

Abstract
Although numerous environmental exposures have been suggested as triggers for pre-clinical autoimmunity, only a few have been confidently linked to autoimmune diseases. For disease associated exposures, the lung is a common site where chronic exposure results in cellular toxicity, tissue damage, inflammation, and fibrosis. These features are exacerbated by exposures to particulate material, which hampers clearance and degradation thus facilitating persistent inflammation. Coincident with exposure and resulting pathological processes is the post-translational modification of self-antigens, which, in concert with the formation of tertiary lymphoid structures containing abundant B cells, is thought to promote the generation of autoantibodies that in some instances demonstrate major histocompatibility complex restriction. Under appropriate gene-environment interactions these responses can have diagnostic specificity. Greater insight into the molecular and cellular requirements governing this process, especially those that distinguish pre-clinical autoimmunity from clinical autoimmune disease, may facilitate determination of the significance of environmental exposures in human autoimmune disease.
Keywords: autoimmunity, innate, adaptive, xenobiotic, inflammation, animal model
INTRODUCTION
The molecular and cellular events that contribute to autoimmune diseases are complex. Nevertheless, many general principles of disease pathogenesis have been elucidated (1), however, the initiating steps that drive auto-reactivity remain for the most part poorly understood. It is for this reason that autoimmune diseases are often described as idiopathic, or of apparent spontaneous origin of unknown cause or mechanism. However, a number of environmental exposures linked to either overt autoimmune disease (2), or to subclinical autoimmunity that manifests solely as cellular infiltrates or autoantibodies (3; 4), have been identified. The role of these environmental exposures as initiating events in autoimmunity has been confirmed in certain induced experimental animal models (5). Accordingly, unlike the idiopathic autoimmunity in autoimmune-prone mice (6; 7), an essential feature of induced models is that the initiating factor is known making it possible to study the temporal and mechanistic course of events that lead to disease.
Among the possible initiating events, epidemiological studies have identified numerous infectious and non-infectious exposures associated with autoimmune diseases (8–11). These can be classified into three broad classes; biological, chemical and physical (8). Biologic agents include infection, dietary constituents, and therapeutics derived from biological components. Chemical agents comprise non-biological exposures that result from occupational, environmental, therapeutic, and lifestyle activities. Physical agents encompass radiation including sunlight, electric and magnetic fields. Autoimmunity and autoimmune diseases associated with infection (12; 13) or with therapeutics, either chemical or biological (14; 15), have been discussed elsewhere and will not be covered here. This review focuses on xenobiotics (chemicals not naturally produced by or present within an organism) that are linked to human autoimmune diseases and their corresponding experimental models used to study mechanisms of disease induction and development. Additional emphasis will focus on xenobiotics that can cause subclinical inflammation and autoimmunity but progress to clinical disease only in susceptible genetic backgrounds. This second group may offer clues to idiopathic autoimmune disease triggers because of evidence that autoimmune diseases are preceded by preclinical phases characterized by inflammatory mediators occurring prior to, or concurrent with, autoantibodies (16–19). Moreover, profiles of inflammatory mediators and autoantibodies can help distinguish between healthy individuals, autoantibody positive healthy individuals, and those with autoimmune disease (17; 18). Thus, a detailed dissection of the innate and adaptive immune sequelae that arise following autoimmunity-promoting xenobiotic exposures will likely contribute significantly to our understanding of the etiopathogenesis of human autoimmunity and autoimmune diseases.
XENOBIOTIC EXPOSURES ASSOCIATED WITH HUMAN AUTOIMMUNITY AND AUTOIMMUNE DISEASES
Although numerous xenobiotics have been suggested as triggers of autoimmunity and autoimmune diseases, definitive association has proven difficult to establish (8) often because of insufficient evidence of exposure to demonstrate causality. Consequently, only a small number of xenobiotics have been strongly implicated in autoimmune diseases. This is exemplified in the findings of a National Institute of Environmental Health Sciences (NIEHS) Expert Panel convened to determine environmental influences on the development of autoimmune diseases (8). Using defined guidelines, associations between individual exposures and autoimmune diseases were classified as “confident” or “likely” based on published evidence. Also identified were a number of exposures where the evidence for causation of autoimmune disease was considered “insufficient” but where exposure leads to features of autoimmunity. Since that review, little has changed to alter the exposure associations. Table 1 lists examples of “confident”, “likely”, or “insufficient” associations that will be discussed.
Table 1.
Xenobiotic Exposures Linked to Human Autoimmunity and Autoimmune Disease.
| Xenobiotic | Exposure | Exposure Site | Autoimmune Diseases or Autoimmunitya | Selected References |
|---|---|---|---|---|
| Confidentb | ||||
| Silica | Particulate | Lungs | SLE, SSc, RA, ANCA-related vasculitis | (8, 20, 25) |
| Smoking | Chemical/Particulate | Lungs | Seropositive RA | (8, 19) |
| Solvents | Liquid/Vapor | Skin/Lungs | SSc | (8, 39) |
| Likelyb | ||||
| Smoking | Particulate | Lungs | SLE, MS | (8) |
| Solvents | Liquid/Vapor | Skin/Lungs | MS | (8, 45) |
| Insufficient evidenceb | ||||
| Mercury | Vapor (gold amalgamation), Lotion (skin cream), Dietary (sea food), Particulate (dental amalgam) | Lungs, Skin, GI tract | autoantibodies, cytokines, nephrotic syndrome | (3, 4, 8) |
| Asbestos | Particulate | Lungs | autoantibodies, atypical rheumatological symptoms | (8, 54, 57) |
Exposures leading to autoimmunity without clinical disease are mercury and asbestos.
Categories of Confident, Likely, and Insufficient Evidence were described by Miller et al (8) essentially as follows.
Confident: Includes exposure disease associations from multiple studies from different populations using different designs; robust evidence of an overall association as identified by high-magnitude risks or the use of high-quality or established exposure assessment methods; evidence of an exposure-response gradient; and/or evidence of effect modification by disease subtype or genetics that supports biologic plausibility.
Likely: collections of research studies missing important elements, such as clarification of the temporal association between exposure and onset of an autoimmune disease, or less consistent results or were based on fewer studies.
Insufficient evidence: reported studies were too limited in design or power to allow conclusions to be drawn.
Silica
Silica, as SiO2, is most commonly found in nature as quartz. It becomes an occupational hazard when inhaled in a particulate crystalline form in workplaces involved in dusty trades such as mining, tunneling, and sand blasting (20–22). Pulmonary deposition of silica particles (<10 μM in diameter) leads to inflammation and fibrotic nodule formation culminating in the respiratory disease silicosis (22). The link between occupational exposure to silica and autoimmune disease, specifically rheumatoid arthritis (RA), was made by Caplan and colleagues (23; 24). Following those initial observations, numerous reports linked silica dust exposure to systemic lupus erythematosus (SLE), RA, systemic sclerosis (SSc), and anti-neutrophil cytoplasmic antibody (ANCA)-related vasculitis (8; 25). Evidence of a breakdown in immune tolerance, including autoantibodies, can occur with silica-exposure in the absence of silicosis (26), and exposure intensity rather than cumulative dose may be important in development of autoimmune disease (25). Recently, autoimmune diseases associated with silicosis have been reported following exposure to dust from the fabrication of artificial stone (>90% silica) (20; 27; 28). Such exposure is associated with acute and accelerated silicosis (20) and incidence of a heterogeneous pattern of autoimmune features and diseases (27; 28). This is similar to autoimmune disease in miners following exposure to very high amounts of silica-containing dust (25; 29; 30). Gene-environment interactions have not been examined for silica-induced autoimmunity, but susceptibility to silicosis is associated with polymorphisms in family with sequence similarity 13 member A (FAM13A) (31) and tumour necrosis factor alpha (TNF-α) (32).
Smoking
Smoking is a well-known risk factor for RA (8; 33), particularly for seropositive RA, which includes either rheumatoid factor (RF) or anti-citrullinated protein antibody (ACPA). Both smoking status and intensity increases risk (19). Another well-known risk factor for RA are certain human leukocyte antigen (HLA) class II alleles which share a conserved amino acid sequence in their antigen binding site called the HLA shared epitope (SE) (34). Meta-analysis has identified significant association between the SE, smoking exposure, and ACPA, suggesting a gene-environment interaction between smoking and HLA linked autoantibody responses in RA (35). Weaker and less consistent associations have been documented between smoking and SLE, multiple sclerosis (MS), and Crohn’s disease (8). Single studies in SLE have also reported gene-environment interactions between smoking and interleukin (IL)-33 (36), integrin subunit alpha M (ITGAM or CD11b) (37), and N-acetyltransferase 2 (NAT2) (38).
Solvents
Several independent meta-analyses have supported an increased risk of SSc with solvent exposure (39; 40). A large number of different solvents have been implicated (39; 41; 42) along with a higher prevalence of disease in males compared to unexposed cohorts (43; 44). Disease severity was also greater in those with exposure (44). Exposure is primarily occupational (39) involving inhalation or dermal adsorption, with greatest risk from aromatic solvents, trichloroethylene (TCE), halogenated solvents, and ketones (40). A less established association has been described for solvent exposure and MS (8; 45) and the gene environment interaction of solvent exposure, smoking and MS-linked HLA genes (46) was found to dramatically increase the risk of MS (47).
Mercury
The association between mercury exposure, autoimmunity and autoimmune diseases has been recently reviewed (3; 4). Mercury exists in elemental, organic and inorganic forms and exposure can occur via occupation, dietary contamination, therapeutic or cosmetic agents, and fossil fuel emissions (3). Although current evidence for mercury induction of autoimmune disease is insufficient (4; 8), mercury exposure is linked to markers of inflammation and autoimmunity (3; 4; 48). Studies of artisanal gold miners and fish consumption in South America identified a significant association between anti-nuclear autoantibodies (ANA) and mercury levels in blood or hair (3; 4), as well as increased levels of inflammatory cytokines with mercury exposure (49; 50). Several other cohorts, including the National Health and Nutrition Examination Survey (NHANES), and the Long Island Study of Seafood Consumption, have produced conflicting data on the relationship between autoantibodies, cytokines and mercury levels (3; 51), but this may reflect the low level of exposure in those groups. Several reports have identified nephrotic syndrome as an outcome of mercury exposure most often from skin whitening/lightening cream use leading to membranous glomerulonephritis and sometimes autoantibodies (3).
