

Abstract
Background:
Lafora disease (LD) is characterized by progressive myoclonus, refractory epilepsy, and cognitive deterioration. This complex neurodegenerative condition is caused by pathogenic variants in EPM2A/EPM2B genes, encoding two essential glycogen metabolism enzymes known as laforin and malin. Long-term follow-up data are lacking. We describe the clinical features and genetic findings of a cohort of 26 Italian patients with a long clinical follow-up.
Methods:
Patients with EPM2A/EPM2B pathogenic variants were identified by direct gene sequencing or gene panels with targeted re-sequencing. Disease progression, motor functions, and mental performance were assessed by a simplified disability scale. Spontaneous/action myoclonus severity was scored by the Magaudda Scale.
Results:
Age range was 12.2–46.2 years (mean:25.53 ± 9.14). Age at disease onset ranged from 10 to 22 years (mean:14.04 ± 2.62). The mean follow-up period was 11.48 ± 7.8 years. Twelve out of the 26 (46%) patients preserved walking ability and 13 (50%) maintained speech. A slower disease progression with preserved ambulation and speech after ≥4 years of follow-up was observed in 1 (11%) out of the 9 (35%) EPM2A patients and in 6 (35%) out of the 17 (65%) EPM2B patients. Follow-up was >10 years in 7 (41.2%) EPM2B individuals, including two harbouring the homozygous p.(D146N) pathogenic variant.
Conclusions:
This study supports an overall worse disease outcome with severe deterioration of ambulation and speech in patients carrying EPM2A mutations. However, the delayed onset of disabling symptoms observed in the EPM2B subjects harbouring the p.(D146N) pathogenic variant suggests that the underlying causative variant may still influence LD severity.
Keywords: Lafora disease, EPM2A, EPM2B, Epilepsy, Progressive myoclonus, Neurodegeneration
1. Introduction
Lafora disease (LD) is a severe autosomal recessive progressive myoclonus epilepsy with onset in early adolescence in otherwise neurologically normal individuals [1]. Initial symptoms rapidly turn into progressive dementia, speech, and motor impairments, eventually leading to respiratory failure and death within a decade [2]. The disease prevalence is around 4 cases per million, but it might be higher due to missed and undiagnosed patients, especially in developing countries [3].
Mutations in two genes located on chromosome 6, EPM2A and EPM2B (MIM #254780), are involved in the pathogenesis of LD, causing deficiency of two fundamental enzymes of glycogen metabolism, laforin and malin. Most pathogenic variants are loss-of-function: splice site, missense, nonsense, and small or large intragenic deletions and insertions [4–8]. As a prototype of glycogen storage disease, LD is characterized by the presence of periodic-acid-Schiff positive intracellular inclusions known as Lafora bodies (LBs) in astrocytes, and neuronal perikarya, dendrites, but not axons [9,10].
Long-term follow-up data in LD patients are lacking. We report on the clinical features and genetic findings in a large cohort of Italian patients, providing a detailed description of the molecular and phenotypic spectrum in this severe condition, as well as investigating genotype-phenotype correlations.
2. Methods
2.1. Patients
Patients with LD were recruited from 14 Italian Epilepsy centres through the collaborative network of the Italian League Against Epilepsy (LICE). Individuals #5, #9, #16, #17a, #17b, #20, #22, and #24 were previously reported [11,12]. The diagnosis of LD was based on the clinical and electrophysiological features, as well as on the identification of typical LBs in skin, liver, or muscle samples. Clinical findings, neurophysiologic features, genetic results, and brain MRI and treatment data were retrospectively collected from medical charts provided by the referring clinicians.
To evaluate disease progression, a simplified disability scale evaluating the residual motor function, cognitive performance (assessed by Montreal Assessment: MoCA), activities of daily living (ADL), and social abilities were used. Scores ranged from 1 to 4 as follows: 1) mild cognitive impairment (MoCA >25), mild gait ataxia (scale 4), preserved ADL, and maintained interpersonal and family interactions; 2) moderate cognitive impairment (MoCA <25), moderate gait ataxia (scale 3 to 2), limited ADL, and preserved but limited social interaction; 3) severe mental impairment (MoCA <10), severe gait ataxia (scale 2 to 1), impaired ADL, and poor social interaction; 4) severe mental impairment (MoCA <10), severe gait ataxia (scale 0), wheelchair-bound or bedridden, no significant ADL, no social interaction, and gastrostomy/tracheostomy.
The severity of spontaneous and action myoclonus was also evaluated using the Magaudda Simplified Myoclonus Rate Scale: 0) no myoclonus; 1) minor myoclonus with no interference with ADL; 2) mild myoclonus with interference with fine movements or speech, but no interference with walking; 3) moderate myoclonus, but preserved ambulation without support; 4) moderate to severe myoclonus with preserved ability to stand and supported ambulation; 5) severe myoclonus with patient wheelchair-bound or bedridden.
The mutational screening was performed using either Next Generation Sequencing (NGS)-based gene panels for epileptic disorders or direct sequencing of EPM2A and EPM2B. Segregation analysis of candidate variants was performed in all families by Sanger sequencing.
2.2. Statistical analysis
Patients were divided into two groups, those harbouring pathogenic variants in EPM2A and those carrying EPM2B mutations. Categorical data were summarised in terms of absolute frequencies and percentages. Quantitative variables were summarised in terms of medians with 1st and 3rd quartiles (1st – 3rd q), as the data were not normally distributed. The normality of the distributions was calculated by the Shapiro-Wilk test. The association between categorical data was evaluated by the Chi-square test or Fisher’s Exact test in case of expected frequencies <5. The comparison of quantitative variables between the 2 groups of patients was evaluated by the Mann-Whitney U test. Non-parametric analysis of variance (Kruskal-Wallis test) was used to assess the relationship between quantitative and categorical polynomial variables. To avoid the “multiple comparison error”, the Bonferroni’s correction was applied, with the P-value indicated as “PB”. The software “Statistica”, release 9 (StatSoft Inc., Tulsa, OK, USA), was used for all univariate and bivariate analyses. The software “Stata”, release 11.0 (StataCorp, College Station, TX, USA), was used to calculate Fisher’s Exact test for tables with more than 2 rows or columns. Spearman’s rank correlation coefficient (rS) was applied to evaluate the statistical dependence between the rankings of two independent variables.
3. Results
3.1. Clinical features of the cohort
Twenty-six patients (16 females and 10 males) with LD from 24 different Italian families were investigated. Age range was 12.2–46.2 years (mean, 25.53 ± 9.14). Age at disease onset ranged from 10 to 22 years (mean, 14.04 ± 2.62) being tonic-clonic or myoclonic seizures the main presenting symptoms, either combined in 30% of the cases (Table 1).
