

ABSTRACT
PUFAs are known to regulate cholesterol synthesis and cellular uptake by multiple mechanisms that do not involve SFAs. Polymorphisms in any of the numerous proteins involved in cholesterol homeostasis, as a result of genetic variation, could lead to higher or lower serum cholesterol. PUFAs are susceptible to lipid peroxidation, which can lead to oxidative stress, inflammation, atherosclerosis, cancer, and disorders associated with inflammation, such as insulin resistance, arthritis, and numerous inflammatory syndromes. Eicosanoids from arachidonic acid are among the most powerful mediators that initiate an immune response, and a wide range of PUFA metabolites regulate numerous physiological processes. There is a misconception that dietary SFAs can cause inflammation, although endogenous palmitic acid is converted to ceramides and other cell constituents involved in an inflammatory response after it is initiated by lipid mediators derived from PUFAs. This article will discuss the many misconceptions regarding how dietary lipids regulate serum cholesterol, the fact that all-cause death rate is higher in humans with low compared with normal or moderately elevated serum total cholesterol, the numerous adverse effects of increasing dietary PUFAs or carbohydrate relative to SFAs, as well as metabolic conversion of PUFAs to SFAs and MUFAs as a protective mechanism. Consequently, dietary saturated fats seem to be less harmful than the proposed alternatives.
Keywords: atherosclerosis, cholesterol regulation, cancer, dietary recommendations, inflammation, lipid peroxidation, palmitic acid, polyunsaturated fatty acids, saturated fats
Statement of Significance: There is a persistent misperception that dietary saturated fats can cause or promote numerous adverse health effects and increase serum total cholesterol and LDL cholesterol. This review attempts to clarify how such misperceptions originated and describes how the dietary alternatives of polyunsaturated oils and processed carbohydrates can be more detrimental to health.
Introduction
The notion that dietary saturated fats raise serum cholesterol originated from the misinterpretation of several studies that showed if confined individuals consumed diets containing fats with mostly SFAs and little or no PUFAs, serum cholesterol was higher compared with when the same individuals consumed diets containing an abundance of PUFAs (1–4). When they consumed diets with a high proportion of MUFAs, serum cholesterol was intermediate. The fact that the high-MUFA diets contained an intermediate amount of PUFAs was generally ignored. One could conclude from such results that dietary saturated fats raise serum cholesterol (5), but the interpretation that PUFAs are responsible for regulating serum cholesterol is generally overlooked.
The emphasis on promoting a low-fat, low-saturated fat diet resulted in a large increase in carbohydrate consumption, and most of that increase was in the form of sugars and highly refined carbohydrates (6, 7). When dietary saturated fats are replaced with carbohydrates, there is no change in serum cholesterol when dietary PUFAs remain unchanged (8), yet replacing saturated fats with carbohydrates, especially refined carbohydrates, gives rise to proatherogenic dyslipidemia (9), as well as low-grade systemic inflammation and insulin resistance, which all increase the risk of cardiovascular disease (10). Changes in dietary saturated fat intake in adults with metabolic syndrome resulted in no significant changes in the proportions of SFAs in any plasma lipid fractions, whereas replacement of dietary saturated fats with carbohydrates showed a direct correlation between carbohydrate intake and proportions of palmitoleate in plasma triglycerides and cholesteryl esters. Palmitoleate in plasma cholesteryl esters, plasma phospholipids, and erythrocyte membranes is a consistent predictor of type 2 diabetes and metabolic syndrome (11).
Overall mortality, and not just coronary deaths, should be the primary concern when considering dietary recommendations for the general public. Several studies from diverse populations have shown that the all-cause death rate is greater for low serum total cholesterol (<180 mg/dL) than for intermediate to moderately high serum cholesterol (180–240 mg/dL) (12–16), particularly in elderly populations (17). It is important to note that even the 2 Finnish cohorts of the Seven Countries Study, which had the highest median serum total cholesterol for all cohorts at >250 mg/dL, had more overall deaths in a subgroup of the lowest 30% of serum cholesterol than there were in the highest 30% of serum cholesterol; the latter subgroup would have had serum total cholesterol well above 250 mg/dL in those 2 cohorts (18). This was true for other cohorts in the Seven Countries Study. So the question of lowering serum cholesterol to increase longevity should be weighed on a case-by-case basis, rather than a blanket recommendation for the general population.
Numerous reports and meta-analyses support the idea that dietary saturated fats have no influence on the incidence of cardiovascular disease, coronary heart disease, type 2 diabetes, or death from all causes (9, 19–22). However, many diet and health authorities continue to stress studies that showed a decrease in serum cholesterol in response to replacing dietary SFAs with PUFAs, and conflate that to mean that dietary saturated fats increase risk of cardiovascular disease and death from coronary heart disease (23). The flaws in the studies used to support the “diet-heart” hypothesis have been documented for years, yet dietary guidelines from many sources continue to push for lower dietary intake of all saturated fats with no substantial scientific support for that policy (22, 24, 25). The fact that overall death rate is not associated with serum cholesterol concentrations, except in the extreme upper concentrations or extreme lower concentrations of serum cholesterol, should render dietary recommendations for the purpose of lowering serum cholesterol in the general population unwarranted. A summary of the potential consequences of replacing dietary saturated fats with refined carbohydrates or polyunsaturated oils is shown in Figure 1.
Figure 1.
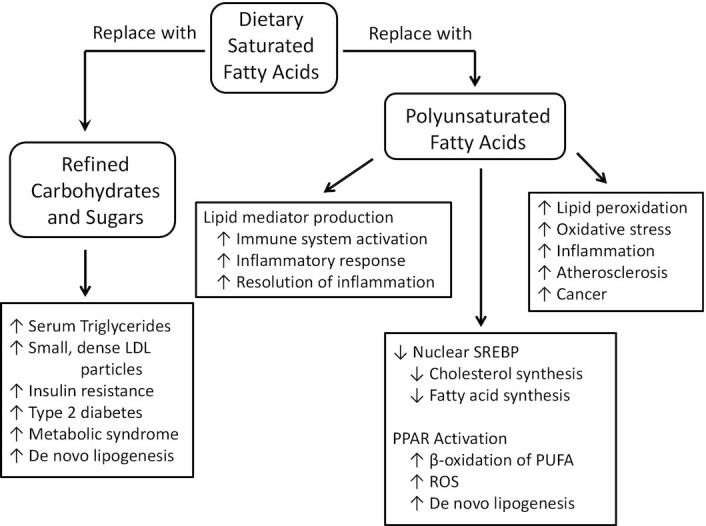
Some potential consequences of replacing dietary saturated fats with refined carbohydrates or polyunsaturated oils. These various physiological and health effects are described in the text. PPAR, peroxisome proliferator-activated receptor; ROS, reactive oxygen species; SREBP, sterol regulatory element-binding protein.