Asbestos
Asbestos refers to a group of naturally occurring silicate minerals broadly classified into the serpentine (chrysotile) and amphibole (crocidolite, amosite, tremolite, anthophyllite, actinolite) groups (52). Amphibole fibers are needle-like and brittle, while serpentine fibers are curved and softer. Exposure may occur via different routes, but inhalation is the most common and can lead to asbestosis as well as malignancy (53). Amphibole asbestos exposure appears to be more closely associated with autoimmunity than the serpentine form (54). Recent analysis of the association between asbestos exposure and autoimmunity (8; 55; 56) concluded that while epidemiological evidence for a causal role is insufficient, it does support a higher than expected risk of systemic autoimmune disease. Additionally, the presentation of rheumatological symptoms can be atypical, making diagnosis of specific autoimmune diseases difficult (57). A caveat is that, particularly with mining, asbestos exposure may also include concomitant exposure to crystalline silica (8). Autoantibodies, especially ANA, have been reported in cohorts exposed to asbestiform amphibole-contaminated vermiculite (Libby, Montana) (58; 59), mined crocidolite (blue asbestos) (Wittenoom, Western Australia), and fluoro-edenite in quarried rock (Biancavilla, Sicily) (60). Studies of the Libby community also found an increased risk of developing connective tissue diseases (61) which is supported by a recent study showing elevated mortality due to systemic autoimmune diseases such as RA, SSc, and SLE (62).
MECHANISMS LEADING TO XENOBIOTIC INDUCED AUTOIMMUNITY AND AUTOIMMUNE DISEASES
While the ability of certain environmental agents to induce autoimmunity is well established (63–66), to what extent idiopathic and induced disease are mediated by common mechanisms is unclear (67; 68). For example, the same agent can induce different autoimmune disorders (i.e., silica-associated RA, SSc and SLE) (8), while multiple agents can produce a similar clinical picture (i.e., different medications leading to similar lupus-like syndromes) (63). Moreover, it is unclear if a xenobiotic-induced response is an exacerbation of underlying disease, a xenobiotic specific response, or a combination of both. This is reflected in the diagnostic criteria for idiopathic autoimmune diseases which are not designed to delineate similarities and/or differences in biological markers that may be useful as diagnostic criteria for xenobiotic-induced autoimmune disease (69).
Pollard et al (70) have argued that xenobiotic exposure leads to tissue damage and the release of damage associated molecular patterns (DAMPs), including nucleic acids, as well as self- and modified self-antigens. This results in the engagement and activation of Toll-like receptors (TLRs) and other innate sensors, which stimulate the production of inflammatory mediators and the induction of inflammation. This innate response promotes presentation of self- and modified self-antigens to non-tolerant lymphocytes, followed by expansion of autoreactive B and T effector cell populations, and the production of autoantibodies. These events often occur at the site of exposure where they can be linked to the development of ectopic or tertiary lymphoid structures (TLS) and/or hypertrophy of secondary lymphoid organs. The expansion of autoreactive lymphocytes and their subsequent migration to target tissues such as the kidney in SLE, or the joints in RA, results in disease. The occurrence of these events following specific xenobiotic exposures are shown in Figure 1 and Table 2 and discussed below.
Figure 1.
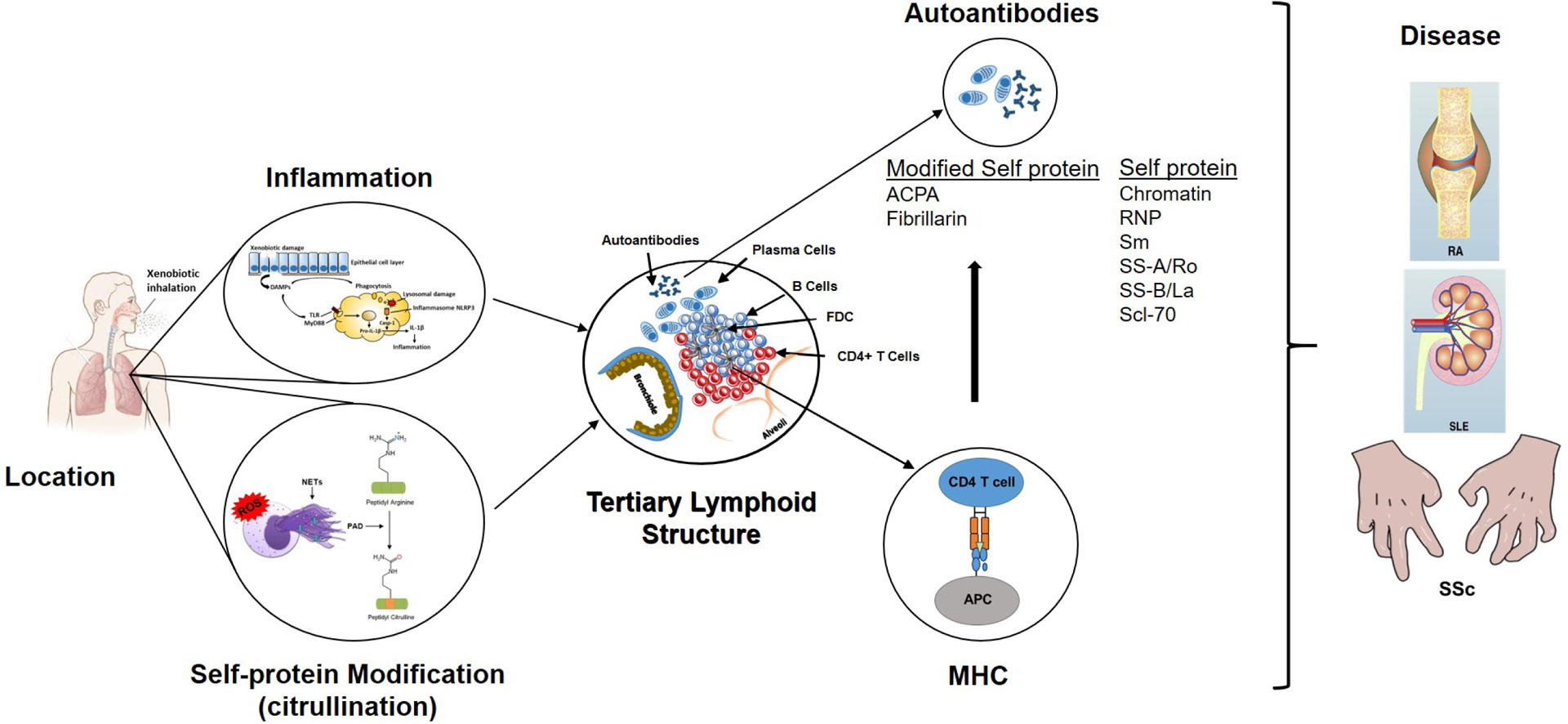
Steps in the development of xenobiotic-induced autoimmunity and autoimmune disease. Information in the figure summarizes the major points discussed in the text as they relate to the seven hypothesized steps leading to autoimmunity or autoimmune disease following xenobiotic exposure. The lungs serve as a common site of xenobiotic exposure. Chronic exposure results in an inflammatory response beginning with cellular and tissue damage, DAMP activation of PRR including TLRs and expression of inflammatory cytokines. Phagocytosis of DAMPS, including particulate xenobiotics, leads to lysosomal damage, inflammasome activation and processing of pro-IL-1β, further enhancing inflammation. Coincident with the inflammatory response are stress response related events, including NETosis and release of PAD enzymes, that leads to post-translational modification of self-proteins, particularly citrullination, and production of neoantigens. The ensuing chronic inflammatory response results in development of TLS comprised of accumulations of B cells within surrounding CD4+ T cells, FDCs and plasma cells. Processing and presentation of self and modified-self proteins, particularly in the context of MHC restriction, leads to autoantibodies. Under appropriate gene-environment interactions these autoantibodies can have diagnostic specificity such as the ACPA response in RA. The culmination of these steps is the development of disease pathology in distant tissues such as the joint in RA, the kidney in SLE and sclerodactyly, a skin manifestation of SSc.
Table 2.
Postulated mechanisms for xenobiotic-induced autoimmunity and autoimmune disease.
| Type of Exposure | Silica | Smoking | Solvents | Mercurya | Asbestos |
| Location | Lung | Lung | Lung/Skin | Lung/Skin/GI Tract | Lung |
| Inflammatory responses | IL-1α, IL-1β Inflammasome MyD88, IFN-γ, IL-17, type I IFN, IRF7 NETosis |
IL-1β Inflammasome IL-6, IL-8, TNF-α MyD88, NETosis |
ND | Endosomal TLRs IL-6, TNF-α, IFN-γ NETosis |
IL-1β Inflammasome |
| Self-Protein Modifications | Citrullination Carbonylation |
Citrullination Carbonylation |
Carbonylation Nitration |
Proteolysis Carbonylation |
Carbonylation Citrullination |
| Tertiary Lymphoid Structure | Yes | Yes | ND | Lymphoid Accumulations | Alveolitis |
| Autoantibodies | ACPA | ACPA | Scl-70 | Fibrillarin | Mesothelial cell |
| MHC association | ND | HLA-SE | HLA-DRB1 | H-2s | ND |
| Diseases | SLE, SSc, RA, ANCA-related vasculitis | Seropositive RA SLE, MS |
SSc | Nephrotic Syndrome | Rheumatological Symptoms |
A comparison of the hypothesized steps leading to autoimmunity or autoimmune disease following exposure to different xenobiotics. Silica, smoking, and solvents exposures are confidently linked to various autoimmune diseases while mercury and asbestos exposures although linked to features of autoimmunity, evidence for causation of autoimmune disease is insufficient.