Table 1.
Table reporting retention of walk and speech capabilities, together with scores at the spontaneous and action myoclonus severity scale for each patient. The disease stage is referred to as the last follow-up.
| Family | ID/Sex | Onset (y) | Follow-up duration (y) | Ambulation | Speech ability | Spontaneous myoclonus: severity | Action myoclonus: severity | Disease stage at last FU | Mutation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | AAG/F | 13 | 4.4 | + | + | 4 | 4 | 3 | EPM2B: c.205C > G (p.P69A) |
| 2a | BV/F | 11 | 1.2 | + | + | 0 | 0 | 0 | EPM2A: c.323G > T (p.R108L) |
| 2b | BV/F | 11 | 7.6 | + | + | 4 | 3 | 3 | EPM2A: c.323G > T (p.R108L) |
| 3 | BG/F | 14 | 8.8 | + | + | 2 | 2 | 2 | EPM2B: c.205C > G (p.P69A) |
| 4 | CE/F | 18 | 26.3 | − | − | NA | NA | 4 | EPM2A: c.712C > T (p.R241Ter); c.835G > A (p.G279S) |
| 5 | DGB/M | 14 | 10.0 | − | − | 5 | 5 | 4 | EPM2A: c.712C > T (p.R241Ter) |
| 6 | DLM/M | 13 | 4.6 | + | + | 3 | 2 | 2 | EPM2B: c.436G > A (p.D146N); c.838G > A (p.E280K) |
| 7 | DPA/F | 13 | 12.3 | − | − | 4 | 4 | 4 | EPM2A: c.243_246del (p. D82RfsTer7) |
| 8 | DDG/M | 16 | 5.3 | + | + | 4 | 4 | 2 | EPM2B: c.436G > A (p.D146N); c.1133 T > C (p.L378P) |
| 9 | DNM/M | 10 | 13.0 | − | − | 5 | 5 | 4 | EPM2A: c.712C > T (p.R241Ter) |
| 10 | FC/F | 14 | 8.9 | − | + | 5 | 4 | 4 | EPM2B: c.205C > G (p.P69A) |
| 11 | FE/F | 15 | 10.0 | − | − | 4 | 4 | 4 | EPM2A: deletion exon 2 |
| 12 | FA/M | 17 | 12.6 | − | + | 5 | 5 | 4 | EPM2A: deletion exon 2 |
| 13 | FM/F | 12 | 19.1 | − | − | NA | NA | 4 | EPM2B: c.612del (p.F204LfsTer28) |
| 14 | GB/F | 11 | 8.1 | − | − | 4 | 5 | 4 | EPM2B: c.205C > G (p.P69A); c.826–829dup (p.A277DfsTer23) |
| 15 | HA/M | 14 | 2.6 | + | + | NA | NA | 2 | EPM2B: c.992del (p.G331EfsTer3); c.1049–1050del (p.E350GfsTer41) |
| 16 | IN/F* | 11 | 19.1 | − | − | 3 | 5 | 4 | EPM2B: c.199G > T (p.E67Ter) |
| 17a | LC/M | 14 | 3.6 | + | + | 3 | 3 | 2 | EPM2B: c.992del (p.G331EfsTer3) |
| 17b | LM/F | 13 | 8.9 | − | − | 5 | 5 | 4 | EPM2B: c.992del (p.G331EfsTer3) |
| 18 | LF/F | 13 | 13.9 | − | − | 5 | 5 | 4 | EPM2B: c.205C > G (p.P69A) |
| 19 | MI/F | 13 | 4.6 (died) | − | − | NA | NA | 4 | EPM2A: c.491 T > G (p.I164S); c.539 T > C (p.L180P) |
| 20 | PL/F* | 15 | 29.3 | + | + | 2 | 3 | 3 | EPM2B: c.436G > A (p.D146N) |
| 21 | PV/M | 16 | 3.1 | + | + | 3 | 3 | 2 | EPM2B: c.468_469delAG (p. G158Rfs); c.205C > G (p.P69A) |
| 22 | RC/F* | 15 | 21.3 | − | − | NA | NA | 4 | EPM2B: c.436G > A (p.D146N); c.838G > A (p.E280K) |
| 23 | RE/F | 22 | 24.2 | + | + | 1 | 3 | 2 | EPM2B: c.436G > A, (p.D146N) |
| 24 | RF/M* | 16 | 17.6 | + | − | 3 | 3 | 3 | EPM2B: c.923A > T (p.D308V) |
ID = identification code; NA = not available; y = year
= previously reported in Franceschetti et al. [12].
Patients with EPM2B mutations showed a mean age at onset of 14.2 ± 2.5 years, whilst the mean age at onset was 13.6 ± 2.9 years in those with EPM2A mutations. Overall, the mean follow-up period was 11.48 ± 7.8 years, being 12.3 ± 8.2 and 9.8 ± 7.0 years in EPM2B and EPM2A patients. Ambulation and speech abilities at the last follow-up were evaluated. Twelve (46%) patients were able to walk independently whereas 14 (54%) lost ambulation after a mean of 13.4 years of follow-up. Thirteen (50%) patients preserved speech, while the other half showed absence of speech at the last follow-up. Thirty-five percent of EPM2B patients showed loss of both walking and speech abilities after a mean of 15.1 years. Isolated loss of ambulation or speech was instead observed in only two patients (#10, #24) at the mean age of 13.3 years from disease onset. As for EPM2A patients, 78% were non-ambulatory and 67% had no speech, with a mean follow-up period of 12.7 years. The mean score of the disease stage was 3.2. Sub-analysis between EPM2A and EPM2B individuals showed a mean disease stage score of 3.4 and 3.0 points, respectively.
The severity mean scores for spontaneous and action myoclonus in 21 patients were 3.5 and 3.7 points, respectively: 3.9 and 3.7 points in EPM2A subjects; 3.4 and 3.6 in EPM2B subjects. Comparison between spontaneous/action myoclonus severity and age at disease onset showed a linear correlation with an rS = −0.44 for spontaneous myoclonus and an rS = −0.61 for action myoclonus (Fig. 1). Tonic-clonic seizures mainly occurred monthly in 70% of cases, 78% of EPM2B patients, and 50% of EPM2A subjects. Weekly or yearly seizures were reported in the remaining cases. Patient #2 experienced a single tonic-clonic seizure. Seventy-seven percent of patients had ataxia, with a mean age at onset of 17.5 years. Severe dementia was also frequent (75% of patients), with a mean onset of 16 ± 1.7 years.
