but also for the coordination of the superquaternary assembly of PKA R1α.
Multiple Concurrent Pathways of PKA C Activation in CNC.
Although the activation of PKA C by PKA R oligomerization appears to play a significant role for both CNC mutations studied here despite their clearly different locations (Fig. 1A and SI Appendix, Fig. S1A), it is clear that this mechanism is not the only viable explanation for all CNC mutations escaping NMD. For instance, A211D is more susceptible to proteolytic digestion compared to wt PKA R1α (SI Appendix, Fig. S4D), which also may lead to enhanced PKA C activation. Alternative models may apply to other CNC mutants different from those investigated here; however, our work presented here sheds light also on each of these mutations. For example, the PKA R1α S145G and R144S CNC mutations have been proposed to perturb quaternary interactions, resulting in less cooperative cAMP-dependent activation and lower EC50 cAMP concentrations compared to wt PKA (18). The enhanced sensitivity to cAMP of the S145G and R144S CNC mutants is consistent with the PKA overactivation phenotype typical of CNC. However, poor solubility was reported for the S145N mutant (18), suggesting that PKA C activation by PKA R oligomerization may play a role also for this CNC mutation. In addition, the agglomerate will not form without the stable intramolecular N3A dimer. Thus, those CNC mutants that occur in the N3A motif, which serves as a central hub not only for the cAMP-dependent allostery but also for oligomerization, are likely to affect the dimer:dimer interfaces that we mapped here. The importance of the N3A motif and its flanking segments for coordinating the interfaces that drive the PKA R1α agglomerates is revealed clearly by the A211D structure (Figs. 5 and 6B; SI Appendix, Fig. S5; and Table 1). In addition, mutation-induced unfolding of CBD-A is effective in destabilizing CBD-B, while in general mutations that induce unfolding of CBD-B have typically little effect on destabilizing CBD-A (53). This notion may explain, in the context of the proposed nonclassical mechanism of PKA C activation by PKA R aggregation, why the majority of CNC PKA R1α mutations are found in CBD-A (16).
These conclusions highlight the importance of considering multiple concurrent mechanisms even when explaining the same disease-related mutation (54–57). Furthermore, additional layers of complexity may need to be considered, as PKA R1α serves as a versatile sensor of specific subcellular metabolic, proteomic, and redox environments (49). Nevertheless, the simple model of noncanonical PKA C activation by PKA R oligomerization proposed here (Fig. 6) offers a viable mechanistic explanation to reconcile the apparent discrepancy between the PKA C overactivation phenotype typical of CNC mutants and the decreased cAMP sensitivity of key CNC PKA R1α mutants. It may also provide an explanation for the recently described LLPS of RIα.
Materials and Methods
Bovine PKA R1α and its deletion constructs were expressed and purified, as previously described (27, 28). Urea unfolding and NMR experiments were implemented as previously discussed (22, 27, 28, 53). Further details are provided in SI Appendix (5, 18, 19, 24, 26–28, 41, 42, 53, 58–71). SI Appendix also includes details on ThT and ANS fluorescence, DLS, and TEM data acquisition, PKA inhibition assay, protein crystallization and data collection, molecular replacement and structure refinement, as well as trypsin digestion.
Supplementary Material
Acknowledgments
We are grateful to Marcia Reid for assistance with the acquisition of TEM images and, together with Dr. R. Maillard, Dr. G. Veglia, Dr. Bryan VanSchouwen, and Dr. Rashik Ahmed, for helpful discussions. Funding was provided by Canadian Institutes of Health Research Grant 389522 (to G.M.), Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2019-05990 (to G.M.), and NIH Grant R35-GM130389 (to S.S.T.). Beamline 822 of the Advanced Light Source, a US Department of Energy Office of Science User Facility under Contract DE-AC02-05CH11231, is supported in part by the Advanced Light Source–ENABLE program funded by NIH–National Institute of General Medical Sciences Grant P30 GM124169-01.
Footnotes
The authors declare no competing interest.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2024716118/-/DCSupplemental.
Data Availability
All relevant data are included in the article and in SI Appendix. The atomic coordinates have been deposited in the Protein Data Bank, https://www.wwpdb.org (PDB ID code 7LZ4) (72).