Includes information from animal studies.
ND, not determined.
Location, location, location.
Although the proposed general mechanism above may not appear dissimilar to that proposed for idiopathic disease (1), significant differences exist in the identity of a triggering exposure and its location. It is immediately noticeable from Table 1 that the lungs are an important site of xenobiotic exposure. Recent reviews have argued that initiation of autoimmunity may occur at mucosal surfaces such as the lung for RA, as a result of smoking (71; 72), or epithelial surfaces such as the skin in SLE, following ultraviolet (UV) light (72). While it is clear that smoking and UV exposures are not the only risk factors for these diseases, they do highlight the growing appreciation of xenobiotic exposure at these body surfaces as a key step in the initiation of autoimmune reactions.
Tissue damage, inflammatory mediators, and inflammation
Inhalation of xenobiotics results in responses by a number of cell types in the lung that can result in cell death, cell activation, and release of reactive oxygen species, peptides, proteases, chemokines and cytokines (73). Inhalation of particulate matter such as crystalline silica, asbestos fibers, and the particulate phase of smoke results in their ingestion by alveolar macrophages and cell activation. Moreover, attempts to clear these degradation resistant particles can lead to frustrated phagocytosis which results in lysosomal damage and release of proteases and inflammatory mediators (73; 74). The cellular stress elicited by these events together with cell death leads to release of DAMPs including heat shock proteins, nucleic acids, and other cellular constituents recognized by pattern recognition receptors (PRR) such as TLRs (74–77). The subsequent innate inflammatory response leads initially to neutrophil recruitment and influx of blood monocytes and lymphocytes which is reflected in increased cellularity of bronchoalveolar lavage fluid (BALF). The inability to clear and/or degrade particulate material deposited in the lung, particularly in the alveoli, leads to persistent tissue damage, chronic inflammation and fibrosis (78). Notably, significant gene-environment interactions are at play in the inflammatory responses to smoking (75), silica (21), and mercury (3), as the levels of responses vary depending on the mouse strain.
IL-1 is a major contributor to the initial pulmonary inflammation induced by exposure to smoke, silica, and asbestos as blockade of IL-1 or deletion of IL-1 receptor significantly diminishes the inflammatory response as well as the production of other proinflammatory cytokines such as IL-6 and TNF-α (74; 79). The generation of IL-1 is determined, in part, by activation of the NACHT, LRR and PYD domains-containing protein 3 (NALP3) inflammasome leading to cleavage of pro-IL-1β and also pro-IL-18 which allows release of mature IL-1β and IL-18 and the amplification of inflammation in the lung (76; 77). In the case of silica-induced lung inflammation, IL-1α (a DAMP, constitutively present in epithelial and mesenchymal cells) is released from alveolar macrophages following cell damage and functions as an ‘alarmin’ preceding the production of inflammatory mediators, including IL-1β, and the infiltration of neutrophils (80; 81).
Lysosomal damage, following subcutaneous injection of inorganic mercury (HgCl2), may be important for early events in mercury-induced inflammation because inhibition of cathepsin B by CA-074 reduces mercury-induced mRNA expression of NLRP3, IL-1β, IL-6 and TNF-α (82). Although It is not known if pulmonary inflammation due to solvents such as TCE requires NLRP3 and IL-1β, dichloroacetyl chloride (DCAC), a reactive metabolite of TCE, has been shown to increase expression of NLRP3 and caspase-1 in the liver (83).
With the exception of mercury-exposure (3), there is little information on the role of innate immunity in the development of xenobiotic-induced autoimmunity. However, there is information about the inflammatory events following smoking (73; 84) and silica exposure (21) that may be relevant. Silica-induced inflammation is dependent on interferon (IFN)-γ, but not IL-12, IL-4, or IL-13. The acute inflammatory response requires IL-17, while chronic inflammation requires type I (IFN and interferon regulatory factor 7 (IRF7). Silica-induced inflammation and fibrosis can be uncoupled because the innate immune responses of inflammation, neutrophil accumulation, IL-1β release, and granuloma formation require myeloid differentiation primary response 88 (MyD88), while development of lung fibrosis (collagen deposition) does not. Smoking induced inflammation is also dependent on MyD88 signaling, and increased proinflammatory cytokine production (IL-1, IL-6, IL-8, TNF-α) leading to accumulation of T helper type 1 (Th1) and Th17 CD4+ T cells (75; 84). In contrast, mercury-induced autoimmunity is dependent on endosomal TLR trafficking and signaling, and IFN-γ (3) while type I IFN, IRF7, and NLRP3 are not required.
An additional step which likely plays a role in development of autoimmunity following xenobiotic exposure is the formation of neutrophil extracellular traps (NETs) during NETosis. NETosis is a form of neutrophil cell death that contributes to pathogenesis in autoimmune diseases such as SLE, RA, and ANCA-associated vasculitis (85). Neutrophils undergo NETosis in response to both non-sterile and sterile stimuli (85) including mercury (3), smoking (84), and silica (86). Many of the cellular constituents externalized during NETosis (e.g. DNA, chromatin) are recognized as autoantigens. Additionally, protein arginine deiminase (PAD) 4, which is involved in the release of decondensed chromatin of neutrophils via citrullination of histones (87), contributes to the production of citrullinated autoantigens and the induction of ACPA in RA. NETosis is therefore an important mechanism for self-protein modification, one of several by which xenobiotic exposure can modify self-proteins.
Self-protein modification
Xenobiotics associated with autoimmunity and autoimmune diseases are typically highly reactive and elicit adverse biological responses leading to oxidative stress, cell death, and inflammation. A common consequence of these reactions is post-translational modification of self-proteins via direct interactions or indirect effects mediated via enzymatic, including proteolytic, reactions as a result of an induced response such as oxidative stress. Smoking results in several different forms of post-translational modifications including citrullination, carbonylation, carbamylation (homocitrullination), and lipid peroxidation (19; 88–90). Silica also induces citrullination via a PAD-dependent mechanism (91), and the generation of reactive oxygen species (ROS) (92) leading to lipid peroxidation (93) and carbonylation (94). Oxidative stress following TCE exposure results in malondialdehyde (MDA)- and 4-hydroxynonenal (HNE)-protein adducts, carbonylation and nitration (95). In addition to its high affinity for protein sulfhydryl groups, mercury exposure also leads to ROS production and protein carbonylation (96). Asbestos exposure is known to lead to protein citrullination (97) and carbonylation (98). An important aspect of these post-translational modifications is that they can alter the immunogenicity of self-proteins, and as discussed below, can lead to autoantibody responses in diseases such as RA (99) and SLE (100).
Tertiary (or ectopic) lymphoid structures
Chronic inflammation is a significant component of many diseases including autoimmune diseases (101), and can be associated with development of tertiary lymphoid structures (TLS) (102). TLS arise at sites of non-resolving inflammation (103; 104) and have been found in target organs of autoimmune diseases (105–107) where they are believed to contribute to the persistence of autoimmunity by providing a microenvironment for survival and maturation of autoreactive cells, particularly B cells (103; 108). TLS contain segregated areas of T and B cells, high endothelial venules, and a CD21+ follicular dendritic cell (FDC) network important for B cell responses (109). Recent evidence supporting the oral mucosal origins of autoimmune diseases (71; 72) provide a basis for the hypothesis that inhalation of autoimmune disease associated xenobiotics leads to lung damage, inflammation, and subsequent development of TLS, providing a niche for self-reactive T and B cells.
Smoke exposure induces a chronic inflammatory response in the lungs, leading to chronic obstructive pulmonary disease (COPD) (88). Smoking is responsible for over 80% of COPD in Western counties. There is extensive evidence for lung TLS in COPD (88). The progression from smoke exposure to the presence of TLS (88) begins with tissue damage and inflammatory mediator recruitment of neutrophils and macrophages that animal experimentation has shown is essential for the inflammatory process. The subsequent appearance of TLS is characterized by aggregates of mainly IgM+IgD− B cells, surrounded by smaller numbers of primarily CD4+ T cells. These follicle-like structures also contain CD21+CD35+ FDCs and nearby CD138+ plasma cells. The severity of COPD, particularly emphysema, is positively associated with B cell follicles, and similar TLS are found in mice following chronic exposure to smoking. Several key mediators, including C-X-C motif chemokine ligand 13 (CXCL13), IL-17A, and B-cell activating factor (BAFF), are required for TLS development in cigarette smoke exposed mice (88). Importantly, smoking, lung inflammation, and the development of TLS are argued to play important roles in the preclinical phases of RA, and establish the lung as an important site for the initiation of seropositive RA (19).
Chronic silicosis is characterized by persistent inflammation and fibrosis leading to interstitial lung disease (21; 22; 110). Similar to the early inflammatory response following smoke exposure, silica-induced inflammation is solely dependent on innate immunity as it can occur independently of T, B, natural killer (NK) T, and NK cells (21). Although lymphocytes accumulate in the lung and draining lymph nodes, TLS have not, to our knowledge, been formally described in human silicosis. However, accumulations of lymphocytes in the lung and tracheobronchial lymph node have been described in experimental studies of crystalline silica exposed healthy C57BL/6 and lupus-prone mice (111–113). The typical appearance is of peribronchiolar or perivascular aggregates of B cells (Figure 2) surrounded by (111), or interspersed with (113), T cells that increase in size with exposure duration (111). Presumptive FDCs (CD21/35+ cells) can also be found. These lymphoid accumulations stain with anti-IgG suggesting the presence of plasma cells (113), which is supported by immunoglobulins including autoantibodies in the BALF (111; 114). This association is further supported by the observation that docosahexaenoic acid (a ω−3 polyunsaturated fatty acid with anti-inflammatory properties) prevents development of silica-induced lung TLS and autoantibodies in BALF (114).
Figure 2.