Fig. 1.
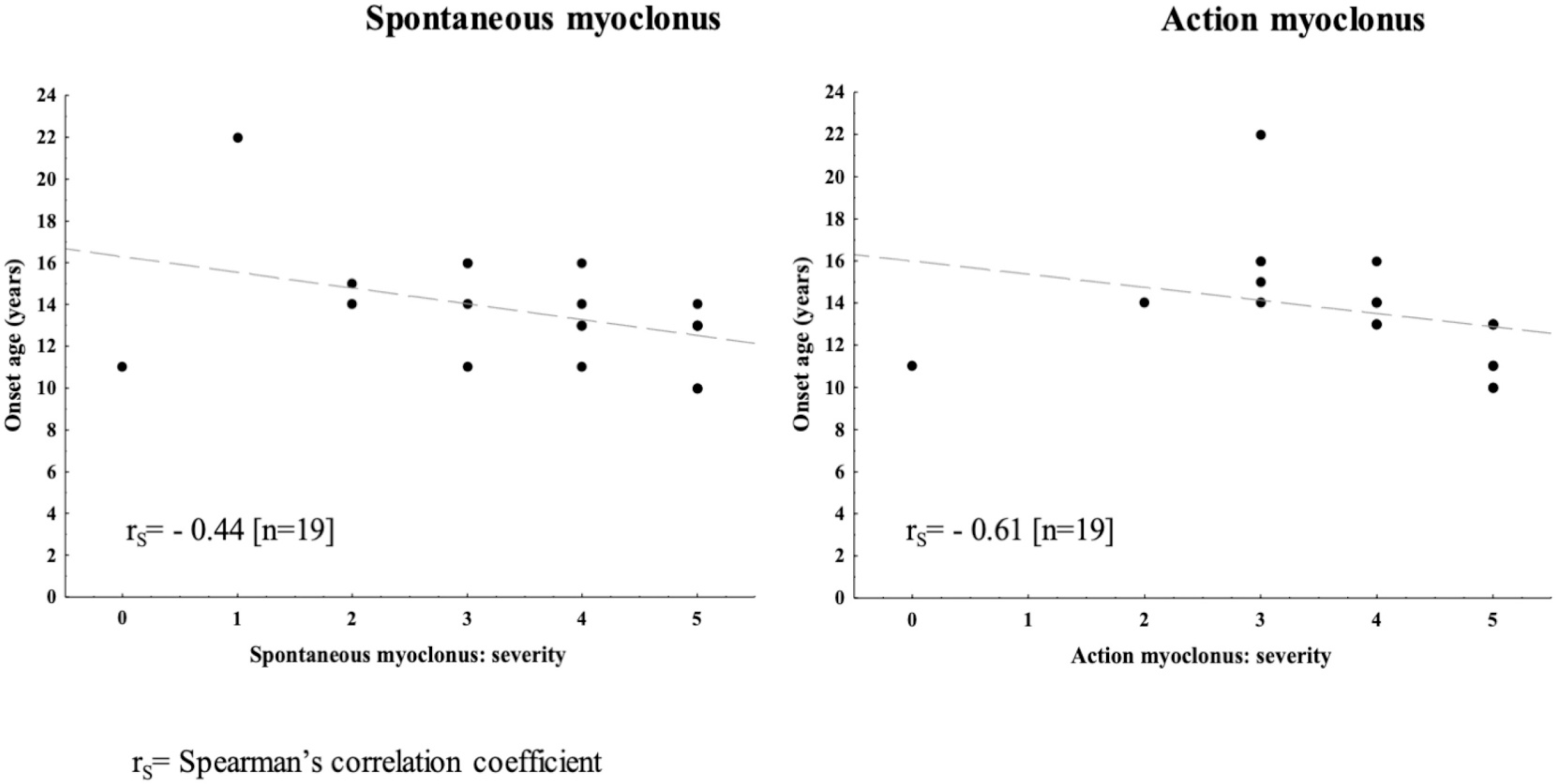
Correlation between age at disease onset and spontaneous or action myoclonus severities.
Brain MRI was normal or revealed slight to moderate cerebellar atrophy in most cases. In one case (#2b), diffuse cortical atrophy after 6.0 years from disease onset was identified. Most individuals manifested refractory epilepsy and myoclonus despite treatment with a combination of antiseizure medications (ASMs), including valproate (VPA), levetiracetam (LEV), carbamazepine, clonazepam, perampanel, and zonisamide. Notably, two patients (#2a, #15) achieved seizure control (isolated or monthly tonic-clonic seizures) with VPA or LEV monotherapy (Supplementary Table).
3.2. Genetic findings
Nine (35%) patients carried pathogenic variants in EPM2A and 17 (65%) in EPM2B (Fig. 2).
Fig. 2.