References
- 1.Linglart A., et al., Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N. Engl. J. Med. 364, 2218–2226 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Mantovani G., Elli F. M., Multiple hormone resistance and alterations of G-protein-coupled receptors signaling. Best Pract. Res. Clin. Endocrinol. Metab. 32, 141–154 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Cho-Chung Y. S., Pepe S., Clair T., Budillon A., Nesterova M., cAMP-dependent protein kinase: Role in normal and malignant growth. Crit. Rev. Oncol. Hematol. 21, 33–61 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Taylor S. S., Ilouz R., Zhang P., Kornev A. P., Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim C., Cheng C. Y., Saldanha S. A., Taylor S. S., PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Ruff E. F., et al., A dynamic mechanism for allosteric activation of Aurora kinase A by activation loop phosphorylation. eLife 7, e32766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lake E. W., et al., Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. U.S.A. 115, E11894–E11903 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olivieri C., et al., Multi-state recognition pathway of the intrinsically disordered protein kinase inhibitor by protein kinase A. eLife 9, e55607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirschner L. S., et al., Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat. Genet. 26, 89–92 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Salpea P., Stratakis C. A., Carney complex and McCune Albright syndrome: An overview of clinical manifestations and human molecular genetics. Mol. Cell. Endocrinol. 386, 85–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamilaris C. D. C., Faucz F. R., Voutetakis A., Stratakis C. A., Carney complex. Exp. Clin. Endocrinol. Diabetes 127, 156–164 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Carney D. N., et al., Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 45, 2913–2923 (1985). [PubMed] [Google Scholar]
- 13.Rhayem Y., et al., Functional characterization of PRKAR1A mutations reveals a unique molecular mechanism causing acrodysostosis but multiple mechanisms causing Carney complex. J. Biol. Chem. 290, 27816–27828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruystens J. G. H., et al., Structure of a PKA RIα recurrent acrodysostosis mutant explains defective cAMP-dependent activation. J. Mol. Biol. 428, 4890–4904 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horvath A., et al., Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Hum. Mutat. 31, 369–379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothenbuhler A., Stratakis C. A., Clinical and molecular genetics of Carney complex. Best Pract. Res. Clin. Endocrinol. Metab. 24, 389–399 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Greene E. L., et al., In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum. Mutat. 29, 633–639 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Bruystens J. G. H., et al., PKA RIα homodimer structure reveals an intermolecular interface with implications for cooperative cAMP binding and Carney complex disease. Structure 22, 59–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dao K. K., et al., The regulatory subunit of PKA-I remains partially structured and undergoes β-aggregation upon thermal denaturation. PloS One 6, e17602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R., Abu-Abed M., Melacini G., Mapping allostery through equilibrium perturbation NMR spectroscopy. J. Am. Chem. Soc. 128, 8406–8407 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Tipping K. W., van Oosten-Hawle P., Hewitt E. W., Radford S. E., Amyloid fibres: Inert end-stage aggregates or key players in disease? Trends Biochem. Sci. 40, 719–727 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leon D. A., Canaves J. M., Taylor S. S., Probing the multidomain structure of the type I regulatory subunit of cAMP-dependent protein kinase using mutational analysis: Role and environment of endogenous tryptophans. Biochemistry 39, 5662–5671 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Cànaves J. M., Leon D. A., Taylor S. S., Consequences of cAMP-binding site mutations on the structural stability of the type I regulatory subunit of cAMP-dependent protein kinase. Biochemistry 39, 15022–15031 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Yin S., Ding F., Dokholyan N. V., Eris: An automated estimator of protein stability. Nat. Methods 4, 466–467 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Das R., et al., cAMP activation of PKA defines an ancient signaling mechanism. Proc. Natl. Acad. Sci. U.S.A. 104, 93–98 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su Y., et al., Regulatory subunit of protein kinase A: Structure of deletion mutant with cAMP binding domains. Science 269, 807–813 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Akimoto M., et al., Mapping the free energy landscape of PKA inhibition and activation: A double-conformational selection model for the tandem cAMP-binding domains of PKA RIα. PLoS Biol. 13, e1002305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akimoto M., et al., Signaling through dynamic linkers as revealed by PKA. Proc. Natl. Acad. Sci. U.S.A. 110, 14231–14236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Escamilla A.-M., Rousseau F., Schymkowitz J., Serrano L., Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22, 1302–1306 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Linding R., Schymkowitz J., Rousseau F., Diella F., Serrano L., A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 342, 345–353 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Milojevic J., Raditsis A., Melacini G., Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism. Biophys. J. 97, 2585–2594 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milojevic J., Esposito V., Das R., Melacini G., Understanding the molecular basis for the inhibition of the Alzheimer’s Abeta-peptide oligomerization by human serum albumin using saturation transfer difference and off-resonance relaxation NMR spectroscopy. J. Am. Chem. Soc. 129, 4282–4290 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Algamal M., Milojevic J., Jafari N., Zhang W., Melacini G., Mapping the interactions between the Alzheimer’s Aβ-peptide and human serum albumin beyond domain resolution. Biophys. J. 105, 1700–1709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Algamal M., et al., Atomic-resolution map of the interactions between an amyloid inhibitor protein and amyloid β (Aβ) peptides in the monomer and protofibril states. J. Biol. Chem. 292, 17158–17168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biancalana M., Koide S., Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gasymov O. K., Glasgow B. J., ANS fluorescence: Potential to augment the identification of the external binding sites of proteins. Biochim. Biophys. Acta 1774, 403–411 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weids A. J., Ibstedt S., Tamás M. J., Grant C. M., Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 6, 24554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahoo B. R., Cox S. J., Ramamoorthy A., High-resolution probing of early events in amyloid-β aggregation related to Alzheimer’s disease. Chem. Commun. 