

Abstract
Chronic graft-versus-host disease (GVHD) results in significant morbidity and mortality, limiting the benefit of allogeneic hematopoietic cell transplantation (HCT). Peripheral blood gene expression profiling of the donor immune repertoire following HCT may provide associated genes and pathways thereby improving the pathophysiologic understanding of chronic GVHD. We profiled 70 patients and identified candidate genes that provided mechanistic insight in the biologic pathways that underlie chronic GVHD. Our data revealed that the dominant gene signature in patients with chronic GVHD represented compensatory responses that control inflammation and included the interleukin-1 decoy receptor, IL-1 receptor type II, and genes that were profibrotic and associated with the IL-4, IL-6 and IL-10 signaling pathways. In addition, we identified three genes that were important regulators of extracellular matrix. Validation of this discovery phase study will determine if the identified genes have diagnostic, prognostic or therapeutic implications.
Keywords: Graft-versus-host-disease, Bone marrow transplantation, Genomic markers, Gene expression
1. Introduction
Chronic graft-versus-host disease (GVHD) is the major late complication after allogeneic hematopoietic cell transplantation (HCT) and approximately half of patients who survive beyond 100 days after transplantation develop chronic GVHD [1,2]. The manifestations of chronic GVHD adversely impact patient quality of life and the three year survival rate for newly diagnosed “favorable” and “poor” risk chronic GVHD is approximately 80% and 40%, respectively [3] and only 50% of patients discontinue immunosuppressive treatment within 5 years after its onset [4,5]. Improvements in supportive care and infection prophylaxis and the introduction of reduced intensity conditioning (RIC) have led to a significantly higher number of patients surviving beyond 100 days after transplant [6–10], and therefore the number of patients at risk for chronic GVHD was estimated to double over a 5 year period [11].
The pathogenesis of chronic GVHD is incompletely understood and although it often follows acute GVHD, it is clinically and biologically distinct, and can occur even in the absence of acute GVHD [12]. Chronic GVHD is predominantly a disease of immune dysregulation with donor T cells mediating chronic alloimmune (directed to recipient tissue histocompatibility antigens) and autoimmune (directed against antigens on both donor and recipient tissue) reactions [13,14]. The activated donor immune response progresses because of attenuated or absent thymic and peripheral mechanisms of clonal deletion and tolerance [15,16]. The pathologic immune response attacks the target tissues of chronic GVHD through direct cellular mechanisms, and inflammatory and sclerosing cytokines, and autoantibody production [17,18].
The diagnosis of chronic GVHD can be somewhat challenging especially when diagnostic features are absent or when the clinical features are confined to internal organs (i.e., lungs) or clinical assessment is hampered by medical co-morbidities. In the more challenging cases biopsy confirmation is essential to the diagnosis [19]. About 10% of patients referred to a chronic GVHD specialist had no biopsy and were incorrectly diagnosed and treated prior to their referral [20]. Furthermore, uniform minimal diagnostic histologic criteria for chronic GVHD have not been established and validated for affected organs, and sampling and technical factors can also contribute to a false negative histologic assessment. Due to the diagnostic challenges in the clinical presentation and the limitations in time and invasiveness of obtaining a tissue biopsy, a recent focus has been to identify surrogate diagnostic markers of chronic GVHD [21–23]. To date, no single gene or pathway has been identified, validated, and introduced into clinical practice for chronic GVHD.
Discovery phase gene expression microarray profiles provide a snapshot of all the transcriptional activity in a biological condition, and facilitate the identification of novel and previously unrecognized functional role of genes. In the current report, we conducted a discovery phase gene expression study and hypothesized that the gene expression profile of the circulating donor immune cell repertoire following allogeneic HCT would highlight a list of candidate genes and pathways associated with the presence of chronic GVHD. Among 70 patient samples assessed in this retrospective cross-sectional, discovery phase study, we identified potential genes that appeared consistent with the pathobiology and cellular basis of chronic GVHD. We used biostatistical methods and repeated the analyses 100 times with random different splits of the confounding gene elimination, training and tests sets, and consistently identified a repertoire of genes that were regulators of excessive inflammatory bioactivity included the interleukin-1 (IL-1) decoy receptor, IL-1 receptor type II (IL-1R2), and genes that were profibrotic and associated with the IL-4, IL-6 and IL-10 signaling pathways. In addition, we identified three genes that were important regulators of extracellular matrix remodeling involved in connective tissue disorders. Our data suggests that the dominant gene signature in patients with established chronic GVHD represent compensatory anti-inflammatory and profibrotic genes. This discovery phase study supports the utility of gene profiling to further the understanding of the molecular pathogenesis of chronic GVHD and following further refinements may represent a useful noninvasive assay in the diagnosis, prognostic assessment, and identification of novel therapeutic approaches in chronic GVHD.
2. Materials and methods
2.1. Study population
The Division of Blood and Marrow Transplantation at Stanford University Medical Center maintains a comprehensive clinical and pathologic database of patients undergoing transplantation on Institutional Review Board approved protocols. Peripheral blood samples for research purposes are routinely obtained from patients at designated time points after transplant following written informed consent. A computer review of the database identified 167 patients between February 13, 2008 and April 24, 2009, who were seen in the outpatient clinic and met the following inclusion criteria: alive beyond 100 days from allogeneic HCT with complete (100%) donor CD3+ T cell chimerism, without disease relapse for greater than 90 days after the time of sample acquisition, and for which research blood samples were available. The exclusion criteria included patients with active viral, fungal, or bacterial infection or who received treatment for documented viral (including viremia), fungal, or bacterial infection within 30 days of sample acquisition. Patients classified in the non-chronic GVHD group had an additional exclusion which was the development of GVHD within 90 days of sample acquisition. From among this group, 68 of 167 patients were conveniently selected for analysis in the current study.
The information in the database is gathered from the chart note following a medical assessment by the primary transplant physician during the clinic visit. A review of the medical records of the 68 patients was performed by two independent investigators to confirm patient inclusion and exclusion criteria, confirm a history of acute and chronic GVHD and its severity, and the presence or absence of chronic GVHD at the time of sample acquisition. The reviewed information also included the immune suppression medication(s), if any, prescribed for GVHD therapy and its dose(s), the peripheral blood leukocyte count and differential, and the status of chimerism and disease. From among the cohort of 68 patients, 3 were excluded due to disease relapse within 90 days of the sample acquisition date, and 2 were excluded because of either mixed chimerism (<100% donor type) or the development of chronic GVHD within 90 days of sample acquisition. The 63 patients were divided into chronic GVHD and non-chronic GVHD groups. The diagnosis and grading of acute [24] and chronic [25] GVHD (including overlap syndrome) followed established criteria and were determined by two independent investigators. Severity scores using the categories of mild, moderate and severe chronic GVHD were also captured according to the consensus document on Guidelines for Global Scoring of Chronic GVHD [19,26]. Chronic GVHD assessment included type of presentation; de novo (no antecedent history of acute GVHD), interrupted (prior history of acute GVHD that resolved yet was later followed by the onset of chronic GVHD), or progressive (prior history of acute GVHD that remained active and developed into chronic GVHD) [25]. Immunosuppressive therapy for the treatment of acute (grades II–IV) and chronic GVHD (extensive) typically consisted of initiating prednisone or solumedrol 1–2 mg/kg/day. Two patients were successfully weaned from immunosuppressive drug therapy to physiologic corticosteroid replacement (prednisone 2.5 mg daily) for greater than 180 days prior to blood sample acquisition, and were considered to have no chronic GVHD for analysis.
2.2. Prospective cohort study population
To mitigate the influence of immune suppression medication on the gene signature we collected a prospective cohort of 7 consecutive patients who presented from July 1, 2011, to September 30, 2011, with new onset chronic GVHD, who fulfilled the following criteria: were beyond 6 months after allogeneic HCT, did not have a history of prior acute or chronic GVHD, and presented with moderate severity de novo chronic GVHD. These patients were free of immune suppression medication at the time of onset of chronic GVHD and at the time of blood sample acquisition. They had 100% donor type among CD3+ T cells, and were free of active bacterial, viral and fungal infection. All signed informed consent for study analysis.
2.3. Sample preparation
Peripheral blood mononuclear cells (PBMCs) from 70 patients were obtained after Ficoll (Amersham, Piscataway, NJ) gradient centrifugation of 20 mL of whole blood collected in heparin containing Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ). Ten to 30 million cells were aliquoted into cryovials containing RPMI with 10% DMSO, and 10% fetal bovine serum and were cryopreserved and stored at −80 °C until thawed for gene expression analysis.