PUFA Oxidation and Lipid Peroxidation
PUFAs are important for production of a wide range of bioactive substances in the body, but like some vitamins and essential minerals, may become toxic when consumed in excess. PUFAs can be oxidized in a spontaneous chemical oxidation process that does not require enzymes and would not be regulated in cells by normal processes. This spontaneous oxidation is initiated by free radicals, reactive oxygen species (ROS), and reactive nitrogen species (RNS), which are constantly being formed in biological systems (26, 27). This free radical oxidation process, which is known as lipid peroxidation, requires molecular oxygen, and produces fatty acid peroxides, reactive carbonyl species such as malondialdehyde, as well as numerous other toxic products. The wide array of ROS, RNS, and toxic organic products formed during lipid peroxidation of PUFAs can cause mutations in DNA, which can lead to cancer (28). Lipid peroxidation can damage cell membranes and lead to cell death. The wide array of ROS and lipid peroxidation products are implicated in oxidative stress, aging, and many diseases (29, 30).
The body has many protective systems to combat lipid peroxidation, including lipid-soluble antioxidants, such as vitamin E, enzymes that eliminate ROS and RNS, and enzymes that metabolize the lipid peroxides to detoxify them (31). Lipid peroxidation of PUFAs in lipoproteins, such as LDLs, results in the oxidized LDLs being removed from the circulation by macrophages lining the arteries, leading to atherosclerosis (32, 33). Although consuming more PUFA-rich vegetable oils can lower serum total cholesterol and LDL cholesterol, it is the PUFAs in LDLs that are susceptible to lipid peroxidation, which destines that lipoprotein and its cholesterol load to the atherosclerotic deposits surrounding the arteries (34). There is also evidence that dietary oxidized PUFAs and oxidized cholesterol can lead to increased atherosclerosis (35). This leads to a conundrum: Consuming PUFAs to lower serum LDL cholesterol can also increase the chances for lipid peroxidation and consequent atherosclerosis. MUFAs are much less susceptible to lipid peroxidation, and SFAs are completely resistant because they do not contain the reactive carbon-carbon double bonds.
The point of this discussion is to stress the fact that PUFAs are not only found in vegetable oils, but also are present at low to moderate concentration in most animal fats as well. Regardless of the source, PUFAs are highly susceptible to lipid peroxidation, which can lead to atherosclerosis, heart disease, cancer, inflammation, and other unhealthy processes (30, 36). Mammals preferentially oxidize PUFAs via the β-oxidation pathway in mitochondria for ATP production, or in peroxisomes to recycle excess PUFAs into SFAs and MUFAs to eliminate a portion of these reactive nutrients and prevent formation of potential toxins, particularly in the fetus and infant (see below) (37, 38). There could be many evolutionary reasons for converting PUFAs into SFAs, cholesterol, and ketones (39). Peroxidation of PUFAs in meats cooked at high temperature (40) or stored in the presence of oxygen (41) could be the reason for any association of meats with adverse health effects, whereas the saturated fat content of that food is less likely to be involved in the mechanisms for disease (42). There are often conflicting data regarding adverse health effects of foods that contain predominantly SFAs, and it is best to use a more holistic approach to dietary recommendations and food choices (22). In addition, some animal fats that are classified as saturated fats contain significant amounts of PUFAs, such as lard, which contains ∼10% linoleic acid (LA), similar to palm oil and olive oil.
Fatty Acids and Serum Lipids
The primary factor that determines whether a person has low, normal, or high concentrations of serum cholesterol is genetic (43, 44). The effect of dietary fats on any individual's serum cholesterol is superimposed on the genetically determined concentration of serum cholesterol (45, 46). Numerous studies have indicated many inconsistencies in dietary responses in relation to specific gene polymorphisms (47), but the complexity of genetic variations and the broad range of gene products involved make this area of exploration rife with unpredictability and controversy. Genetic polymorphisms have been studied for several specific apolipoproteins, lipoprotein receptors, fatty acid–binding proteins, lipoprotein lipase, and other proteins involved in lipid transport (48). It is clear that focusing on apolipoproteins and their receptors is only a small aspect of the serum cholesterol picture.
The biochemical processes that determine serum cholesterol concentrations are the rate of cholesterol synthesis, especially in liver, and metabolic consumption to form numerous steroid products in the body. The expression of genes that code for enzymes that synthesize cholesterol and other lipids is regulated by a group of transcription factors known as sterol regulatory element-binding proteins (SREBPs) (49). When cholesterol concentrations are low in a cell, a series of events take place to increase the nuclear presence of SREBPs (SREBP1a and SREBP2) to promote the expression of genes for cholesterol synthesis, as well as its uptake from circulating lipoproteins (50). Insulin can also induce an increase in nuclear SREBP1 (both SREBP1a and SREBP1c), which will result in expression of genes for synthesis of fatty acids, cholesterol, and other lipids when carbohydrates are consumed in excess and stored as fat. When cholesterol concentrations are high in the cell, events occur to decrease the presence of SREBPs in the nucleus, which diminishes cholesterol synthesis and uptake from the circulation. The presence of PUFAs can decrease the amount of nuclear SREBP1 to decrease the synthesis of fatty acids, cholesterol, and other lipids, with long-chain ω-3 PUFAs showing the strongest activity in this capacity (51). SFAs and MUFAs do not have any influence on these control mechanisms.
Another class of fatty acid–activated transcription factors that promote gene expression are peroxisome proliferator-activated receptors (PPARs). Three different PPARs have been identified: PPARα, PPARβ/δ, and PPARγ. These nuclear receptors are structurally similar, but have distinct ligand-binding properties, functions, and tissue distributions (52). All PPARs bind fatty acids, with a strong preference for long-chain PUFAs and relatively little activity with SFAs and MUFAs. PPARα is present in many tissues that rely heavily on oxidative metabolism, such as brown adipose tissue, heart muscle, skeletal muscle, and liver (53). Selectively activating PPARα with a specific agonist (WY-14,643) increased liver mass relative to body weight in rats, and decreased serum total cholesterol, HDL cholesterol, and triglycerides compared with control animals fed the same standard chow diet (54). In addition, β-oxidation occurred at a higher rate in liver, although liver contents of SFAs and MUFAs were higher, whereas PUFA content was lower, indicating a preferential oxidation of PUFAs in peroxisomes with subsequent de novo lipogenesis in PPARα agonist–treated rats compared with controls.
Treatment of rats with a selective agonist for PPARγ (rosiglitazone) had relatively little effect on liver but a significant effect on lipid metabolism in adipose tissue, resulting in lower serum triglycerides and no significant changes in serum cholesterol compared with control rats (54). In view of the fact that PPARs exhibit a strong response with PUFAs and little or no response to MUFAs and SFAs, it has become clear that PUFAs, particularly DHA and other longer-chain PUFAs, can lower serum cholesterol via PPARα. In addition, PUFA activation of PPARγ can elicit the beneficial reduction of serum triglycerides, increase adipose tissue storage of lipids, and decrease release of free fatty acids from adipose tissue. This latter mechanism explains the beneficial effects of longer-chain ω-3 PUFAs on insulin sensitivity and glucose metabolism.
Consequently when PUFAs are abundant as a result of increased dietary intake, the liver enzymes for β-oxidation of excess PUFAs in peroxisomes will help to limit the amount of PUFAs in the body to prevent excessive lipid peroxidation. In addition, many PUFA metabolites, such as eicosanoids, endocannabinoids, and the oxidized derivatives 9- and 13-hydroxyoctadecadienoic acids, will also activate PPARα in the nucleus (53). These metabolites and oxidation products of PUFAs would be an indicator of sufficient PUFAs present in body tissues to satisfy their need for synthesis of bioactive signaling agents and the excess should be eliminated. The main point of this discussion is that PUFAs, rather than SFAs or MUFAs, are responsible for regulating cholesterol metabolism, and consequently serum cholesterol and lipoprotein concentrations. The key factor is the level of PUFAs in the diet, which can influence the mechanisms for cholesterol synthesis, uptake, and distribution. Changes in the amount of SFAs or MUFAs consumed in the diet will have little or no influence on these mechanisms.