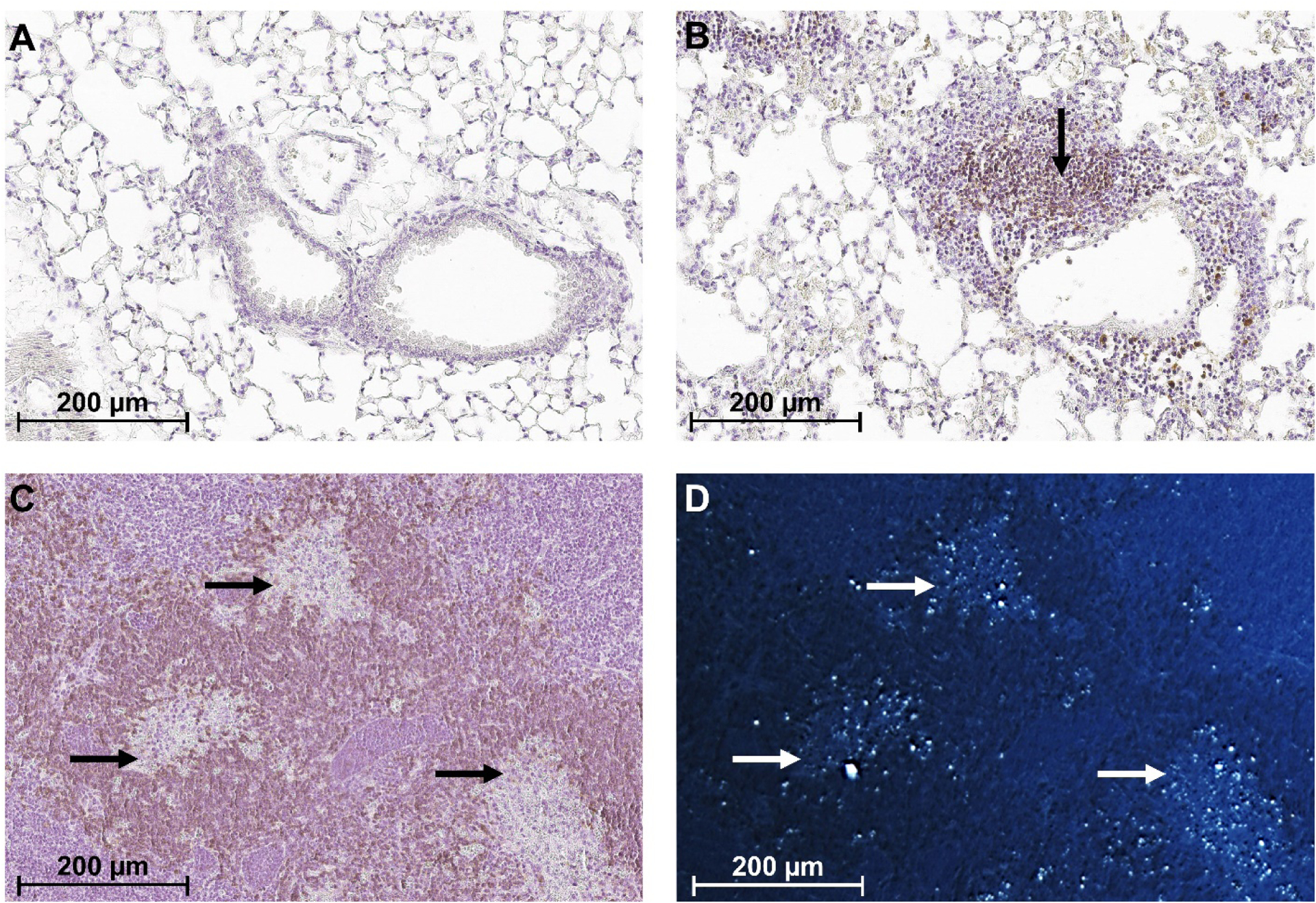
B cell aggregates in the lung and lymph node of silica exposed mice. Representative images of lung sections from Diversity Outbred (DO) mice 12 weeks following one 50 μl transoral instillation of (A) PBS or (B) 5 mg of crystalline silica (Min-U-Sil 5). IHC staining indicates B220+ B cells (black arrow in B) surrounding blood vessel in silica, but not vehicle, exposed mice. Tracheobronchial lymph node of silica exposed DO mouse imaged using (C) bright-field and (D) polarized microscopy shows areas of cell death and/or granulomas (black arrows in C) containing silica (white arrows in D) surrounded by B cells. B220+ cells are stained with DAB (brown) and hematoxylin (purple) was used as the counterstain.
Although mercury vapor is a major occupational hazard during artisanal small-scale gold mining (48), development of TLS in the lung has not been described in humans. However, dental amalgam-associated oral lichenoid lesions of the oral mucosa were found to show circumscribed lymphoid follicle-like structures containing B cells within a CD3+ T cell infiltrate (115). Dental amalgam constituents were suggested to be causally involved, although the mechanism is unknown. Subcutaneous injection of HgCl2 in mice results in skin induration with marked expansion of the dermis and subcutaneous tissue by neutrophils, macrophages, proliferating fibroblasts and lymphocytes (116) but the presence of TLS has yet to be observed. Transoral mercury exposure leads to multiple accumulations of lymphoid-like cells in the lungs, especially adjacent to bronchioles (70), but whether these are TLS or simple inflammatory lymphoid cell aggregates has not been established.
Exposure to asbestos can lead to chronic inflammation and asbestosis, another form of lung disease characterized by interstitial fibrosis (53). Lung biopsy has shown alveolitis consisting of accumulations of macrophages and lymphocytes (117) but unequivocal evidence for TLS following asbestos exposure has not been described in human or experimental animal studies. This likely reflects the greater focus on outcomes such as fibrosis and malignancy, diagnoses made years after initial exposure. A similar focus on silicotic nodules as a diagnostic marker, years after initial exposure, may also explain the lack of information on TLS in human silicosis. The presence of lung TLS following silica exposure in experimental animals, however, has been documented (111; 114) and are present after shorter exposure times which do not allow extensive development of fibrosis or fibrotic nodules (118). Additionally, the apparent need for lymphocytes in the development of fibrosis (119) is consistent with the formation of TLS early in the disease process. The close association of lung TLS and autoimmunity in other forms of pulmonary fibrosis (120) is further evidence of a linkage between these features.
The above observations clearly support the hypothesis that xenobiotic exposures can lead to the presence of lymphoid accumulations and TLS in the lung, however the role of pulmonary TLS in the development of autoimmunity and autoimmune disease remains unclear. Accordingly, it is undecided if lung TLS are pathogenic for autoimmunity or an unrelated response to infection or fibrosis (88; 120). One feature that points to a pathological component is the presence of plasma cells within TLS and autoantibodies in BALF that characterize a particular exposure, such as ACPA production in RA subsets associated with smoking or silica exposure.
Autoantibodies
Autoantibodies are a hallmark of idiopathic autoimmune diseases with ANA being a characteristic of systemic autoimmune diseases such as SLE, SSc and RA. As xenobiotic exposure can typically exacerbate both ANA and autoimmune disease in a wide range of autoimmune-prone mice, it can be argued that genetically susceptible backgrounds are generally sensitive to the immunostimulation provided by exposure (70). In addition, xenobiotic exposure can elicit other self-antigen specificities, particular to the type of exposure (Table 3). The similarities and differences in these responses argues that highly specific responses, can be of diagnostic utility and could yield new mechanistic insights. However there are significant gaps in our current understanding of the spectrum of autoantibody specificities that can be ascribed to xenobiotics, particularly for human exposure.
Table 3.
Autoantibodies in Xenobiotic-Induced Autoimmunity and Autoimmune Disease.
| Xenobiotic | Autoimmune Diseases or Autoimmunitya | Idiopathicb | Xenobiotic Humanc | Xenobiotic Experimentalc | Selected References |
|---|---|---|---|---|---|
| Confident | |||||
| Silica | SLE, SSc, RA |
ANA DNA, Sm, RNP, SS-A/Ro, SS-B/La Centromere, DNA topoisomerase 1 RF ACPAd |
ANA DNA SS-A/Ro SS-B/La Centromere, DNA topoisomerase 1 ACPA |
ANA Sm RNP dsDNA RF |
(21, 25, 30, 111,112,113) (25, 29) (123) |
| Smoking | Seropositive RA | RF ACPA |
ACPA | (19) | |
| Solvents | SSc | Scl-70 | Scl-70 |
MDA HNE ssDNA dsDNA |
(126) (95) |
| Insufficient evidence | |||||
| Mercury | autoantibodies, cytokines, nephrotic syndrome | NA | ANA | ANA Fibrillarin Chromatin |
(3) |
| Asbestos | autoantibodies, atypical rheumatological symptoms | NA | ANA dsDNA RNP SS-A/Ro52 Scl-70 Mesothelial cell |
ANA dsDNA SS/A-Ro52 Mesothelial cell |
(127) |
Note: This table is not a comprehensive listing of all autoantibodies found in a particular idiopathic autoimmune disease. Rather it is a comparison of autoantibody responses found with a specific xenobiotic exposure and whether the same response has been identified in idiopathic disease.
Exposures leading to autoimmunity without clinical disease are mercury and asbestos.
Idiopathic disease means no known association with an environmental exposure.
Antibodies in humans or experimental animal models following exposure.
In italics, antibodies against post-translational protein modification.
Abbreviations: NA, not applicable.
COPD, even in the absence of overt autoimmune disease, is associated with autoantibodies against many cell and tissue components including ANA. Furthermore, oxidative stress-induced protein modifications including carbonylation or oxidation of protein side chains (88), leads to anti-carbonylated protein antibodies whose titers correlate with the severity of COPD (121). A more significant protein modification for autoimmunity is the citrullination of β- and γ-actins, enolase, fibrinogen, filaggrin and vimentin, most likely mediated by smoking-induced increases in PAD-2 and 4 in the lungs. Consequently, conversion of protein arginine residues to citrulline (89) provides the autoantigens for the induction of ACPAs. Sparks and Karlson (22) have reviewed the association of smoking with preclinical stages of RA and the evidence that the lungs are critical for the development of RA, particularly seropositive RA which includes the ACPA+ subset. Although COPD patients without RA can be ACPA positive, the diagnostic significance of ACPAs was considerably more important in the context of RA. Furthermore, a study of 12,950 twins in Sweden found that while non-genetic factors (i.e. environment, lifestyle) are important in ACPA development, the presence of the SE had a significant impact on determining which ACPA-positive individuals developed RA (122). Another study of newly diagnosed, untreated RA patients showed that BALF had higher levels of ACPA compared to serum, and that adaptive immune cells, including B cells, were more likely to be present in bronchial biopsies of early RA patients with anti-cyclic citrullinated peptide (anti-CCP) antibodies (19). This suggests, indirectly, that the lungs are a site of ACPA production.
