

Abstract
Increased brain microvascular permeability and disruption of blood-brain barrier (BBB) function are among hallmarks of several acute neurodegenerative disorders, including stroke. Numerous studies suggest the involvement of bradykinin (BK), neurotensin (NT) and substance P (SP) in BBB impairment and oedema formation after stroke; however, there is paucity of data in regard to the direct effects of these peptides on the brain microvascular endothelial cells (BMECs) and BBB. The present study aimed to evaluate the direct effects of BK, NT and SP on the permeability of BBB in an in vitro model based on human induced pluripotent stem cell (iPSC)-derived BMECs. Our data indicate that all three peptides increase BBB permeability in a concentration-dependent manner in an in vitro model formed from two different iPSC lines (CTR90F and CTR65M) and widely used hCMEC/D3 human BMECs. The combination of BK, NT and SP at a sub-effective concentration also resulted in increased BBB permeability in the iPSC-derived model indicating potentiation of their action. Furthermore, we observed abrogation of BK, NT and SP effects with pretreatment of pharmacological blockers targeting their specific receptors. Additional mechanistic studies indicate that the short-term effects of these peptides are not mediated through alteration of tight-junction proteins claudin-5 and occludin, but likely involve redistribution of F-actin and secretion of vascular endothelial growth factor. This is the first experimental study to document the increased permeability of the BBB in response to direct action of NT in an in vitro model. In addition, our study confirms the expected but not well-documented, direct effect of SP on BBB permeability and adds to the well-recognised actions of BK on BBB. Lastly, we demonstrate that peptidase neurolysin can neutralise the effects of these peptides on BBB, suggesting potential therapeutic implications.
Keywords: blood-brain barrier, oedema formation, microvascular permeability, neuropeptide, peptidase neurolysin
1 |. INTRODUCTION
Stroke is one of the major acute neurodegenerative disorders, constituting the fifth cause of death and the leading cause of acquired disability in the USA.1 The majority of stroke events are caused by occlusion of a cerebral artery, followed by secondary injury cascades involving glutamate-mediated excitotoxicity, oxidative stress, blood-brain barrier (BBB) hyper-permeability, brain oedema, neurogenic inflammation and ultimately neuronal cell death. Importantly, the maintenance of BBB and its subsequent disruption during and after ischaemic stroke have been recognised as central for resistance to ischaemic injury and its expansion.2–5 The contribution of neurone-derived signalling molecules on the maintenance and disruption of the BBB during ischaemic stroke remains the focus of ongoing studies. In particular, the contribution of neuropeptides, which represent the largest group of signalling molecules with pleiotropic functions,6 remains understudied. The possible contribution of neuropeptides in the response of BBB to ischaemic injury is of critical importance because neuropeptides are commonly considered as the ‘language of the stressed nervous system’, and their expression is especially increased following various stressors.7
In particular, three neuropeptides, bradykinin (BK), neurotensin (NT) and substance P (SP), as well as their cognate signalling pathways, have been identified as critical players in pathogenic mechanisms of ischaemic stroke.8,9 For example, BK has been reported to increase BBB permeability via down-regulation of claudin-5,10 whereas activation of NT receptor via pharmacological agonists has been reported as stroke neuroprotective via pharmacologically-induced hypothermia.11 On the other hand, SP has been reported to increase BBB permeability,12 whereas elevated SP serum level in stroke patients was associated with poor prognosis of survival.13 Despite this, the direct effect of these neuropeptides on the BBB integrity remains poorly documented.
Notably, these neuropeptides are the main substrates of neurolysin (Nln), a peptidase that degrades and inactivates all three of these peptides.9 Nln is a monomeric metalloendopeptidase with an approximate size of 75 kDa that is highly expressed in the human brain.14,15 Initially described as a peptidase capable of cleaving NT, this enzyme has been also shown to degrade BK and SP.16,17 Nln has been primarily described as a cytosolic protein but, depending on a cell type, it can also be found as a secreted or membrane-bound enzyme.18,19 Notably, expression of membrane-bound Nln is up-regulated following focal cerebral ischaemia in vitro and in vivo.20,21 Overexpression of Nln in mice prior to ischaemia (using an adeno-associated virus vector) was shown to improve stroke outcomes, whereas inhibition of Nln by agaricoglyceride A (a small molecule Nln inhibitor) resulted in a worsening of stroke injury.9 Importantly, the observed improvement of stroke outcomes by Nln overexpression in this study was associated with decreased levels of BK, NT and SP, whereas aggravation of injury in response to Nln inhibition, was associated with increased levels of these neuropeptides.
Because the direct effects of NT, BK and SP on the brain capillary endothelial cells and BBB function are not fully understood, the present study aimed to evaluate the direct effects of NT, BK and SP on the permeability of BBB in an in vitro model formed from human induced pluripotent stem cell (iPSC)-derived brain microvascular endothelial cells.22–26 Our data indicate that all three peptides increase the permeability of BBB in a concentration-dependent manner in an in vitro model formed from two different iPSC lines (CTR90F and CTR65M). Notably, these effects were similar to that of a widely used in vitro BBB model based on hCMEC/D3 human brain microvascular endothelial cells. Combination of NT, BK and SP at a sub-effective concentration also resulted in increased BBB permeability in the iPSC-derived model. Furthermore, we observed abrogation of NT, BK and SP effects with pretreatment of pharmacological blockers targeting their specific receptors or in the presence of recombinant Nln (rNln). Our preliminary mechanistic studies indicate that the short-term effects of these peptides are not mediated through alteration of tight-junction proteins claudin-5 and occludin, but likely involve the redistribution of F-actin and the secretion of vascular endothelial growth factor (VEGF). This is the first experimental study to document increased permeability of BBB in response to direct action of NT in an in vitro model. In addition, our study confirms the expected but not well-known, direct effect of SP on BBB permeability in the same in vitro model, and adds to the well-recognised actions of BK on BBB. Lastly, the observed ability of peptidase Nln to neutralise the effects of all three peptides on BBB suggests that this peptidase may serve as a therapeutic target.
2 |. MATERIALS AND METHODS
2.1 |. Cell culture
hCMEC/D3 immortalised human brain microvascular cell line (RRID: CVCL_U985) was purchased from EMD Millipore (Burlington, MA, USA) and maintained in accordance with the manufacturer’s instructions. This cell line was originally isolated from a female patient suffering from temporal lobe epilepsy.27 CTR90F28 (IPS(IMR90)-c4; RRID: CVCL_C437) and CTR65M (ND41865, RRID: CVCL_Y837) human iPSCs were obtained from WiCell (Madison, WI, USA) and from Coriell Institute of Medical Research (Camden, NJ, USA) repositories, respectively. Cells were maintained and differentiated as described previously.25 Undifferentiated iPSCs were seeded at a density of 2 × 104 cells cm−2 and maintained in Essential-8 Medium (Life Technologies, ThermoFisher, Waltham, MA, USA) and grown on a Matrigel-coated (Trevigen, Gaithersburg, MD, USA) tissue culture plastic surface (TCPS) for 5 days prior to differentiation. For differentiation we followed the protocol developed by Lippmann et al.29 Differentiation was achieved by growing undifferentiated iPSCs in unconditioned media (Dulbecco’s modified Eagle’s medium/F12 with 15 mmol L−1 Hepes supplemented with 20% KO serum replacement, 1% minimum essential medium non-essential amino acids and 0.5% Glutamax I [ThermoFisher] and 0.1 mmol L−1 β-mercaptoethanol [Sigma-Aldrich, St Louis, MO, USA]) for 6 days, with medium changed daily. After 6 days, iPSC-derived brain microvascular endothelial cells (BMECs)s were incubated in EC+/+ (EC serum free medium; ECSFM; ThermoFisher), supplemented with 1% platelet-poor derived plasma serum (PDS; ThermoFisher), 20 μg mL−1 human basic fibroblast growth factor [R&D Systems, Minneapolis, MN, USA] and 10 μmol/L all-trans retinoic acid [Sigma-Aldrich]) for 2 days. Maturing BMECs were enzymatically dissociated by Accutase® (Corning Inc., Corning, NY, USA) and seeded on TCPS and Transwell® inserts (Corning Inc.; polyester membrane, 0.4 μm pore size) coated with collagen from human placenta (Sigma-Aldrich) and fibronectin from bovine plasma (Sigma-Aldrich) at concentrations of 80 and 20 μg cm−2, respectively. Cells were allowed to recover for 24 hours and were incubated in EC−/− (supplemented with 1% PDS) for another 24 hours. All experiments were carried out by day 10 of differentiation.
The average transendothelial electrical resistance (TEER) values found in CTR90F and CTR65M BMECs under resting conditions were 1054 and 1387 Ω‧cm2, whereas hCMEC/D3 cells had average of 20.83 Ω‧cm2. Likewise, under basal conditions, fluorescein permeability (Pe, expressed as 10−4 cm min−1) was 0.21 and 0.18 in CTR90F and CTR65M BMECs and 5.22 in hCMEC/D3 cells.
2.2 |. Barrier function and co-culture experiments
BMEC monolayer tightness was measured by assessing TEER using an EVOM STX2 chopstick electrode (World Precision Instruments, Sarasota, FL, USA). TEER was measured in three different locations on the insert, averaged, and subtracted from values obtained in blank inserts. TEER was measured prior the experiment (to establish the baseline of each insert), followed by measurements at 15, 30, 45 and 60 minutes during the experiment. Paracellular permeability was assessed by adding 1 μmol L−1 fluorescein solution (SigmaAldrich) in the donor (top) chamber. Diffusion of fluorescein across monolayers was measured by sampling 100 μL of the receiver (bottom) chamber every 15 minutes for 60 minutes. Fluorescence was measured using a Synergy MX2 plate reader (Bio-Tek Instruments, Winooski, VT, USA).
2.3 |. Recombinant neurolysin
Recombinant Nln was produced in house as described previously.16 In brief, we used a high-expressing Escherichia coli, clone which was transformed by a plasmid construct for N-terminal polyhistidine-tagged recombinant rat Nln.30 Recombinant Nln was purified to homogeneity (> 95%) from the soluble pool of E. coli lysate using Ni-NTA affinity (Ni-NTA Superflow; Qiagen, Venlo, The Netherlands) and size-exclusion chromatography (Superdex 200 Increase 10/300 GL column in AKTA Purifier FPLC system; GE Healthcare, Chicago, IL, USA). Purification of Nln was followed by removal of endotoxins (endotoxin removal magnetic beads; Miltenyi Biotec, Bergisch Gladbach, Germany) and confirmation of its catalytic activity using a quenched fluorescent substrate.9,18 Small volume aliquots of Nln (1–2 mg mL−1) were kept frozen in −80°C and used within 3–4 months after isolation.
2.4 |. Neuropeptides and pharmacological treatment
NT, SP and BK were purchased from Phoenix Peptides (Burlingame, CA), dissolved in autoclaved water to obtain 5 or 10 mmol L−1 stock solution and kept frozen at −20°C until use. Neuropeptides were freshly diluted in EC−/− (with 1% PDS) to obtain the desired concentrations and immediately added to the apical chamber. Similarly, the cells in control wells, which were not treated with either of the peptides, received fresh EC−/−. In experiments involving Nln, neuropeptides were co-incubated in presence of Nln for 10 minutes at 37°C. Following incubation, the mixture was added on the apical chamber of the Transwell® inserts. In experiments involving selective pharmacological antagonists of BK, NT or SP receptors, cells were pre-treated with pharmacological inhibitors dissolved in EC−/− for 1 hour. After 1 hour of pre-incubation, the medium on the apical chamber was replaced with fresh medium containing both a neuropeptide and an antagonist. Selective antagonists used in this study were icatibant acetate (B2 bradykinin receptor antagonist; Tocris, Bio-Techne, Minneapolis, MN, USA), des-Arg9-[Leu8]-Bradykinin (B1 bradykinin receptor antagonist; Phoenix Peptides), SR48692 (NT1 neurotensin receptor antagonist; Tocris, Bio-Techne), Aprepitant and L 760 735 (NK1 receptor antagonists; Tocris, Bio-Techne).