2.4. Microarray target preparation, hybridization, and gene expression profiling
Total RNA was isolated from PBMC samples using the RNeasy mini kit (Qiagen). RNA concentration was measured using NanoDrop® ND-1000 (NanoDrop Technologies, Wilmington, DE) and the integrity of RNA was assessed with the Agilent 2100 Bioanalyzer using RNA Nano Chips (Agilent Technologies, Santa Clara, CA). RNA was stored in RNase-free water at −80 °C. For hybridization onto Agilent Whole Human Genome 4 × 44K 60mer oligonucleotide arrays (G4112F, Agilent Technologies, Palo Alto, CA), 100 ng of total RNA was used in the Agilent LIRAK PLUS, two-color Low RNA input Linear Amplification method, according to the manufacturer’s instructions. Briefly, total RNA was reverse transcribed into complimentary DNA (cDNA) using T7-promotor primer and MMLV reverse transcriptase. The cDNA was transcribed into complimentary RNA (cRNA), during which it was fluorescently labeled by incorporation of cyanine (Cy) 5-CTP (target samples) or Cy3-CTP. After purification, using the RNeasy mini kit (Qiagen), cRNA yield and Cy incorporation efficiency (specific activity) into the cRNA were determined using a NanoDrop Spectrophotometer (NanoDrop Technologies). cRNAs with a yield of >825 ng and a specific activity of 8–20 pmol/μg were selected for further processing. Equal amounts of the target and reference control sample (825 ng) were competitively hybridized onto Agilent Whole 4 × 44K Human oligonucleotide arrays in a hybridization oven at 65 °C for 17 h. Slides were washed according to the manufacturer’s instructions with washing buffers and finally dipped in Stabilization and Drying Solution (Agilent Technologies) to protect them from environmental ozone. The arrays were scanned on an Agilent scanner and further processed using Agilent Feature Extraction Software.
2.5. Microarray analysis
We used the Agilent 44K array and a cut-off for absolute value of log2red channel/green channel >0.5 for at least one array; data was processed and normalized in GeneSpring GX7 (Agilent Technologies, Redwood, CA).
2.6. Analysis for potential confounding variables
We examined the correlation between the classification score from PAM and potential clinical confounding variables that included length in days of date of sample acquisition relative to transplant date, donor–recipient relationship (related and unrelated), type of conditioning regimen (full-dose versus reduced intensity conditioning), recipient age, donor gender and type of cancer (myeloid and lymphoid) based on linear or logistic regression when appropriate. We repeated the PAM analysis and adjusted for length in days of sample time acquisition relative to transplant date, the type of conditioning regimen and donor–recipient relationship, which were selected a priori as potentially important confounders.
2.7. Biological pathway analysis
The probes on Agilent platform were re-annotated using AILUN [27] and mapped to get the most recent NCBI gene identifiers for the analysis. Ingenuity Pathways Analysis software (http://www.ingenuity.com/, Ingenuity Systems, Inc., Redwood City, CA) and Pathway Express [28] were used to assess the functional composition of genes and related regulatory networks for the minimum significant gene classifiers [29]. Data was deposited at http://www.ncbi.nlm.nih.gov/projects/geo (GSE23924).
3. Results
3.1. Patient and sample characteristics
Table 1 summarizes the clinical features of 63 patients (34 with myeloid malignancies, 27 with lymphoid malignancies, and 2 with bone marrow failure disorders) included in this cross-sectional, retrospective, series and of the prospective 7 patient cohort with newly diagnosed chronic GVHD not yet on immune suppression medication. At the time of peripheral blood collection for gene expression analysis, a median of 424 days post-HCT, 28 patients had no evidence of chronic GVHD and were weaned from immune suppression medication prior to blood sample acquisition (Table 2). Among the 35 patients with a history of chronic GVHD; 20 patients (32%) had de novo chronic GVHD, 6 patients (10%) had interrupted chronic GVHD, and 9 patients (14%) had progressive chronic GVHD. The prospective cohort of 7 patients had new onset moderate severity de novo (2/7) and interrupted (5/7) chronic GVHD and were not on immune suppression drugs at the time of sample acquisition which was a median of 266 days following HCT (range of 177–561 days) (Table 2). At the time of blood sample acquisition, all 44 patients with chronic GVHD had diagnostic oral chronic GVHD features with lichen planus-like changes on the buccal mucosa. Patients with chronic GVHD also had diagnostic skin manifestations that included poikiloderma, and/or lichen planus-like, morphea-like, or sclerosis-like lesions.
Table 1.
Patient and disease characteristics.
| Retrospective patient data set | Prospective, newly-diagnosed cGvHD | |||
|---|---|---|---|---|
| All patients | cGvHDa | Non-cGvHDa | ||
| Characteristics | n = 63 | n = 35 | n = 28 | n = 7 |
| Age, years | ||||
| Median (and range) | 50 (19–64) | 45 (19–62) | 51 (26–64) | 44 (31–58) |
| Gender, no. (%) | ||||
| Male | 31 (49.2) | 15 (42.9) | 16 (57.1) | 6 (85.7) |
| Female | 32 (50.8) | 20 (57.1) | 12 (42.9) | 1 (13.3) |
| Diagnosis - no. (%) | ||||
| Lymphoid | 27 (41.3) | 14 (40.0) | 17 (60.7) | 4 (57.1) |
| Prior autologous transplantation | 4 | 3 | 1 | 1 |
| Myeloid | 34 (54.0) | 23 (65.7) | 12 (42.9) | 3 (42.9)) |
| Anemia (Aplastic Anemia or Diamond-Blackfan Anemia) | 2 (3.2) | 1 (2.9) | 1 (3.6) | 0 |
| Donor - no. (%) | ||||
| Sibling | 41 (65.1) | 22 (62.9) | 19 (67.9) | 2 (28.6) |
| Unrelated | 22 (34.9) | 13 (37.1) | 9 (32.1) | 5 (71.4) |
| Conditioning - no. (%) | ||||
| Reduced-intensity conditioning | 25 (39.7) | 9 (25.7) | 16 (57.1) | 2 (28.6) |
| Myeloablative | 38 (60.3) | 26 (74.3) | 12 (42.9) | 5 (71.4) |
| Acute GvHD - no. (%) | ||||
| None | 46 (73.0) | 20 (57.1) | 26 (92.9) | 2 (28.6) |
| Yes | 17 (27.0) | 15 (42.9) | 2 (7.1) | 5 (71.4) |
| Grade 1 | 1 | 1 | 0 | 2 |
| Grade 2 | 5 | 4 | 1 | 3 |
| Grade 3 | 9 | 8 | 1 | 0 |
| Grade 4 | 2 | 2 | 0 | 0 |
2 patients with a history of mild severity cGvHD who were weaned from immunosuppressive medications yet maintained on 5 mg every-other-day of prednisone for >180 days at the time of sample acquisition were considered non-GvHD.
Table 2.
Patient laboratory and cGvHD characteristics at sample.
| Retrospective patient data set | Prospective, newly-diagnosed cGvHD | |||
|---|---|---|---|---|
| Characteristics | All patients | cGvHDa | Non-cGvHDa | |
| n = 63 | n = 35 | n = 28 | n = 7 | |
| Post-transplant interval, days Median (and range) | 475 (131–3,445) | 570 (131–3,445) | 326 (189–2,065) | 266 (177–561) |
| White blood count at sample, cells/μL Median (and range) | 6.3 (2.0–11.7) | 7.1 (2.0–11.5) | 5.7 (2.4–11.7) | 6.0 (2.2–9.7) |
| Differential, neutrophil, % Median (and range) | 61.1 (3.8–94.2) | 73.2 (39.8–94.2) | 52.2 (3.8–82.7) | 59.8 (5.4–90.5) |
| Differential, lymphocyte, % Median (and range) | 25.2 (2.9–85.1) | 20 (2.9–49.9) | 34.8 (8.6–85.1) | 26.3 (3.0–84.9) |
| Differential, monocyte, % Median (and range) | 8.9 (1.1–18.3) | 7.8 (1.1–15.4) | 9.8 (3–18.3) | 9.0 (1.9–17.1) |
| Chronic GvHD at sample, n (%) | ||||
| None | 26 (41.3) | 0 | 26 (92.9) | 0 |
| De novo | 21 (33.3) | 20 (57.1) | 1 (3.6) | 2 (28.6) |
| Mild/moderate/severe | 10/10/1 | 9/10/1 | 1/0/0 | 0/2/0 |
| Active/inactive | 20/1 | 20/0 | 0/1a | 0/2 |
| Interrupted | 7 (11.1) | 6 (17.1) | 1 (3.6) | 5 (71.4) |
| Mild/moderate/severe | 1/6/0 | 0/5/0 | 1/0/0 | 0/5/0 |
| Active/inactive | 6/1 | 5/0 | 0/1a | 0/5 |
| Progressive | 9 (14.3) | 9 (25.7) | 0 | 0 |
| Mild/moderate/severe | 0/7/2 | 0/7/2 | 0/0/0 | 0/0/0 |
| Active/inactive | 9/0 | 9/0 | 0 | 0 |
| Immunosuppression | ||||
| None | 26 (41.3) | 0 | 26 (92.9) | 7b |
| One | 12 (19.0) | 10 (28.6) 2 (7.1)a | 0 | |
| Two | 20 (31.7) | 20 (57.1) | 0 | 0 |
| Three or >three | 5 (7.9) | 5 (14.3) | 0 | 0 |
2 patients with a history of mild severity cGvHD who were weaned from immunosuppressive medications yet maintained on 5mg every-other-day of prednisone for N180 days at the time of sample acquisition were considered non-GvHD.