Some studies that compared olive oil (SFA:MUFA:PUFA ratio ∼15:75:10) with palm oil (SFA:MUFA:PUFA ∼50:40:10) found no difference in serum cholesterol concentrations of young, healthy human volunteers. Palm oil diets had much more SFAs than olive oil diets; however, because the diets contained the same amount of PUFAs, serum cholesterol did not change with these changes in dietary SFA (55, 56). It is estimated that ≤6–8 g/d PUFAs (8–10% of total fatty acids; 2–3% of energy) in the diet would be sufficient to satisfy normal requirements for production of bioactive products without causing adverse health effects. In addition, a ratio of ω-6/ω-3 PUFAs near 1 or 2, but certainly <5 would be optimum for good health (57, 58).
Importance of Palmitic Acid in Development and Physiology
Palmitic acid (PA) is the major SFA in the human diet and in the human body. The importance of PA in the development of the human fetus and infant was reviewed by the late Sheila Innis (59), who devoted much of her research to the nutrients in mammalian milk. The human baby is one of the fattest of all mammals at normal gestational birth; the adipose fat is 45–50% palmitic acid, and the baby continues to add fat during the early months of life under normal circumstances. A human baby will have ∼2% PUFAs in adipose tissue at birth. Innis raised concerns that infants fed formulas with lipids coming from vegetable oils accumulated ≤26% LA in adipose tissue, mostly replacing palmitic acid (60), whereas LA increased to 6–8% in breast-fed infants after 7 mo (61). Mammary glands produce predominantly SFAs, regardless of the mother's dietary fat intake, and Innis raised the question of the importance of milk fatty acid composition with respect to developmental biology, and pointed out the broader implication for proper nutritional care for infants and children (59).
The unique composition of triglycerides in mammalian milk (human as well as ruminant milk) compared with vegetable oils provides clues regarding the importance of PA to infant development. Palmitate in the sn-2 position of glycerol of milk triglycerides improves absorption from the intestinal tract (62). The structure as well as composition of milk triglycerides ensures that newborn infants absorb an abundance of PA in the early stages of development, which is most likely evolutionarily advantageous. When infants were fed formula consisting of vegetable triglycerides, the proportion of LA increased dramatically to ≤46% of fatty acids in adipose tissue by 10 mo of age (63). Although increasing PUFA intake in infants can marginally lower serum cholesterol at 1 y of age (64), such manipulations in the PUFA content of infant formula and diets could have longer-term consequences that do not seem to have been followed in those cohorts.
PA is a major fatty acid in brain phospholipids, constituting 45–55% in phosphatidylcholine fractions (65). PA is used by cells to form sphingolipids, which with cholesterol and palmitoylated proteins, are important components of lipid rafts that facilitate endocytosis in caveolae, as well as numerous cell signaling pathways (66). PA is the major fatty acid in lung surfactant, and palmitoylethanolamide is a lipid mediator of intra- and intercellular signals in many cells and tissues (67).
Perhaps the greatest tragedy regarding dietary recommendations is for parents to give their children low-fat or skim milk rather than full-fat milk. A recent review indicated that there is no evidence that full-fat milk consumption leads to obesity or cardiometabolic risk in children and adolescents (68). A recent study found that higher milk fat consumption in 3-y-old Latino children was associated with significantly lower odds for severe obesity in that population (69). Another study found significantly less adiposity at age 13 for children in the highest quartile of dairy fat consumption compared with the lowest quartile (70). Milk consumption by children and adolescents decreased by 38% between 1977 and 1996, whereas sweetened beverage consumption increased by 215% and consumption of fruit drinks increased by 189% (71). It seems the dietary recommendation to decrease milk fat consumption has caused parents to allow or even encourage children to consume more sweetened beverages under the false impression that they might be healthier for their children.
Fatty Acids and Inflammation
Inflammation can arise from an acute immune response to insult or injury, causing pain and discomfort, but generally subsides when the pathogenic insult is brought under control and injured tissue is repaired. A more subtle form of inflammation, known as low-grade systemic inflammation, occurs when immune cells infiltrate adipose tissue, especially visceral adipose tissue in obese individuals (72). Chronic systemic inflammation can lead to insulin resistance, type 2 diabetes, metabolic syndrome, atherosclerosis, and a variety of other metabolic disruptions and unhealthy conditions.
Inflammatory responses are initiated by a wide range of lipid mediators, including many eicosanoids formed from arachidonic acid (AA), the major ω-6 long-chain PUFA in membrane phospholipids of immune cells (73). The proinflammatory eicosanoids include: 5-hydroxyeicosatetraenoic acid, PGE2, and leukotriene B4 (LTB4), among others. A similar array of proinflammatory eicosanoids are formed from EPA, but these are generally considered to be less provoking than the ω-6 derivatives from AA with regard to an immune response (74). Proresolving eicosanoids are formed from these two 20-carbon PUFAs, such as lipoxin A4 and resolvin E1, among others, which actively participate in abatement of the inflammatory response. There are additional proresolving lipid mediators formed from DHA, which include resolvin D1, protectin D1, and maresin, among others (75).
The roles of PUFA-derived lipid mediators in an inflammatory response have been reviewed (73, 76, 77). Nonsteroidal anti-inflammatory drugs (NSAIDs) are effective at suppressing an acute inflammatory response because they inhibit cyclooxygenase enzymes that produce the precursors for many of the bioactive eicosanoids in the PG and thromboxane branch of eicosanoids. Steroidal anti-inflammatory drugs can be even more effective by suppressing inflammation that is resistant to NSAID treatment because they inhibit the release of long-chain PUFAs from cell membranes and diminish production of all eicosanoids, including the lipoxygenase products and the LT branch of proinflammatory agents. The eicosanoids are upstream lipid mediators, which initiate a cascade of events to release cytokines and other factors in an immune response. Peptide cytokines are generally measured as markers of inflammation because they have much longer half-lives than the lipid mediators. The point here is that it takes very little AA to trigger an immune response via its conversion to bioactive eicosanoids.
Dietary LA shows a nonlinear correlation with liver AA in rats fed fat-free background diets (78). A study in humans consuming high or low levels of LA showed a positive linear correlation with LA, but no significant changes in AA in neutrophils and plasma lipids. In that study, supplementation with fish oil (1.6 g/d EPA, 0.3 g/d DHA) resulted in reduced AA in neutrophil phospholipids (79). A review of 36 articles that studied changes in dietary LA intake and changes in tissue AA (erythrocytes and plasma/serum phospholipids) concluded that large variations in LA consumption have no significant effect on AA concentrations in those tissues (80). A recent review concluded that AA consumption can increase AA in membrane phospholipids of peripheral blood mononuclear cells (lymphocytes and monocytes) that are involved in inflammation, whereas changes in LA consumption showed no significant effect on AA in those cells (81). One would conclude from the above information that cells, particularly immune cells, regulate the synthesis of AA from LA and maintain a relatively constant concentration of AA in membrane phospholipids when humans consume a diet with ≥2% of energy from LA. Typical LA consumption by humans consuming a Western diet is closer to 6% of energy (82). However, EPA and DHA can reduce the concentration of AA in membrane phospholipids when they are consumed in the diet.