2.5 |. Immunofluorescence microscopy
Following exposure to 1 µmol L−1 BK, NT or SP for 60 minutes, the cells were quickly washed with ice-cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Fisher Scientific, Waltham, MA, USA) for 5 minutes and blocked for 30 minutes (10% goat serum and 0.2% Triton-X100 in PBS). Then, the BMEC monolayers were incubated with mouse-anti human claudin-5 (dilution 1:100, ThermoFisher; RRID: AB_2533200) or mouse-anti human occludin (dilution 1:100, ThermoFisher; AB_2533101) antibodies overnight at 4°C. Primary antibodies were detected using a goat-anti mouse IgG Alexa Fluor® 555-conjugated secondary anti-body (ThermoFisher). In experiments involving F-actin, freshly fixed BMEC monolayers were stained with 0.1 mg mL−1 fluorescein isothiocyanate-conjugated phalloidin (Sigma-Aldrich) for 30 minutes. All images were acquired using a DMi8 inverted epifluorescence microscope (Leica Microsystems, Wetzlar, Germany). Images were analysed using IMAGEJ (NIH, Bethesda, MD, USA).
2.6 |. VEGF ELISA
Confluent BMEC monolayers grown on 24-well plates were treated with 1 µmol L−1 BK, NT or SP for 60 minutes followed by collection of the cell culture medium. The latter were briefly centrifuged and supernatants were immediately frozen and stored at −80°C. The amount of VEGF was measured using Quantikine® ELISA kit for human VEGF (R&D Systems). The medium of cells not exposed to either of peptides served as negative control in these experiments.
2.7 |. Statistical analysis
All experiments were performed using at least three independent biological replicates (three independent passages and cell differentiation). As a result of variability in recorded TEER values between experiments, the average TEER value of untreated cells (ie control group) within each experiment was arbitrarily set to 100% and was used to normalise the TEER values of other experimental groups. The difference in TEER values (ΔTEER) was obtained by calculating slope of the line for each experiment using linear regression model in PRISM, version 8.0 (GraphPad Software Inc., San Diego, CA, USA) and multiplying by the duration of the experiment (60 minutes). Likewise, permeability (Pe) for fluorescein was calculated using the clearance slopes for each 60-minute experiment as described previously.25 Statistical analysis was performed using two-tailed unpaired t test and one- or two-way ANOVA complemented by Dunnett’s post-hoc test (PRISM, version 8.0). Data from time-course experiments were analysed using repeated measures two-way ANOVA followed by Dunnett’s post-hoc test. Output from the statistical analyses of all experiments/figures is provided in the Supporting information (File S1). Data are presented as the mean ± SD. P < 0.05 was considered statistically significant.
3 |. RESULTS
3.1 |. iPSC-derived BMECs monolayers are responsive to BK, NT and SP
The effects of BK, NT and SP on the barrier function of hCMEC/D3 and iPSC-derived BMEC monolayers were first assessed at a concentration of 1 µmol L−1 (Figure 1), as used by other researchers in various in vitro studies.31,32 Treatment of hCMEC/D3 monolayers with 1 µmol L−1 BK, NT or SP resulted in a slight but statistically not significant decrease in TEER values compared to control cells (time × treatment F12,32 = 0.969, P = 0.496) within the first 30 minutes of treatment (Figure 1A). However, this decrease was less apparent by 45 and 60 minutes of the experiment. By contrast, the first group of BMEC monolayers derived from CTR90F iPSCs (CTR90F-BMECs), showed a more profound decrease in TEER values following treatment with the peptides (time × treatment F12,32 = 10.58, P < 0.05). In particular, in the presence of 1 µmol L−1 NT, the effect was sustained for at least 60 minutes (q = 5.42, df = 2.39, P < 0.05) (Figure 1A). Furthermore, the second group of BMEC monolayers derived from CTR65M iPSCs (CTR65M-BMECs), showed a statistically significant decrease in TEER values (time × treatment F12,48 = 11.42, P < 0.05) after BK (q = 5.41, df = 3.15, P < 0.05 at 60 minutes) but not NT (q = 1.37, df = 5.52, P > 0.05 at 60 minutes) treatments. A substantial decrease in TEER values was also observed after treatment of these cells with 1 µmol L−1 SP (q = 4.79, df = 3.18, P < 0.05 at 60 minutes) (Figure 1A). To confirm the observed changes in TEER values in these experiments, we also conducted parallel assessment of fluorescein permeability in the same Transwells® (Figure 1B). Treatment with 1 µmol L−1 NT or SP yielded an approximately two-fold albeit statistically non-significant increase in permeability of fluorescein in hCMEC/D3 monolayers (q = 2.00, df = 8, P > 0.05 and q = 1.45, df = 8, P > 0.05), whereas BK treatment resulted in a two-fold statistically significant increase in fluorescein permeability in both CTR90F and CTR65M-derived BMECs (q = 4.40, df = 8, P < 0.05 and q = 2.76, df = 12, P < 0.05, respectively). Notably, treatment with 1 µmol L−1 NT resulted in a statistically significant increase in permeability of fluorescein in CTR90F-BMECs (q = 3.14, df = 8, P < 0.05), whereas CTR65M-BMECs showed an apparent increase in permeability following incubation with 1 µmol L−1 SP (q = 2.54, df = 12, P = 0.06).
FIGURE 1.
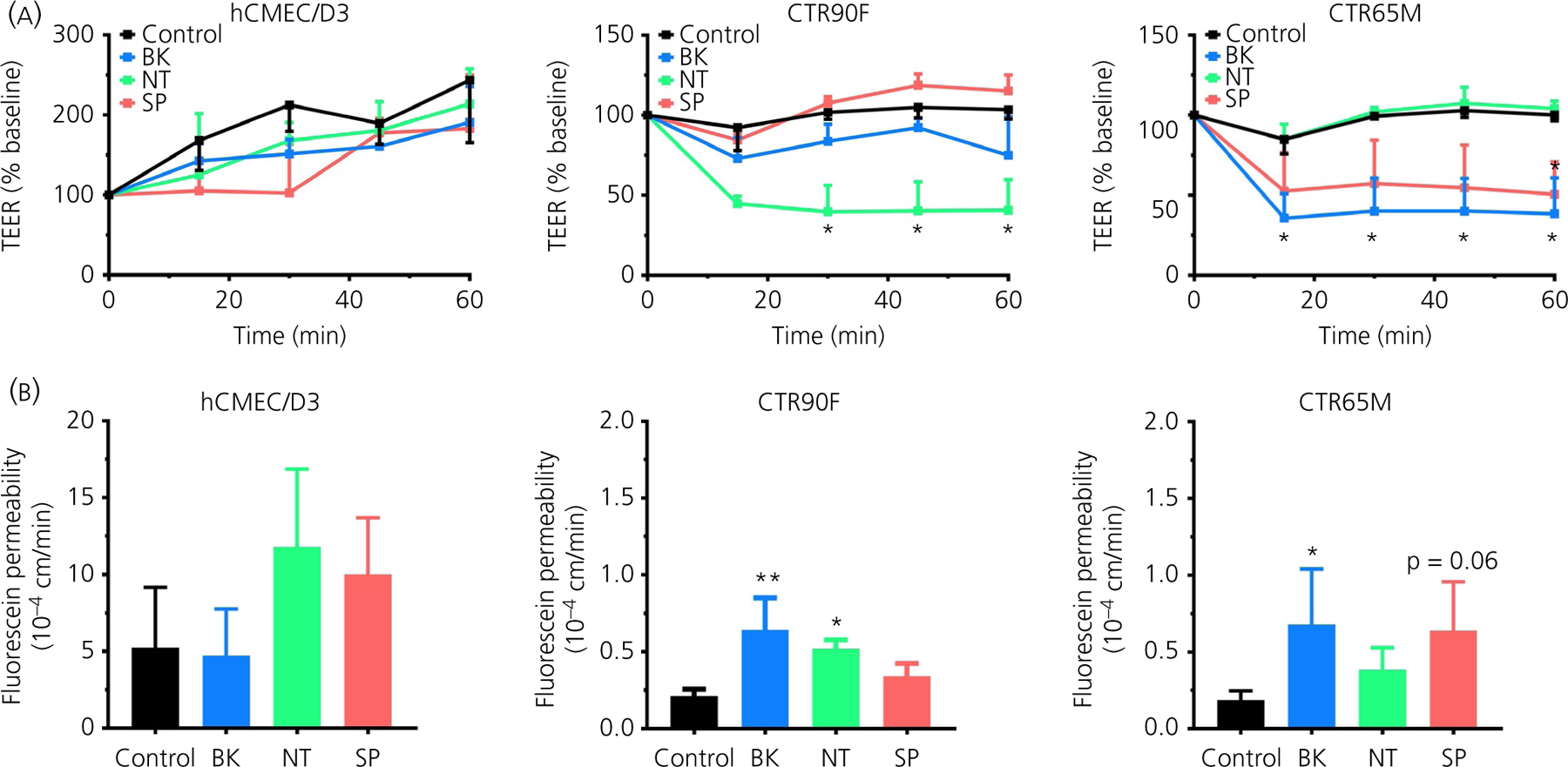
Bradykinin (BK), neurotensin (NT) and substance P (SP) increase the barrier permeability in hCMEC/D3 and induced pluripotent stem cell (iPSC)-derived brain microvascular endothelial cells (BMECs). Changes in the barrier function were assessed in real-time in hCMEC/D3 and two iPSC-derived BMECs (CTR90F and CTR65M). Sampling for transendothelial electrical resistance (TEER) measurements (A) and paracellular permeability of fluorescein (B) were carried out every 15 minutes for 60 minutes. Baseline TEER was measured immediately before the start of treatments to normalise the experimental data. The apical compartment conditioned medium was replaced with fresh EC−/− containing nothing additional (control), 1 µmol L−1 BK (blue), 1 µmol L−1 NT (green) or 1 µmol L−1 SP (red) (n = 3 or 4 per group; *P < 0.05, **P < 0.01 compared to control group)
3.2 |. Concentration-dependent effects of BK, NT and SP on BBB integrity
In this set of experiments different concentrations of BK, NT and SP (0.1–1.0 µmol L−1) were used to determine their effects on the barrier function of iPSC-derived BMECs (Figure 2). Both CTR90F and CTR65M-derived BMECs showed unique TEER response to varying concentrations of BK, NT and SP (Figure 2A, B). In CTR90F-derived BMECs, BK or SP treatment resulted in transient and non-significant changes of TEER, whereas a statistically significant decrease was observed with 1 µmol L−1 NT (time × concentration F12,64 = 4.20, P < 0.05; q = 4.26, df = 3.71 at 60 minutes) (Figure 2A). By contrast, concentration-dependent decrease in TEER values was observed in CTR65M-derived BMECs treated with BK (time × concentration F12,64 = 3.88, P < 0.05; q = 4.54, df = 4.08 at 60 minutes) or SP (time × concentration F12,60 = 2.97, P < 0.05; q = 3.86, df = 4.33 at 60 minutes), with a statistically significant drop at a concentration of 1 µmol L−1 (Figure 2B). In addition to time-course evaluation of changes in TEER values (Figure 2A, B), we also calculated the difference in TEER (ΔTEER) during 60-minute experimental period (Figure 2C), similar to the extrapolation method used to determine paracellular permeability (Pe) values (Figure 2D). Here, we report similar outcomes and observed statistically significant decrease in ΔTEER in CTR90F-derived BMECs following 1 µmol L−1 NT treatment (q = 4.96, df = 26, P < 0.01). Likewise, we observed statistically significant decrease in ΔTEER in CTR65M-BMECs following 1 µmol L−1 BK (q = 4.88, df = 30, P < 0.01) or SP (q = 4.03, df = 30, P < 0.01) treatment. Similar to the previous set of experiments, here too, a parallel evaluation of fluorescein permeability was carried out in the same Transwells® (Figure 2D). In CTR90F-derived BMECs, a statistically significant increase in fluorescein permeability was observed for BK and NT at a concentration of 1 µmol L−1 (q = 6.90, df = 26, P < 0.01 and q = 5.02, df = 26, P < 0.05, respectively). In CTR65M-BMECs statistically significant increase in permeability of fluorescein was observed following treatment with 1 µmol L−1 BK (q = 4.75, df = 30, P < 0.01) and 0.5 and 1 µmol L−1 SP (q = 4.45, df = 30, P < 0.01 and q = 4.38, df = 30, P < 0.01, respectively). Similar to the first set of experiments (Figure 1B), 1 µmol L−1 NT showed a small but statistically non-significant increase in permeability of fluorescein in CTR65M-BMECs (q = 1.97, df = 30, P > 0.05) (Figure 2D).