Consistent with the appearance of autoantibodies in preclinical phases of autoimmune diseases, silicosis can be associated with autoantibodies, including ANA, in the absence of connective tissue disease (21). Autoimmune disease-related specificities such as anti-DNA, anti-Sjögrens syndrome-A/Ro (anti-SS-A/Ro), anti-SS-B/La, anti-centromere, and anti-DNA topoisomerase 1 have been documented in silica-exposed individuals with and without a diagnosable disease such as SLE or SSc. Patients with silica-associated SSc have a greater prevalence of anti-DNA topoisomerase 1 autoantibodies, but fewer patients with silica-associated SSc and SLE had high titer ANA (>1:1,280) than in idiopathic disease. In comparison, autoimmune- and nonautoimmune-prone mice exposed to silica exhibited a broader range of ANA specificities that often included anti-ribonucleoprotein (RNP) and anti-Smith (Sm) (111–113). Silica exposure is also a risk factor for ACPA-positive RA, and this risk increases when coupled with smoking (123; 124). The mechanism of silica-mediated citrullination appears similar to smoking as in vitro exposure of cells to SiO2 nanoparticles induces protein citrullination together with an increase in PAD enzyme activity (91). The finding that silica-exposed Diversity Outbred (DO) mice develop anti-CCP3 antibodies (Mayeux and Pollard, unpublished) also supports this possibility.
Exposure to TCE is also associated with ANA positivity (125). In SSc, solvent exposure including TCE is more likely to be associated with anti-Scleroderma (Scl)-70 autoantibodies (126). Animal experimentation supports the potential of solvents such as TCE to exacerbate autoimmunity in autoimmune prone mice (95; 125), however evidence of scleroderma-like disease is lacking. TCE exposure of autoimmune prone female MRL+/+ mice is associated with anti-MDA- and anti-HNE-protein antibodies together with ANA, anti-ssDNA, and anti-dsDNA-antibodies possibly related to TCE-mediated oxidative stress (95).
Mercury also exacerbates autoantibody responses in lupus prone mice, as well as inducing autoantibodies in non-autoimmune prone strains. In the majority of strains tested, the specificities are primarily ANAs that target anti-chromatin and other anti-nuclear specificities (3). Mercury exposure, similar to many of the aforementioned xenobiotics, can lead to oxidative stress and carbonylation of proteins, including nuclear proteins (96), however antibodies against such modified proteins have not been demonstrated. Instead, it is the affinity of mercuric ions for sulfhydryl groups that has led to our understanding of its most characteristic response, the development of major histocompatibility complex (MHC) restricted antibodies to the nucleolar protein fibrillarin (3). Although mercury binds fibrillarin, human and mouse anti-fibrillarin autoantibodies do not bind a fibrillarin-mercury conjugate. Rather, it is mercury-induced cell death and proteolysis that leads to generation of a fibrillarin fragment that is immunogenic (3). Thus, phagocytosis and proteolysis of cellular material following mercury-induced cell death may be another primary source of neoantigenic determinants for self-reactive T lymphocytes following xenobiotic exposure.
For asbestosis, most of our understanding of autoantibodies comes from exposure to asbestiform amphibole-contaminated vermiculite in the community of Libby, Montana. The profile of ANA specificities in individuals from this area includes anti-dsDNA,-RNP,-SS-A/Ro52, and -Scl-70 (127). Autoantibodies against mesothelial cells have also been observed but anti-CCP and RF are uncommon (57). In C57BL/6 mice asbestos induces ANA including anti-dsDNA and anti-SS-A/Ro52 as well as mesothelial cell autoantibodies (127). ANA has also been induced in rats, but asbestos did not exacerbate disease in two rat models of arthritis (128). Oxidative stress is also a prominent feature in the initiation of asbestos-induced inflammation (129) and it is likely that there are protein modifications or autoimmune responses against modified self-antigens similar to silica.
As the discussion above reveals, autoantibody induction, especially ANA, by xenobiotics is a common observation, but examination of the antigenic specificities often reveals subtle, and sometimes marked, differences between types of exposures (e.g. ACPA in silica but not asbestos exposure) (Table 3). Differences can also be observed between species (e.g. humans versus experimental animals). This is not unexpected given the differences in physical and chemical properties among xenobiotics as well as their biological reactivity and the influence of genetic background. Apart from the linkage of smoking and/or silica exposure to protein citrullination and the development of ACPA, little has been done to examine the role of xenobiotic-induced self-protein modification in xenobiotic-induced autoantibody responses and autoimmune disease. This remains a missed opportunity because such modifications have been shown to generate immunogenic moieties whose cognate antibodies can facilitate epitope spreading and accelerate disease (19; 130).
The MHC link
Associations with the MHC locus, or HLA, constitute the strongest risk factors for autoimmune diseases (131). MHC associations also contribute to xenobiotic-induced autoimmunity particularly the regulation of CD4+ T cells and autoantibody responses. The most convincing association is between smoking, the HLA SE, and ACPA. The HLA SE consists of a common amino acid sequence at positions 70–74 of the HLA-DRB1 molecule with smoking increasing RA susceptibility in those carrying the HLA-DRB1 SE (35). Linkage between HLA-DRB1*15, and smoking increases the risk of MS, and this is enhanced when combined with solvent exposure (47). However, any associated antibody response remains to be identified. The anti-fibrillarin response in SSc is linked to HLA-DRB1 alleles (132), and the possibility of increased exposure to mercury (133). This is an interesting probability because the anti-fibrillarin response in mercury exposed mice is MHC linked (3), unlike responses to other nuclear antigens such as chromatin which are not restricted to mercury-induced autoimmunity. In silica-exposed miners, anti-Scl-70 positivity is more strongly associated with DRB1*0300 and DQB1*0201 alleles than in idiopathic SSc (134). In Japanese silicosis patients HLA-DQB1-0402 is more common in anti-DNA topoisomerase I positive patients than anti-DNA topoisomerase I negative patients or healthy controls (135).
These observations hint at the possibility that post-translational modification of self-antigens and MHC restriction are important factors in xenobiotic-induced autoantibody responses. This is best exemplified by the relationship between the SE, smoking and ACPA+ RA. In a similar vein, xenobiotic-induced cell death and proteolysis appears to play an important role in the mercury-induced MHC-restricted anti-fibrillarin response. A more concerted effort is needed to identify relationships between MHC and autoantibody responses following xenobiotic-exposure, and to determine the nature of the MHC antigens, and the contribution of autoantibodies to disease pathogenesis.
Disease complexity
As noted earlier, apart from epidemiological studies of exposure and disease association, diagnosis of autoimmune diseases does not discriminate between idiopathic and xenobiotic induced disease (69). Additional issues stem from poorly performed cohort studies, as well as the timeframe, often in years, between exposure and diagnosis of disease. Comorbidities, such as COPD, silicosis or asbestosis, although important clues in identifying a possible trigger for autoimmunity, also add to the complexity of understanding disease mechanisms.
The complexity of disease features that may be present in an exposed population is typified by screening of more than 6,000 community members in Libby, Montana (57). This community has a history of mining and use of asbestiform amphibole contaminated vermiculite, and almost 14% have been diagnosed with an autoimmune disease. The evolution of autoimmune diseases in these patients is atypical, with mixed diagnostic criteria suggesting that there is not one underlying autoimmune disease associated with the exposure. Moreover, the 1:1 male-to-female ratio is highly unusual given the prominent female predisposition of systemic autoimmune diseases such as SLE, SSc, and Sjögren’s Syndrome (SS) (66). Other studies also reveal differences in exposure related clinical features. Male miners, exposed to silica dust, with SLE had considerably less arthritis and photosensitivity compared with idiopathic SLE, and reduced prevalence of discoid lesions. However, the control group was not matched by age or sex, thus it is unclear if the differences reflect silica exposure or sex and/or age differences (21). In a more carefully controlled study, silica-exposed SLE patients were found to have reduced prevalence of anemia and leukopenia compared to idiopathic disease (21).
An important pathogenesis issue requiring resolution is the mechanism by which inflammation and autoreactive TLS development in an exposure site such as the lung leads to disease pathology in distant organs such as the joint in RA or kidney in SLE (72; 136; 137). Several hypotheses have been proposed including the existence of shared antigenic targets between the lungs and target tissues, epitope spreading to target specific antigen(s), and immune complex deposition in target tissues (137). Citrullinated proteins are found in the RA joint, suggesting that antigenic targets exist but it still remains unclear how events in the lung lead to pathogenesis in a target tissue such as the joint (136). This is one of the many mechanistic issues that remain to be clarified concerning xenobiotic-induced autoimmune disease.
CONCLUSION
An important commonality of xenobiotic exposures associated with human autoimmunity and autoimmune diseases is the lung as the major site of exposure. Another important feature is that the exposures are often protracted leading to persistent tissue damage. The most disease relevant exposures are also particulate material, which hampers clearance and degradation thereby facilitating a state of persistent inflammation. The failure to resolve this chronic inflammatory response allows the appearance of TLS characterized by aggregates of B and T cells which may become sites of autoreactivity. Coincident with these steps are stress response related events that lead to post-translational modification of self-proteins and the production of autoantibodies against these neoantigens. Under appropriate gene-environment interactions these responses can have diagnostic specificity such as the ACPA response in seropositive RA mediated by the HLA SE. Numerous questions still remain concerning the disease relevance not only of the xenobiotic-induced chronic inflammatory response but also the pathogenic role of pulmonary TLS particularly as to how they relate to the development of autoreactivity against modified self-proteins. Greater insight into the molecular and cellular requirements that govern this process, especially those that might distinguish pre-clinical autoimmunity from clinical autoimmune disease, may allow determination of the significance of xenobiotic exposures in human autoimmune disease.
