

Abstract
Pulmonary capillary hemangiomatosis (PCH) is a rare cause of pulmonary hypertension (PH) of unknown etiology resulting from pulmonary capillary proliferation. Clinically, PCH is seen in young adults with equal sex predilection and rarely reported familial predisposition. PCH’s main clinical presentations are progressive dyspnea, fatigue, hemoptysis, palpitations, and later irreversible pulmonary hypertension and right-sided heart failure. Hereby, we report three PCH cases, each case presented with a peculiar presentation with a comprehensive literature review highlighting etiology, clinical presentations, diagnostic modalities and pathology in establishing a diagnosis, current treatment options, and prognosis of PCH. In conclusion, defining PCH as the underlying cause of PH is of utmost importance as most medications used for PH are ineffective in PCH. Vasodilators should be avoided due to the increased risk of pulmonary oedema. Pathological examination of the lung is still considered the most definitive diagnostic tool, yet it is associated with complications risk. High-Resolution Computed Tomography (HRCT) chest is currently considered the cornerstone non-invasive modality for the diagnosis of PH. So far, no definitive treatment of PCH excluding lung transplantation with preliminary promising results with angiogenesis Inhibitors. PCH carries a very poor prognosis with a median survival of 3 years from the time of diagnosis.
Keywords: Pulmonary capillary hemangiomatosis (PCH), pulmonary veno-occlusive disease (PVOD), pulmonary artery hypertension (PAH), right-sided heart failure, high-resolution computed tomography (HRCT)
Background
Pulmonary capillary hemangiomatosis (PCH) is a rare vascular pulmonary disease characterized by abnormal pulmonary capillary angiogenesis that infiltrates peri-bronchial, peri-vascular interstitium and lung parenchyma. Typically, PCH patients manifest with progressive PH with subsequent development of right-sided heart failure [1]. We report three cases of PH caused by PCH highlighting the clinical presentation, diagnosis, management and fate of each case. Informed consent was acquired either from the patients or the next in kin and it was also approved by the local ethics committee.
Case (1)
A 37-year-old male presented with a 5-year history of dyspnea grade II with no associated orthopnea or paroxysmal nocturnal dyspnea (PND) and a 3-year history of bilateral progressive lower limb oedema and marked abdominal enlargement. On Clinical examination, he had an icteric tinge with bilateral congested neck veins with severe ascites, tender hepatomegaly and soft bilateral lower limb oedema till upper thigh. X-ray of his chest showed cardiomegaly. Ultrasound abdomen revealed marked liver cirrhosis with a dilated inferior vena cava (IVC). Transthoracic Echocardiography (TTE) (Supplementary Video 1) demonstrated dilated both right ventricle (RV) and right atrium (RA) and reduced RV systolic function with crescent-shaped RA thrombus and severe tricuspid regurgitation (TR), dilated main pulmonary artery (MPA) and branches and a moderate amount of pericardial effusion. HRCT chest with pulmonary angiography (PA) showed centrilobular diffuse small ground glass pulmonary nodules affecting both lungs with no lobar predominance such radiological features are classical for PCH. So, long-lasting PH and right-sided heart failure was explained by PCH. He was discharged on warfarin and diuretics with scheduled follow-up, but he died a year after diagnosis (Figures 1 and 2).
Figure 1.
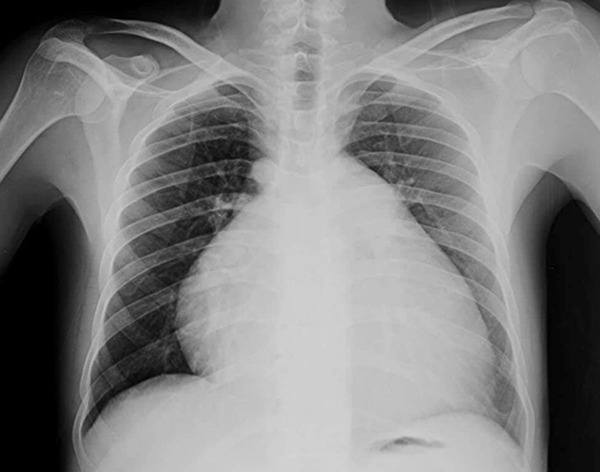
Chest X ray: Marked cardiomegaly with flask shaped heart.