FIGURE 2.
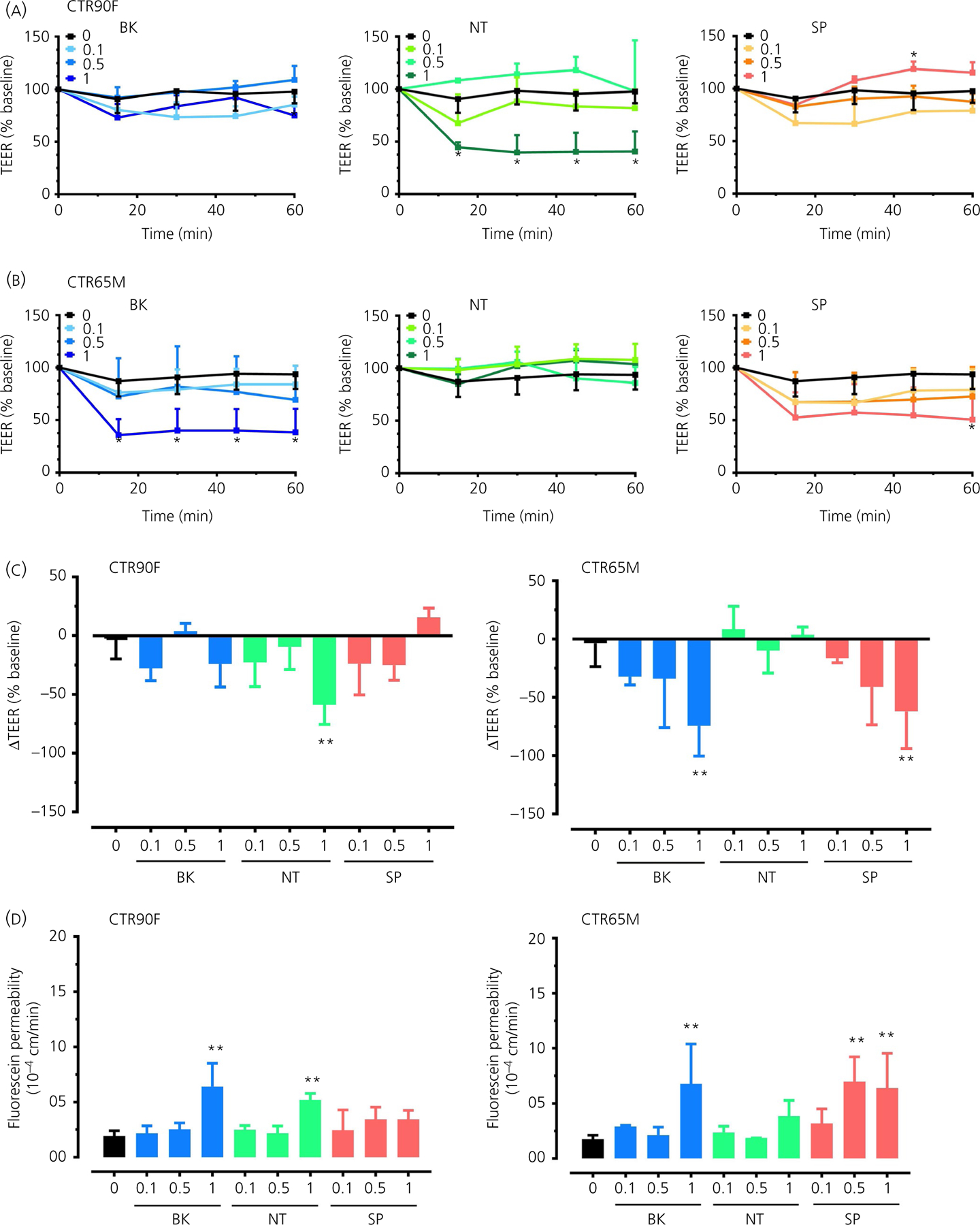
Induced pluripotent stem cell (iPSC)-derived brain microvascular endothelial cells (BMECs) show a concentration-dependent response to bradykinin (BK), neurotensin (NT) and substance P (SP). Concentration-dependent (0.1, 0.5 and 1 µmol L−1) effect of BK (blue), NT (green) and SP (red) on transendothelial electrical resistance (TEER) values in CTR90F (A) and CTR65M (B) iPSC-BMECs, with effects being observed as early as 15 minutes after treatment. Difference in TEER values (ΔTEER) measured in CTR90F and CTR65M BMECs in response to different concentrations of the peptides (C). Fluorescein permeability measured in the iPSC-derived BMECs monolayers (D) (n = 9 for no peptide, ie ‘0’ group and 3–4 for all other groups; *P < 0.05, **P < 0.01 compared to no peptide group)
3.3 |. BK, NT or SP do not acutely alter tight junction complexes
To provide basic mechanistic insight on the observed effects of BK, NT and SP on barrier function, in a separate set of experiments, we assessed expression of tight junction proteins claudin-5 and occludin in CTR90F- and CTR65M-derived BMECs after 60 minutes of incubation with 1 µmol L−1 BK, NT or SP (Figure 3). No substantial changes were observed in relative expression (immunoreactivity) or continuity (continuous cell border localisation) of claudin-5 (Figure 3A) or occludin (Figure 3B) after 60 minutes of exposure to either of the peptides. In addition, we also evaluated the distribution of F-actin under the same experimental conditions (Figure 3C). By contrast to control conditions where F-actin is mostly present in the form of an actin belt lining the cell borders, discrete but notable appearance of actin stress fibres were observed in CTR90F-BMECs following treatment with 1 µmol L−1 NT, as well as in CTR65M-BMECs following treatment with 1 µmol L−1 BK or SP (Figure 3C).
FIGURE 3.
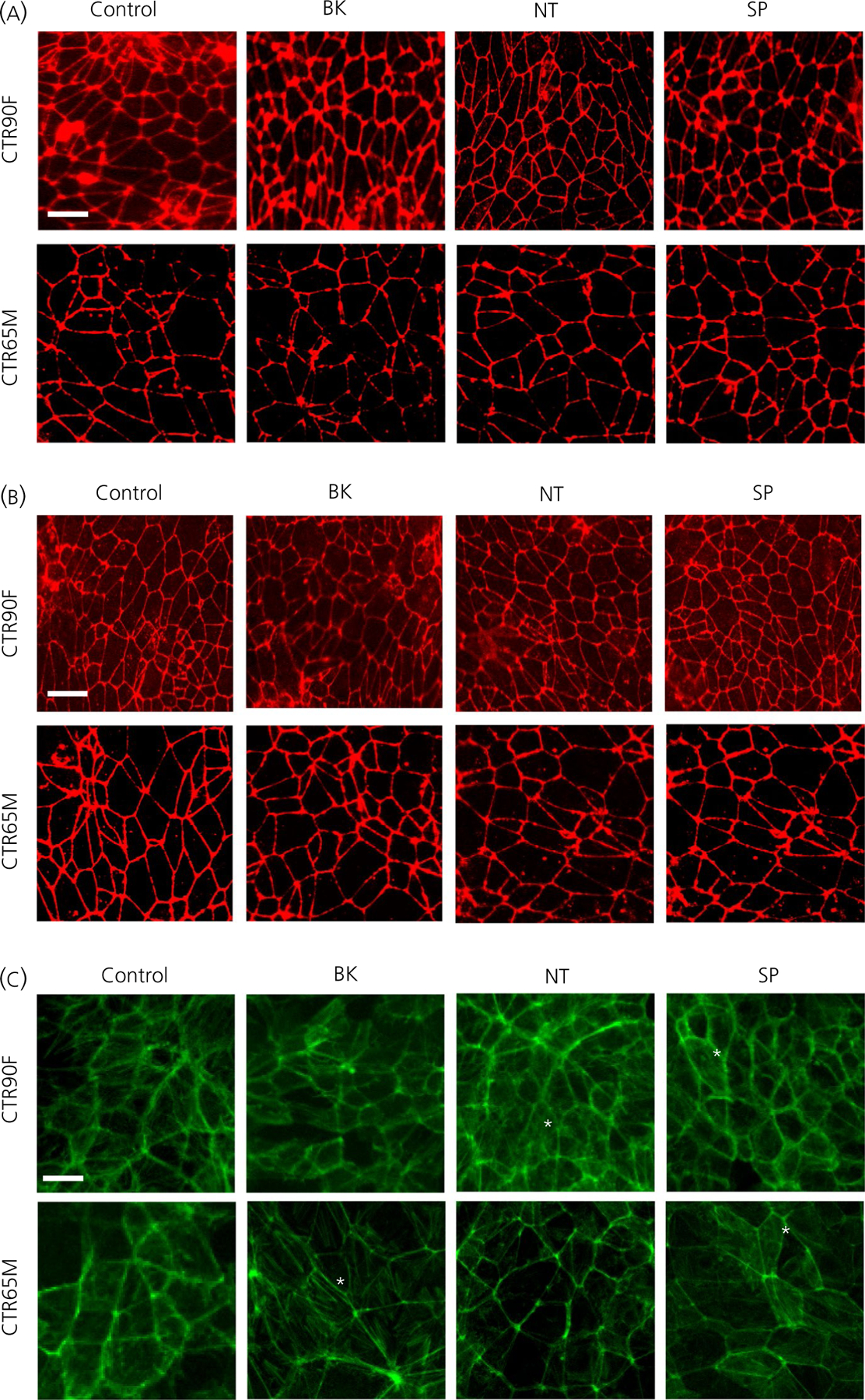
The effect of bradykinin (BK), neurotensin (NT) and substance P (SP) on tight junction complexes and actin cytoskeleton. Representative micrograph images of claudin-5 (A), occludin (B) and F-actin (C) localization in induced pluripotent stem cell-derived brain microvascular endothelial cells exposed to 1 µmol L−1 BK, NT or SP for 60 min. Note the subtle change in F-actin immunopositive staining and delocalisation from the cell border into the cytosol (asterisks). Scale bar = 20 µm
3.4 |. BK, NT or SP induce secretion of VEGF from iPSC-derived BMECs
To further explore the potential mechanism of the observed BK, NT and SP action, in this set of experiment we evaluated the effect of all three peptides (at 1 µmol L−1, 60 minutes of exposure) on the secretion of VEGF (Figure 4). Interestingly, all three peptides resulted in a two- to four-fold increase in VEGF levels compared to control cells. Both NT and SP induced statistically significant increase in secreted VEGF levels in CTR90F-BMECs (q = 2.82, df = 20, P < 0.05 and q = 4.19, df = 20, P < 0.01, respectively) and CTR65M-BMECs (q = 3.57, df = 20, P < 0.01 and q = 3.67, df = 20, P < 0.01, respectively), whereas the effect of BK was apparent but statistically non-significant (q = 2.26, df = 20, P = 0.08 for CTR90F-BMECs and q = 1.96, df = 20, P > 0.05 for CTR65M-BMECs). Notably, in these experiments CTR90F-BMECs showed somewhat more profound response to the peptides than CTR65M-BMECs.
FIGURE 4.