SUMMARY POINTS.
Several environmental exposures, including smoking, silica dust, and solvents, have been confidently linked to autoimmune diseases. Other exposures, such as mercury and asbestos, result in autoimmunity including autoantibodies but with insufficient evidence of clinical disease. The latter may be important for identifying factors required for progression from pre-clinical to clinically significant autoimmune diseases.
The lungs are an important site of exposure to environmental agents linked to autoimmune diseases. The association of smoking and silica dust exposure to pre-clinical features of rheumatoid arthritis supports the growing appreciation that mucosal and epithelial cell surfaces are key sites in the initiation of autoimmune reactions.
IL-1 and the inflammasome are major contributors to early pulmonary inflammation mediated by the innate immune system following exposure to smoke, silica, and asbestos. Toll-like receptor signaling via MyD88, activation of Th1 CD4+ T cells and interferons also appear important for the subsequent adaptive immune response.
Post-translational modification of self-proteins is a shared feature of xenobiotic exposure. Several different mechanisms are operative but the most clinically important is citrullination.
The chronic inflammation of the lung following environmental exposure can result in formation of tertiary (or ectopic) lymphoid structures. These are comprised of focal areas of T and B cells and follicular dendritic cells that could provide a microenvironment for survival and maturation of autoreactive cells including autoantibody producing B cells.
Antibody responses in environmental-induced autoimmunity and autoimmune diseases include autoantibody responses against modified self-proteins. Of these, the anti-citrullinated protein antibodies found in seropositive rheumatoid arthritis following smoking or silica dust exposure have clinical and diagnostic significance.
The major histocompatibility complex (MHC) locus, or human leukocyte antigen (HLA), is linked to autoantibody responses accompanying environmental exposures. This is best exemplified by the association between the HLA-shared epitope, smoking, and anti-citrullinated protein antibodies. This association also exemplifies the importance of gene-environment interactions in environmental-induced autoimmunity.
FUTURE ISSUES.
Are tertiary lymphoid structures (TLS) a common feature of all chronic environmental exposures in the lung, or are they restricted to exposures associated with autoimmune diseases?
Do environmental exposure induced TLS only provide a B cell survival niche, or are they important in differentiation and expansion of autoreactive T and B cells?
What post-translational protein modifications occur following environmental exposures, do they elicit pathogenic autoantibody responses, and do they have diagnostic potential?
Are major histocompatibility (MHC) restrictions common to environmental-induced autoantibody responses against post-translationally modified antigens?
Can greater understanding of the gene-environment interactions of environmental-induced autoimmunity help discriminate pre-clinical autoimmunity from clinical autoimmune disease?
Can studies of the exposome, microbiome and epigenetics help us understand the mechanisms responsible for environmental-induced autoimmunity?
ACKNOWLEDGMENTS
The authors acknowledge the important contributions of many investigators in the field, only some of whom are mentioned here because of space constraints. Where possible we cite recent reviews that include a more thorough listing of relevant literature. Research was supported by the Extramural Research Program of the National Institutes of Health (NIH R01ES029581 and R01ES022625 to K.M.P., R01AI136492 and R01AI144070 to D.H.K., and a postdoctoral fellowship from T32 AI007244 to J.M.M.). P.H. was supported by the Swedish Research Council - Medicine, the County Council of Ostergotland, and Linköping University.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Theofilopoulos AN, Kono DH, Baccala R. 2017. The multiple pathways to autoimmunity. Nat Immunol 18:716–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parks CG, Miller FW, Pollard KM, Selmi C, Germolec D, et al. 2014. Expert panel workshop consensus statement on the role of the environment in the development of autoimmune disease. International journal of molecular sciences 15:14269–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollard KM, Cauvi DM, Toomey CB, Hultman P, Kono DH. 2019. Mercury-induced inflammation and autoimmunity. Biochim Biophys Acta Gen Subj [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crowe W, Allsopp PJ, Watson GE, Magee PJ, Strain JJ, et al. 2017. Mercury as an environmental stimulus in the development of autoimmunity - A systematic review. Autoimmun Rev 16:72–80 [DOI] [PubMed] [Google Scholar]
- 5.Germolec D, Kono DH, Pfau JC, Pollard KM. 2012. Animal models used to examine the role of the environment in the development of autoimmune disease: Findings from an NIEHS Expert Panel Workshop. J Autoimmun 39:285–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celhar T, Fairhurst AM. 2017. Modelling clinical systemic lupus erythematosus: similarities, differences and success stories. Rheumatology (Oxford) 56:i88–i99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Y, Sanam S, Kate K, Mohan C. 2015. Animal models of lupus and lupus nephritis. Curr Pharm Des 21:2320–49 [DOI] [PubMed] [Google Scholar]
- 8.Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, et al. 2012. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun 39:259–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gulati G, Brunner HI. 2018. Environmental triggers in systemic lupus erythematosus. Semin Arthritis Rheum 47:710–7 [DOI] [PubMed] [Google Scholar]
- 10.Van Loveren H, Vos JG, Germolec D, Simeonova PP, Eijkemanns G, McMichael AJ. 2001. Epidemiologic associations between occupational and environmental exposures and autoimmune disease: report of a meeting to explore current evidence and identify research needs. International journal of hygiene and environmental health 203:483–95 [DOI] [PubMed] [Google Scholar]
- 11.Parks CG, de Souza Espindola Santos A, Barbhaiya M, Costenbader KH. 2017. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract Res Clin Rheumatol 31:306–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ercolini AM, Miller SD. 2009. The role of infections in autoimmune disease. Clin Exp Immunol 155:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Getts DR, Chastain EM, Terry RL, Miller SD. 2013. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev 255:197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez-De-Lis M, Retamozo S, Flores-Chavez A, Kostov B, Perez-Alvarez R, et al. 2017. Autoimmune diseases induced by biological agents. A review of 12,731 cases (BIOGEAS Registry). Expert Opin Drug Saf 16:1255–71 [DOI] [PubMed] [Google Scholar]
- 15.Rubin RL. 2015. Drug-induced lupus. Expert Opin Drug Saf 14:361–78 [DOI] [PubMed] [Google Scholar]
- 16.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, et al. 2003. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 349:1526–33 [DOI] [PubMed] [Google Scholar]
- 17.Lu R, Munroe ME, Guthridge JM, Bean KM, Fife DA, et al. 2016. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slight-Webb S, Lu R, Ritterhouse LL, Munroe ME, Maecker HT, et al. 2016. Autoantibody-Positive Healthy Individuals Display Unique Immune Profiles That May Regulate Autoimmunity. Arthritis & rheumatology 68:2492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparks JA, Karlson EW. 2016. The Roles of Cigarette Smoking and the Lung in the Transitions Between Phases of Preclinical Rheumatoid Arthritis. Curr Rheumatol Rep 18:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes H, Goh NSL, Leong TL, Hoy R. 2019. Silica-associated lung disease: An old-world exposure in modern industries. Respirology 24:1165–75 [DOI] [PubMed] [Google Scholar]
- 21.Pollard KM. 2016. Silica, Silicosis, and Autoimmunity. Frontiers in immunology 7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung CC, Yu IT, Chen W. 2012. Silicosis. Lancet 379:2008–18 [DOI] [PubMed] [Google Scholar]
- 23.Caplan A 1953. Certain unusual radiological appearances in the chest of coal-miners suffering from rheumatoid arthritis. Thorax 8:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miall WE, Caplan A, Cochrane AL, Kilpatrick GS, Oldham PD. 1953. An epidemiological study of rheumatoid arthritis associated with characteristic chest x-ray appearances in coal-workers. Br Med J 2:1231–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parks CG, Conrad K, Cooper GS. 1999. Occupational exposure to crystalline silica and autoimmune disease. Environ Health Perspect 107 Suppl 5:793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brilland B, Beauvillain C, Mazurkiewicz G, Rucay P, Roquelaure Y, et al. 2019. T Cell Dysregulation in Non-silicotic Silica Exposed Workers: A Step Toward Immune Tolerance Breakdown. Frontiers in immunology 10:2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shtraichman O, Blanc PD, Ollech JE, Fridel L, Fuks L, et al. 2015. Outbreak of autoimmune disease in silicosis linked to artificial stone. Occup Med (Lond) 65:444–50 [DOI] [PubMed] [Google Scholar]
- 28.Turner MT, Samuel SR, Silverstone EJ, Yates DH. 2019. Silica Exposure and Connective Tissue Disease: An Under-Recognised Association in Three Australian Artificial Stone Workers. Am J Respir Crit Care Med [DOI] [PubMed] [Google Scholar]
- 29.Conrad K, Mehlhorn J. 2000. Diagnostic and prognostic relevance of autoantibodies in uranium miners. Int Arch Allergy Immunol 123:77–91 [DOI] [PubMed] [Google Scholar]
- 30.Conrad K, Mehlhorn J, Luthke K, Dorner T, Frank KH. 1996. Systemic lupus erythematosus after heavy exposure to quartz dust in uranium mines: clinical and serological characteristics. Lupus 5:62–9 [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Yu Y, Wu S, Sang L, Wang X, et al. 2018. The rs2609255 polymorphism in the FAM13A gene is reproducibly associated with silicosis susceptibility in a Chinese population. Gene 661:196–201 [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Peng LL, Ji XL, Yang HB, Zha RS, Gui GP. 2019. Tumor necrosis factor gene polymorphisms are associated with silicosis: a systemic review and meta-analysis. Biosci Rep 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baka Z, Buzas E, Nagy G. 2009. Rheumatoid arthritis and smoking: putting the pieces together. Arthritis Res Ther 11:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowes J, Barton A. 2008. Recent advances in the genetics of RA susceptibility. Rheumatology (Oxford) 47:399–402 [DOI] [PubMed] [Google Scholar]