56, 4627–4639 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox S. J., et al., High-throughput screening at the membrane interface reveals inhibitors of Amyloid-β. Biochemistry 59, 2249–2258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rousseau F., Schymkowitz J., Serrano L., Protein aggregation and amyloidosis: Confusion of the kinds? Curr. Opin. Struct. Biol. 16, 118–126 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Conchillo-Solé O., et al., AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8, 65 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu T.-W., et al., Two PKA RIα holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc. Natl. Acad. Sci. U.S.A. 116, 16347–16356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J., Brown S., Xuong N.-H., Taylor S. S., RIalpha subunit of PKA: A cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure 12, 1057–1065 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Wu J., Jones J. M., Nguyen-Huu X., Ten Eyck L. F., Taylor S. S., Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry 43, 6620–6629 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Shell J. R., Lawrence D. S., Proteolytic regulation of the mitochondrial cAMP-dependent protein kinase. Biochemistry 51, 2258–2264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Seisdedos H., Villegas J. A., Levy E. D., Infinite assembly of folded proteins in evolution, disease, and engineering. Angew. Chem. Int. Ed. Engl. 58, 5514–5531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker-Gray R., Stengel F., Gold M. G., Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. U.S.A. 114, 10414–10419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang-Hartwich Y., Bingham J., Garofalo F., Alvero A. B., Mor G., Detection of p53 protein aggregation in cancer cell lines and tumor samples. Methods Mol. Biol. 1219, 75–86 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Byun J. A., et al., Allosteric pluripotency as revealed by protein kinase A. Sci. Adv. 6, eabb1250 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J. Z., et al., Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell 182, 1531–1544.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mathieu C., Pappu R. V, Taylor J. P., Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 370, 56–60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao Y., et al., Activation of PKA via asymmetric allosteric coupling of structurally conserved cyclic nucleotide binding domains. Nat. Commun. 10, 3984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNicholl E. T., Das R., SilDas S., Taylor S. S., Melacini G., Communication between tandem cAMP binding domains in the regulatory subunit of protein kinase A-Iα as revealed by domain-silencing mutations. J. Biol. Chem. 285, 15523–15537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng F., Liang H., Butte A. J., Eng C., Nussinov R., Personal mutanomes meet modern oncology drug discovery and precision health. Pharmacol. Rev. 71, 1–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lei J., et al., Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA. FASEB J. 33, 4225–4235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyken S. E., Fulton D. B., Andreotti A. H., Rescue of the aggregation prone Itk Pleckstrin Homology domain by two mutations derived from the related kinases, Btk and Tec. Protein Sci. 21, 1288–1297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levytskyy R. M., et al., In vivo consequences of disrupting SH3-mediated interactions of the inducible T-cell kinase. J. Signal Transduct. 2012, 694386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moleschi K. J., Akimoto M., Melacini G., Measurement of state-specific association constants in allosteric sensors through molecular stapling and NMR. J. Am. Chem. Soc. 137, 10777–10785 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Otwinowski Z., Minor W., Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 60.Storoni L. C., McCoy A. J., Read R. J., Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Cowtan K., Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 470–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schröder G. F., Levitt M., Brunger A. T., Super-resolution biomolecular crystallography with low-resolution data. Nature 464, 1218–1222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Brunger A. T., Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Winn M. D., et al., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winn M. D., Isupov M. N., Murshudov G. N., Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M., PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993). [Google Scholar]
- 68.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Frousios K. K., Iconomidou V. A., Karletidi C.-M., Hamodrakas S. J., Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 9, 44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Badireddy S., et al., Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: Conformational selection highlights a new concept in allosteric inhibitor design. Mol. Cell. Proteomics 10, 004390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Das R., et al., Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J. Biol. Chem. 284, 23682–23696 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Del Rio J., Wu J., Taylor S. S., Crystal structure of A211D mutant of Protein Kinase A RIa subunit, a Carney Complex mutation. Protein Data Bank. https://www.rcsb.org/structure/7LZ4. Deposited 8 March 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Del Rio J., Wu J., Taylor S. S., Crystal structure of A211D mutant of Protein Kinase A RIa subunit, a Carney Complex mutation. Protein Data Bank. https://www.rcsb.org/structure/7LZ4. Deposited 8 March 2021.
Supplementary Materials
Data Availability Statement
All relevant data are included in the article and in SI Appendix. The atomic coordinates have been deposited in the Protein Data Bank, https://www.wwpdb.org (PDB ID code 7LZ4) (72).