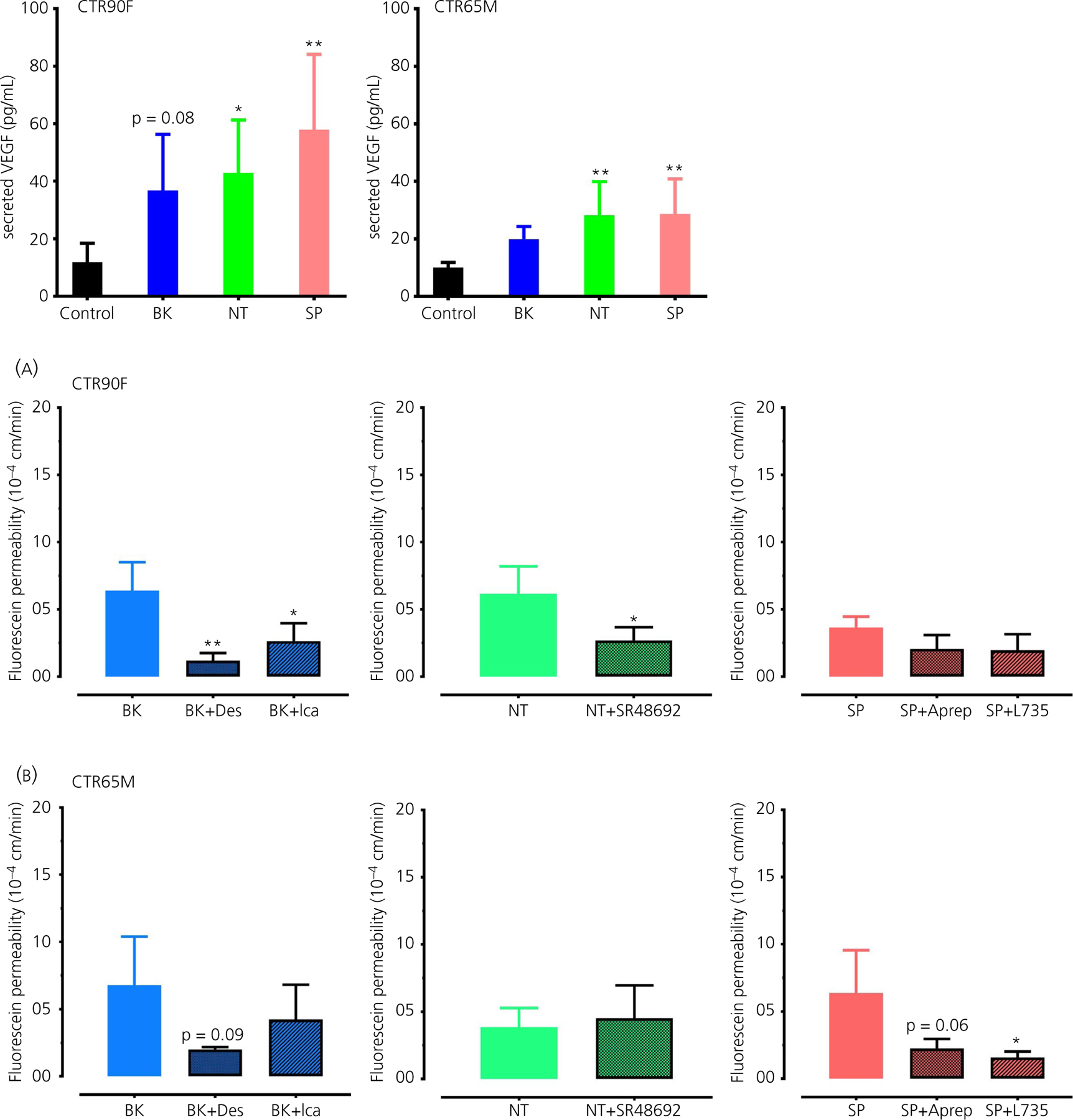
Bradykinin (BK), neurotensin (NT) and substance P (SP) induce secretion of vascular endothelial growth factor (VEGF) in brain microvascular endothelial cells (BMECs). The cell culture medium of induced pluripotent stem cell-derived BMECs exposed to 1 µmol L−1 BK, NT or SP for 60 min was used to measure the level of VEGF using an enzyme-linked immunosorbent assay (n = 6 per group; *P < 0.05, **P < 0.01 compared to control [ie, no peptide treatment] group)
3.5 |. The effect of BK, NT and SP on barrier function is reversed by pharmacological blockers
To confirm that the observed effects of BK, NT and SP on BMEC monolayers are mediated through their specific receptors, the cells were pre-incubated with selective pharmacological antagonists followed by evaluation of BK, NT and SP effects (at a concentration of 1 µmol L−1) on the permeability of fluorescein (Figure 5). Treatment with BK B1 receptor antagonist des-Arg9-[Leu8]-BK (Des, at 30 µmol L−1) resulted in a statistically significant decrease in BK-induced fluorescein permeability in CTR90F (q = 4.32, df = 6, P < 0.01) but not in CTR65M-derived BMECs (q = 2.28, df = 7, P = 0.09). Similarly, BK B2 receptor antagonist icatibant acetate (Ica, at 30 µmol L−1) significantly prevented BK-induced permeability of fluorescein in CTR90F-BMEC monolayers (q = 3.13, df = 6, P < 0.05), whereas only an apparent effect was observed in CTR65M monolayers in response to this blocker (q = 1.21, df = 7, P > 0.05) (Figure 5). Treatment of CTR90F-BMEC monolayers with NT1 receptor antagonist SR48692 (at 30 µmol L−1) significantly blocked the effect of NT on fluorescein permeability (t = 2.69, df = 5, P < 0.05); however, this effect was absent in CTR65M-BMEC monolayers (t = 0.45, df = 5, P > 0.05) (Figure 5). Lastly, neurokinin 1 receptor (NK1) antagonists Aprepitant and L-760735 (Aprep and L735, respectively, both at 30 µmol L−1) showed a modest but statistically non-significant decrease in permeability of fluorescein in response to SP in CTR90F-BMECs (q = 2.07, df = 7, P > 0.05 and q = 2.21, df = 7, P > 0.05) and more profound effects in CTR65M-BMECs (q = 2.55, df = 7, P = 0.06 and q = 2.97, df = 7, P < 0.03) (Figure 5).
FIGURE 5.
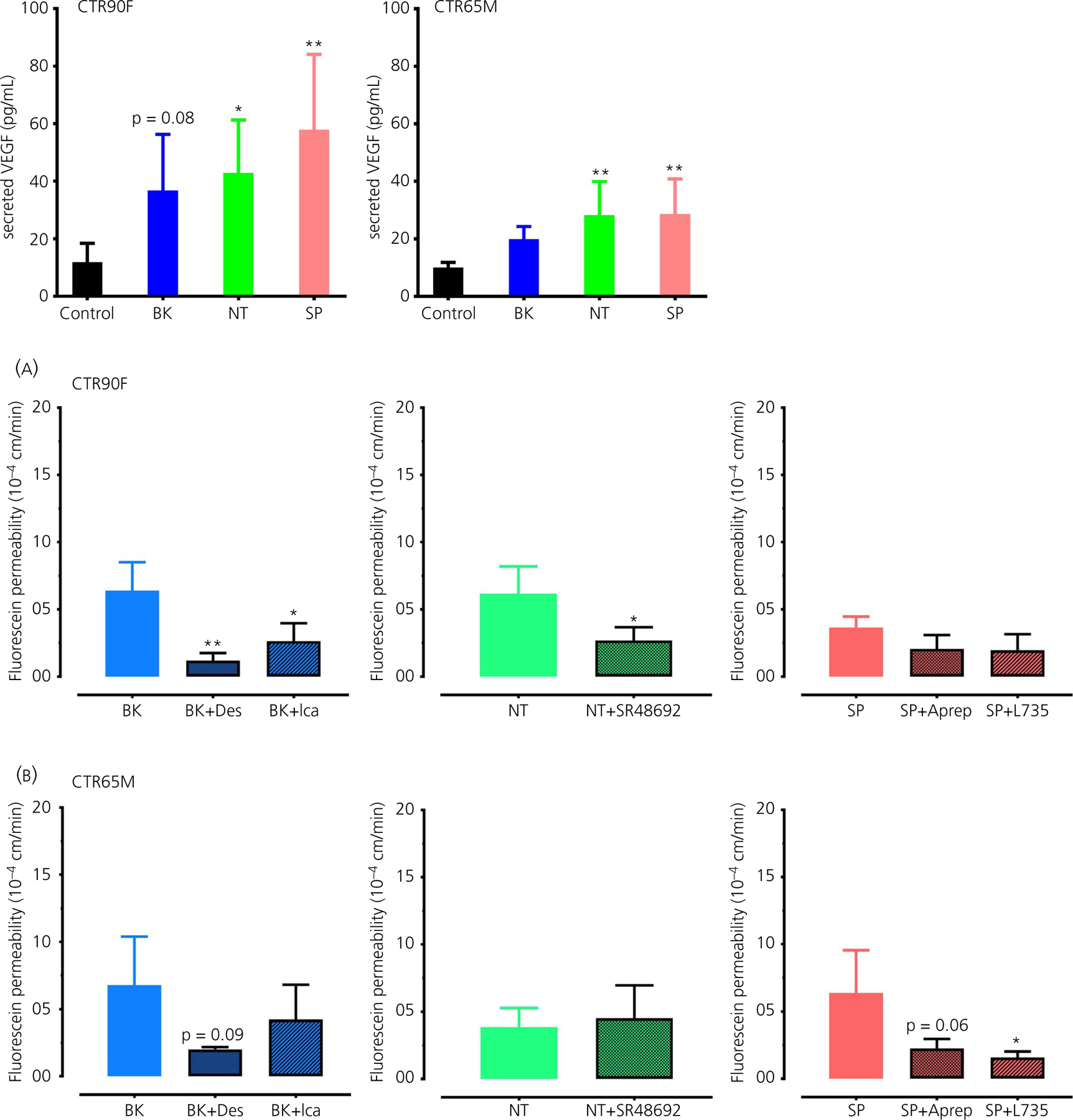
The effects of bradykinin (BK), neurotensin (NT) and substance P (SP) on barrier function are mediated through their respective receptors. CTR90F (A) and CTR65M (B) brain microvascular endothelial cells were pre-treated with pharmacological antagonists of BK B1 (Des), B2 (Ica), NT NT1 (SR48692) or SP NK1 (Aprep or L735) receptor antagonists followed by evaluation of the effects of BK, NT and SP on fluorescein permeability (n = 3–4 per group; *P < 0.05, **P < 0.01; compared to the respective peptide treatment alone; Des, des-Arg9-[Leu8]-BK; Ica, icatibant acetate; Aprep, Aprepitant; L735, L-760735)
3.6 |. Combined effect of BK, NT and SP on barrier function
Because BK, NT and SP did not show a significant effect on barrier function below a concentration of 1 µmol L−1, in this set of experiments, BMEC monolayers were treated with the mixture of all three peptides at concentrations of 0.1 or 0.5 µmol L−1 followed by evaluation of TEER and fluorescein permeability (Figure 6). The combined effect of all three peptides was concentration-dependent with a maximum ΔTEER decrease observed at the concentration of 0.5 µmol L−1, in both CTR90F (q = 3.41, df = 17, P < 0.01 at 0.5 µmol L−1) (Figure 6A) and CTR65M-derived (q = 4.91, df = 13, P < 0.01 at 0.5 µmol L−1) (Figure 6B) BMECs (approximate 35% and 75% decrease in ΔTEER, respectively). We also report statistically significant increase in fluorescein permeability at a concentration of 0.5 µmol L−1 in both CTR90F (q = 7.10, df = 13, P < 0.01 at 0.5 µmol L−1) and CTR65M-derived monolayers (q = 8.93, df = 14, P < 0.01 at 0.5 µmol L−1) (Figure 6A, B). Notably, this effect was more accentuated in CTR65M-BMECs than in CTR90F-BMECs because an approximately eight-fold increase was observed in CTR65M-BMECs in contrast to an approximately 2.5-fold increase reported in CTR90F-BMECs.