- 35.Lee YH, Bae SC, Song GG. 2014. Gene-environmental interaction between smoking and shared epitope on the development of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: a meta-analysis. International journal of rheumatic diseases 17:528–35 [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Xie L, Qin H, Liang J, Yang Y, et al. 2019. Interaction between IL-33 Gene Polymorphisms and Current Smoking with Susceptibility to Systemic Lupus Erythematosus. J Immunol Res 2019:1547578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf BJ, Ramos PS, Hyer JM, Ramakrishnan V, Gilkeson GS, et al. 2018. An Analytic Approach Using Candidate Gene Selection and Logic Forest to Identify Gene by Environment Interactions (G × E) for Systemic Lupus Erythematosus in African Americans. Genes (Basel) 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiyohara C, Washio M, Horiuchi T, Tada Y, Asami T, et al. 2009. Cigarette smoking, N-acetyltransferase 2 polymorphisms and systemic lupus erythematosus in a Japanese population. Lupus 18:630–8 [DOI] [PubMed] [Google Scholar]
- 39.Rubio-Rivas M, Moreno R, Corbella X. 2017. Occupational and environmental scleroderma. Systematic review and meta-analysis. Clin Rheumatol 36:569–82 [DOI] [PubMed] [Google Scholar]
- 40.Zhao JH, Duan Y, Wang YJ, Huang XL, Yang GJ, Wang J. 2016. The Influence of Different Solvents on Systemic Sclerosis: An Updated Meta-analysis of 14 Case-Control Studies. J Clin Rheumatol 22:253–9 [DOI] [PubMed] [Google Scholar]
- 41.Walecka I, Roszkiewicz M, Malewska A. 2018. Potential occupational and environmental factors in SSc onset. Ann Agric Environ Med 25:596–601 [DOI] [PubMed] [Google Scholar]
- 42.Ingegnoli F, Ughi N, Mihai C. 2018. Update on the epidemiology, risk factors, and disease outcomes of systemic sclerosis. Best Pract Res Clin Rheumatol 32:223–40 [DOI] [PubMed] [Google Scholar]
- 43.De Decker E, Vanthuyne M, Blockmans D, Houssiau F, Lenaerts J, et al. 2018. High prevalence of occupational exposure to solvents or silica in male systemic sclerosis patients: a Belgian cohort analysis. Clin Rheumatol 37:1977–82 [DOI] [PubMed] [Google Scholar]
- 44.Marie I, Menard JF, Duval-Modeste AB, Joly P, Dominique S, et al. 2015. Association of occupational exposure with features of systemic sclerosis. J Am Acad Dermatol 72:456–64 [DOI] [PubMed] [Google Scholar]
- 45.Barragan-Martinez C, Speck-Hernandez CA, Montoya-Ortiz G, Mantilla RD, Anaya JM, Rojas-Villarraga A. 2012. Organic solvents as risk factor for autoimmune diseases: a systematic review and meta-analysis. PLoS One 7:e51506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bell JS, DeLuca GC. 2018. Genes, smoking, and organic solvent exposure: An alarming cocktail for MS risk. Neurology 91:199–200 [DOI] [PubMed] [Google Scholar]
- 47.Hedstrom AK, Hossjer O, Katsoulis M, Kockum I, Olsson T, Alfredsson L. 2018. Organic solvents and MS susceptibility: Interaction with MS risk HLA genes. Neurology 91:e455–e62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibb H, O’Leary KG. 2014. Mercury exposure and health impacts among individuals in the artisanal and small-scale gold mining community: a comprehensive review. Environ Health Perspect 122:667–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nyland JF, Fillion M, Barbosa F Jr, Shirley DL, Chine C, et al. 2011. Biomarkers of methylmercury exposure immunotoxicity among fish consumers in Amazonian Brazil. Environ Health Perspect 119:1733–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gardner RM, Nyland JF, Silva IA, Ventura AM, de Souza JM, Silbergeld EK. 2010. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: a cross-sectional study. Environ Res 110:345–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Monastero RN, Karimi R, Nyland JF, Harrington J, Levine K, Meliker JR. 2017. Mercury exposure, serum antinuclear antibodies, and serum cytokine levels in the Long Island Study of Seafood Consumption: A cross-sectional study in NY, USA. Environ Res 156:334–40 [DOI] [PubMed] [Google Scholar]
- 52.Sporn TA. 2011. Mineralogy of asbestos. Recent Results Cancer Res 189:1–11 [DOI] [PubMed] [Google Scholar]
- 53.Solbes E, Harper RW. 2018. Biological responses to asbestos inhalation and pathogenesis of asbestos-related benign and malignant disease. J Investig Med 66:721–7 [DOI] [PubMed] [Google Scholar]
- 54.Pfau JC. 2018. Immunotoxicity of asbestos. Current Opinion in Toxicology 10:1–7 [Google Scholar]
- 55.Bunderson-Schelvan M, Pfau JC, Crouch R, Holian A. 2011. Nonpulmonary outcomes of asbestos exposure. J Toxicol Environ Health B Crit Rev 14:122–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfau JC, Serve KM, Noonan CW. 2014. Autoimmunity and asbestos exposure. Autoimmune Dis 2014:782045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diegel R, Black B, Pfau JC, McNew T, Noonan C, Flores R. 2018. Case series: rheumatological manifestations attributed to exposure to Libby Asbestiform Amphiboles. J Toxicol Environ Health A 81:734–47 [DOI] [PubMed] [Google Scholar]
- 58.Pfau JC, Sentissi JJ, Weller G, Putnam EA. 2005. Assessment of autoimmune responses associated with asbestos exposure in Libby, Montana, USA. Environ Health Perspect 113:25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marchand LS, St-Hilaire S, Putnam EA, Serve KM, Pfau JC. 2012. Mesothelial cell and anti-nuclear autoantibodies associated with pleural abnormalities in an asbestos exposed population of Libby MT. Toxicol Lett 208:168–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ledda C, Caltabiano R, Loreto C, Cina D, Senia P, et al. 2018. Prevalence of anti-nuclear autoantibodies in subjects exposed to natural asbestiform fibers: a cross-sectional study. J Immunotoxicol 15:24–8 [DOI] [PubMed] [Google Scholar]
- 61.Noonan CW, Pfau JC, Larson TC, Spence MR. 2006. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ Health Perspect 114:1243–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larson TC, Williamson L, Antao VC. 2020. Follow-Up of the Libby, Montana Screening Cohort: A 17-Year Mortality Study. J Occup Environ Med 62:e1–e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rubin RL. 2005. Drug-induced lupus. Toxicology 209:135–47 [DOI] [PubMed] [Google Scholar]
- 64.Pollard KM. 2015. Environment, autoantibodies, and autoimmunity. Frontiers in immunology 6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pollard KM, Kono DH. 2013. Requirements for innate immune pathways in environmentally induced autoimmunity. BMC medicine 11:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pollard KM. 2012. Gender differences in autoimmunity associated with exposure to environmental factors. J Autoimmun 38:J177–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pollard KM, Hultman P, Kono DH. 2010. Toxicology of autoimmune diseases. Chem Res Toxicol 23:455–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gualtierotti R, Biggioggero M, Penatti AE, Meroni PL. 2010. Updating on the pathogenesis of systemic lupus erythematosus. Autoimmun Rev 10:3–7 [DOI] [PubMed] [Google Scholar]
- 69.Miller FW, Pollard KM, Parks CG, Germolec DR, Leung PS, et al. 2012. Criteria for environmentally associated autoimmune diseases. J Autoimmun 39:253–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pollard KM, Christy JM, Cauvi DM, Kono DH. 2018. Environmental Xenobiotic Exposure and Autoimmunity. Curr Opin Toxicol 10:15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, et al. 2018. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol 14:542–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pentony P, Duquenne L, Dutton K, Mankia K, Gul H, et al. 2017. The initiation of autoimmunity at epithelial surfaces: a focus on rheumatoid arthritis and systemic lupus erythematosus. Discov Med 24:191–200 [PubMed] [Google Scholar]
- 73.Wong J, Magun BE, Wood LJ. 2016. Lung inflammation caused by inhaled toxicants: a review. Int J Chron Obstruct Pulmon Dis 11:1391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franklin BS, Mangan MS, Latz E. 2016. Crystal Formation in Inflammation. Annu Rev Immunol 34:173–202 [DOI] [PubMed] [Google Scholar]
- 75.Brusselle GG, Joos GF, Bracke KR. 2011. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378:1015–26 [DOI] [PubMed] [Google Scholar]
- 76.Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, et al. 2017. Inflammasomes in the lung. Mol Immunol 86:44–55 [DOI] [PubMed] [Google Scholar]
- 77.Sayan M, Mossman BT. 2016. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol 13:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore BB, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. 2013. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 49:167–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. 2008. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dinarello CA. 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281:8–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rabolli V, Badissi AA, Devosse R, Uwambayinema F, Yakoub Y, et al. 2014. The alarmin IL-1alpha is a master cytokine in acute lung inflammation induced by silica micro- and nanoparticles. Part Fibre Toxicol 11:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toomey CB, Cauvi DM, Hamel JC, Ramirez AE, Pollard KM. 2014. Cathepsin B regulates the appearance and severity of mercury-induced inflammation and autoimmunity. Toxicol Sci 142:339–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang H, Wang G, Ansari GAS, Khan MF. 2018. Trichloroethene metabolite dichloroacetyl chloride induces apoptosis and compromises phagocytosis in Kupffer Cells: Activation of inflammasome and MAPKs. PLoS One 13:e0210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strzelak A, Ratajczak A, Adamiec A, Feleszko W. 2018. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int J Environ Res Public Health 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gupta S, Kaplan MJ. 2016. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12:402–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Cao X, Liu Y, Zhao Y, Herrmann M. 2018. Neutrophil Extracellular Traps Formation and Aggregation Orchestrate Induction and Resolution of Sterile Crystal-Mediated Inflammation. Frontiers in immunology 9:1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wong SL, Wagner DD. 2018. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J:fj201800691R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Polverino F, Seys LJ, Bracke KR, Owen CA. 2016. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol 311:L687–L95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anderson R, Meyer PW, Ally MM, Tikly M. 2016. Smoking and Air Pollution as Pro-Inflammatory Triggers for the Development of Rheumatoid Arthritis. Nicotine Tob Res 18:1556–65 [DOI] [PubMed] [Google Scholar]