Fish oil supplements and ω-3 PUFAs have received the most attention with regard to dietary fats influencing arthritis symptoms, such as morning stiffness, pain, grip strength, swollen or tender joints, and reduction in the use of NSAIDs (83). One meta-analysis found significant reduction in patient-reported symptoms and use of NSAIDs with ω-3 fish oil consumption, whereas physician assessments showed no statistically significant clinical improvements (84). Study designs often have limitations, such as short duration, inappropriate placebo or control, low dose for ω-3 fish oil supplements, and not recommending a reduction in the use of ω-6 PUFA intake. Another meta-analysis of 10 randomized controlled trials found that supplementation with >2.7 g ω-3 long-chain PUFA per day for ≥3 mo duration consistently and significantly reduced NSAID consumption (85). Clinical evaluation of arthritic symptoms showed a trend toward improvement but again were not statistically significant relative to placebo controls.
There have been several recent reviews and meta-analyses regarding dietary ω-3 PUFA supplements or fish consumption effects on arthritic inflammation (86–88). The general consensus seems to be that ω-3 PUFA supplements can result in a decrease in the quantity of NSAIDs taken to relieve pain and inflammation, but there is no overall significant effect with regard to several clinical markers of inflammation. Because some eicosanoids from EPA can be proinflammatory, albeit less so than those from AA, the differences in the degree of inflammation can be subtle. EPA can suppress the production of AA metabolites by competing with AA for incorporation into cell membranes and is considered a poorer substrate for cyclooxygenase and PG production. EPA is a relatively good substrate for lipoxygenase and LT production, although LTB5 from EPA exhibited much less activity than LTB4 from AA with regard to activation of leukocytes (89). Another drawback of many studies in humans is that participants might not be advised to decrease their consumption of ω-6 PUFA-rich vegetable oils while taking ω-3 PUFA supplements, and consequently continue to have a relatively large ω-6/ω-3 PUFA ratio in their overall diet.
A few studies of arthritic models in rats compared dietary saturated fats with polyunsaturated oils or supplementation with oils after arthritis was induced. When a diet containing corn oil (high in LA) was compared with beef tallow (low in essential fatty acids, EFAs), and a fish oil diet (high in ω-3 PUFAs), the corn oil diet strongly exacerbated adjuvant-induced arthritis in rats, whereas the beef tallow diet resulted in relatively little inflammation, and rats fed the fish oil diet showed an intermediate level of inflammation (90). When rats were fed an EFA-deficient diet, they showed much less adjuvant-induced inflammation compared with animals fed a control diet, but the inflammatory response was restored when rats fed the EFA-deficient diet were given a corn oil supplement after adjuvant treatment (91). Rats fed an EFA-deficient diet starting with the day of adjuvant treatment had 87% less edema in the hind foot pads compared with control rats, and edema increased when the animals on the EFA-deficient diets were given a dose of 273 mg/d LA after adjuvant treatment (92). Another study found that dietary fish oil increased inflammation relative to beef tallow for collagen-induced arthritis in rats, indicating that the ω-3 PUFAs in fish oil are proinflammatory relative to SFAs (93). These studies indicate that minimizing dietary PUFAs was beneficial in reducing arthritic inflammation in animal models. The fact that ω-3 EPA produces eicosanoids that generally have similar, albeit less potent, actions relative to ω-6 eicosanoids from AA, would explain the in vivo results of these animal studies. It is interesting that dietary DHA, but not a combination of DHA with EPA, reduced collagen-induced inflammation in rats (94), whereas oral administration of monoacylglycerol derivatives of EPA decreased the severity of adjuvant-induced arthritis in rats more than the monoacylglycerol DHA derivative (95).
There has been much discussion in the literature of the role of SFAs, particularly PA, in inflammation. There seems to be widespread misunderstanding of how PA is involved in an immune response, which stems from a study that found high concentrations (200 μM) of SFAs, but not MUFAs or PUFAs, could increase the release of inflammatory cytokines when the fatty acids were added to mouse macrophages in cell culture. PA has low solubility in water (0.2 μM), so fatty acid–free BSA was used to suspend the high concentration of fatty acids in the cell culture medium (96). The response to added PA in the cell culture medium was dependent on Toll-like receptor 4 (TLR4) activation in the macrophages. The authors tested the fatty acid–free BSA for LPS and found it to be present but negligible. Another study found that 100 μM PA added to culture media with 2% fatty acid–free BSA did not give a significant change in inflammatory cytokines, although 200 μM PA did give a significant increase (97). This level of BSA is several fold higher than albumin found in human blood, as is the concentration of PA needed to produce a significant increase in inflammatory cytokines. A later study showed that commercial fatty acid–free BSA is contaminated with lipopeptide, which activates another inflammatory TLR, TLR2 (98). Murumalla et al. (99) found that SFAs do not activate TLR2 or TLR4.
The mechanism by which TLR4 becomes activated is complex and involves dimerization of a TLR4-MD2 complex through linkage with LPS and further interaction with other membrane proteins, including myeloid differentiation factor 88 (MyD88) and CD14, to stimulate inflammatory cytokine production (100). It is unlikely that free SFAs would accomplish the same molecular interactions to activate TLR4. Membrane microdomains known as lipid rafts are involved in this process, and it has been suggested that PA augments an activated TLR4 response because it is a precursor for ceramides that constitute lipid rafts (101, 102). Ceramides are also necessary for TLR4-induced insulin resistance, which is often a consequence of low-grade inflammation arising from obesity in mice (103). It must be emphasized that the PA for ceramide synthesis is endogenous and dietary intake will have little or no influence.
Fetuin-A is a glycoprotein secreted predominantly by liver that carries free fatty acids in the blood and has been shown to activate TLR4 (104). Serum fetuin-A is high in obese diabetic humans, as well as in high-fat-diet–induced insulin-resistant mice and genetically obese (ob/ob) mice. Fetuin-A and TLR4 are both necessary to produce high-fat-diet–induced insulin resistance in mice (104). Fetuin-A was known from the mid-20th century to be an abundant protein in fetal calf serum (105). Fetal calf serum was used in the cell culture studies described above that showed SFAs augment TLR4-dependent increases in inflammatory cytokine production by macrophages. It is likely that fetuin-A rather than LPS was mediating TLR4 activation when SFAs were added to the culture medium in the studies described above. Little is currently known about the mechanisms that influence levels of fetuin-A in the body (106).
Dietary Fats and Cancer
Early studies found that high-fat diets in laboratory animals could increase the incidence of spontaneous and chemically induced cancers (107). It was later found that chemically induced mammary tumors in rats were augmented by dietary polyunsaturated vegetable oil compared with saturated fats in coconut oil (108). However, an epidemiological study of the incidence and rate of mortality from breast cancer in various countries correlated with total dietary fat, rather than with any particular type of fat, that is, saturated compared with polyunsaturated (109). A recent review has described how high-fat diets can increase cancer incidence in laboratory animals, with low-grade systemic inflammation arising from the high-fat diets suggested as a contributing factor (110). The role of eicosanoids in carcinogenesis, cell proliferation, cell migration, and angiogenesis has been reviewed (111). It is generally believed that ω-3 PUFAs from fish oils can suppress the effects of ω-6 PUFA derivatives by displacing AA in cell membranes and competing with AA for metabolism to eicosanoids, in addition to the ω-3 eicosanoids often having less potent actions compared with their ω-6 counterparts. It is estimated that 20% of cancer deaths could be due to inflammation and chronic infections. There is strong evidence to support the use of anti-inflammatory drugs, both cyclooxygenase and lipoxygenase inhibitors, as anticancer drugs (112).