FIGURE 6.
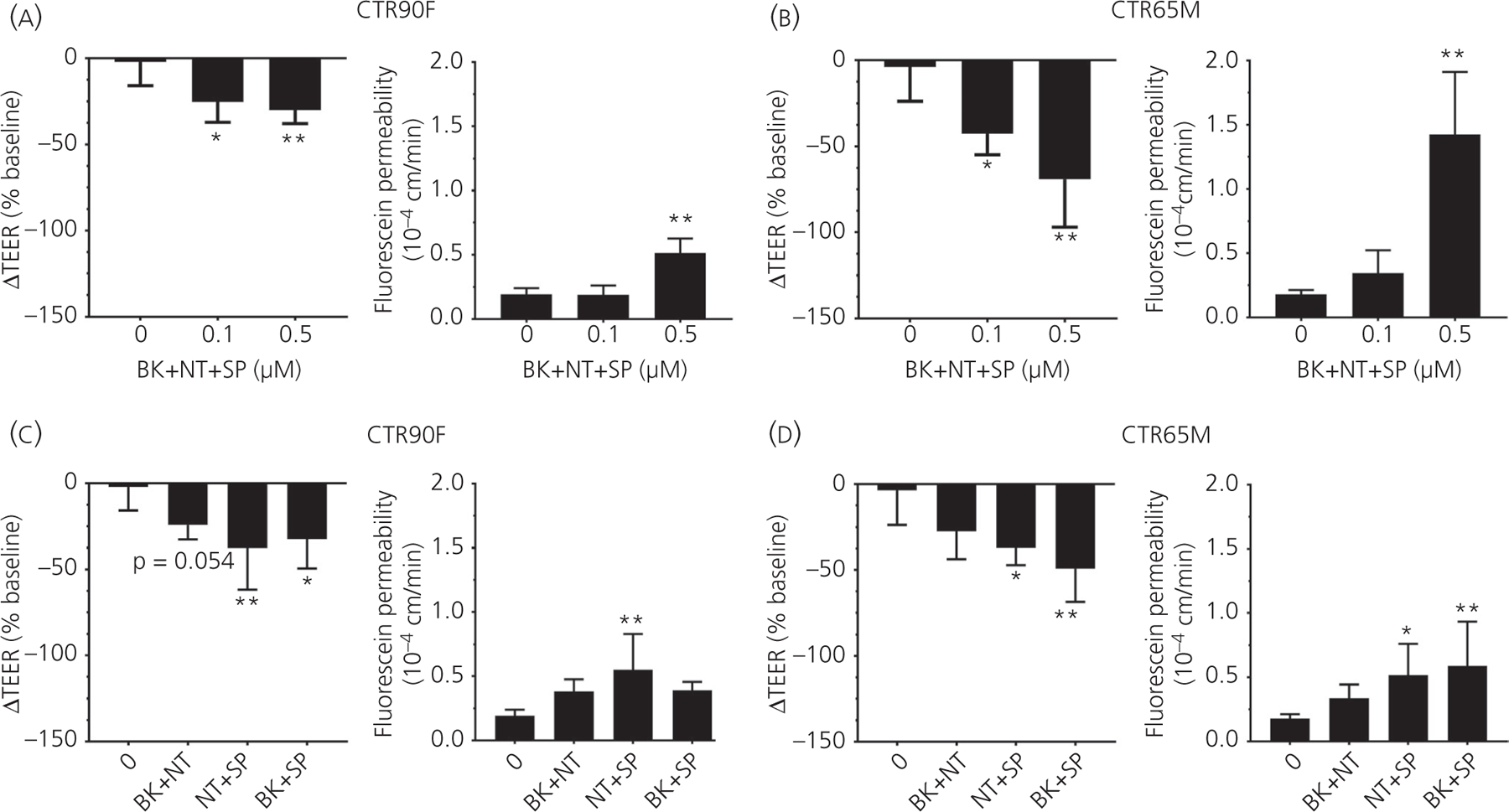
A combination of bradykinin (BK), neurotensin (NT) and substance P (SP) results in a potentiated effect. CTR90F (A) and CTR65M (B) brain microvascular endothelial cells (BMECs) were treated with a cocktail of BK, NT and SP at sub-effective concentrations (0.1 and 0.5 µmol L−1) followed by transendothelial electrical resistance (TEER) and fluorescein permeability measurements. To dissect the contribution of each peptide CTR90F (C) and CTR65M (D) BMECs were treated with a cocktail of BK + NT, NT + SP or BK + SP (each peptide at a concentration of 0.5 µmol L−1) followed by TEER and fluorescein permeability measurements (n = 9–13 for no peptide, ie ‘0’ group and 3–5 for all other groups; *P < 0.05, **P < 0.01 compared to no peptide treatment)
In a separate set of experiments, we also tested the effect of BK + NT, NT + SP, BK + SP (each peptide at a concentration of 0.5 µmol L−1) on the barrier function of BMEC monolayers (Figure 6C, D). Here, NT + SP and BK + SP combinations resulted in statistically significant decrease in ΔTEER values in CTR90F-BMECs (30%−35% decrease), with combination of NT with SP having somewhat stronger effect (q = 4.0, df = 20, P < 0.01). Similarly, NT + SP and BK + SP combinations resulted in statistically significant decrease of ΔTEER values in CTR65M-BMEC by 33%–50% (q = 3.08, df = 17, P < 0.05 and q = 4.54, df = 17, P < 0.01, respectively). Fluorescein permeability studies indicated an approximate two-fold increase in permeability, with the NT + SP combination being the most potent in CTR90F-BMECs (q = 4.49, df = 16, P < 0.01). In CTR65M-BMECs both NT + SP (q = 2.81, df = 17, P < 0.05) and BK + SP (q = 3.65, df = 17, P < 0.01) combinations showed a statistically significant effect.
3.7 |. Effect of Nln on barrier function
It has been suggested that the neuroprotective function of Nln is in part associated with degradation of its peptide substrates that affect integrity of the BBB8,9; however, the direct effect of Nln on barrier function remains undocumented. To address this question, a concentration-dependent effect of rNln on the permeability of iPSC-derived BMECs was evaluated (Figure 7). We did not observe a statistically significant effect on the barrier integrity of BMECs measured by ΔTEER (Figure 7A) and fluorescein permeability (Figure 7B) in the presence of rNln at concentrations of 0.1 and 0.3 µg mL−1. However, a small but statistically significant change in fluorescein permeability was observed with 1 µg mL−1 rNln in both CTR90F (q = 4.05, df = 15, P < 0.01) and CTR65M-BMECs (q = 3.75, df = 11, P < 0.01). Based on these observations, a concentration of 0.3 µg mL−1 rNln was used in subsequent experiments.
FIGURE 7.
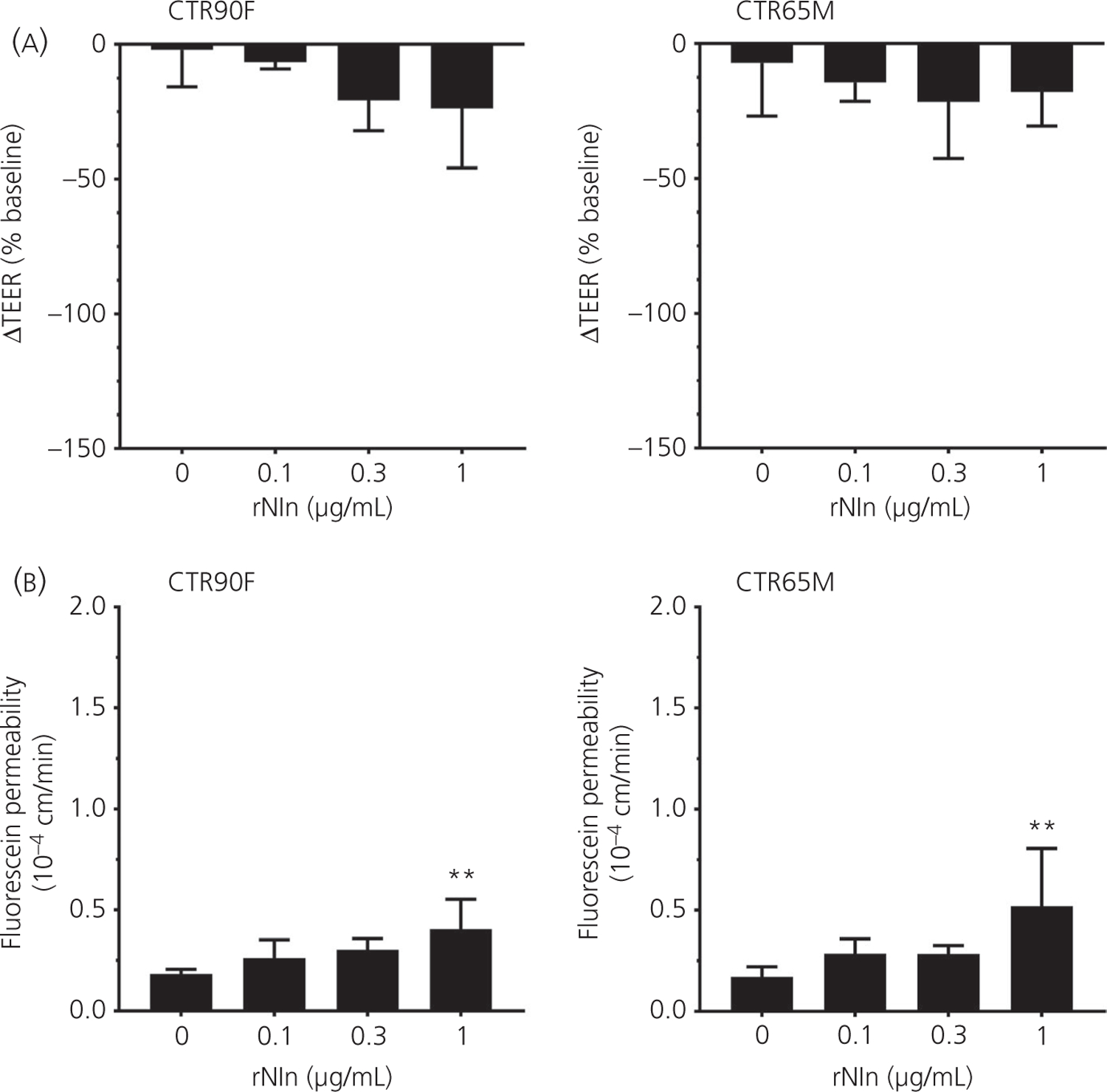
Human recombinant neurolysin has a negligible effect on barrier function. CTR90F and CTR65M brain microvascular endothelial cells were treated with different concentrations of recombinant neurolysin (rNln) (0.1–1 µg mL−1) followed by transendothelial electrical resistance (TEER) (A) and fluorescein permeability (B) measurements (n = 6–13 for no rNln, ie, ‘0’ group and 3–4 for all other groups; **P < 0.01 compared to no rNln group)
3.8 |. Nln blunts the effect of vasoactive peptides on barrier function
In this set of experiments, the ability of Nln to enzymatically degrade BK, NT and SP, and prevent their effects on integrity of BBB was evaluated (Figure 8). Here, the peptides (at a final concentration of 1 µmol L−1) were pre-incubated with 0.3 µg mL−1 rNln at 37°C for 10 minutes, followed by incubation with BMEC monolayers and evaluation of their barrier function using ΔTEER (Figure 8A) and fluorescein permeability (Figure 8B) measurements. rNln pre-treatment resulted in statistically significant blunting of NT-induced ΔTEER decrease in CTR90F-derived BMECs (t = 4.05, df = 4, P < 0.05), whereas, in CTR65M-BMECs, rNln statistically significantly reversed the effects of BK and SP on ΔTEER (t = 7.83, df = 6, P < 0.01 and t = 4.75, df = 6, P < 0.01, respectively). Fluorescein permeability measurements in the same cells were in line with our expectations indicating significant reduction of BK and NT, and a trend for SP-induced fluorescein permeability in CTR90F-derived BMECs after rNln pretreatment (t = 3.23, df = 4, P < 0.05; t = 7.16, df = 4, P < 0.01 and t = 2.23, df = 4, P = 0.08, respectively). Similar observations were found in CTR65M-BMECs, where rNln pretreatment significantly attenuated the effect of BK (t = 2.69, df = 6, P < 0.05), whereas the effects of NT and SP were evident but not statistically significant (t = 2.15, df = 7, P = 0.06 and t = 2.08, df = 6, P = 0.08, respectively).