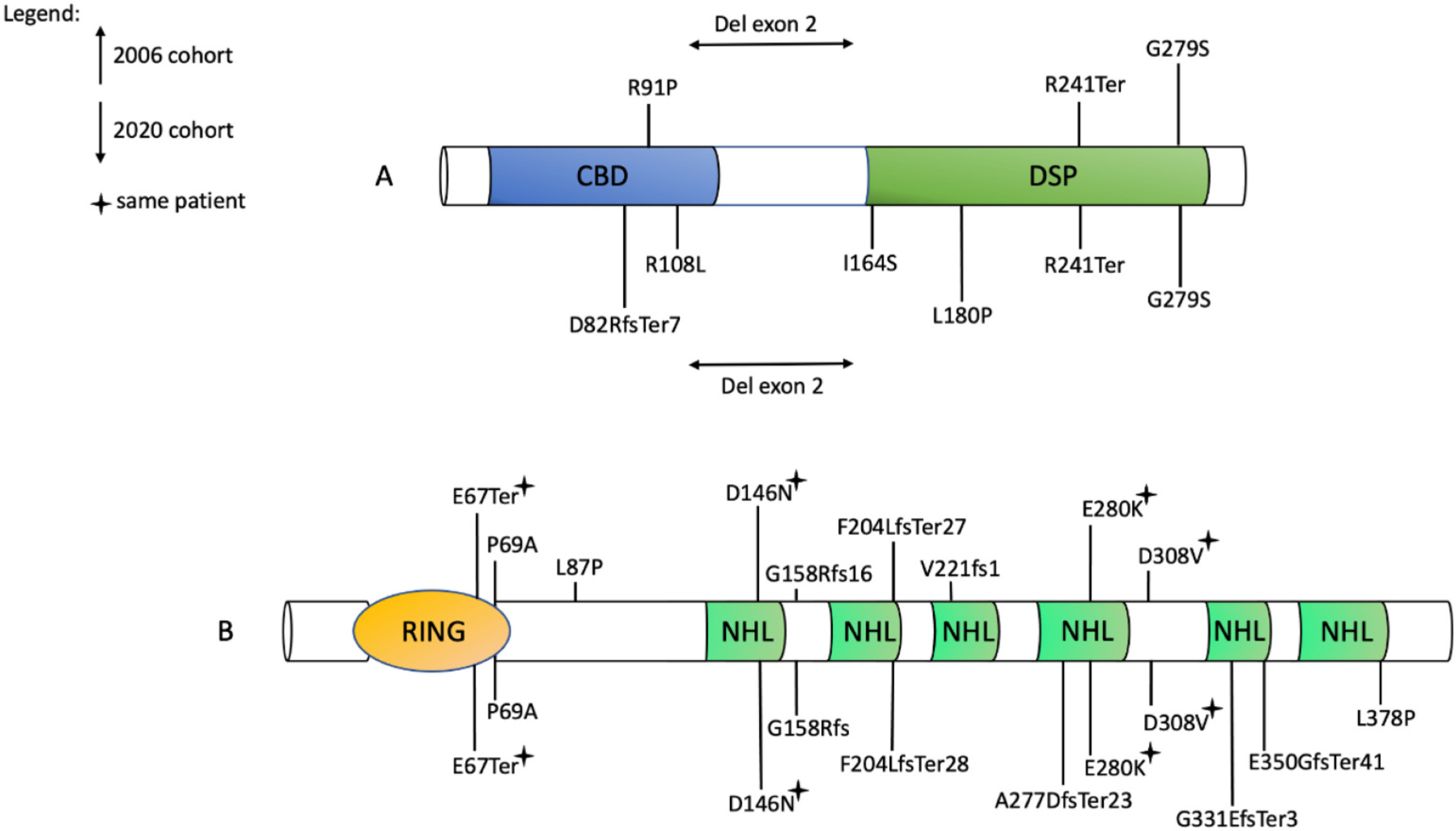
LD causing mutations in our cohort and that of 2006.
A) Laforin. CBD, carbohydrate-binding domain; DSP, dual-specificity phosphatase. B) Malin. RING, E2 interacting domain; NHL, protein interaction domain. The six NHL repeats are typical of E2 ubiquitin ligases [13]. Recurrent mutations are marked with a “star”.
Compound heterozygous variants were detected in 2 (22%) and 6 (35%) patients in the EPM2A and EPM2B groups. The remaining individuals were found to harbour homozygous variants. Seven distinct mutations (four missense, one truncating, one frameshift, and one exonic deletion) in EPM2A and nine different mutations in EPM2B (five missense, one truncating, and three frameshift) were identified. Three novel pathogenic variants were detected, including the p.(D82RfsTer7) in EPM2A and the p.(F204LfsTer28) and p.(A277DfsTer23) in EPM2B. The p.(P69A) and p.(D146N) mutations were the most frequent, being detected in 6 (35%) and 5 (29%) EPM2B patients, respectively. In patients #11 and #12, a deletion involving the exon 2 of EPM2A was identified.
3.3. Genotype-phenotype correlations
In the EPM2B group, the follow-up period was >10 years in 7 (41.2%) patients, including two subjects (#20, #23) homozygous for the p.(D146N) pathogenic variant. Patient #23 showed a remarkably late-onset (22 years) of symptoms and a very long follow-up (24.2 years) with a disease stage of 2. A moderate to severe disease stage was observed in patients with the homozygous mutation p.(P69A) (4 patients). In the EPM2A group, the follow-up period was >10 years in 4 (44%) patients: #4, harbouring the compound heterozygous p. (R241Ter) and p.(G279S) pathogenic variants; #9, carrying the homozygous p.(R241Ter) pathogenic variant; #7, homozygous for the p. (D82RfsTer7) pathogenic variant; #12, harbouring the exon 2 deletion.
The mean spontaneous myoclonus scores were 4.5 [4,5] [n = 6] and 3 [3,4] [n = 13] points in the EPM2A and EPM2B groups, respectively. Six (35.3%) EPM2B patients had severe spontaneous myoclonus. Patients #1 and #21 (harbouring the p.P69A) showed severe spontaneous myoclonus after <5 years since disease onset. Patients #20 and #23, carrying the homozygous p.(D146N), showed mild spontaneous myoclonus after >20 years of follow-up. Three patients showed severe action myoclonus: patient #1 (p.P69A), patient #17a (p.G331EfsTer3), and patient #21 (p.P69A). Patient #3 (p.P69A) showed mild action myoclonus after 8.8 years of follow-up.
Mean disease stage score was 3.5 ± 1.5 points in the EPM2A group, whereas the mean score was 3.0 ± 0.9 points in EPM2B patients. Disease stage was severe in eight (88.9%) EPM2A subjects and mild in only 1 case (11%). One EPM2A patient (#2b), carrying the homozygous p. (R108L) pathogenic variant, showed a disease stage score of 3 points after 9 years of follow-up. Seven out of 9 (77.8%) EPM2A patients lost ambulation and 6 (66.7%) speech ability. Instead, patient #2b remained able to walk and speak after 9 years from disease onset. Seven (41.2%) EPM2B patients lost both ambulation and speech. Loss of ambulation occurred <10 years since disease onset in patient #10 (p.P69A), patient #14 (p.P69A; A277DfsTer23), and patient #17b (p.G331EfsTer3). However, patient #10 preserved speech. Three EPM2B patients preserved ambulation after >20 years from disease onset: #20, (p.D146N); #22, (p.D146N; p.E280K); #24 (p.D308V).
Ataxia was observed in 11 out of 14 (78.6%) EPM2B patients, with a mean age at onset of 17 years. The EPM2B pathogenic variants p.(P69A) and p.(D146N) were associated with both early (#21, #23) and late-onset ataxia (#8, #10), as well as the absence of ataxia after ≥5 years from disease onset (#14, #20). Particularly, patient #20 did not show any ataxia after 29 years of follow-up. In the EPM2A group, ataxia occurred in 5 out of 6 (83.3%) patients, with a mean age at onset of 16 years. Deletions involving the exon 2 of EPM2A were associated with ataxia within the first year of follow-up, whereas delayed ataxia onset was observed in the patient with the p.(R241Ter) pathogenic variant (#5).