All patients had peripheral blood sampling for gene expression prior to initiating immunosuppression.
3.2. Confounding gene elimination screen, training and test set populations
In order to understand the genes potentially contributing to chronic GVHD, our first analytic objective was to develop a classification rule to distinguish chronic GVHD from non-GVHD patients based on expression levels of selected genes. The data set consisted of 7 newly diagnosed chronic GVHD patients who were not yet on immune suppression medicine, 35 chronic GVHD patients on immune suppression medicine and 28 non-GVHD controls not on immune suppression medication. We constructed three data sets as shown in Fig. 1: a confounding gene elimination screen set that consisted of 7 new GVHD patients and 14 chronic GVHD patients; a training set that consisted of 21 chronic GVHD patients and 21 non-GVHD controls and a test set that consisted of 7 new GVHD patients and 7 non-GVHD controls. The only overlap among the three data sets was the 7 new GVHD patients who appeared in both the confounding gene elimination screen and test set.
Figure 1.
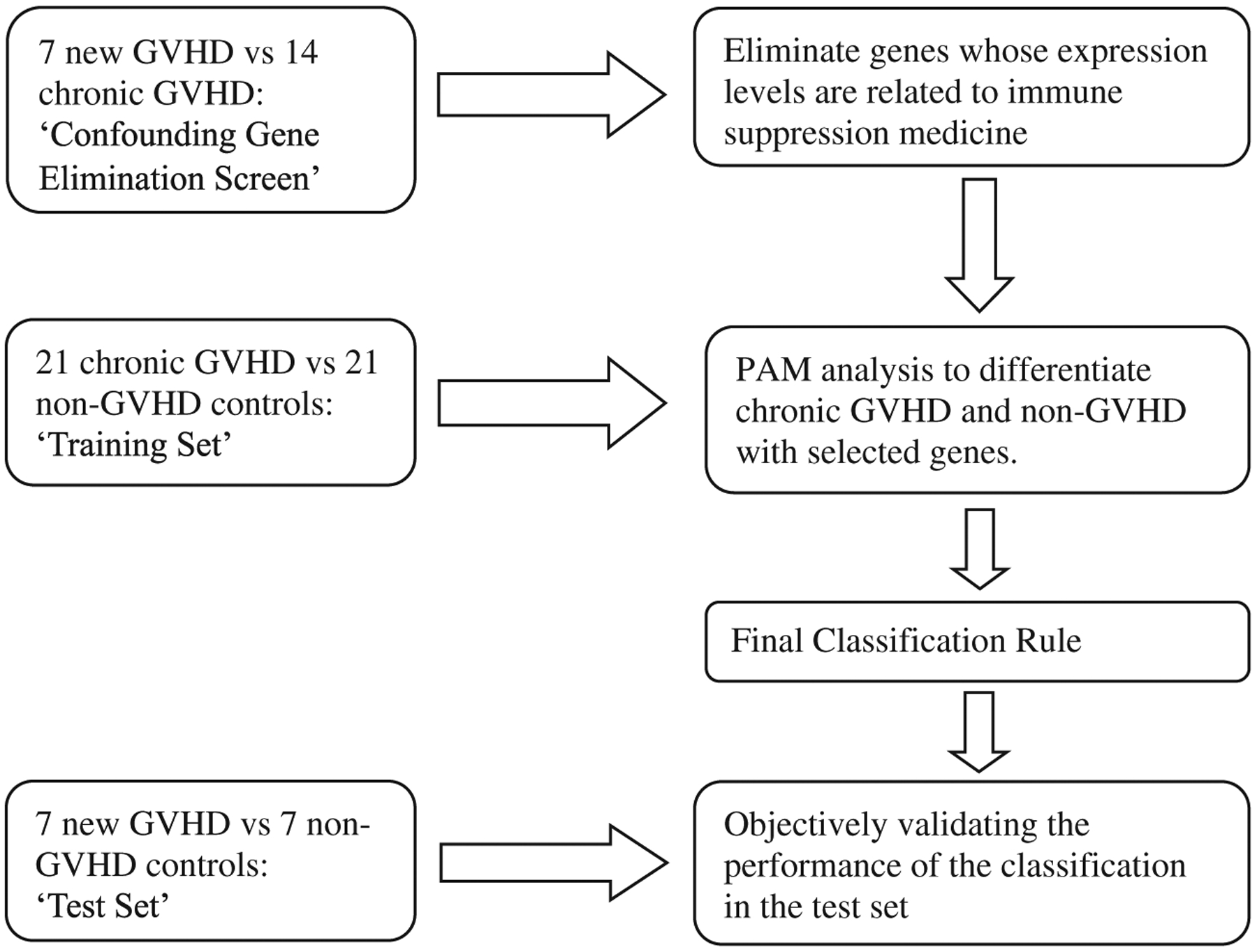
Study design. Retrospective banked samples from 63 patients (35 with chronic GVHD and 28 non-GVHD) and prospectively obtained samples from 7 patients with new onset chronic GVHD satisfied study inclusion and exclusion criteria for gene expression profiling. We constructed three data sets: a ‘confounding gene elimination screen’ set that consisted of the 7 new GVHD patients and 14 chronic GVHD patients; a ‘training set’ that consisted of 21 chronic GVHD patients and 21 non-GVHD controls and a ‘test set’ that consisted of the 7 new GVHD patients and 7 non-GVHD controls. In the confounding gene elimination screen, we compared gene expression levels in patients with GVHD on immune suppression medicine to those not yet on immune suppression medicine and disregarded genes whose t-test statistics were beyond the interval of [−1,1]. In the training set, we applied the PAM method to discriminate GVHD from non-GVHD using the selected probe sets identified from the screening step. We evaluated the performance of the classification rule developed from the training set to differentiate chronic GVHD patients from non-GVHD patients in the test set. We repeated the entire analysis 100 times using different random splits of patient samples in the screening, training and test sets and recorded the frequency of the selected genes from the PAM classifiers to determine reproducibility.
In the confounding gene elimination screen, we compared gene expression levels in patients with chronic GVHD on immune suppression medicine to those with chronic GVHD not yet on immune suppression medicine using a two-sample t-test. We eliminated 27k genes whose t-test statistics were beyond the interval of [−1,1] as differences in expression could potentially be related to the presence or absence of immune suppression medication. After the elimination screen we found that 17,208 genes remained whose t-test statistics were within the [−1,1] interval. The gene elimination screen was not used to identify genes that discriminate GVHD from non-GVHD patients. The latter identification was done in the training set.
In the training set, that consisted of 42 samples (21 patients with chronic GVHD compared to 21 non-GVHD patients) we applied the Prediction Analysis of Microarray (PAM) [30]5 method, an algorithm based on nearest shrunken centoids, to discriminate GVHD from non-GVHD using the 17,208 probes that remained following the confounding gene elimination screen. Within the training set, 20 rounds of cross validation were performed using different partitions to determine the optimal threshold level in PAM. We found a minimum misclassification error of 6 samples was achieved with probe sets that contained 10 or 12 nested genes, as shown in Fig. 2. The 6 samples were found in the chronic GVHD group, and were misclassified as non-GVHD because the probability scores were less than 50% (Fig. 2). All non-GVHD patients were correctly classified. In contrast, an average misclassification error of 21 (50%) was expected if we applied a random classification rule. The gene names of the 12 probe set are listed in Table 3, which were also the same genes in the 10 probe set plus two additional ones. The expression values of genes in Tables 3 were significantly higher in the chronic GVHD group compared to non-GVHD patients.
Figure 2.