- 90.Caramori G, Ruggeri P, Di Stefano A, Mumby S, Girbino G, et al. 2018. Autoimmunity and COPD: Clinical Implications. Chest 153:1424–31 [DOI] [PubMed] [Google Scholar]
- 91.Mohamed BM, Verma NK, Davies AM, McGowan A, Crosbie-Staunton K, et al. 2012. Citrullination of proteins: a common post-translational modification pathway induced by different nanoparticles in vitro and in vivo. Nanomedicine (Lond) 7:1181–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fubini B, Hubbard A. 2003. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med 34:1507–16 [DOI] [PubMed] [Google Scholar]
- 93.Castranova V, Vallyathan V. 2000. Silicosis and coal workers’ pneumoconiosis. Environ Health Perspect 108 Suppl 4:675–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Petrache Voicu SN, Dinu D, Sima C, Hermenean A, Ardelean A, et al. 2015. Silica Nanoparticles Induce Oxidative Stress and Autophagy but Not Apoptosis in the MRC-5 Cell Line. International journal of molecular sciences 16:29398–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khan MF, Wang G. 2018. Environmental Agents, Oxidative Stress and Autoimmunity. Curr Opin Toxicol 7:22–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boatti L, Rapallo F, Viarengo A, Marsano F. 2017. Toxic effects of mercury on the cell nucleus of Dictyostelium discoideum. Environ Toxicol 32:417–25 [DOI] [PubMed] [Google Scholar]
- 97.Mohamed BM, Boyle NT, Schinwald A, Murer B, Ward R, et al. 2018. Induction of protein citrullination and auto-antibodies production in murine exposed to nickel nanomaterials. Scientific reports 8:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ogasawara Y, Ishii K. 2010. Exposure to chrysotile asbestos causes carbonylation of glucose 6-phosphate dehydrogenase through a reaction with lipid peroxidation products in human lung epithelial cells. Toxicol Lett 195:1–8 [DOI] [PubMed] [Google Scholar]
- 99.Carubbi F, Alunno A, Gerli R, Giacomelli R. 2019. Post-Translational Modifications of Proteins: Novel Insights in the Autoimmune Response in Rheumatoid Arthritis. Cells 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF. 2010. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 62:2064–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, et al. 2019. Chronic inflammation in the etiology of disease across the life span. Nat Med 25:1822–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bombardieri M, Lewis M, Pitzalis C. 2017. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol 13:141–54 [DOI] [PubMed] [Google Scholar]
- 103.Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. 2016. Ectopic Lymphoid Structures: Powerhouse of Autoimmunity. Frontiers in immunology 7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin C, Mohanta S, Maffia P, Habenicht AJ. 2017. Editorial: Tertiary Lymphoid Organs (TLOs): Powerhouses of Disease Immunity. Frontiers in immunology 8:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jones GW, Jones SA. 2016. Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology 147:141–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vu Van D, Beier KC, Pietzke LJ, Al Baz MS, Feist RK, et al. 2016. Local T/B cooperation in inflamed tissues is supported by T follicular helper-like cells. Nature communications 7:10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pitzalis C, Jones GW, Bombardieri M, Jones SA. 2014. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 14:447–62 [DOI] [PubMed] [Google Scholar]
- 108.Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. 2018. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Frontiers in immunology 9:1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Corsiero E, Delvecchio FR, Bombardieri M, Pitzalis C. 2019. B cells in the formation of tertiary lymphoid organs in autoimmunity, transplantation and tumorigenesis. Curr Opin Immunol 57:46–52 [DOI] [PubMed] [Google Scholar]
- 110.Kawasaki H 2015. A mechanistic review of silica-induced inhalation toxicity. Inhal Toxicol 27:363–77 [DOI] [PubMed] [Google Scholar]
- 111.Foster MH, Ord JR, Zhao EJ, Birukova A, Fee L, et al. 2019. Silica Exposure Differentially Modulates Autoimmunity in Lupus Strains and Autoantibody Transgenic Mice. Frontiers in immunology 10:2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mayeux JM, Escalante GM, Christy JM, Pawar RD, Kono DH, Pollard KM. 2018. Silicosis and Silica-Induced Autoimmunity in the Diversity Outbred Mouse. Frontiers in immunology 9:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bates MA, Brandenberger C, Langohr I, Kumagai K, Harkema JR, et al. 2015. Silica Triggers Inflammation and Ectopic Lymphoid Neogenesis in the Lungs in Parallel with Accelerated Onset of Systemic Autoimmunity and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. PLoS One 10:e0125481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bates MA, Akbari P, Gilley KN, Wagner JG, Li N, et al. 2018. Dietary Docosahexaenoic Acid Prevents Silica-Induced Development of Pulmonary Ectopic Germinal Centers and Glomerulonephritis in the Lupus-Prone NZBWF1 Mouse. Frontiers in immunology 9:2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Larsson A, Warfvinge G. 1998. Immunohistochemistry of ‘tertiary lymphoid follicles’ in oral amalgam-associated lichenoid lesions. Oral diseases 4:187–93 [DOI] [PubMed] [Google Scholar]
- 116.Cauvi DM, Cauvi G, Toomey CB, Jacquinet E, Pollard KM. 2017. From the Cover: Interplay Between IFN-gamma and IL-6 Impacts the Inflammatory Response and Expression of Interferon-Regulated Genes in Environmental-Induced Autoimmunity. Toxicol Sci 158:227–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rom WN, Travis WD. 1992. Lymphocyte-macrophage alveolitis in nonsmoking individuals occupationally exposed to asbestos. Chest 101:779–86 [DOI] [PubMed] [Google Scholar]
- 118.Green FH, Vallyathan V, Hahn FF. 2007. Comparative pathology of environmental lung disease: an overview. Toxicol Pathol 35:136–47 [DOI] [PubMed] [Google Scholar]
- 119.Huaux F 2007. New developments in the understanding of immunology in silicosis. Curr Opin Allergy Clin Immunol 7:168–73 [DOI] [PubMed] [Google Scholar]
- 120.Hoyne GF, Elliott H, Mutsaers SE, Prele CM. 2017. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol Cell Biol 95:577–83 [DOI] [PubMed] [Google Scholar]
- 121.Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, et al. 2011. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184:796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hensvold AH, Magnusson PK, Joshua V, Hansson M, Israelsson L, et al. 2015. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis 74:375–80 [DOI] [PubMed] [Google Scholar]
- 123.Stolt P, Yahya A, Bengtsson C, Kallberg H, Ronnelid J, et al. 2010. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis 69:1072–6 [DOI] [PubMed] [Google Scholar]
- 124.Yahya A, Bengtsson C, Larsson P, Too CL, Mustafa AN, et al. 2013. Silica exposure is associated with an increased risk of developing ACPA-positive rheumatoid arthritis in an Asian population: evidence from the Malaysian MyEIRA case-control study. Mod Rheumatol [DOI] [PubMed] [Google Scholar]
- 125.Cooper GS, Makris SL, Nietert PJ, Jinot J. 2009. Evidence of autoimmune-related effects of trichloroethylene exposure from studies in mice and humans. Environ Health Perspect 117:696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nietert PJ, Sutherland SE, Silver RM, Pandey JP, Knapp RG, et al. 1998. Is occupational organic solvent exposure a risk factor for scleroderma? Arthritis Rheum 41:1111–8 [DOI] [PubMed] [Google Scholar]
- 127.Pfau JC. 2018. Immunotoxicity of asbestos. Current Opinion in Toxicology 10:1–7 [Google Scholar]
- 128.Salazar KD, Copeland CB, Luebke RW. 2012. Effects of Libby amphibole asbestos exposure on two models of arthritis in the Lewis rat. J Toxicol Environ Health A 75:351–65 [DOI] [PubMed] [Google Scholar]
- 129.Liu G, Cheresh P, Kamp DW. 2013. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol 8:161–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kurien BT, Scofield RH. 2008. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev 7:567–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. 2017. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol 18:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sharif R, Fritzler MJ, Mayes MD, Gonzalez EB, McNearney TA, et al. 2011. Anti-fibrillarin antibody in African American patients with systemic sclerosis: immunogenetics, clinical features, and survival analysis. J Rheumatol 38:1622–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arnett FC, Fritzler MJ, Ahn C, Holian A. 2000. Urinary mercury levels in patients with autoantibodies to U3-RNP (fibrillarin). J Rheumatol 27:405–10 [PubMed] [Google Scholar]
- 134.Baur X, Rihs HP, Altmeyer P, Degens P, Conrad K, et al. 1996. Systemic sclerosis in German uranium miners under special consideration of autoantibody subsets and HLA class II alleles. Respiration; international review of thoracic diseases 63:368–75 [DOI] [PubMed] [Google Scholar]
- 135.Ueki A, Isozaki Y, Tomokuni A, Ueki H, Kusaka M, et al. 2001. Different distribution of HLA class II alleles in anti-topoisomerase I autoantibody responders between silicosis and systemic sclerosis patients, with a common distinct amino acid sequence in the HLA-DQB1 domain. Immunobiology 204:458–65 [DOI] [PubMed] [Google Scholar]
- 136.Fert-Bober J, Darrah E, Andrade F. 2020. Insights into the study and origin of the citrullinome in rheumatoid arthritis. Immunol Rev 294:133–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Demoruelle MK, Wilson TM, Deane KD. 2020. Lung inflammation in the pathogenesis of rheumatoid arthritis. Immunol Rev 294:124–32 [DOI] [PubMed] [Google Scholar]