Lipid peroxides, ROS, and oxidative stress are associated with inflammation and are frequently invoked in tumor promotion by causing a switch in cellular metabolism (113). One of the major switches in metabolism in cancer cells is toward glycolysis as the major pathway for ATP production. This makes tumor cells more reliant on carbohydrate as an energy source. Conditions that increase carbohydrate availability, such as insulin resistance and diabetes, can promote tumor growth (114). Animal studies have shown that caloric restriction, ketogenic diets, and fasting can diminish cancer progression and metastasis (115). Although there is much interest in the use of ketogenic diets for cancer therapy in humans, the clinical evidence regarding ketogenic diets and tumor formation and progression is lacking (116). One problem might be the level of ω-6 PUFAs consumed in a ketogenic diet, which could have an adverse effect in terms of cancer therapy. The complexity of human dietary evaluation and manipulation in a free-living population makes it difficult to draw conclusions regarding dietary constituents and diseases.
Studies attempting to elucidate the role of dietary fats, whether total fats, saturated fats, or polyunsaturated oils, have been inconsistent with regard to cancer. It appears that a high caloric intake can promote cancer and cell proliferation, but evidence that cancer cells shift metabolism toward glycolysis for energy production, indicates that dietary carbohydrates would have a greater influence than dietary saturated fats on tumor promotion. Sugar, or more specifically fructose, is known to cause increased serum triglycerides, insulin resistance, and hyperglycemia (117), all of which would promote cancer.
Conclusions
Whether dietary saturated fat raises serum cholesterol or whether dietary PUFAs lower serum cholesterol might appear to be a moot point to some individuals. It is important to recognize that the focus on decreasing dietary fats resulted in a large increase in carbohydrate consumption, which has had undesirable health consequences as illustrated by the dramatic rise in obesity, type 2 diabetes, metabolic syndrome, and associated diseases. In addition, replacing saturated fats with unsaturated oils could have exacerbated diseases associated with lipid peroxidation and oxidative stress. It should not be surprising that numerous studies have tried to decipher the effects of specific dietary components on a broad range of risk factors and health effects only to obtain inconsistent results. When studying a free-living population it is impossible to separate the effects of any given dietary component from the myriad of other constituents of the diet, their respective impacts on some physiological phenomenon, and the gene–dietary constituent interactions. Because the genetic makeup of each individual is different, each can respond to any given dietary alteration in a somewhat different way (118). It can also be misleading to study manipulations of a particular line of cells in culture to understand how those cells will respond to a similar manipulation in an intact organism.
The various ways in which PUFAs, especially the ω-6 PUFAs found in vegetable oils, can augment inflammation and exacerbate a wide range of diseases associated with inflammation have been discussed here. The data are quite consistent in most animal studies, although often less convincing when dealing with humans. A major drawback in human studies is that humans generally consume relatively large amounts of ω-6 PUFAs, so any intervention that attempts to alter the amounts of PUFAs in the diet can make very little difference in the amounts of ω-6 PUFAs stored in adipose tissue or in membrane lipids. There has been some success in this respect when ω-3 PUFAs are substituted for ω-6 PUFAs in the diet. This requires more than merely supplementing the diet with ω-3 PUFAs, because dietary levels of ω-6 PUFAs are generally quite high and modest amounts of ω-3 PUFAs added to that will not be sufficient to displace the ω-6 PUFAs that are already in the body and continue to be added in the diet. Generally, it would require decreasing ω-6 vegetable oils to very low levels when supplementing with ω-3 PUFAs to see a significant effect. In addition, it is likely to take more than a month or two on a low ω-6 PUFA diet to deplete the substantial stores of LA that can be in adipose tissue as a result of a lifetime of consuming a Western diet.
Lipid peroxidation can cause oxidative stress and vice versa, and the role of these phenomena in several diseases is well documented (28, 30, 119). It is inappropriate to assign adverse effects to “dietary” saturated fats, because SFAs are chemically stable, synthesized from other nutrients in the body (notably carbohydrates and PUFAs), and are generally maintained within certain limits in most tissues according to physiological control mechanisms. On the other hand, PUFAs are unstable to chemical oxidation and their oxidation products are harmful in a variety of ways. PUFAs also form powerful signaling agents that can initiate inflammation, which can have dire health consequences, as described above. Many of the oxidized metabolites of PUFAs, especially ω-3 PUFAs, can also resolve inflammation. If saturated fats are replaced by carbohydrates in the diet, there would be no significant improvement in serum cholesterol, and it can result in a more atherogenic lipoprotein profile. When looking at much of the data in the context of known biochemical and physiological mechanisms, it appears that saturated fats are less harmful than the common alternatives.
Acknowledgments
The sole author was responsible for all aspects of this manuscript.
Notes
The author reported no funding received for this study.
Author disclosures: The author reports no conflicts of interest.
Perspective articles allow authors to take a position on a topic of current major importance or controversy in the field of nutrition. As such, these articles could include statements based on author opinions or point of view. Opinions expressed in Perspective articles are those of the author and are not attributable to the funder(s) or the sponsor(s) or the publisher, Editor, or Editorial Board of Advances in Nutrition. Individuals with different positions on the topic of a Perspective are invited to submit their comments in the form of a Perspectives article or in a Letter to the Editor.
Abbreviations used: AA, arachidonic acid; EFA, essential fatty acid; LA, linoleic acid; LT, leukotriene; NSAID, nonsteroidal anti-inflammatory drug; PA, palmitic acid; PPAR, peroxisome proliferator-activated receptor; RNS, reactive nitrogen species; ROS, reactive oxygen species; SREBP, sterol regulatory element-binding protein; TLR, Toll-like receptor.