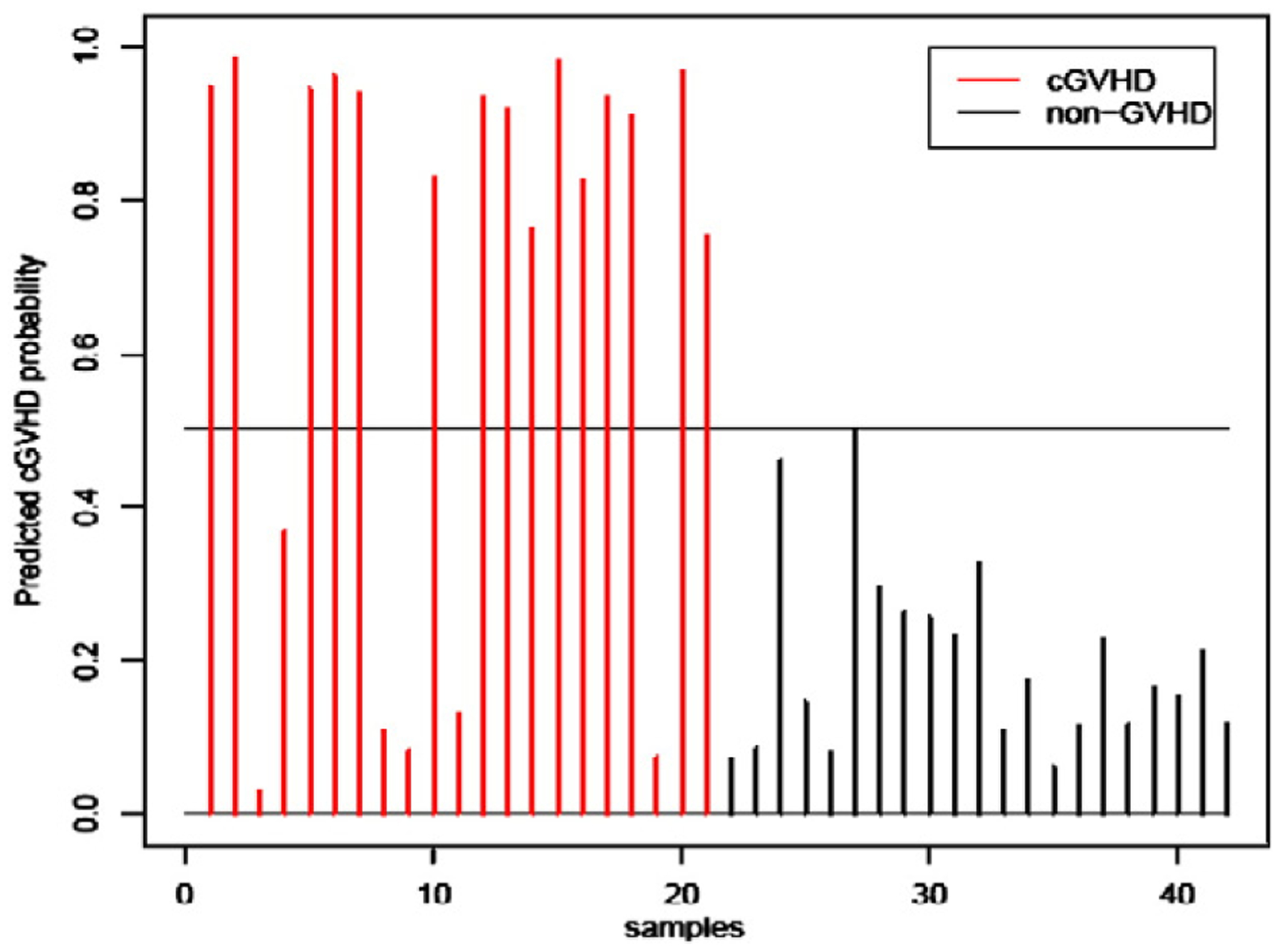
Chronic GVHD ‘training set’. We applied the Prediction Analysis of Microarray (PAM) method to a ‘training set’ to discriminate 21 GVHD patients from 21 non-GVHD using the 17,208 probes that remained from the confounding gene elimination screen and found a minimum misclassification error of 6 samples was achieved with a probe set that contained 10 or 12 genes. Shown is the cross-validated performance of classifier of the 10 genes from PAM. (A) Fifteen of the 21 chronic GVHD patients were correctly scored (red). (B) All 21 non-GVHD patients were correctly scored as no GVHD (black). Near identical results were observed with the performance of the 12 gene probe set (not shown).
Table 3.
Twelve probe set.
| Probe name | Gene symbol | GVHD | Non-GVHD | p-value (t-test) | Foldchange |
|---|---|---|---|---|---|
| A_24_P251866a | ADAMTS2 | 1.499 | 0.0175 | 2.50E - 07 | 85.66 |
| A_23_P321307a | ADAMTS2 | 1.995 | 0.0144 | 2.90E - 06 | 138.54 |
| A_23_P79398a | IL1R2 | 2.251 | 0.005 | 8.10E - 06 | 450.2 |
| A_24_P63019a | IL1R2 | 2.735 | 0.497 | 8.40E-06 | 5.5 |
| A_32_P142521 | BCAT1 | 1.736 | 0.433 | 1.80E - 04 | 4.01 |
| A_23_P162449 | SRGAP1 | −0.975 | −2.156 | 1.00E - 04 | |
| A_23_P213615a | ADAMTS3 | 0.299 | −0.835 | 1.60E - 04 | |
| A_23_P259071 | AREG | −0.77 | −2.307 | 1.20E - 03 | |
| A_23_P123848 | DAP2IP | −0.997 | −1.911 | 5.80E-05 | |
| A_24_P154037 | IRS2 | 1.404 | 0.587 | 6.95E-05 | 2.39 |
| A_23_P131676 | CXCR7 | −1.644 | −2.679 | 0.002 | |
| A_23_P162668 | CPM | 2.273 | 1.442 | 0.0063 | 1.58 |
AREG: Amphiregulin; BCAT1: Branched chain aminotransferase 1; CPM: Carboxypeptidase M; CXCR7: C-X-C chemokine receptor type 7; DAB2IP: DOC-2/DAB2 interactive protein; IRS2: Insulin receptor substrate 2; SRGAP1: Slit-Robo GTPase 1.
Appears in Table 1.
We next evaluated the performance of the classification rule developed from the training set to differentiate chronic GVHD patients from non-GVHD patients in a 14 patient test set and correctly predicted 3 new chronic GVHD and all 7 non-GVHD controls when the 10 probe set was used (Fig. 3); similar results were observed when the 12 probe set was used (data not shown).
Figure 3.
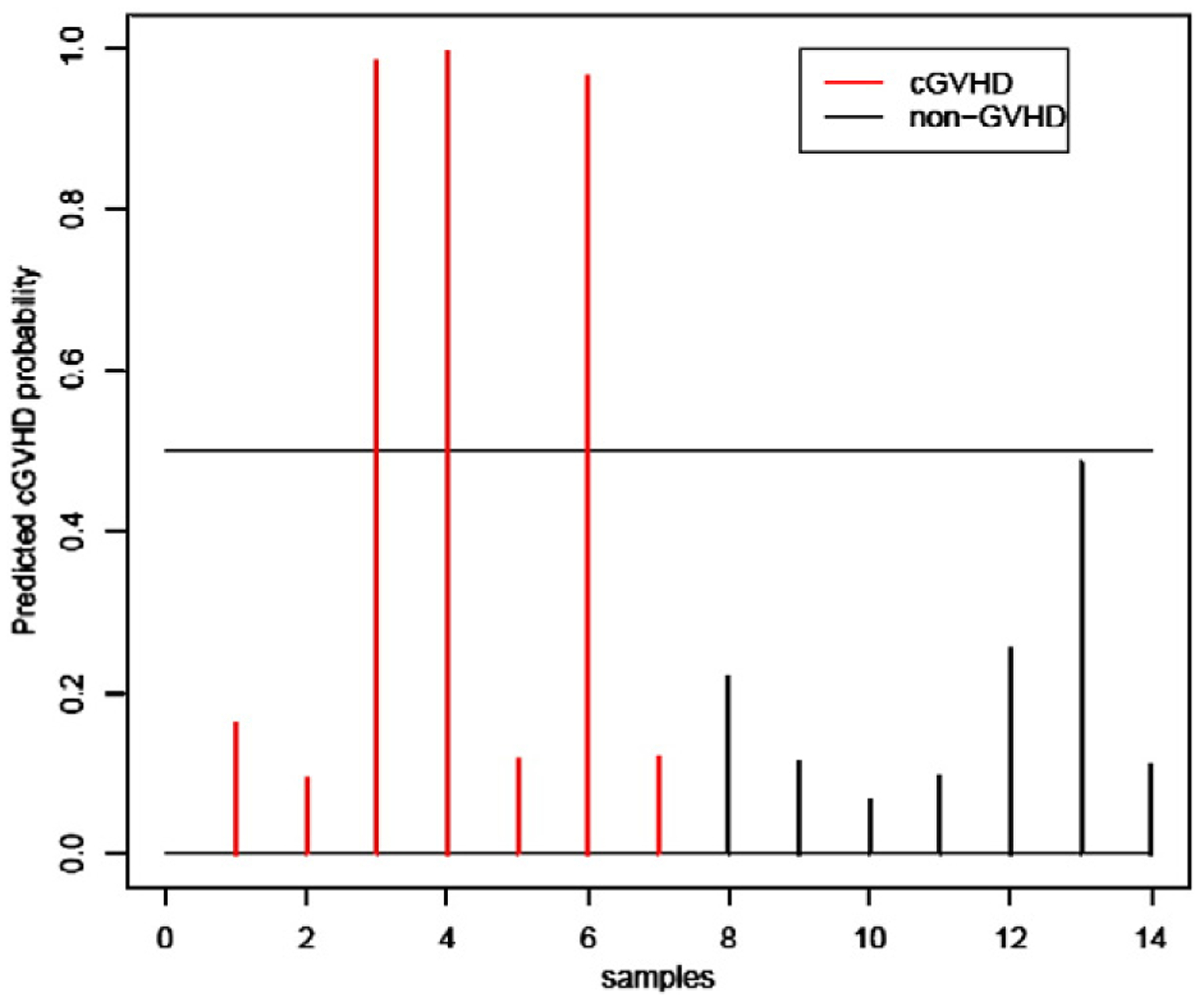
Chronic GVHD ‘test set’ sample prediction. Performance of the 10 probe set for predicted probabilities of the 14 samples in the test set. (A) Three of the 7 new onset chronic GVHD patients were correctly predicted (red). (B) All 7 non-GVHD patients were correctly predicted as no GVHD (black).