FIGURE 8.
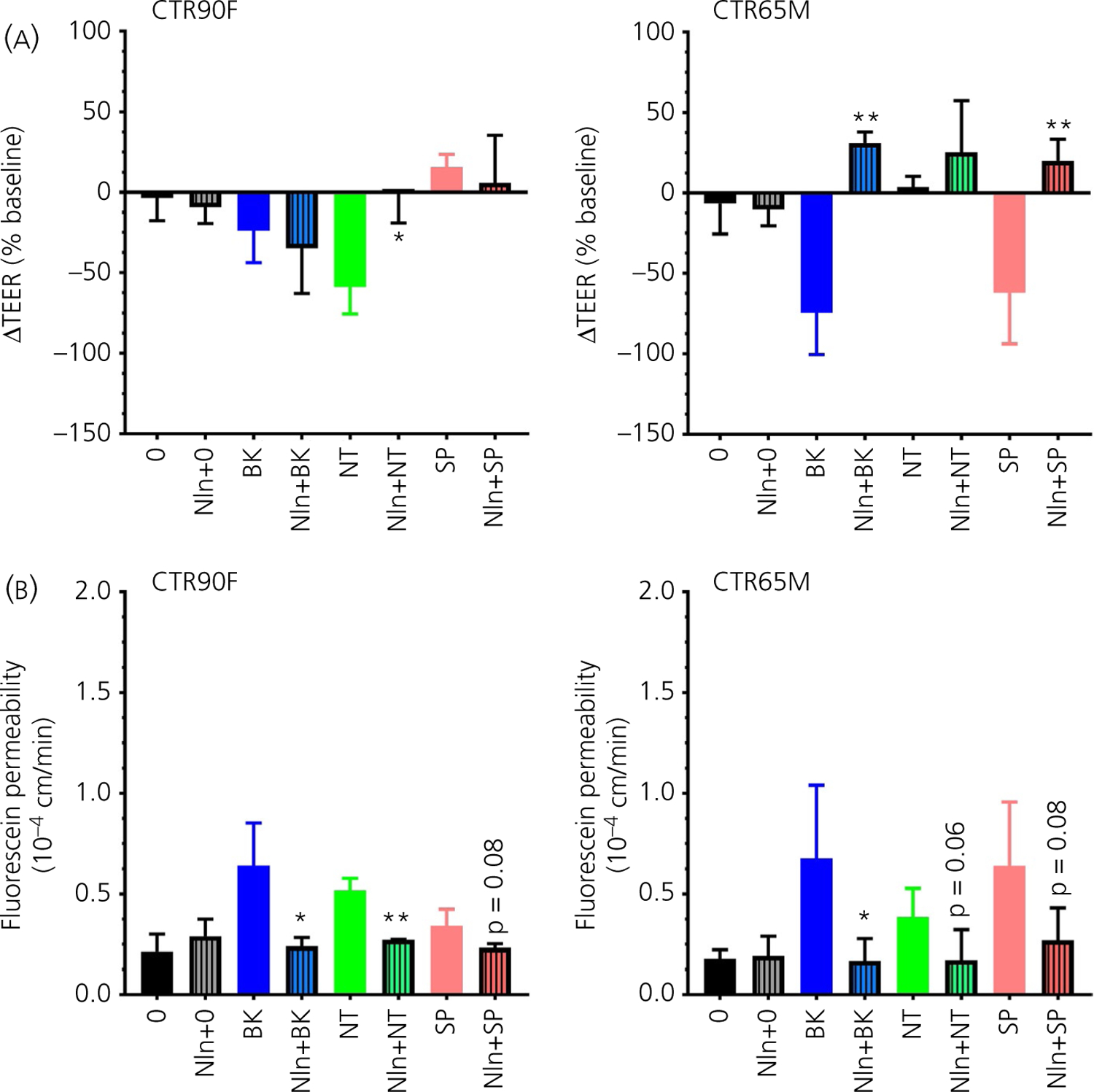
Human recombinant neurolysin neutralises the effects of bradykinin (BK), neurotensin (NT) and substance P (SP) on barrier function. The peptides were pre-treated with rNln (hashed bars) followed by evaluation of their effects on transendothelial electrical resistance (TEER) (A) and fluorescein permeability (B) in CTR90F and CTR65M brain microvascular endothelial cells (n = 14–17 for no peptide, ie, ‘0’ group and 3–6 for all other groups; *P < 0.05, **P < 0.01 compared to the respective peptide alone; ie, plain bar, group)
4 |. DISCUSSION
In the present study, we assessed the direct effect of Nln substrates BK, NT and SP on the permeability of BBB in vitro using human stem cell-derived, brain microvascular endothelial cells (iPSC-derived BMECs) in culture. Our observations indicate that all three peptides enhance BBB permeability in iPSC-derived BMECs similar to that of hCMEC/D3 cells (Figure 1), a widely used human brain microvascular endothelial cell line.27,33–41 The observed effects of BK, NT and SP on the permeability of BBB in iPSC-derived BMECs were concentration-dependent, showing a pronounced effect at 1 µmol L−1 and marginal or no effects at concentrations of 0.1 and 0.5 µmol L−1 (Figure 2). These concentrations are higher than physiological concentrations of BK, NT and SP found in vivo; however, they are within the same range as in the majority of in vitro studies evaluating effects of biological peptides in cell culture, including in vitro BBB models.31,32,42–444 In this context, it is important to note that the presence of 1% PDS (platelet-poor derived plasma serum) in EC−/− could have contributed to degradation of the peptides and hence to attenuation of their effects in our experiments. We did not investigate this factor systematically, although we do speculate that, in the absence of serum and accompanying proteolytic enzymes, the observed effects of BK, NT and SP might have been more profound.
To evaluate selectivity of BK, NT and SP effects on BBB permeability, in a separate set of experiments, we used small molecule antagonists of B1, B2, NT1 and NK1 receptors to block the respective receptors of these peptides (Figure 5). As expected, selective blockade of BK, NT or SP-induced BBB permeability was observed by pharmacological antagonists of their cognate receptors. Here, we also assessed the combined effect of these peptides on the barrier function of iPSC-derived BMECs and found BBB hyperpermeability in the presence of BK, NT and SP applied together at a sub-effective, concentration (ie 0.5 µmol L−1) (Figure 6). To obtain more insight about these effects, we also tested the outcome of two peptide combinations (BK + NT, NT + SP and BK + SP at 0.5 µmol L−1) on the barrier function of iPSC-derived BMECs. The differential responses of CTR90F- and CRT65M-derived BMECs, as well as the variability of TEER vs fluorescein permeability measurements, make it hard to conclusively interpret our observations. However, the current data indicate that a combination of these peptides leads to more profound effects compared to any of the peptides alone, and that, at least in case of CRT65M-derived BMECs, all three peptides together result in higher permeability than any of the peptide pairs alone. Our observations do not clarify whether these cooperative effects are additive or synergetic and whether they translate to enhanced intracellular signalling, and therefore additional experiments are required to address these important questions. Notably, this experimental condition imitates situations at which BK, NT and SP are available concurrently and may potentiate each other’s biological effects. Elevated levels of all three peptides were reported in experimental models of ischaemic stroke45–48 and traumatic brain injury,49–51 as well as in patients suffering these conditions.13,52 Severity and subsequent mortality associated with ischaemic stroke and traumatic brain injury have been linked to the elevated levels of BK, NT and SP in these studies. Furthermore, a number of experimental studies utilising small molecule antagonists of BK, NT or SP receptors have reported cerebroprotective, including anti-oedematous, effects of these agents in ischaemic stroke and/or traumatic brain injury models.53–55
To obtain more mechanistic understanding about the effects of these peptides on BBB, we also assessed their effects on integrity of tight junction complexes and F-actin (Figure 3). We did not document substantial changes in the distribution or expression of claudin-5 and occludin in the iPSC-derived BMECs in response to 1 µmol L−1 BK, NT or SP 60 minutes after treatment, although the possibility of such alterations at later time points cannot be excluded. In parallel, we also evaluated cytoskeletal protein F-actin in these experiments and observed subtle changes in distribution and appearance of actin stress fibres in response to the peptides. This observation suggests possible cell retractions in response to the peptides, which within the context of BBB disruption are known to proceed alteration of tight junction complexes and involve activation of ROCK/MLC signalling.56
Additionally, we evaluated the effect of BK, NT and SP on the secretion of VEGF from iPSC-derived BMECs (60 minutes of exposure at 1 µmol L−1) and found enhanced secretion of the growth factor in response to all three peptides (Figure 4). Notably, stimulated secretion of VEGF within such short period of time has been reported by other research groups.57–60 Furthermore, we are not the first group to document increased secretion of VEGF in response to BK,61–63 NT42,64 and SP65,66 from endothelial or other cells. At the moment, it is difficult to state whether the noted signalling molecules are within the same pathway and downstream of BK, NT or SP receptors. However, our observations warrant future studies that aim to systematically determine the molecular mechanisms governing the actions of these peptides on BBB.
Because BK, NT and SP are endogenous substrates of peptidase Nln, in another set of experiments, we investigated the ability of rNln to prevent the effects of these peptides on the permeability of BBB in iPSC-derived BMECs. For this, first, the direct effect of rNln on BBB permeability was studied, revealing that 1 µg mL−1 rNln, but not lower concentrations, induced a slight but statistically significant increase in permeability of fluorescein through iPSC-derived BMECs (Figure 7). The latter was in contrast to our expectation because we anticipated a lack of any effect of rNln on BBB permeability, similar to our previous observation in mixed primary astrocyte and neuronal co-cultures.16 The observed effect of rNln on the permeability of BBB cannot be explained by hydrolytic processing of known, extracellular substrates of Nln.8 However, one cannot exclude existence of other extracellular substrates of Nln that are responsible for rNln-induced permeability of BBB. Additionally, it is possible that trace amount of endotoxins (approximately 3 mEU µg−1 protein) available in our rNln preparation could be responsible for the observed effect.16 Based on these results we used 0.3 µg mL−1 rNln to investigate its ability to prevent the effects of BK, NT and SP on the permeability of BBB. Substantial attenuation of BK, NT and SP-induced fluorescein permeability in iPSC-derived BMECs was observed (Figure 8), indicating the ability of Nln to hydrolyse and inactivate all three peptides.