References
- 1. Bronte-Stewart B, Antonis A, Eales L, Brock JF. Effects of feeding different fats on serum-cholesterol level. Lancet. 1956;270:521–6. [DOI] [PubMed] [Google Scholar]
- 2. Malmros H, Wigand G. The effect on serum-cholesterol of diets containing different fats. Lancet. 1957;273:1–7. [DOI] [PubMed] [Google Scholar]
- 3. Keys A, Anderson JT, Grande F. Prediction of serum-cholesterol responses of man to changes in fats in the diet. Lancet. 1957;273:959–66. [DOI] [PubMed] [Google Scholar]
- 4. Hegsted DM, McGandy RB, Myers ML, Stare FJ. Quantitative effects of dietary fat on serum cholesterol in man. Am J Clin Nutr. 1965;17:281–95. [DOI] [PubMed] [Google Scholar]
- 5. Mensink RP, Zock PL, Kester AD, Katan MB. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr. 2003;77:1146–55. [DOI] [PubMed] [Google Scholar]
- 6. Austin GL, Ogden LG, Hill JO. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am J Clin Nutr. 2011;93:836–43. [DOI] [PubMed] [Google Scholar]
- 7. Lawrence GD. The low-fat lie: rise of obesity, diabetes and inflammation. Irvine (CA): Universal Publishers;2019. [Google Scholar]
- 8. Siri PW, Krauss RM. Influence of dietary carbohydrate and fat on LDL and HDL particle distributions. Curr Atheroscler Rep. 2005;7:455–9. [DOI] [PubMed] [Google Scholar]
- 9. Siri-Tarino PW, Sun Q, Hu FB, Krauss RM. Saturated fat, carbohydrate, and cardiovascular disease. Am J Clin Nutr. 2010;91:502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuipers RS, de Graaf DJ, Luxwolda MF, Muskiet MH, Dijck-Brouwer DA, Muskiet FA. Saturated fat, carbohydrates and cardiovascular disease. Neth J Med. 2011;69:372–8. [PubMed] [Google Scholar]
- 11. Volk BM, Kunces LJ, Freidenreich DJ, Kupchak BR, Saenz C, Artistizabal JC, Fernandez ML, Bruno RS, Maresh CM, Kraemer WJet al. . Effects of step-wise increases in dietary carbohydrate on circulating saturated fatty acids and palmitoleic acid in adults with metabolic syndrome. PLoS One. 2014;9:e113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kwon D, Yi J-J, Ohrr H, Yi S-W. Total cholesterol and mortality from ischemic heart disease and overall cardiovascular disease in Korean adults. Medicine (Baltimore). 2019;98:e17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yi S-W, Yi J-J, Ohrr H. Total cholesterol and all-cause mortality by sex and age: a prospective cohort study among 12.8 million adults. Sci Rep. 2019;9:1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kirihara Y, Hamazaki K, Hamazaki T, Ogushi Y, Tsuji H, Shirasaki S. The relationship between total blood cholesterol levels and all-cause mortality in Fukui City, and meta-analysis of this relationship in Japan. J Lipid Nutr. 2008;17:67–78. [Google Scholar]
- 15. Schatz IJ, Kamal M, Yano K, Chen R, Rodriguez BL, Curb JD. Cholesterol and all-cause mortality in elderly people from the Honolulu Heart Program: a cohort study. Lancet. 2001;358:351–5. [DOI] [PubMed] [Google Scholar]
- 16. Jacobs D, Blackburn H, Higgins M, Reed D, Iso H, McMillan G, Neaton J, Nelson J, Potter J, Rifkind Bet al. . Report of the Conference on low blood cholesterol: mortality associations. Circulation. 1992;86:1046–60. [DOI] [PubMed] [Google Scholar]
- 17. Petersen LK, Christensen K, Kragstrup J. Lipid-lowering treatment to the end? A review of observational studies and RCTs on cholesterol and mortality in 80+-year olds. Age Ageing. 2010;39:674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keys A. Seven countries: a multivariate analysis of death and coronary heart disease. Cambridge (MA): Harvard University Press;1980. [Google Scholar]
- 19. de Souza RJ, Mente A, Maroleanu A, Cozma AI, Ha V, Kishibe T, Uleryk E, Budylowski P, Schunemann H, Beyene Jet al. . Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ. 2015;351:h3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu Y, Bo Y, Liu Y. Dietary total fat, fatty acids intake, and risk of cardiovascular disease: a dose-response meta-analysis of cohort studies. Lipids Health Dis. 2019;18:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Astrup A, Dyerberg J, Elwood P, Hermansen K, Hu FB, Jakobsen MU, Kok FJ, Krauss RM, Lecerf JM, LeGrand Pet al. . The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: where does the evidence stand in 2010?. Am J Clin Nutr. 2011;93:684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Astrup A, Magkos F, Bier DM, Brenna JT, de Oliveira Otto MC, Hill JO, King JC, Mente A, Ordovas JM, Volek JSet al. . Saturated fats and health: a reassessment and proposal for food-based recommendations: JACC state-of-the-art review. J Am Coll Cardiol. 2020;76:844–57. [DOI] [PubMed] [Google Scholar]
- 23. Kris-Etherton PM, Krauss RM. Public health guidelines should recommend reducing saturated fat consumption as much as possible: YES. Am J Clin Nutr. 2020;112:13–8. [DOI] [PubMed] [Google Scholar]
- 24. Krauss RM, Kris-Etherton PM. Public health guidelines should recommend reducing saturated fat consumption as much as possible: NO. Am J Clin Nutr. 2020;112:19–24. [DOI] [PubMed] [Google Scholar]
- 25. Heileson JL. Dietary saturated fat and heart disease: a narrative review. Nutr Rev. 2020;78:474–85. [DOI] [PubMed] [Google Scholar]
- 26. Lawrence G. The fats of life: essential fatty acids in health and disease. Piscataway (NJ): Rutgers University Press;2010. [Google Scholar]
- 27. Di Meo S, Venditti P. Evolution of the knowledge of free radicals and other oxidants. Oxid Med Cell Longev. 2020;2020:9829176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. [DOI] [PubMed] [Google Scholar]
- 29. Davies JMS, Cillard J, Friguet B, Cadenas E, Cadet J, Cayce R, Fishmann A, Liao D, Bulteau AL, Derbre Fet al. . The oxygen paradox, the French paradox, and age-related diseases. Geroscience. 2017;39:499–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spiteller G. Lipid peroxidation in aging and age-dependent diseases. Exp Gerontol. 2001;36:1425–57. [DOI] [PubMed] [Google Scholar]
- 31. Vendemiale G, Grattagliano I, Altomare E. An update on the role of free radicals and antioxidant defense in human disease. Int J Clin Lab Res. 1999;29:49–55. [DOI] [PubMed] [Google Scholar]
- 32. Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, BJ Ansell, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour Net al. . The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. [DOI] [PubMed] [Google Scholar]
- 33. Zhong S, Li L, Shen X, Li Q, Xu W, Wang X, Tao Y, Yin H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic Biol Med. 2019;144:266–78. [DOI] [PubMed] [Google Scholar]
- 34. Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88:1785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Staprans I, Pan XM, Rapp JH, Feingold KR. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol Nutr Food Res. 2005;49:1075–82. [DOI] [PubMed] [Google Scholar]
- 36. Bitorina AV, Oligschlaeger Y, Shiri-Sverdlov R, Theys J. Low profile high value target: the role of OxLDL in cancer. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:158518. [DOI] [PubMed] [Google Scholar]
- 37. Sheaff Greiner RC, Zhang Q, Goodman KJ, Giussani DA, Nathanielsz PW, Brenna JT. Linoleate, alpha-linolenate, and docosahexaenoate recycling into saturated and monounsaturated fatty acids is a major pathway in pregnant or lactating adults and fetal or infant rhesus monkeys. J Lipid Res. 1996;37:2675–86. [PubMed] [Google Scholar]