3.3. Analysis for confounding variables
A limitation to this cross-sectional, retrospective study is the heterogeneity of the patient population and the influence of unmeasured covariates. We therefore assessed the correlation between the classification score from PAM for potential confounding variables that included length in days of sample acquisition date relative to the transplant date, recipient age, donor gender, disease histology (myeloid or lymphoid malignancy), whether the donor was related or unrelated to the recipient, and type of conditioning regimen (full dose or reduced intensity conditioning). We calculated the PAM-based gene score for the probability of GVHD in the 42 patient test set and found that the gene score was significantly lower in patients who were related with their donor (p = 0.049) or who received RIC (p = 0.025). In contrast, the gene score was not significantly associated with the length in days of sample acquisition date relative to the transplant date (p = 0.098), recipient age (p = 0.116), donor gender (p = 0.894) and type of cancer (p = 0.240).
3.4. Validation analysis
We repeated PAM and adjusted for the length in days of sample acquisition date relative to the transplant date, the type of conditioning regimen and donor–recipient relationship, and observed a result similar to that from the unadjusted analysis. Specifically, with a gene score based on the following probes; “A_23_P321307”, “A_24_P63019”, “A_23_P79398” and “A_24_P251866”, PAM achieved a minimum cross-validated misclassification error rate of 9 among the test set cohort of 42 samples (Fig. 4). The adjusted gene score was not significantly associated with any of the confounding factors, which confirmed that the identified genes were informative to GVHD status beyond the influence of confounding factors. It is noteworthy that the four probes were identical to the top four probes from the unadjusted PAM analysis. Lastly, we repeated the entire analysis 100 times and used different random splits of the 70 samples in the confounding gene elimination screen, training and test sets and obtained similar results with each run. The probe sets with top frequencies are listed in Table 4. The top 4 probes identified the ADAMTS 2 gene that encodes a disintegrin and metalloproteinase, and the IL-1R2 gene that encodes an IL-1 decoy receptor.
Figure 4.
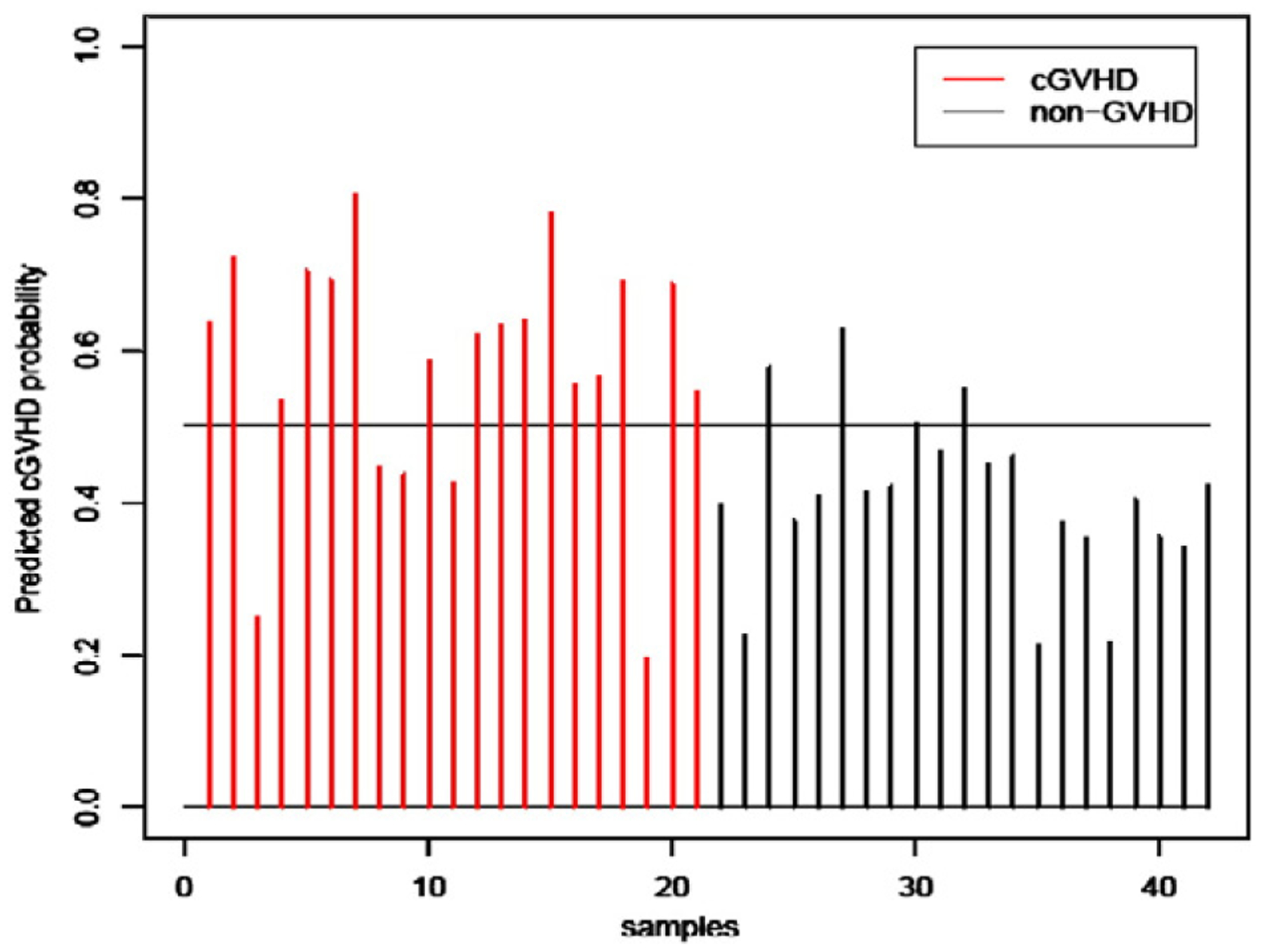
Cross-validated performance based on 4 genes from adjusted PAM in the training set of 42 samples. (A) Sixteen of the 21 chronic GVHD patients were correctly scored (red). (B) Seventeen of the 21 non-GVHD patients were correctly scored as no GVHD (black).
Table 4.
Probe set and biologic gene function.
| Probe set | Frequency (%) | Entrez gene ID | Gene symbol | Entrez gene name | Function |
|---|---|---|---|---|---|
| A_23_P321307a | 94 | 9509 | ADAMTS2 | A disintegrin and metalloproteinase with thrombospondin motifs 2 | Immunological disease, extracellular matrix modification |
| A_24_P63019a | 92 | 7850 | IL1R2 | Interleukin 1 receptor, type II | Cell signaling, immune cell trafficking, hematological and lymphoid system development and function |
| A_23_P79398a | 92 | 7850 | IL1R2 | – | – |
| A_24_P251866a | 75 | 9509 | ADAMTS2 | – | – |
| A_23_P213615a | 42 | 9508 | ADAMTS3 | A disintegrin and metalloproteinase with thrombospondin motifs 3 | Immunological disease, extracellular matrix modification |
| A_23_P145965 | 34 | 8460 | TPST1 | Tyrosylprotein sulfotransferase 1 | Immunological disease, immune cell trafficking, hematological and lymphoid system development and function |
| A_23_P126540a | 30 | 9508 | ADAMTS3 | – | – |
| A_23_P93562a | 27 | 27,244 | SESN1 | Sestrin 1 | Cellular growth and proliferation, immune cell trafficking, hematological and lymphoid system development and function |
| A_23_P60627a | 26 | 247 | ALOX15B | Arachidonate 15-liopxygenase, type B | metabolism, fatty acid production |
| A_24_P154037b | 24 | 8660 | IRS2 | Insulin receptor substrate 2 | Cell cycle, humoral immune response, immune cell trafficking, hematological and lymphoid system development and function |
| A_23_P259071b | 24 | 374 | AREG | Amphiregulin | Cellular growth and proliferation, immunological disease |
| A_23_P8640 | 22 | 2852 | GPER | G protein-coupled estrogen receptor 1 | Cellular growth and proliferation, cell signaling, immune cell trafficking, hematological and lymphoid system development and function |
| A_23_P87528 | 19 | 586 | BCAT1 | Branched chain aminotransferase 1 | Cellular growth and proliferation, amino acid metabolism |
| A_23_P131676b | 19 | 57,007 | CXCR7 | C-X-C chemokine receptor type 7 | Immune cell trafficking, immune system development and function |
| A_23_P34345 | 18 | 7412 | VCAM1 | Vascular cell adhesion molecule 1 | Immune cell trafficking, cell signaling |
| A_23_P146233 | 18 | 4023 | LPL | Lipoprotein lipase | Fatty acid metabolism |
| A_24_P52921 | 17 | 586 | BCAT1 | – | |
| A_23_P162668b | 17 | 1368 | CPM | Carboxypeptidase M | Hematologic and lymphoid system development and function, cell signaling, immune cell trafficking |
In the 10 probe set.