It is important to note that BK, NT and SP are the most studied substrates of Nln in the context of neurological disorders. Nln (neurolysin; EC3.4.24.16) is a zinc metallopeptidase containing a His-Glu-X-X-His domain, which is maximally active at neutral pH and is widely distributed in both central and peripheral nervous system.14,15 Nln functions to regulate the levels of several extracellular neuropeptides, including neurotensin, bradykinin, substance P, angiotensin I and II, metamorphamide, dynorphin A (1–8) and haemopressin.17,67,68 These substrate peptides are hydrolysed and inactivated by Nln, except angiotensin I, metamorphamide and dynorphin A (1–8), which are hydrolysed and converted into angiotensin-(1–7), Met- and Leu-enkephalins, respectively.20 Notably, in a recent in vivo study, the function of Nln was linked to the actions of BK, NT and SP after ischaemic stroke.9 In that previous study, inhibition of Nln with a specific pharmacological inhibitor led to increased levels of BK, NT and SP in mice after stroke and this was accompanied by worsened outcomes such as increased infarct and oedema volumes, BBB disruption, inflammatory cytokines, and neurological and motor deficit. In a reverse approach, Nln was overexpressed in the mouse brain before stroke using an adeno-associated virus vector and decreased levels of BK, NT and SP were observed. These mice were dramatically more resilient to stroke injury compared to control animals, which did not overexpress Nln in their brain. As a result of methodological limitations with respect to simultaneously detecting/quantifying multiple endogenous peptides, the other peptide substrates of Nln were not tracked, although detailed insights regarding experimental and clinical evidence related to pathological actions of BK, NT and SP in a stroke setting were provided. With regard to the present study in iPSC-derived BMECs, the effect of these three peptides on vascular permeability is of particular interest, which likely contributes to the neuroprotective function of Nln in a stroke setting. Numerous in vivo studies have reported the ability of BK, NT and SP to increase microvascular permeability, upon intravenous/-arterial administration, in different vascular beds/tissues, including mucosal tissues and skin,69,70 gastrointestinal tract,71,72 joints,73 lungs74,75 and dura mater.76,77 These observations highlight the direct effect of these peptides on endothelial cells because the peptides were administered i.v./-arterially in these experimental studies. It is important to note that a number of other studies have reported increased microvascular permeability after local/extravascular administration of BK, NT or SP,78 and several of these studies linked the effect of the peptides to mast cell degranulation.79–82
Regarding brain microvascular permeability, BK is the most studied peptide out of these three neuropeptides that are known to enhance permeability of the BBB in various in vitro and in vivo settings.44,83,84 This effect is primarily mediated through bradykinin B2 receptor in brain capillary endothelial cells and was the basis for the development of selective B2 agonists for clinical evaluation and use in controlled therapeutic opening of the BBB.85 Notably, the role of BK, mediated through both B1 and B2 receptors, in the pathogenesis of BBB opening and oedema formation has been well-documented in a number of acute neurodegenerative disorders, including stroke and traumatic brain injury.48,51,86 Only one study has reported the ability of SP to increase BBB permeability in intact brain microvascular endothelial cells in culture32; however, there are several in vivo studies that have demonstrated an integral role of SP in pathogenesis of increased BBB permeability and oedema formation during stroke and traumatic brain injury.50,87,88 Lastly, NT is the least studied peptide in this group regarding its direct effects on the permeability of BBB under physiological or pathophysiological conditions, with no published studies documenting this effect.
Another interesting point about our observations is the differential effects of BK, NT and SP in CTR90F and CTR65M-derived monolayers, where CTR90F-derived BMECs responded more strongly to BK and NT, whereas, in CTR65M-derived BMECs, BK and SP had more profound effects. The reason(s) for this differential effect remain unclear, although potential differences in cells lines are indicated, comprising an important question for the overall field of stem cell/iPSC research. One potential reason for these observations could be the differential expression of BK, NT and SP receptors in these cells as determined by preliminary cDNA microarray analysis (see Supporting information, Figure S1). This question is the subject of our ongoing mechanistic, molecular biological studies and is in line with published studies documenting the differential response of CTR90F and CTR65M-derived BMECs to oxygen-glucose deprivation and VEGF secretion.22,25
In summary, this is the first experimental study to document an increased permeability of BBB in response to the direct action of NT in an in vitro model. In addition, our study confirms the expected but not well-documented, direct effect of SP on BBB permeability in the same in vitro model, and adds to the well-recognised actions of BK on BBB. Our study also demonstrates that peptidase Nln can prevent the effects of BK, NT and SP on BBB. The latter may have clinical implications for pathophysiological conditions (eg acute neurodegeneration) at which all three of these peptides act concurrently/synergistically to increase BBB/microvascular permeability. Under such conditions, Nln-mediated modulation of BK, NT and SP may be more efficacious than the use of selective antagonists to block the respective receptors of these peptides. These questions are currently being investigated in our laboratory,89 and mechanistic, disease-based information should emerge revealing the potential of Nln, in comparison with pharmacological antagonists, with respect to mitigating the effects of BK, NT and SP on the impairment of BBB under pathophysiological conditions.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
AA and VTK contributed equally to the experimental design and manuscript writing. AA and IP conducted the experiments, VTK prepared recombinant neurolysin. We thank Dr Srihidhi Jayaraman for her help with the preparation of the enzyme. The authors have no conflicts of interests to disclose. This study was funded through TTUHSC institutional funds to AA and a NIH grant (1R01NS106879).
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Number: 1R01NS106879
Footnotes
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jne.12931.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY
The data that support the findings of our study will be made available upon reasonable request.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. [DOI] [PubMed] [Google Scholar]
- 3.van Bruggen N, Thibodeaux H, Palmer JT, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts J, de Hoog L, Bix GJ. Mice deficient in endothelial alpha5 integrin are profoundly resistant to experimental ischemic stroke. J Cereb Blood Flow Metab. 2017;37:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eiden LE, Gundlach AL, Grinevich V, et al. Regulatory peptides and systems biology: A new era of translational and reverse-translational neuroendocrinology. J Neuroendocrinol. 2020;32:e12844. [DOI] [PubMed] [Google Scholar]
- 7.Hokfelt T, Bartfai T, Bloom F. Neuropeptides: opportunities for drug discovery. Lancet Neurol. 2003;2:463–472. [DOI] [PubMed] [Google Scholar]
- 8.Karamyan VT. Peptidase neurolysin is an endogenous cerebroprotective mechanism in acute neurodegenerative disorders. Med Hypotheses. 2019;131:109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayaraman S, Al Shoyaib A, Kocot J, et al. Peptidase neurolysin functions to preserve the brain after ischemic stroke in male mice. J Neurochem. 2020;153:120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nokkari A, Abou-El-Hassan H, Mechref Y, et al. Implication of the Kallikrein-Kinin system in neurological disorders: Quest for potential biomarkers and mechanisms. Prog Neurogibol. 2018;165–167: 26–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue TF, Ding X, Ji J, et al. PD149163 induces hypothermia to protect against brain injury in acute cerebral ischemic rats. J Pharmacol Sci. 2017;135:105–113. [DOI] [PubMed] [Google Scholar]
- 12.Annunziata P, Cioni C, Santonini R, Paccagnini E. Substance P antagonist blocks leakage and reduces activation of cytokine-stimulated rat brain endothelium. J Neuroimmunol. 2002;131:41–49. [DOI] [PubMed] [Google Scholar]
- 13.Lorente L, Martin MM, Almeida T, et al. Serum levels of substance P and mortality in patients with a severe acute ischemic stroke. Int J Mol Sci. 2016;17:991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Checler F, Ferro ES. Neurolysin: From Initial Detection to Latest Advances. Neurochem Res. 2018;43:2017–2024. [DOI] [PubMed] [Google Scholar]
- 15.Checler F, Barelli H, Dauch P, Dive V, Vincent B, Vincent JP. Neurolysin: purification and assays. Methods Enzymol. 1995;248:593–614. [DOI] [PubMed] [Google Scholar]
- 16.Wangler NJ, Jayaraman S, Zhu R, et al. Preparation and preliminary characterization of recombinant neurolysin for in vivo studies. J Biotechnol. 2016;234:105–115. [DOI] [PubMed] [Google Scholar]
- 17.Mentlein R, Dahms P. Endopeptidases 24.16 and 24.15 are responsible for the degradation of somatostatin, neurotensin, and other neuropeptides by cultivated rat cortical astrocytes. J Neurochem. 1994;62:27–36. [DOI] [PubMed] [Google Scholar]
- 18.Wangler NJ, Santos KL, Schadock I, et al. Identification of membrane-bound variant of metalloendopeptidase neurolysin (EC 3.4.24.16) as the non-angiotensin type 1 (Non-AT1), non-at2 angiotensin binding site. J Biol Chem. 2012;287:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Distinct properties of neuronal and astrocytic endopeptidase 3.4.24.16: a study on differentiation, subcellular distribution, and secretion processes. J Neurosci. 1996;16(16):5049–5059. https://pubmed.ncbi.nlm.nih.gov/8756435/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rashid M, Wangler NJ, Yang L, et al. Functional up-regulation of endopeptidase neurolysin during post-acute and early recovery phases of experimental stroke in mouse brain. J Neurochem. 2014;129:179–189. [DOI] [PubMed] [Google Scholar]
- 21.Rashid M, Arumugam TV, Karamyan VT. Association of the novel non-AT1, non-AT2 angiotensin binding site with neuronal cell death. J Pharmacol Exp Ther. 2010;335:754–761. [DOI] [PubMed] [Google Scholar]
- 22.Page S, Raut S, Al-Ahmad A. Oxygen-glucose deprivation/reoxygenation-induced barrier disruption at the human blood-brain barrier is partially mediated through the HIF-1 pathway. Neuromolecular Med. 2019;21:414–431. [DOI] [PubMed] [Google Scholar]
- 23.Albekairi TH, Vaidya B, Patel R, et al. Brain delivery of a potent opioid receptor agonist, biphalin during ischemic stroke: role of organic anion transporting polypeptide (OATP). Pharmaceutics. 2019;11;467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Ahmad AJ, Patel R, Palecek SP, Shusta EV. Hyaluronan impairs the barrier integrity of brain microvascular endothelial cells through a CD44-dependent pathway. J Cereb Blood Flow Metab. 2019;39:1759–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel R, Page S, Al-Ahmad AJ. Isogenic blood-brain barrier models based on patient-derived stem cells display inter-individual differences in cell maturation and functionality. J Neurochem. 2017;142:74–88. [DOI] [PubMed] [Google Scholar]
- 26.Page S, Munsell A, Al-Ahmad AJ. Cerebral hypoxia/ischemia selectively disrupts tight junctions complexes in stem cell-derived human brain microvascular endothelial cells. Fluids Barriers CNS. 2016;13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weksler BB, Subileau EA, Perriere N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. [DOI] [PubMed] [Google Scholar]
- 28.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- 29.Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown CK, Madauss K, Lian W, Beck MR, Tolbert WD, Rodgers DW. Structure of neurolysin reveals a deep channel that limits substrate access. Proc Natl Acad Sci U S A. 2001;98:3127–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JT, Lin YL, Chen TL, Tai YT, Chen CY, Chen RM. Ketamine alleviates bradykinin-induced disruption of the mouse cerebrovascular endothelial cell-constructed tight junction barrier via a calcium-mediated redistribution of occludin polymerization. Toxicology. 2016;368–369:142–151. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez PL, Jiang S, Fu Y, Avraham S, Avraham HK. The proinflammatory peptide substance P promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int J Cancer. 2014;134:1034–1044. [DOI] [PubMed] [Google Scholar]