Figure 2.
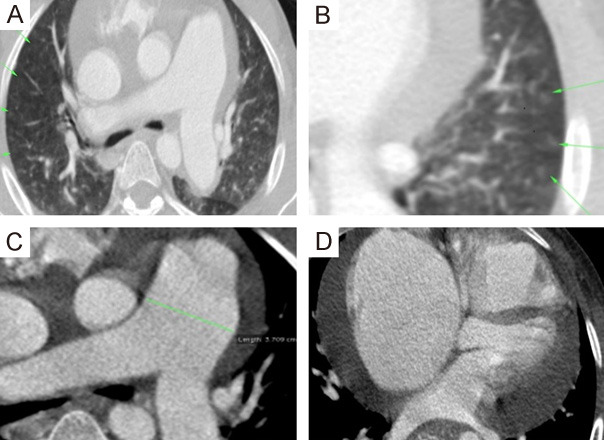
MDCT chest with PA. A, B. Axial and zoomed axial lung window CT showing numerous bilateral uniform sized sub-centimeter centrilobular ground glass nodules (arrows). C. Axial CT mediastinal window image shows dilated central pulmonary arteries. D. Axial CT mediastinal window image shows associated moderate amount pericardial effusion … Features suggestive of PCH.
Case (2)
A 19-year-old male with negative previous medical or surgical history was referred to our institute for evaluation of progressive exertional dyspnea with no orthopnea or PND and effort intolerance for the past 6 months. On clinical examination, a pansystolic murmur was best heard over the tricuspid area. TTE revealed marked RV hypertrophy (RVH) and evident dilatation with severe TR (PG = 170 mmHg) with pulmonary hypertension (ESPAP = 190 mmHg) (Supplementary Video 2). HRCT chest with PA was done proving that PCH was the cause of severe PH. After patient counseling regarding lung biopsy to confirm the diagnosis, he refused to do any invasive procedures. He was discharged on diuretics, anticoagulation, and strict advice to avoid vasodilator therapies and strenuous activities with scheduled close follow-up visits but after 6 months he developed a sudden attack of chest pain, cyanosis and palpitations and died before even reaching the hospital (Figure 3).
Figure 3.
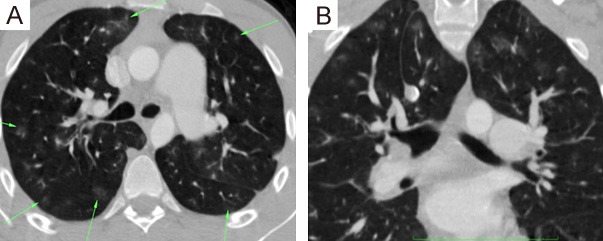
MDCT chest with PA: (A, B) Axial and coronal lung window CT showing numerous bilateral uniform sized sub-centimeter centrilobular ground glass nodules (arrows).
Case (3)
A 36-year-old female patient was referred to our medical facility complaining of exertional dyspnea, hemoptysis and symptoms of right-sided heart failure. Her primary physician reported that she was treated from long-standing PH and right-sided heart failure for years and that she was admitted with unexplained recurrent pulmonary oedema after the initiation of vasodilator therapies. HRCT chest with PA confirmed that PCH was the cause of her severe PH and recurrent pulmonary oedema after vasodilator therapies. Unfortunately, she died 6 months after diagnosis (Figure 4).
Figure 4.