In the 13 probe set.
3.5. Biological pathways analysis
To identify the biological pathways and functional groups enriched by the differentially expressed loci, the frequently identified candidate genes beyond the top four listed in Table 4 were entered into the Ingenuity Pathway Analysis database and the Database for Annotation, Visualization and Integrated Discovery. The predominant biological pathways and function identified were immune cell trafficking and hematological and lymphoid system development and function (Table 4; (http://www.ingenuity.com/, Redwood city, CA). The cluster of the up-regulated genes was used in the Kyoto Encyclopedia of Genes and Genomes (KEGG) [29] Pathway Analysis component and revealed sharing with IL-10, IL-6 and IL-4 signaling.
4. Discussion
In this cross-sectional, retrospective discovery phase series we used whole genome wide analysis of peripheral blood mononuclear cells from 70 allogeneic HCT transplant recipients coupled with biostatistical analysis and identified several candidate genes associated with chronic GVHD. Consistent with the clinical presentation and pathobiology that underlie chronic GVHD, the candidate genes identified in this study attenuated the bioactivity of proinflammatory IL-1 via a decoy receptor, converged through profibrotic immune response pathways, or were direct regulators of extracellular matrix remodeling. The genes highlighted in the present study have not been previously associated with GVHD and represent novel pathways for further study.
The proinflammatory cytokine interleukin-1 (IL-1) is central in inflammation and in the pathogenesis of inflammatory diseases such as rheumatoid arthritis and systemic lupus erythematosus [31] and also is a recognized proinflammatory cytokine mediating acute GVHD in animal models [32]. Two distinct IL-1 receptors (IL-1R) exist: the type 1 receptor (IL-1R1) that is important for transmitting the inflammatory effects of IL-1 [33] and, the type II receptor (IL-1R2) that functions as a non-signaling decoy receptor to prevent excessive IL-1 bioactivity [33]. Up-regulation of IL-1R2 represents a mechanism by which IL-1 bioactivity is attenuated [34,35]. In the current study we used an adjusted PAM to account for potential confounding variables and repeated the analysis 100 times with different random splits of the samples and consistently found significantly increased IL-1R2 expression in the classification rule for chronic GVHD. The finding of increased IL1R2 expression in chronic GVHD patients compared to non-GVHD patients is consistent with an acquired attenuation response to pathologic inflammatory processes. In other discovery phase gene expression association studies of patients with allo- and auto-immune reactivity, an attenuation response was also reported. In small bowel transplant recipients, a discovery phase PCR based gene signature study evaluated 280 selected immune, inflammation and apoptosis related genes and identified over-expression of IL-1R2 as associated with cellular rejection [36]. In another study, whole blood gene expression was evaluated with a commercially available Genechip oligonucleotide array and established a four gene panel that included elevated IL-1R2 expression as a blood based biomarker that associated with inflammatory ulcerative colitis but not non-inflammatory diarrhea [37]. Over-expression of IL1-R2 was reported in the inflammatory arthritides and not the non-inflammatory type in a discovery phase genome wide association study of blood mononuclear cells obtained from patients with systemic lupus erythematosus (SLE) and non-inflammatory arthritis [38]. We consider the finding of significantly increased IL-1R2 expression in more than 90 of the 100 repeated classification rules for chronic GVHD, combined with the aggregate biomarker literature, support the contention that upregulation of this gene can be considered evidence that chronic GVHD is associated with an attenuation response.
Other genes consistently identified in the chronic GVHD classification rule in the current discovery phase study were over-expression of ADAMTS2, and ADAMTS3. Although these genes are not directly related to pathologic immune activation per se, their over-expression may have biological relevance to chronic GVHD. The ADAMTS designates a family of secreted enzymes that have a key role in extracellular matrix integrity, degradation and turn over [37]. Specifically, the ADAMTS2 and ADAMTS3 genes are grouped among the pro-collagen modifying enzymes that regulate the clipping of collagen molecules to allow their assembly into fibrils outside cells and impart structure to connective tissue. Mutations in ADAMTS2 have been implicated in inherited connective tissue disorders like Ehrlos-Danlos [39] whereas increased levels of ADAMTS2 expression have been described as part of the pathologic lesion in patients with fixed flexion deformities of Dupuytren’s disease [38,40]. The over-expression of ADAMTS2, and ADAMTS3, in chronic GVHD patients compared to non-GVHD patients has suggestive biological relevance to the pathophysiology of the skin manifestations associated with chronic GVHD.
In addition to skin changes, oral lichen planus (OLP) is a ‘diagnostic’ clinical feature sufficient to establish a diagnosis of chronic GVHD [19]. The AREG gene, an EGFR ligand, is a key mitogen for keratinocytes in OLP, and is highly expressed by infiltrating mononuclear cells in the lamina propria in OLP lesions but not by mononuclear cells in normal oral mucosa [41]. Our finding of AREG up-regulation in chronic GVHD patients compared to non-GVHD patients suggests this too may be a relevant marker of active disease. The observation of AREG up-regulation in the current report is in keeping with a smaller study that used a commercially available 114 set gene array and compared 7 chronic GVHD patients to 7 non-chronic GVHD patients following RIC allogeneic HCT [21]. In this other report significant up-regulation of AREG was also identified in the chronic GVHD patient group [21].
GVHD is mediated in part by a cascade of pro-inflammatory pathways [8]. We evaluated the other candidate genes (other than IL1R2 and ADAMTS2, and ADAMTS3 from Table 4) frequently observed in the GVHD classification rule using database resources that highlight cell function and biological systems from large-scale molecular level information generated by high throughput technologies. The analyses revealed immune cell trafficking, and IL-4, IL-6 and IL-10 signaling as having a central role in the biologic function among the frequently identified candidate genes. IL-4, and IL-10 are considered GVHD blocking signals [42] and are controllers of inflammation and promote tolerance [43,44]. IL-4 and IL-6 help control inflammation in part by promoting matrix remodeling and regulating pro-fibrotic aspects of immune activation [45]. Our finding of upregulated genes that converged through IL-4, IL-6 and IL-10 signaling pathways suggest that the dominant signature associated with chronic GVHD represented a compensatory gene response. It remains to be determined how much in advance of a clinical diagnosis of chronic GVHD is the compensatory gene response set in play.
The current report highlights a challenge in designing discovery phase gene expression association studies using a cross-sectional method and archived samples. The main potential weakness in the current study was the comparison of gene expression in chronic GVHD patients on immune suppressive drugs to patients without chronic GVHD and without immune suppressive drugs. Chronic GVHD patients on immune suppression medications were also treated with a mixture of antifungal, antiviral, and anti-bacterial prevention medicine, and had lower absolute blood lymphocyte counts compared to non-GVHD patients not on immune suppression medication (data not shown). It is possible that the classification rule for distinguishing the two groups reflected, in part, the presence or absence of immune suppression medication and infection prevention drugs, or the imbalance in myeloid to lymphoid cells, rather than the presence of absence of chronic GVHD. To mitigate this selection bias we first compared a prospective cohort of drug-free newly diagnosed chronic GVHD patients to patients with chronic GVHD on immune suppression and anti-infection medicine and removed differentially expressed genes. We made the assumption that the differential gene expression between these two cohorts was due, at least in part, to the medicine, and otherwise we considered the groups of chronic GVHD patients similar with regards to genes that promote and/or maintain chronic GVHD. Thereafter, with genes removed that were possibly related to medication, we compared GVHD patients on medication to drug free non-GVHD patients. Three other groups of investigators also used a retrospective, cross-sectional approach and compared patients with chronic GVHD to non-GVHD patients [21,22,46]. These studies contained from 8 to 21 patients yet no attempt was made to control for immune suppression medication or other medication or confounding variables.