- 33.Al-Ahmad AJ. Comparative study of expression and activity of glucose transporters between stem cell-derived brain microvascular endothelial cells and hCMEC/D3 cells. Am J Physiol Cell Physiol. 2017;313:C421–C429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacob A, Potin S, Saubamea B, et al. Hypoxia interferes with aryl hydrocarbon receptor pathway in hCMEC/D3 human cerebral microvascular endothelial cells. J Neurochem. 2015;132:373–383. [DOI] [PubMed] [Google Scholar]
- 35.Artus C, Glacial F, Ganeshamoorthy K, et al. The Wnt/planar cell polarity signaling pathway contributes to the integrity of tight junctions in brain endothelial cells. J Cereb Blood Flow Metab. 2014;34:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eigenmann DE, Xue G, Kim KS, Moses AV, Hamburger M, Oufir M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS. 2013;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urich E, Lazic SE, Molnos J, Wells I, Freskgard PO. Transcriptional profiling of human brain endothelial cells reveals key properties crucial for predictive in vitro blood-brain barrier models. PLoS One. 2012;7:e38149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dauchy S, Miller F, Couraud PO, et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol. 2009;77:897–909. [DOI] [PubMed] [Google Scholar]
- 39.Tai LM, Reddy PS, Lopez-Ramirez MA, et al. Polarized P-glycoprotein expression by the immortalised human brain endothelial cell line, hCMEC/D3, restricts apical-to-basolateral permeability to rhodamine 123. Brain Res. 2009;1292:14–24. [DOI] [PubMed] [Google Scholar]
- 40.Cucullo L, Couraud PO, Weksler B, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. 2008;28:312–328. [DOI] [PubMed] [Google Scholar]
- 41.Poller B, Gutmann H, Krahenbuhl S, et al. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J Neurochem. 2008;107:1358–1368. [DOI] [PubMed] [Google Scholar]
- 42.Alysandratos KD, Asadi S, Angelidou A, et al. Neurotensin and CRH interactions augment human mast cell activation. PLoS One. 2012;7:e48934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel AB, Theoharides TC. Methoxyluteolin Inhibits Neuropeptide-stimulated Proinflammatory Mediator Release via mTOR Activation from Human Mast Cells. J Pharmacol Exp Ther. 2017;361(3): 462–471. [DOI] [PubMed] [Google Scholar]
- 44.Easton AS, Abbott NJ. Bradykinin increases permeability by calcium and 5-lipoxygenase in the ECV304/C6 cell culture model of the blood-brain barrier. Brain Res. 2002;953:157–169. [DOI] [PubMed] [Google Scholar]
- 45.Chen YH, Chiang YH, Ma HI. Analysis of spatial and temporal protein expression in the cerebral cortex after ischemia-reperfusion injury. J Clin Neurol. 2014;10:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Bree L, Zhang F, Schiffmann SN, Halleux P, Mailleux P, Vanderhaeghen JJ. Homolateral cerebrocortical changes in neuropeptide and receptor expression after minimal cortical infarction. Neuroscience. 1995;69:847–858. [DOI] [PubMed] [Google Scholar]
- 47.Turner RJ, Blumbergs PC, Sims NR, Helps SC, Rodgers KM, Vink R. Increased substance P immunoreactivity and edema formation following reversible ischemic stroke. Acta Neurochir Suppl. 2006;96:263–266. [DOI] [PubMed] [Google Scholar]
- 48.Groger M, Lebesgue D, Pruneau D, et al. Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:978–989. [DOI] [PubMed] [Google Scholar]
- 49.Elliott MB, Tuma RF, Amenta PS, Barbe MF, Jallo JI. Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a model of traumatic brain injury. J Neurotrauma. 2011;28:973–981. [DOI] [PubMed] [Google Scholar]
- 50.Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1388–1398. [DOI] [PubMed] [Google Scholar]
- 51.Trabold R, Eros C, Zweckberger K, et al. The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2010;30:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunz M, Nussberger J, Holtmannspotter M, Bitterling H, Plesnila N, Zausinger S. Bradykinin in blood and cerebrospinal fluid after acute cerebral lesions: correlations with cerebral edema and intracranial pressure. J Neurotrauma. 2013;30:1638–1644. [DOI] [PubMed] [Google Scholar]
- 53.Antonelli T, Tomasini MC, Fournier J, et al. Neurotensin receptor involvement in the rise of extracellular glutamate levels and apoptotic nerve cell death in primary cortical cultures after oxygen and glucose deprivation. Cereb Cortex. 2008;18:1748–1757. [DOI] [PubMed] [Google Scholar]
- 54.Albert-Weissenberger C, Siren AL, Kleinschnitz C. Ischemic stroke and traumatic brain injury: the role of the kallikrein-kinin system. Prog Neurobiol. 2013;101–102:65–82. [DOI] [PubMed] [Google Scholar]
- 55.Sorby-Adams AJ, Marcoionni AM, Dempsey ER, Woenig JA, Turner RJ. The role of neurogenic inflammation in blood-brain barrier disruption and development of cerebral oedema following acute central nervous system (CNS) injury. Int J Mol Sci. 2017;18:1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi Y, Zhang L, Pu H, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016;7:10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grutzkau A, Kruger-Krasagakes S, Baumeister H, et al. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206. Mol Biol Cell. 1998;9:875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryzhov S, McCaleb JL, Goldstein AE, Biaggioni I, Feoktistov I. Role of adenosine receptors in the regulation of angiogenic factors and neovascularization in hypoxia. J Pharmacol Exp Ther. 2007;320:565–572. [DOI] [PubMed] [Google Scholar]
- 59.Shi CS, Huang TH, Lin CK, et al. VEGF Production by Ly6C+high Monocytes Contributes to Ventilator-Induced Lung Injury. PLoS One. 2016;11:e0165317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiriakidis S, Andreakos E, Monaco C, Foxwell B, Feldmann M, Paleolog E. VEGF expression in human macrophages is NF-kappaB-dependent: studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J Cell Sci. 2003;116:665–674. [DOI] [PubMed] [Google Scholar]
- 61.Knox AJ, Corbett L, Stocks J, Holland E, Zhu YM, Pang L. Human airway smooth muscle cells secrete vascular endothelial growth factor: up-regulation by bradykinin via a protein kinase C and prostanoid-dependent mechanism. Faseb J. 2001;15:2480–2488. [DOI] [PubMed] [Google Scholar]
- 62.Kawamura A, Miura SI, Matsuo Y, Tanigawa H, Saku K. Azelnidipine, not amlodipine, induces secretion of vascular endothelial growth factor from smooth muscle cells and promotes endothelial tube formation. Cardiol Res. 2014;5:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu HS, Wang SW, Chang AC, et al. Bradykinin promotes vascular endothelial growth factor expression and increases angiogenesis in human prostate cancer cells. Biochem Pharmacol. 2014;87:243–253. [DOI] [PubMed] [Google Scholar]
- 64.Vasiadi M, Therianou A, Alysandratos KD, et al. Serum neurotensin (NT) is increased in psoriasis and NT induces vascular endothelial growth factor release from human mast cells. Br J Dermatol. 2012;166:1349–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asadi S, Alysandratos KD, Angelidou A, et al. Substance P (SP) induces expression of functional corticotropin-releasing hormone receptor-1 (CRHR-1) in human mast cells. J Invest Dermatol. 2012;132:324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Theoharides TC, Zhang B, Kempuraj D, et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci U S A. 2010;107:4448–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rioli V, Gozzo FC, Heimann AS, et al. Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J Biol Chem. 2003;278:8547–8555. [DOI] [PubMed] [Google Scholar]
- 68.Shrimpton CN, Smith AI, Lew RA. Soluble metalloendopeptidases and neuroendocrine signaling. Endocr Rev. 2002;23:647–664. [DOI] [PubMed] [Google Scholar]
- 69.Inoue H, Nagata N, Koshihara Y. Involvement of tachykinin receptors in oedema formation and plasma extravasation induced by substance P, neurokinin A, and neurokinin B in mouse ear. Inflamm Res. 1996;45:316–323. [DOI] [PubMed] [Google Scholar]
- 70.Gyorfi A, Fazekas A, Irmes F, Rosivall L. Effect of substance P administration on vascular permeability in the rat oral mucosa and sublingual gland. J Periodontal Res. 1995;30:181–185. [DOI] [PubMed] [Google Scholar]
- 71.Harper SL, Barrowman JA, Kvietys PR, Granger DN. Effect of neurotensin on intestinal capillary permeability and blood flow. Am J Physiol. 1984;247:G161–G166. [DOI] [PubMed] [Google Scholar]
- 72.Figini M, Emanueli C, Grady EF, et al. Substance P and bradykinin stimulate plasma extravasation in the mouse gastrointestinal tract and pancreas. Am J Physiol. 1997;272:G785–G793. [DOI] [PubMed] [Google Scholar]
- 73.Cambridge H, Brain SD. Mechanism of bradykinin-induced plasma extravasation in the rat knee joint. Br J Pharmacol. 1995;115:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rioux F, Kerouac R, St-Pierre S. Peripheral vasodilation and plasma extravasation are part of the mechanism of neurotensin-induced hypotension in the anesthetized rat. Neuropeptides. 1983;3:355–365. [DOI] [PubMed] [Google Scholar]
- 75.Gitter BD, Bruns RF, Howbert JJ, et al. Pharmacological characterization of LY303870: a novel, potent and selective nonpeptide substance P (neurokinin-1) receptor antagonist. J Pharmacol Exp Ther. 1995;275:737–744. [PubMed] [Google Scholar]
- 76.Ghabriel MN, Lu MX, Leigh C, Cheung WC, Allt G. Substance P-induced enhanced permeability of dura mater microvessels is accompanied by pronounced ultrastructural changes, but is not dependent on the density of endothelial cell anionic sites. Acta Neuropathol. 1999;97:297–305. [DOI] [PubMed] [Google Scholar]
- 77.Dux M, Messlinger K. Histological demonstration of increased vascular permeability in the dura mater of the rat. Microsc Res Tech. 2001;53:229–231. [DOI] [PubMed] [Google Scholar]
- 78.Norman MU, Lew RA, Smith AI, Hickey MJ. Metalloendopeptidases EC 3.4.24.15/16 regulate bradykinin activity in the cerebral micro-vasculature. Am J Physiol Heart Circ Physiol. 2003;284:H1942–H1948. [DOI] [PubMed] [Google Scholar]
- 79.Donelan J, Boucher W, Papadopoulou N, et al. Corticotropin-releasing hormone induces skin vascular permeability through a neurotensin-dependent process. Proc Natl Acad Sci USA. 2006;103:7759–7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miller LA, Cochrane DE, Carraway RE, Feldberg RS. Blockade of mast cell histamine secretion in response to neurotensin by SR 48692, a nonpeptide antagonist of the neurotensin brain receptor. Br J Pharmacol. 1995;114:1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Theoharides TC, Singh LK, Boucher W, et al. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology. 1998;139:403–413. [DOI] [PubMed] [Google Scholar]
- 82.Yano H, Wershil BK, Arizono N, Galli SJ. Substance P-induced augmentation of cutaneous vascular permeability and granulocyte infiltration in mice is mast cell dependent. J Clin Invest. 1989;84:1276–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bartus RT, Elliott P, Hayward N, Dean R, McEwen EL, Fisher SK. Permeability of the blood brain barrier by the bradykinin agonist, RMP-7: evidence for a sensitive, auto-regulated, receptor-mediated system. Immunopharmacology. 1996;33:270–278. [DOI] [PubMed] [Google Scholar]
- 84.Mackic JB, Stins M, Jovanovic S, Kim KS, Bartus RT, Zlokovic BV. Cereport (RMP-7) increases the permeability of human brain microvascular endothelial cell monolayers. Pharm Res. 1999;16:1360–1365. [DOI] [PubMed] [Google Scholar]
- 85.Emerich DF, Dean RL, Osborn C, Bartus RT. The development of the bradykinin agonist labradimil as a means to increase the permeability of the blood-brain barrier: from concept to clinical evaluation. Clin Pharmacokinet. 2001;40:105–123. [DOI] [PubMed] [Google Scholar]
- 86.Sang H, Qiu Z, Cai J, et al. Early Increased Bradykinin 1 Receptor Contributes to Hemorrhagic Transformation After Ischemic Stroke in Type 1 Diabetic Rats. Transl Stroke Res. 2017;8:597–611. [DOI] [PubMed] [Google Scholar]
- 87.Sorby-Adams AJ, Leonard AV, Hoving JW, et al. NK1-r antagonist treatment comparable to decompressive craniectomy in reducing intracranial pressure following stroke. Front Neurosci. 2019;13:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turner RJ, Helps SC, Thornton E, Vink R. A substance P antagonist improves outcome when administered 4 h after onset of ischaemic stroke. Brain Res. 2011;1393:84–90. [DOI] [PubMed] [Google Scholar]
- 89.Karamyan VT. The role of peptidase neurolysin in neuroprotection and neural repair after stroke. Neural Regen Res. 2020;16:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of our study will be made available upon reasonable request.