Dementia occurred at 3.3 ± 2.0 years and 2.4 ± 1.5 years after disease onset in EPM2A and EPM2B patients, respectively. Dementia occurred in 10 out of 15 (66.7%) EPM2B patients, with a mean age at onset of 17 years. In the EPM2A group, 83% of patients showed dementia, with a mean age at onset of 15 years.
Interestingly, the two patients (#7, #13) harbouring the novel homozygous mutations in EPM2A and EPM2B showed a long follow-up (mean, 15.7 years) with a disease stage of 4 suggesting long-survival. Likewise, patient #14 compound heterozygous for p.(P69A) and the new pathogenic variant p.(A277DfsTer23) did show long follow-up.
Overall, EPM2B patients showed a milder disease stage. In particular, patients #20, #23, and #24 showed a slower disease progression, with a follow-up period nearly to 20 years. No EPM2A patient in our cohort showed such a prolonged course. EPM2B patients also showed, on average, more preserved motor and cognitive functions. In particular, 10 out of 17 (59%) EPM2B patients showed moderate to severe disease stage, and just 70% of them lost speech or ambulation. A similar disease course was instead observed in eight (89%) EPM2A patients, with seven (88%) and six (75%) of them losing speech and ambulation, respectively. Remarkably, not all EPM2B patients showed a mild disease course. Of note, an earlier disease onset correlated with the early loss of ambulation in the EPM2B group, whereas a correlation was not observed between preserved ability to ambulate and later disease onset (P = 0.013) (Fig. 3).
Fig. 3.
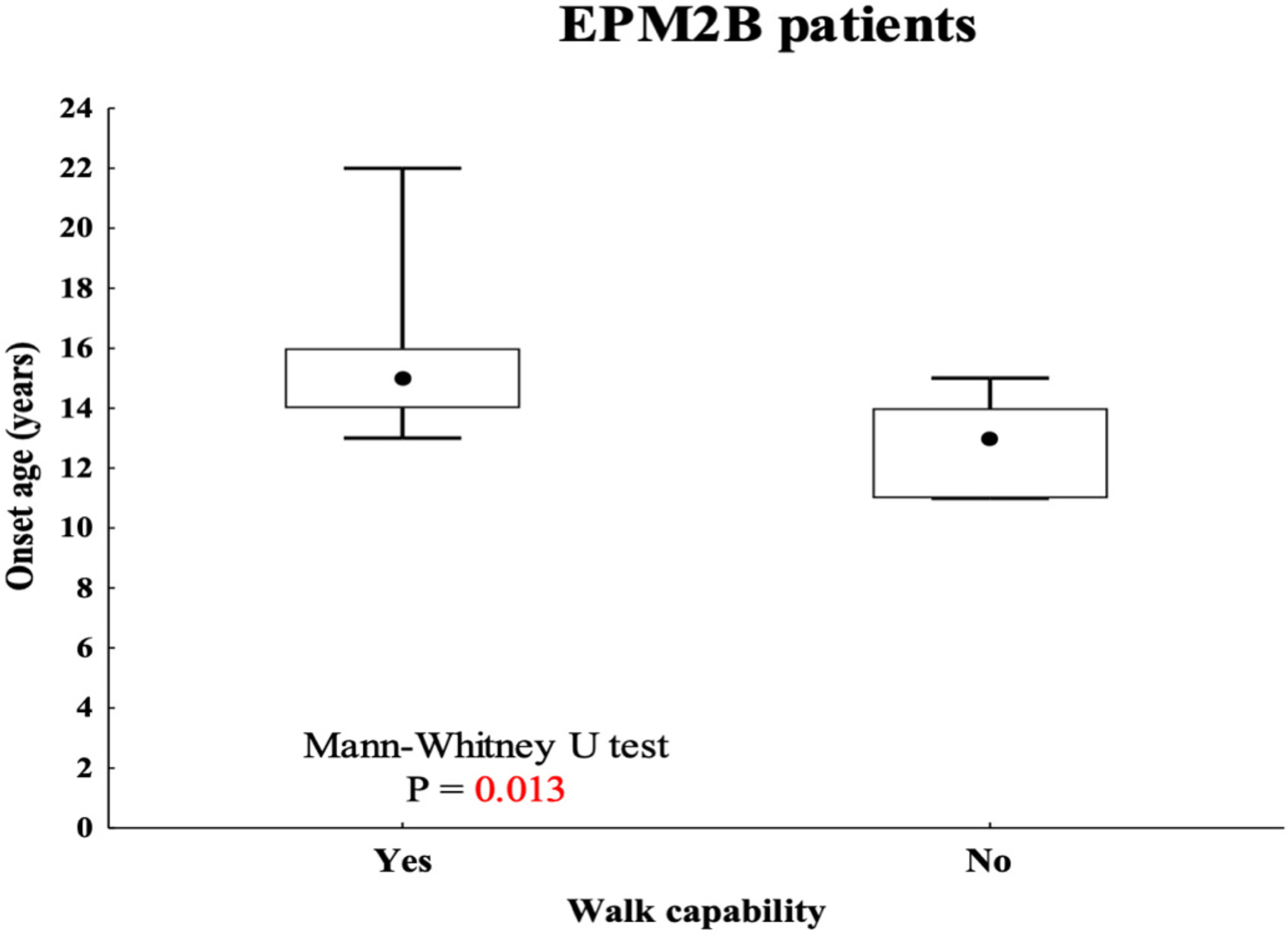
Correlation between onset ages and walking capability in EPM2B patients.
Specific missense mutations in EPM2A and EPM2B were associated with delayed disease evolution. Patients carrying the EPM2B p.(D146N) mutation (#8, #20, #22, #23) showed mild disease stage and myoclonus severity, with a follow-up period of 5 (#8) and 20 (#20, #22, #23) years. Moreover, patient #20 and #23 preserved speech and ambulation. Similarly, the p.(P69A) mutation in EPM2B was associated with preserved ambulation and speech ability in patient #3 after 9 years from disease onset. The EPM2A missense mutation p.(R108L) was associated with milder disease stage: patient #2b preserved walk and speech capabilities after 8 years of follow-up.
No significant differences emerged between EPM2A and EPM2B mutations concerning disease duration (P = 0.89), presence of ataxia (P = 1.0) and dementia (P = 0.62), disease stage (P = 0.07), absence of ambulation (P = 0.11) or speech (P = 0.41), and spontaneous (P = 0.28) or action (P = 0.44) myoclonus severity. However, earlier disease onset was observed in the EPM2A group, with a value of P = 0.039 (Table 2). Furthermore, patients with EPM2A pathogenic variants (rS = 0.29) showed a faster disease progression as compared to EPM2B subjects (rS = 0.44) (Fig. 4).