The classification score from PAM was also assessed for correlation with additional confounding variables. It is noteworthy that we did not find a correlation between the gene scores and the length of time of sample acquisition after transplant in chronic and control non-chronic GVHD patients. In another study, 4 candidate plasma-derived, soluble chronic GVHD markers were evaluated in children, and 3 showed no change in plasma levels with time after transplant in the control non-GVHD patients [47]. Likewise, soluble BAFF, which was identified as the most significant GVHD marker in the study, was equally increased in early and late time points after transplant in the chronic GVHD patients [47]. Taken together, there appears little evidence, at present, to require the need for temporarily matching control non-GVHD with chronic GVHD patients. Instead the lack of correlation between gene scores and the length of time after transplant in patients with and without chronic GVHD, support the current study design in which patients at 6 months were grouped together with those at 20 months. It is possible that after a diagnosis of new onset chronic GVHD the gene pattern may take weeks to months to develop, and this may be why individuals with established chronic GVHD used in the training set might not have had the same pattern as the newly diagnosed chronic GVHD patients. In the current report, only 3 of 7 new onset chronic GVHD patients showed the gene pattern, suggesting that the pattern may be a product of both chronic inflammation and use of immunosuppressive drugs. It is also reasonable to consider that some genes that underlie chronic GVHD may be expressed in a time dependent manner, or that their expression level may be influenced by the tissue affected by chronic GVHD. Yet this type of analysis would require a sufficiently large number of patients, longitudinally collected samples at designated post-transplant time points, and correlation with the clinical presentation of disease, which is beyond the scope of this discovery phase study.
Among other possible cofounding variables, we found the gene score for the probability of chronic GVHD significantly higher in patients who were unrelated with their donor, or who received a full dose transplant preparative regimen. It is reasonable to consider that recipients of unrelated donors grafts or patients who undergo full dose conditioning had immune dysregulation that might overlap to some degree with GVHD. When we adjusted for these variables the adjusted PAM-based gene score with the top 4 probes resulted in a low misclassification rate of 9 among 42 test set samples which confirmed these genes were informative for GVHD status.
It has become clear that no one cytokine is responsible for polarizing donor immune cells to one specific GVHD phenotype. Rather, GVHD represents a complex imbalance between donor derived Th-1, Th-2 and Th17 cells with other effector T and B cell subsets and antigen presenting cell subsets, all driven by a combination of cytokines and antigen stimulators [48,49]. Discovery phase studies using high through put analysis of blood mononuclear cells comparing GVHD from non-GVHD patients has the potential to identify candidate molecular pathways involved in graft-versus host reactions by screening vast numbers of genes. In the current discovery phase study we used expression profiling of peripheral blood mononuclear cells combined with biostatical analyses and identified several candidate genes that associated with chronic GVHD. The genes and signaling pathways highlighted in the current analysis suggest that compensatory responses that control inflammation are involved with profibrotic matrix remodeling, and represent the GVHD “gene footprint”. We did not identify the pro-inflammatory genes that initiate chronic GVHD before the compensatory responses occur. Upregulation of the pro-inflammatory genes may occur before manifestations of chronic GVHD and perhaps only in selected cell subsets and this is the subject of continuing investigations, and beyond the scope of the current study. The identified compensatory genes have biological relevance to clinical manifestations of chronic GVHD and may further the understanding of its pathogenesis and treatment. This study raises the possibility that compensatory responses to the inflammatory GVHD immune reactions may be engaged before the signs of GVHD become clinically apparent. Perhaps intervention with medication to suppress GVHD inflammation in patients with a compensatory gene signature might prevent the subsequent development of the diagnostic features of chronic GVHD. The candidate genes from discovery phase studies ultimately require prospective trials that use targeted gene specific methods (such as real–time PCR) to confirm that dysregulated expression is associated with disease.
Acknowledgments
This work was supported by NIH grants R01 AI085024, P01 CA049605, and P01 HL075462, and a Stanford Cancer Center Support Grant 1PO30CA124435-01. H.E.K is supported as an American Society of Hematology Scholar, Leukemia and Lymphoma Society Fellow, and Department of Defense Post-doctoral Fellow.
Abbreviations:
- GVHD
graft-versus-host disease
- HCT
allogeneic hematopoietic cell transplantation
- IL-1
interleukin-1
- IL-1R2
IL-1 receptor type II
- RIC
reduced intensity conditioning
- PBMC
peripheral blood mononuclear cells
- cDNA
complimentary DNA
- cRNA
complimentary RNA
- PAM
Prediction Analysis of Microarray
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- SLE
systemic lupus erythematosus
- OLP
oral lichen planus
References
- [1].Akpek G, Zahurak ML, Piantadosi S, Margolis J, Doherty J, Davidson R, Vogelsang GB, Development of a prognostic model for grading chronic graft-versus-host disease, Blood 97 (2001) 1219–1226. [DOI] [PubMed] [Google Scholar]
- [2].Lee SJ, Klein JP, Barrett AJ, Ringden O, Antin JH, Cahn JY, Carabasi MH, Gale RP, Giralt S, Hale GA, Ilhan O, McCarthy PL, Socie G, Verdonck LF, Weisdorf DJ, Horowitz MM, Severity of chronic graft-versus-host disease: association with treatment-related mortality and relapse, Blood 100 (2002) 406–414. [DOI] [PubMed] [Google Scholar]
- [3].Akpek G, Lee SJ, Flowers ME, Pavletic SZ, Arora M, Lee S, Piantadosi S, Guthrie KA, Lynch JC, Takatu A, Horowitz MM, Antin JH, Weisdorf DJ, Martin PJ, Vogelsang GB, Performance of a new clinical grading system for chronic graft-versus-host disease: a multicenter study, Blood 102 (2003) 802–809. [DOI] [PubMed] [Google Scholar]
- [4].Arora M, Burns LJ, Davies SM, Macmillan ML, Defor TE, Miller WJ, Weisdorf DJ, Chronic graft-versus-host disease: a prospective cohort study, Biol. Blood Marrow Transplant 9 (2003) 38–45. [DOI] [PubMed] [Google Scholar]
- [5].Stewart BL, Storer B, Storek J, Deeg HJ, Storb R, Hansen JA, Appelbaum FR, Carpenter PA, Sanders JE, Kiem HP, Nash RA, Petersdorf EW, Moravec C, Morton AJ, Anasetti C, Flowers ME, Martin PJ, Duration of immunosuppressive treatment for chronic graft-versus-host disease, Blood 104 (2004) 3501–3506. [DOI] [PubMed] [Google Scholar]
- [6].Kohrt HE, Turnbull BB, Heydari K, Shizuru JA, Laport GG, Miklos DB, Johnston LJ, Arai S, Weng WK, Hoppe RT, Lavori PW, Blume KG, Negrin RS, Strober S, Lowsky R, TLI and ATG conditioning with low risk of graft-versus-host disease retains anti-tumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors, Blood 114 (2009) 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kantarjian H, O’Brien S, Cortes J, Wierda W, Faderl S, Garcia-Manero G, Issa JP, Estey E, Keating M, Freireich EJ, Therapeutic advances in leukemia and myelodysplastic syndrome over the past 40 years, Cancer 113 (2008) 1933–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ferrara JLM, Levine JE, Reddy P, Holler E, Graft-versus-host disease, Lancet 373 (2009) 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ho VT, Aldridge J, Kim HT, Cutler C, Koreth J, Armand P, Antin JH, Soiffer RJ, Alyea EP, Comparison of Tacrolimus and Sirolimus (Tac/Sir) versus Tacrolimus, Sirolimus, and mini-methotrexate (Tac/Sir/MTX) as acute graft-versus-host disease prophylaxis after reduced-intensity conditioning allogeneic peripheral blood stem cell transplantation, Biol. Blood Marrow Transplant 15 (2009) 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, Wadleigh M, DeAngelo DJ, Stone RM, Sakamaki H, Appelbaum FR, Dohner H, Antin JH, Soiffer RJ, Cutler C, Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials, JAMA 301 (2009) 2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Appelbaum FR, Haematopoietic cell transplantation as immunotherapy, Nature 411 (2001) 385–389. [DOI] [PubMed] [Google Scholar]
- [12].Pavletic SZ, Carter SL, Kernan NA, Henslee-Downey J, Mendizabal AM, Papadopoulos E, Gingrich R, Casper J, Yanovich S, Weisdorf D, Influence of T-cell depletion on chronic graft-versus-host disease: results of a multicenter randomized trial in unrelated marrow donor transplantation, Blood 106 (2005) 3308–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Anderson BE, McNiff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ, Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ, Blood 105 (2005) 2227–2234. [DOI] [PubMed] [Google Scholar]