- 38. Cunnane SC, Menard CR, Likhodii SS, Brenna JT, Crawford MA. Carbon recycling into de novo lipogenesis is a major pathway in neonatal metabolism of linoleate and alpha-linolenate. Prostaglandins Leukot Essent Fatty Acids. 1999;60:387–92. [DOI] [PubMed] [Google Scholar]
- 39. Cunnane SC, Ryan MA, Nadeau CR, Bazinet RP, Musa-Veloso K, McCloy U. Why is carbon from some polyunsaturates extensively recycled into lipid synthesis?. Lipids. 2003;38:477–84. [DOI] [PubMed] [Google Scholar]
- 40. Brown ED, Morris VC, Rhodes DG, Sinha R, Levander OA. Urinary malondialdehyde-equivalents during ingestion of meat cooked at high or low temperatures. Lipids. 1995;30:1053–6. [DOI] [PubMed] [Google Scholar]
- 41. Conchillo A, Ansorena D, Astiasaran I. Combined effect of cooking (grilling and roasting) and chilling storage (with and without air) on lipid and cholesterol oxidation in chicken breast. J Food Prot. 2003;66:840–6. [DOI] [PubMed] [Google Scholar]
- 42. Lawrence GD. Dietary fats and health: dietary recommendations in the context of scientific evidence. Adv Nutr. 2013;4:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Namboodiri KK, Kaplan EB, Heuch I, Elston RC, Green PP, Rao DC, Laskarzewski P, Glueck CJ, Rifkind BM. The Collaborative Lipid Research Clinics Family Study: biological and cultural determinants of familial resemblance for plasma lipids and lipoproteins. Genet Epidemiol. 1985;2:227–54. [DOI] [PubMed] [Google Scholar]
- 44. Hamsten A, Iselius L, Dahlen G, de Faire U. Genetic and cultural inheritance of serum lipids, low and high density lipoprotein cholesterol and serum apolipoproteins A-I, A-II and B. Atherosclerosis. 1986;60:199–208. [DOI] [PubMed] [Google Scholar]
- 45. Ordovas JM, Corella D. Genetic variation and lipid metabolism: modulation by dietary factors. Curr Cardiol Rep. 2005;7:480–6. [DOI] [PubMed] [Google Scholar]
- 46. Beynen AC, Katan MB, Van Zutphen LF. Hypo- and hyperresponders: individual differences in the response of serum cholesterol concentration to changes in diet. Adv Lipid Res. 1987;22:115–71. [DOI] [PubMed] [Google Scholar]
- 47. Ordovas JM. Pharmacogenetics of lipid diseases. Hum Genomics. 2004;1:111–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lopez-Miranda J, Williams C, Lairon D. Dietary, physiological, genetic and pathological influences on postprandial lipid metabolism. Br J Nutr. 2007;98:458–73. [DOI] [PubMed] [Google Scholar]
- 49. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DeBose-Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. 2018;43:358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jump DB. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr Opin Lipidol. 2008;19:242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int J Mol Sci. 2019;20:5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Georgiadi A, Kersten S. Mechanisms of gene regulation by fatty acids. Adv Nutr. 2012;3:127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strand E, Lysne V, Grinna ML, Bohov P, Svardal A, Nygard O, Berge RK, Bjorndal B. Short-term activation of peroxisome proliferator-activated receptors alpha and gamma induces tissue-specific effects on lipid metabolism and fatty acid composition in male Wistar rats. PPAR Res. 2019;2019:8047627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choudhury N, Tan L, Truswell AS. Comparison of palmolein and olive oil: effects on plasma lipids and vitamin E in young adults. Am J Clin Nutr. 1995;61:1043–51. [DOI] [PubMed] [Google Scholar]
- 56. Ng TK, Hayes KC, DeWitt GF, Jegathesan M, Satgunasingam N, Ong AS, Tan D. Dietary palmitic and oleic acids exert similar effects on serum cholesterol and lipoprotein profiles in normocholesterolemic men and women. J Am Coll Nutr. 1992;11:383–90. [DOI] [PubMed] [Google Scholar]
- 57. Gibson RA, Neumann MA, Lien EL, Boyd KA, Tu WC. Docosahexaenoic acid synthesis from alpha-linolenic acid is inhibited by diets high in polyunsaturated fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2013;88:139–46. [DOI] [PubMed] [Google Scholar]
- 58. Simopoulos AP. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother. 2002;56:365–79. [DOI] [PubMed] [Google Scholar]
- 59. Innis SM. Palmitic acid in early human development. Crit Rev Food Sci Nutr. 2016;56:1952–9. [DOI] [PubMed] [Google Scholar]
- 60. Boersma ER. Changes in fatty-acid composition of body fat before and after birth in Tanzania: an international comparative study. Br Med J. 1979;1:850–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Farquharson J, Cockburn F, Patrick WA, Jamieson EC, Logan RW. Effect of diet on infant subcutaneous tissue triglyceride fatty acids. Arch Dis Child. 1993;69:589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Innis SM. Dietary triacylglycerol structure and its role in infant nutrition. Adv Nutr. 2011;2:275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Widdowson EM, Dauncey MJ, Gairdner DMT, Jonxis JHP, Pelikan-Fiipkova M. Body fat of British and Dutch infants. Br Med J. 1975;1:653–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lapinleimu H, Viikari J, Jokinen E, Salo P, Routi T, Leino A, Rönnemaa T, Seppänen R, Välimäki I, Simell O. Prospective randomised trial in 1062 infants of diet low in saturated fat and cholesterol. Lancet. 1995;345:471–6. [DOI] [PubMed] [Google Scholar]
- 65. Martinez M, Mougan I. Fatty acid composition of human brain phospholipids during normal development. J Neurochem. 1998;71:2528–33. [DOI] [PubMed] [Google Scholar]
- 66. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carta G, Murru E, Banni S, Manca C. Palmitic acid: physiological role, metabolism and nutritional implications. Front Physiol. 2017;8:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. O'Sullivan TA, Schmidt KA, Kratz M. Whole-fat or reduced-fat dairy product intake, adiposity, and cardiometabolic health in children: a systematic review. Adv Nutr. 2020;11:928–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Beck AL, Heyman M, Chao C, Wojcicki J. Full fat milk consumption protects against severe childhood obesity in Latinos. Prev Med Rep. 2017;8:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bigornia SJ, LaValley MP, Moore LL, Northstone K, Emmett P, Ness AR, Newby PK. Dairy intakes at age 10 years do not adversely affect risk of excess adiposity at 13 years. J Nutr. 2014;144:1081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nielsen SJ, Popkin BM. Changes in beverage intake between 1977 and 2001. Am J Prev Med. 2004;27:205–10. [DOI] [PubMed] [Google Scholar]
- 72. Trim W, Turner JE, Thompson D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol. 2018;9:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Calder PC. n–3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr. 2006;83(6 Suppl):1505S–19S. [DOI] [PubMed] [Google Scholar]
- 75. Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2015;1851:397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raphael W, Sordillo LM. Dietary polyunsaturated fatty acids and inflammation: the role of phospholipid biosynthesis. Int J Mol Sci. 2013;14:21167–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Saini RK, Keum YS. Omega-3 and omega-6 polyunsaturated fatty acids: dietary sources, metabolism, and significance—a review. Life Sci. 2018;203:255–67. [DOI] [PubMed] [Google Scholar]