Table 2.
Summary of the patients’ cohort and EPM2A/EPM2B correlations.
| EPM2A mutations (N=9) | EPM2B mutations (N=17) | p value | |
|---|---|---|---|
| Gender: Male | 3 (33.3%) | 6 (35.3%) | 1.00a |
| Female | 6 (66.7%) | 11 (64.7%) | |
| Onset: Epilepsy | 3/9 (50.0%) | 6/9 (42.8%) | 1.00a |
| Myoclonus | 1/5 (16.7%) | 4/5 (28.6%) | |
| Epilepsy and myoclonus | 2/6 (33.3%) | 4/6 (26.6%) | |
| Median [1st-3rd q] | Median [1st-3rd q] | ||
| Age at disease onset (y) | 13 (11–13) | 14.5 (13–16) [n=16] | 0.039b |
| Age at last evaluation (y) | 23 (18.6–24) | 22.8 (19.1–31.1) | 0.83b |
| Follow-up duration (y) | 10 (7.6–12.6) | 8.9 (4.8–19.1) [n=16] | 0.89b |
N = number; q = quartile; y = years.
P: Fisher’s Exact test.
P: Mann-Whitney U test.
Fig. 4.
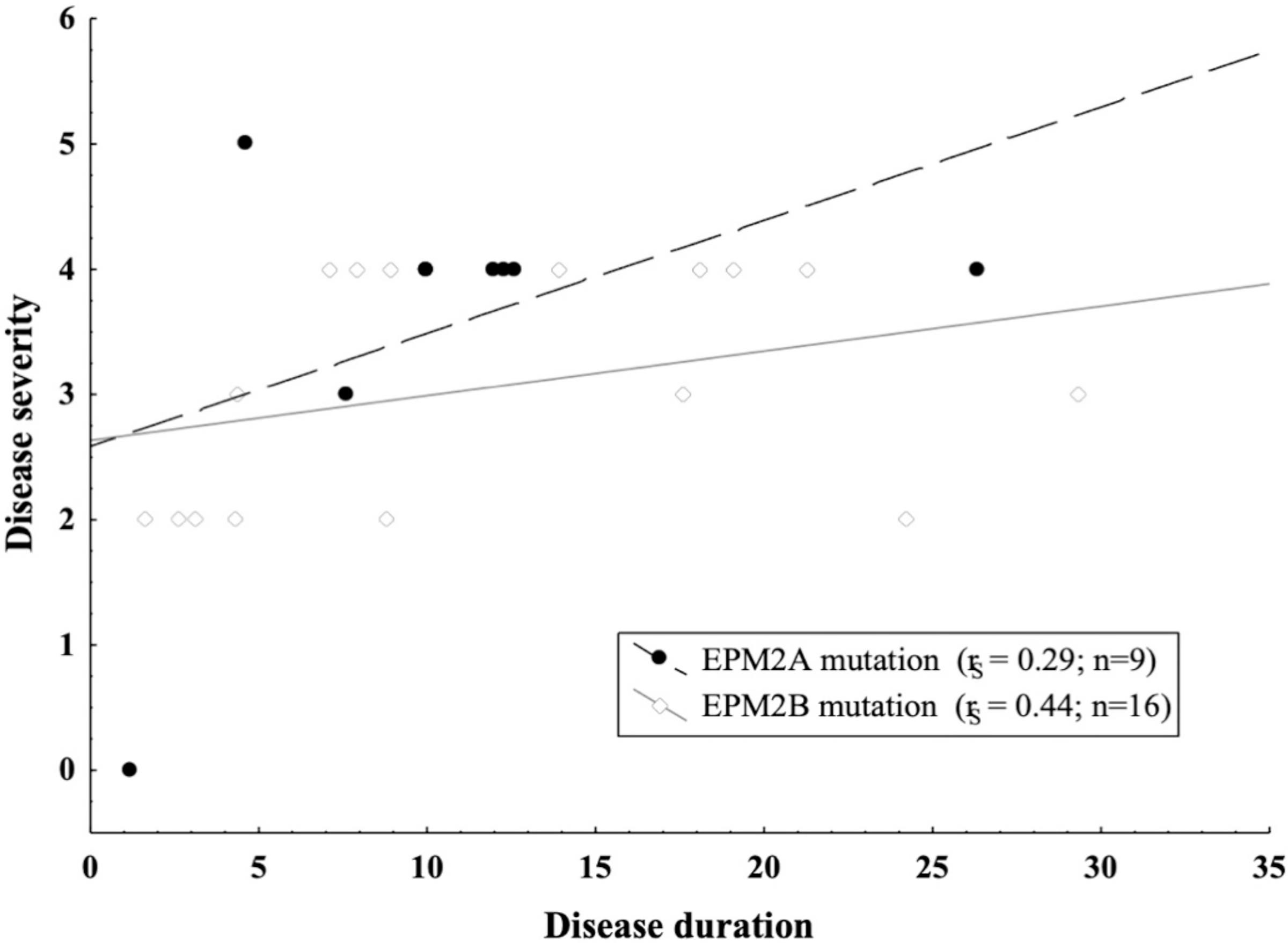
Relationship between disease severity and disease duration in the EPM2A and EPM2B patients. Legend: rS = Spearman’s correlation coefficient; n = number.
4. Discussion
LD is a well-known but extremely complex neurological condition and definite correlations between the underlying genetic mutations and phenotypic features remain to be elucidated [14–17]. In our cohort of Italian LD patients, EPM2A mutations were associated with a more severe disease course, whereas EPM2B patients generally have milder disease course and longer survival. The type of genetic mutation also appears to influence the disease course, as highlighted by the milder phenotype observed in the patient harbouring the missense mutation p. (D146N) in EPM2B (#20). Eventually, the extension of the deletion may impact disease severity, since multiexonic and whole gene deletions have been associated with a more severe phenotype [18]. However, we did not identify large deletions in EPM2A or EPM2B in our cohort, making it difficult to speculate on specific genotype-phenotype correlations. However, the eight patients harbouring small intragenic deletions (three for EPM2A and five for EPM2B) did not show a particularly severe phenotype in comparison to other affected individuals.
Noteworthy, Italian patients showed a higher prevalence of EPM2B mutations [19], which may in part indicate a founder effect for some of these pathogenic variants in this population. However, no predominant mutation could be identified, likely due to the highly heterogeneous genetic background. In line with previous reports, the p.(P69A) and p. (D146N) were the most common EPM2B mutations, whereas the p. (R241Ter) and exon 2 deletion were frequent in the EPM2A group [12].