- [14].Shlomchik WD, Lee SJ, Couriel D, Pavletic SZ, Transplantation’s greatest challenges: advances in chronic graft-versus-host disease, Biol. Blood Marrow Transplant 13 (2007) 2–10. [DOI] [PubMed] [Google Scholar]
- [15].Weinberg K, Blazar BR, Wagner JE, Agura E, Hill BJ, Smogorzewska M, Koup RA, Betts MR, Collins RH, Douek DC, Factors affecting thymic function after allogeneic hematopoietic stem cell transplantation, Blood 97 (2001) 1458–1466. [DOI] [PubMed] [Google Scholar]
- [16].Zorn E, Kim HT, Lee SJ, Floyd BH, Litsa D, Arumugarajah S, Bellucci R, Alyea EP, Antin JH, Soiffer RJ, Ritz J, Reduced frequency of FOXP3+ CD4 + CD25+ regulatory T cells in patients with chronic graft-versus-host disease, Blood 106 (2005) 2903–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Svegliati S, Olivieri A, Campelli N, Luchetti M, Poloni A, Trappolini S, Moroncini G, Bacigalupo A, Leoni P, Avvedimento EV, Gabrielli A, Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease, Blood 110 (2007) 237–241. [DOI] [PubMed] [Google Scholar]
- [18].Banovic T, MacDonald KP, Morris ES, Rowe V, Kuns R, Don A, Kelly J, Ledbetter S, Clouston AD, Hill GR, TGF-beta in allogeneic stem cell transplantation: friend or foe? Blood 106 (2005) 2206–2214. [DOI] [PubMed] [Google Scholar]
- [19].Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, Martin P, Chien J, Przepiorka D, Couriel D, Cowen EW, Dinndorf P, Farrell A, Hartzman R, Henslee-Downey J, Jacobsohn D, McDonald G, Mittleman B, Rizzo JD, Robinson M, Schubert M, Schultz K, Shulman H, Turner M, Vogelsang G, Flowers ME, National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report, Biol. Blood Marrow Transplant 11 (2005) 945–956. [DOI] [PubMed] [Google Scholar]
- [20].Jacobsohn DA, Montross S, Anders V, Vogelsang GB, Clinical importance of confirming or excluding the diagnosis of chronic graft-versus-host disease, Bone Marrow Transplant. 28 (2001) 1047–1051. [DOI] [PubMed] [Google Scholar]
- [21].Poloni A, Sartini D, Emanuelli M, Trappolini S, Mancini S, Pozzi V, Costantini B, Serrani F, Berardinelli E, Renzi E, Olivieri A, Leoni P, Gene expression profile of cytokines in patients with chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation with reduced conditioning, Cytokine 53 (2011) 376–383. [DOI] [PubMed] [Google Scholar]
- [22].Lai P, Weng J, Lu Z, Guo R, Luo C, Wu S, Ling W, Geng S, Du X, Gene expression profiling-based identification of CD28 and PI3K as new biomarkers for chronic graft-versus-host disease, DNA Cell Biol. 30 (2011) 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Paczesny S, Levine JE, Braun TM, Ferrara JL, Plasma biomarkers in graft-versus-host disease: a new era? Biol. Blood Marrow Transplant 15 (2009) 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, Lerner KG, Thomas ED, Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors, Transplantation 18 (1974) 295–304. [DOI] [PubMed] [Google Scholar]
- [25].Lee SJ, Vogelsang G, Flowers ME, Chronic graft-versus-host disease, Biol. Blood Marrow Transplant 9 (2003) 215–233. [DOI] [PubMed] [Google Scholar]
- [26].Filipovich AH, Diagnosis and manifestations of chronic graft-versus-host disease, Best Pract. Res. Clin. Haematol 21 (2008) 251–257. [DOI] [PubMed] [Google Scholar]
- [27].Chen R, Li L, Butte AJ, AILUN: reannotating gene expression data automatically, Nat. Methods 4 (2007) 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Khatri P, Bhavsar P, Bawa G, Draghici S, Onto-Tools: an ensemble of web-accessible, ontology-based tools for the functional design and interpretation of high-throughput gene expression experiments, Nucleic Acids Res. 32 (2004) W449–W456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M, KEGG for representation and analysis of molecular networks involving diseases and drugs, Nucleic Acids Res. 38 (2010) D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tibshirani R, Hastie T, Narasimhan B, Chu G, Diagnosis of multiple cancer types by shrunken centroids of gene expression, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaijzel EL, Bayley JP, van Krugten MV, Smith L, van de Linde P, Bakker AM, Breedveld FC, Huizinga TW, Verweij CL, Allele-specific quantification of tumor necrosis factor alpha (TNF) transcription and the role of promoter polymorphisms in rheumatoid arthritis patients and healthy individuals, Genes Immun. 2 (2001) 135–144. [DOI] [PubMed] [Google Scholar]
- [32].Hill GR, Teshima T, Gerbitz A, Pan L, Cooke KR, Brinson YS, Crawford JM, Ferrara JL, Differential roles of IL-1 and TNF-alpha on graft-versus-host disease and graft versus leukemia, J. Clin. Invest 104 (1999) 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dunne A, O’Neill LA, The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense, Sci. STKE 2003 (2003) re3. [DOI] [PubMed] [Google Scholar]
- [34].Lang D, Knop J, Wesche H, Raffetseder U, Kurrle R, Boraschi D, Martin MU, The type II IL-1 receptor interacts with the IL-1 receptor accessory protein: a novel mechanism of regulation of IL-1 responsiveness, J. Immunol 161 (1998) 6871–6877. [PubMed] [Google Scholar]
- [35].Giri JG, Wells J, Dower SK, McCall CE, Guzman RN, Slack J, Bird TA, Shanebeck K, Grabstein KH, Sims JE, et al. , Elevated levels of shed type II IL-1 receptor in sepsis. Potential role for type II receptor in regulation of IL-1 responses, J. Immunol 153 (1994) 5802–5809. [PubMed] [Google Scholar]
- [36].Asaoka T, Island ER, Tryphonopoulos P, Selvaggi G, Moon J, Tekin A, Amador A, Levi DM, Garcia J, Smith L, Nishida S, Weppler D, Tzakis AG, Ruiz P, Characteristic immune, apoptosis and inflammatory gene profiles associated with intestinal acute cellular rejection in formalin-fixed paraffin-embedded mucosal biopsies, Transpl. Int 24 (2011) 697–707. [DOI] [PubMed] [Google Scholar]
- [37].Le Goff C, Cormier-Daire V, The ADAMTS(L) family and human genetic disorders, Hum. Mol. Genet 20 (2011) R163–R167. [DOI] [PubMed] [Google Scholar]
- [38].Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS, Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis, Development 133 (2006) 1587–1596. [DOI] [PubMed] [Google Scholar]
- [39].Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapiere CM, Prockop DJ, Nusgens BV, Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene, Am. J. Hum. Genet 65 (1999) 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Weng FL, Pancoska C, Patel AM, Fatal graft-versus-host disease presenting as fever of unknown origin in a pancreas-after-kidney transplant recipient, Am. J. Transplant 8 (2008) 881–883. [DOI] [PubMed] [Google Scholar]
- [41].Kumagai K, Horikawa T, Gotoh A, Yamane S, Yamada H, Kobayashi H, Hamada Y, Suzuki S, Suzuki R, Up-regulation of EGF receptor and its ligands, AREG, EREG, and HB-EGF in oral lichen planus, Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod 110 (2010) 748–754. [DOI] [PubMed] [Google Scholar]
- [42].Foley JE, Mariotti J, Ryan K, Eckhaus M, Fowler DH, Th2 cell therapy of established acute graft-versus-host disease requires IL-4 and IL-10 and is abrogated by IL-2 or host-type antigen-presenting cells, Biol. Blood Marrow Transplant 14 (2008) 959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sad S, Mosmann TR, Interleukin (IL) 4, in the absence of antigen stimulation, induces an anergy-like state in differentiated CD8+ TC1 cells: loss of IL-2 synthesis and autonomous proliferation but retention of cytotoxicity and synthesis of other cytokines, J. Exp. Med 182 (1995) 1505–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, de Waal Malefyt R, de Vries JE, Roncarolo MG, High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells, J. Exp. Med 179 (1994) 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lafyatis R, Farina A, New insights into the mechanisms of innate immune receptor signalling in fibrosis, Open Rheumatol. J 6 (2012) 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Oh SJ, Cho SB, Park SH, Piao CZ, Kwon SM, Kim I, Yoon SS, Kim BK, Park EK, Kang JJ, Yang SJ, Lee WJ, Yoo CH, Hwang S, Kim SH, Kim JH, Park S, Cell cycle and immune-related processes are significantly altered in chronic GVHD, Bone Marrow Transplant. 41 (2008) 1047–1057. [DOI] [PubMed] [Google Scholar]
- [47].Fujii H, Cuvelier G, She K, Aslanian S, Shimizu H, Kariminia A, Krailo M, Chen Z, McMaster R, Bergman A, Goldman F, Grupp SA, Wall DA, Gilman AL, Schultz KR, Biomarkers in newly diagnosed pediatric-extensive chronic graft-versus-host disease: a report from the Children’s Oncology Group, Blood 111 (2008) 3276–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Coghill JM, Sarantopoulos S, Moran TP, Murphy WJ, Blazar BR, Serody JS, Effector CD4+ T cells, the cytokines they generate, and GVHD: something old and something new, Blood 117 (2011) 3268–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nishimori H, Maeda Y, Teshima T, Sugiyama H, Kobayashi K, Yamasuji Y, Kadohisa S, Uryu H, Takeuchi K, Tanaka T, Yoshino T, Iwakura Y, Tanimoto M, Synthetic retinoid Am80 ameliorates chronic graft-versus-host disease by down-regulating Th1 and Th17, Blood 119 (2012) 285–295. [DOI] [PubMed] [Google Scholar]