- 78. Mohrhauer H, Holman RT. The effect of dose level of essential fatty acids upon fatty acid composition of the rat liver. J Lipid Res. 1963;4:151–9. [PubMed] [Google Scholar]
- 79. James MJ, Gibson RA, D'Angelo M, Neumann MA, Cleland LG. Simple relationships exist between dietary linoleate and the n–6 fatty acids of human neutrophils and plasma. Am J Clin Nutr. 1993:497–500. [DOI] [PubMed] [Google Scholar]
- 80. Rett BS, Whelan J. Increasing dietary linoleic acid does not increase tissue arachidonic acid content in adults consuming Western-type diets: a systematic review. Nutr Metab. 2011;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. 2018;132:41–8. [DOI] [PubMed] [Google Scholar]
- 82. Jandacek RJ. Linoleic acid: a nutritional quandary. Healthcare (Basel). 2017;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hurst S, Zainal Z, Caterson B, Hughes CE, Harwood JL. Dietary fatty acids and arthritis. Prostaglandins Leukot Essent Fatty Acids. 2010;82:315–8. [DOI] [PubMed] [Google Scholar]
- 84. Goldberg RJ, Katz J. A meta-analysis of the analgesic effects of omega-3 polyunsaturated fatty acid supplementation for inflammatory joint pain. Pain. 2007;129:210–23. [DOI] [PubMed] [Google Scholar]
- 85. Lee Y-H, Bae S-C, Song G-G. Omega-3 polyunsaturated fatty acids and the treatment of rheumatoid arthritis: a meta-analysis. Arch Med Res. 2012;43:356–62. [DOI] [PubMed] [Google Scholar]
- 86. Petersson S, Philippou E, Rodomar C, Nikiphorou E. The Mediterranean diet, fish oil supplements and rheumatoid arthritis outcomes: evidence from clinical trials. Autoimmun Rev. 2018;17:1105–14. [DOI] [PubMed] [Google Scholar]
- 87. Gioxari A, Kaliora AC, Marantidou F, Panagiotakos DP. Intake of omega-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: a systematic review and meta-analysis. Nutrition. 2018;45:114–24. e4. [DOI] [PubMed] [Google Scholar]
- 88. Alunno A, Nikiphorou E, Philippou E, Daien C, Wiek D, Kouloumas M, Cutolo M. Nutrition in RMDs: is it really food for thought? Focus on rheumatoid arthritis. BMC Rheumatol. 2020;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Moncada S, Salmon JA. Leukocytes and tissue injury: the use of eicosapentaenoic acid in the control of white cell activation. Prog Lipid Res. 1986;25:563–6. [PubMed] [Google Scholar]
- 90. Lawrence GD. Effect of dietary lipids on adjuvant-induced arthritis in rats. Nutr Res. 1990;10:283–90. [Google Scholar]
- 91. Denko CW. Modification of adjuvant inflammation in rats deficient in essential fatty acids. Agents Actions. 1976;6:636–41. [DOI] [PubMed] [Google Scholar]
- 92. Chinn KS, Welsch DJ, Salsgiver WJ, Mehta A, Raz A, Obukowicz MG. Modulation of adjuvant-induced arthritis by dietary arachidonic acid in essential fatty acid-deficient rats. Lipids. 1997;32:979–88. [DOI] [PubMed] [Google Scholar]
- 93. Prickett JD, Trentham DE, Robinson DR. Dietary fish oil augments the induction of arthritis in rats immunized with type II collagen. J Immunol. 1984;132:725–9. [PubMed] [Google Scholar]
- 94. Olson MV, Liu YC, Dangi B, Paul Zimmer J, Salem Jr N, Nauroth JM. Docosahexaenoic acid reduces inflammation and joint destruction in mice with collagen-induced arthritis. Inflamm Res. 2013;62:1003–13. [DOI] [PubMed] [Google Scholar]
- 95. Morin C, Blier PU, Fortin S. Eicosapentaenoic acid and docosapentaenoic acid monoglycerides are more potent than docosahexaenoic acid monoglyceride to resolve inflammation in a rheumatoid arthritis model. Arthritis Res Ther. 2015;17:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27:84–91. [DOI] [PubMed] [Google Scholar]
- 98. Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. 2009;29:1944–9. [DOI] [PubMed] [Google Scholar]
- 99. Murumalla RK, Gunasekaran MK, Padhan JK, Bencharif K, Gence L, Festy F, Cesari M, Roche R, Hoareau L. Fatty acids do not pay the toll: effect of SFA and PUFA on human adipose tissue and mature adipocytes inflammation. Lipids Health Dis. 2012;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maeshima N, Fernandez RC. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol. 2013;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wong SW, Kwon M-J, Choi AMK, Kim H-P, Nakahira K, Hwang DH. Fatty acids modulate toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chang ZQ, Lee SY, Kim HJ, Kim JR, Kim SJ, Hong IK, Oh BC, Choi CS, Goldberg IJ, Park TS. Endotoxin activates de novo sphingolipid biosynthesis via nuclear factor kappa B-mediated upregulation of Sptlc2. Prostaglandins Other Lipid Mediat. 2011;94:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Holland WL, Bikman BT, Wang L-P, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MRet al. . Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–85. [DOI] [PubMed] [Google Scholar]
- 105. Lieberman I, Lamy F, Ove P. Nonidentity of fetuin and protein growth (flattening) factor. Science. 1959;129:43–4. [DOI] [PubMed] [Google Scholar]
- 106. Robinson KN, Teran-Garcia M. From infancy to aging: biological and behavioral modifiers of Fetuin-A. Biochimie. 2016;124:141–9. [DOI] [PubMed] [Google Scholar]
- 107. Tannenbaum A. The genesis and growth of tumors. III. Effects of high fat diets. Cancer Res. 1942;2:468–75. [Google Scholar]
- 108. Carroll KK. Dietary fats and cancer. Am J Clin Nutr. 1991;53:1064S–7S. [DOI] [PubMed] [Google Scholar]
- 109. Carroll KK, Braden LM, Bell JA, Kalamegham R. Fat and cancer. Cancer. 1986;58:1818–25. [DOI] [PubMed] [Google Scholar]
- 110. Duan Y, Zeng L, Zheng C, Song B, Li F, Kong X, Xu K. Inflammatory links between high fat diets and diseases. Front Immunol. 2018;9:2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gomes RN, Felipe da Costa S, Colquhoun A. Eicosanoids and cancer. Clinics (Sao Paulo). 2018;73:e530s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Andrisic L, Dudzik D, Barbas C, Milkovic L, Grune T, Zarkovic N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018;14:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bose S, Allen AE, Locasale JW. The molecular link from diet to cancer cell metabolism. Mol Cell. 2020;78:1034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lv M, Zhu X, Wang H, Wang F, Guan W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: a systematic review and meta-analysis. PLoS One. 2014;9:e115147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Erickson N, Boscheri A, Linke B, Huebner J. Systematic review: isocaloric ketogenic dietary regimes for cancer patients. Med Oncol. 2017;34:72. [DOI] [PubMed] [Google Scholar]
- 117. Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang Wet al. . Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gibney ER. Personalised nutrition – phenotypic and genetic variation in response to dietary intervention. Proc Nutr Soc. 2020;79:236–45. [DOI] [PubMed] [Google Scholar]
- 119. Gueraud F, Tache S, Steghens J-P, Milkovic L, Borovic-Sunjic S, Zarkovic N, Gultier E, Naud N, Helies-Toussaint C, Pierre Fet al. . Dietary polyunsaturated fatty acids and heme iron induce oxidative stress biomarkers and a cancer promoting environment in the colon of rats. Free Radic Biol Med. 2015;83:192–200. [DOI] [PubMed] [Google Scholar]