Clinical onset appeared to be earlier in EPM2A patients, with a younger age at onset of myoclonic and tonic-clonic seizures. Neither dystonia nor parkinsonism was observed, although previously reported in other studies [20–22]. Moreover, no early-onset of learning disabilities, suggestive for an atypical form of LD was observed in our cohort [23]. Mean latency of 2 years between disease onset and the diagnosis of dementia was observed in both EPM2A and EPM2B patients. However, on average, subjects harbouring EPM2A mutations were diagnosed with dementia 1 year before EPM2B-mutated patients. The observation that patients in the EPM2B group had a longer follow-up duration and different EPM2B mutations were associated with different follow-up periods suggests the existence of possible correlations. In particular, a slower disease progression might be associated with specific mutations in EPM2A/EPM2B or even with different types of genetic mutations within the same gene.
Long-term follow-up of LD patients is extremely rare due to the severely progressive disease course. Furthermore, the combination of underlying genetic factors and the quality of available care strongly impacts the frequency of disease complications, influencing the overall life expectancy of affected individuals. Accordingly, only a few studies on small LD cohorts with a mean follow-up over 10 years have been reported in the literature so far [24–27]. According to the rapid disease progression, MoCA assessment timelines need to be pointful and shared through the scientific community to achieve a reliable characterization of the clinical course [16]. Based on follow-up duration and disease stage, we observed that there may be a more aggressive disease in EPM2A patients. Indeed, a higher prevalence of loss of speech and ambulation, as well as a higher disease stage at the last follow-up, were observed in this group. Patients with EPM2B mutations exhibited instead a longer follow-up period (more than 5 years) and a milder disease stage. Interestingly, 4 of these subjects were also included in the study published in 2006 [12], suggesting a prolonged disease course, whereas no subject carrying EPM2A mutations was reported. Genetic mutations of protein targeting to glycogen (PTG, a protein modulating glycogen synthesis) were believed to contribute to the milder phenotype in LD [28], but their role was not confirmed by a subsequent study showing significant PTG mutation in only 1/6 mild LD patients [25]. None of the EPM2A patients in the current cohort showed such a comparably long survival. In particular, two out of the four previously reported patients preserved ambulation and exhibited a low disease stage after more than 20 years of follow-up. Moreover, patients harbouring the p.(D146N) pathogenic variant presented with a milder disease course in two heterozygous (#8, #22) and two homozygous cases (#20, #23). Of note, the same mutation was associated with later disease onset, around the age of 20 years, according to the literature [26–30]. Finally, hydrogen 1 (1H) MR spectroscopy may prove effective to detect metabolic changes such as the reduction in the (N-acetylaspartate) NAA/creatine ratio in the frontal cortex, cerebellum, and basal ganglia of LD patients as compared to healthy controls. Conversely, structural MRI is usually less sensitive to detect brain involvement in these patients [31]. These findings suggest imaging studies may be suitable to monitor the disease progression; however, such correlations between MRI data and the LD course were not possible due to a lack of longitudinal MRI data in our cohort.
5. Conclusions
We report a large cohort of Italian LD patients, focusing on the possible genotype-phenotype correlations and specifically aiming to dissect disease outcomes with distinct genetic mutations. Our observations support earlier disease onset and faster disease progression with severe deterioration of ambulation and speech in EPM2A individuals. Conversely, later disease onset and delayed progression were confirmed in EPM2B patients [7,29,32]. Specific gene mutations in EPM2B, such as the p.(D146N), appeared to be associated with delayed onset of disabling symptoms [26], suggesting that disease course might be also influenced by the underlying causative genetic mutation itself. Some limitations can be recognized in our study, including the small number of patients carrying EPM2A mutations and the limited duration of follow-up in some cases (#2a, #15), requiring further evaluations in the future. The effort towards the collection of large case series will play a relevant role in providing insights into genotype-phenotype correlations in different countries, expanding our knowledge on the pathogenic mechanisms underlying LD, eventually allowing the development of targeted therapies.
Supplementary Material
Acknowledgments
We acknowledge the families for their participation in this study. A special thanks to A.I.LA. (Associazione Italiana Lafora; http://www.lafora.it/). This work was developed within the framework of the DINOGMI Department of Excellence of MIUR 2018–2022 (legge 232 del 2016).
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations:
- LD
Lafora disease
- LBs
Lafora Bodies
- ADL
Activities of daily living
- MoCA
Montreal Cognitive Assessment
- ASMs
Antiseizure medications
- VPA
Valproate
- LEV
Levetiracetam
Footnotes
Declaration of Competing Interest
P.S. has received speaker fees and participated at advisory boards for Biomarin, Zogenyx, GW Pharmaceuticals, and has received research funding by ENECTA BV, GW Pharmaceuticals, Kolfarma srl., Eisai. The other authors do not report any conflict of interest.
Ethical publication statement
This study has been performed following the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Family members or parents gave written informed consent.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jns.2021.117409.
References
- [1].Turnbull J, Tiberia E, Striano P, et al. , Lafora disease, Epileptic Disord. 18 (S2) (2016) 38–62, 10.1684/epd.2016.0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nitschke F, Ahonen SJ, Nitschke S, et al. , Lafora disease – From pathogenesis to treatment strategies, Nat. Rev. Neurol. 14 (10) (2018) 606–617, 10.1038/s41582-018-0057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Minassian BA, Lafora’s disease: towards a clinical, pathologic, and molecular synthesis, Pediatr. Neurol. 25 (1) (2001) 21–29, 10.1016/S0887-8994(00)00276-9. [DOI] [PubMed] [Google Scholar]
- [4].Serratosa MJ, Delgado-Escueta VA, Posada I, et al. , The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q, Hum. Mol. Genet. 4 (9) (1995) 1657–1663, 10.1093/hmg/4.9.1657. [DOI] [PubMed] [Google Scholar]
- [5].Minassian BA, Lee JR, Herbrick JA, et al. , Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy, Nat. Genet. 20 (1998) 171–174, 10.1038/2470 (october). [DOI] [PubMed] [Google Scholar]
- [6].Chan EM, Young EJ, Ianzano L, et al. , Mutations in NHLRC1 cause progressive myoclonus epilepsy, Nat. Genet. 35 (2) (2003) 125–127, 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- [7].Chan EM, Bulman DE, Paterson AD, et al. , Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22, J. Med. Genet. 40 (9) (2003) 671–675, 10.1136/jmg.40.9.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].García-Gimeno MA, Knechtet E, Sanz P, Lafora disease: a Ubiquitination-related pathology, Cells. 7 (8) (2018) 87, 10.3390/cells7080087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cavanagh JB, Corpora-amylacea and the family of polyglucosan diseases, Brain Res. Rev. 29 (2–3) (1999) 265–295, 10.1016/S0165-0173(99)00003-X. [DOI] [PubMed] [Google Scholar]
- [10].Rubio-Villena C, Viana R, Bonet J, et al. , Astrocytes: new players in progressive myoclonus epilepsy of Lafora type, Hum. Mol. Genet. 27 (7) (2018) 1290–1300, 10.1093/hmg/ddy044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].d’Orsi G, Lalla A, Palumbo O, et al. , The presenting symptoms of lafora disease: an electroclinical and genetic study in five apulian (southern Italy) families, Seizure. 83 (2020. October 27) 145–153, 10.1016/j.seizure.2020.10.022. [DOI] [PubMed] [Google Scholar]
- [12].Franceschetti S, Gambardella A, Canafoglia L, et al. , Clinical and genetic findings in 26 Italian patients with Lafora disease, Epilepsia. 47 (3) (2006) 640–643, 10.1111/j.15281167.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- [13].Gentry MS, et al. , Lafora disease offers a unique window into neuronal glycogen metabolism, J. Biol. Chem. 293 (19) (2018) 7117–7125, 10.1074/jbc.R117.803064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brenner D, Baumgartner T, von Spiczak S, et al. , Genotypes and phenotypes of patients with Lafora disease living in Germany, Neurol. Res. Pract. 1 (2019) 34, 10.1186/s42466-019-0040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gentry MS, Afawi Z, Armstrong DD, et al. , The 5th International Lafora Epilepsy Workshop: basic science elucidating therapeutic options and preparing for therapy in the clinic, Epilepsy Behav. 103 (Pt A) (2020), 106839, 10.1016/j.yebeh.2019.106839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Singh S, Sethi I, Franceschetti S, et al. , Novel NHLRC1 mutations and genotype-phenotype correlations in patients with Lafora’s progressive myclonic epilepsy, J. Med. Genet. 43 (9) (2006. September), e48, 10.1136/jmg.2005.039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lohi H, Turnbull J, Zhao XC, et al. , Genetic diagnosis in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls, Neurology. 68 (13) (2007. March 27) 996–1001, 10.1212/01.wnl.0000258561.02248.2f. [DOI] [PubMed] [Google Scholar]
- [18].Kecmanović M, Jović N, Cukić M, et al. , Lafora disease: severe phenotype associated with homozygous deletion of the NHLRC1 gene, J. Neurol. Sci. 325 (1–2) (2013. February 15) 170–173, 10.1016/j.jns.2012.12.006. [DOI] [PubMed] [Google Scholar]
- [19].Franceschetti S, Michelucci R, Canafoglia L, et al. , Progressive myoclonic epilepsies: definitive and still undetermined causes, Neurology. 82 (5) (2014) 405–411, 10.1212/WNL.0000000000000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ragona F, Canafoglia L, Castellotti B, et al. , Early Parkinsonism in a Senegalese girl with Lafora disease, Epileptic Disord. 22 (2) (2020. April 1) 233–236, 10.1684/epd.2020.1150. [DOI] [PubMed] [Google Scholar]
- [21].Yildiz EP, Yesil G, Ozkan MU, et al. , A novel EPM2A mutation in a patient with Lafora disease presenting with early parkinsonism symptoms in childhood, Seizure 51 (2017. October) 77–79, 10.1016/J.seizure.2017.07.011. [DOI] [PubMed] [Google Scholar]
- [22].Gökdemir S, Cağlayan H, Kızıltan M, et al. , Presentation of an unusual patient with Lafora disease, Epileptic Disord. 14 (1) (2012. March) 94–98, 10.1684/epd.2012.0489. [DOI] [PubMed] [Google Scholar]
- [23].Ganesh S, Delgado-Escueta AV, Suzuki T, et al. , Genotype-phenotype correlations for EPM2A mutations in Lafora’s progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype, Hum. Mol. Genet. 11 (11) (2002. May 15) 1263–1271, 10.1093/hmg/11.11.1263. [DOI] [PubMed] [Google Scholar]
- [24].Kecmanović M, Jović N, Keckarević-Marković M, et al. , Clinical and genetic data on Lafora disease patients of Serbian/Montenegrin origin, Clin. Genet. 89 (1) (2016. January) 104–108, 10.1111/cge.12570. [DOI] [PubMed] [Google Scholar]
- [25].Ferlazzo E, Canafoglia L, Michelucci R, et al. , Mild Lafora disease: clinical, neurophysiologic, and genetic findings, Epilepsia. 55 (12) (2014. December) e129–e133, 10.1111/epi.12806. [DOI] [PubMed] [Google Scholar]
- [26].Baykan B, Striano P, Gianotti S, et al. , Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation, Epilepsia. 46 (10) (2005) 1695–1697, 10.1111/j.1528-1167.2005.00272.x. [DOI] [PubMed] [Google Scholar]
- [27].Yoshimura I, Kaneko S, Yoshimura N, Murakami T, Long-term observations of two siblings with Lafora disease treated with zonisamide, Epilepsy Res. 46 (3) (2001. September) 283–287, 10.1016/s0920-1211(01)00282-0. [DOI] [PubMed] [Google Scholar]
- [28].Guerrero R, Vernia S, Sanz R, et al. , A PTG variant contributes to a milder phenotype in Lafora disease, PLoS One 6 (6) (2011), e21294, 10.1371/journal.pone.0021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gómez-Abad C, Gómez-Garre P, et al. , Lafora disease due to EPM2B mutations: a clinical and genetic study, Neurology. 64 (6) (2005) 982–986, 10.1212/01.WNL.0000154519.10805.F7. [DOI] [PubMed] [Google Scholar]
- [30].Lanoiselée HM, Genton P, Lesca G, et al. , Are c.436G>A mutations less severe forms of Lafora disease? A case report, epilepsy Behav, Case Rep. 2 (2014. January 19) 19–21, 10.1016/j.ebcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Villanueva V, Alvarez-Linera J, Gómez-Garre P, et al. , MRI volumetry and proton MR spectroscopy of the brain in Lafora disease, Epilepsia. 47 (4) (2006. April) 788–792, 10.1111/j.1528-1167.2006.00526.x. [DOI] [PubMed] [Google Scholar]
- [32].Zutt R, Drost G, et al. , Unusual course of Lafora disease, Epilepsia Open. 1 (3–4) (2016) 136–139, 10.1002/epi4.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.