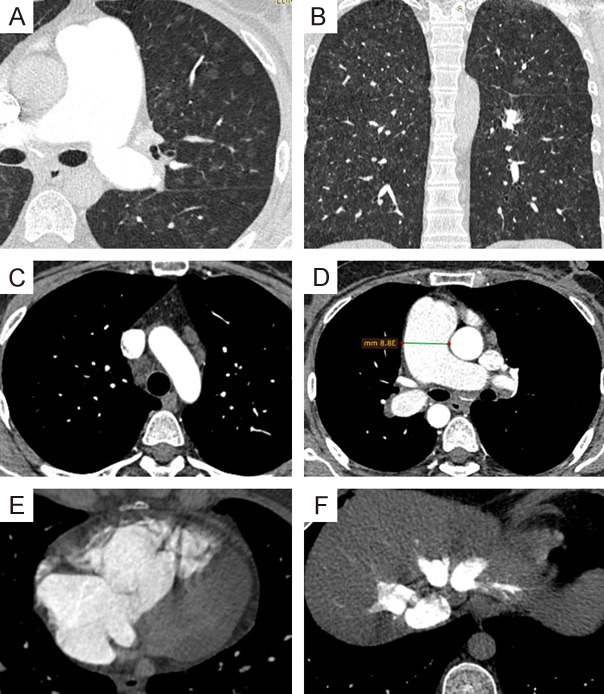
MDCT chest with PA: (A, B) Zoomed axial and coronal lung window CT showing numerous bilateral uniform sized sub-centimeter centrilobular ground glass nodules. (C) Axial CT mediastinal window image shows enlarged mediastinal lymph nodes. (D) Axial CTA images showing dilated central pulmonary arteries. (E, F) Axial CTA images showing dilated right cardiac chambers as well as congested hepatic veins with reflux … Features suggest PCH.
Discussion
Hereby, we describe a case series of PCH with a comprehensive literature review presenting with a wide diversity of symptoms ranging from long-standing right-sided HF, exertional dyspnea, hemoptysis and pulmonary oedema after vasodilator initiation. The diagnosis was based on clinical presentations and classical radiological features using HRCT chest with PA. Although our patients were followed-up closely, they died within a year of diagnosis.
Overview
Previously, pulmonary hypertension (PH) was divided into 2 groups: (1) primary pulmonary hypertension or (2) secondary pulmonary hypertension according to the presence or absence of identifiable etiologies or risk factors [2]. During successive PH world meetings, [3,4] clinical classification was established in order to identify different classes of PH which have the same pathological findings, hemodynamics or management. Five PH groups were determined:
● Group 1: PAH.
● Group 2: PH due to left-sided heart diseases.
● Group 3: PH due to chronic lung diseases.
● Group 4: chronic thromboembolic pulmonary hypertension (CTEPH).
● Group 5: PH due to idiopathic multifactorial mechanisms.
According to the updated classification of PH, PCH and PVOD are classified as a subcategory of group 1 PAH [4]. PCH is a rare etiology of PH characterized by a widespread proliferation of pulmonary capillaries within the alveolar walls [5], unlike PVOD which is caused by extensive vascular obstructive process originating from pulmonary venules and small veins. Wagenvoort et al, was the first to describe PCH in a 71-year-old woman complaining of progressive dyspnea and hemoptysis. He discovered a characteristic proliferation of capillary-like channels in lung tissue that appeared to be distinct from the classical findings described in PAH or PVOD, leading to the identification of a new entity, PCH [6].
Risk factors and genetics
So far, no identifiable risk factors for PCH are recognized. Several case reports have described the characteristic PCH pathological lesion in patients with Takayasu arteritis, Kartagener syndrome, scleroderma, hypertrophic cardiomyopathy, and systemic lupus erythematosus [7-9]. Unlike cases of PVOD, current reports don’t link PCH to the exposure to chemotherapy, organic solvents, or stem cell transplant [10]. Langleben et al, [11] described a case series of familial PCH suggesting autosomal recessive inheritance. Best et al, [12] identified Eukaryotic translation initiation factor 2α kinase 4 (EIF2AK4) mutations in familial PCH in 2 brothers and in 2 of 10 cases with sporadic PCH. Also, Eyries et al, [13] discovered 11 of causative EIF2AK4 mutations in patients with familial and sporadic PVOD. So, the current guidelines agreed to advise patients with PVOD and PCH about genetic testing and counseling [14,15].
Pathology
The hallmark of PCH is the presence of aberrant proliferation of capillary vessels at least 2 layers thick [6]. This multiple layered capillaries can differentiate PCH from congested capillaries or atelectasis [11]. Grossly it appears as multiple, red-brown, congested, odematous ill-defined centrilobular nodular lesions without significant fibrosis. Histopathological description shows a patchy parenchymal involvement often forming nodules with capillary proliferation lined by bland-looking endothelium with intra-alveolar hemosiderin-laden macrophages, small areas of acute or old hemorrhage, hemosiderosis. Small pulmonary arteries with intimal thickening/medial hypertrophy [16]. High-power microscopy can demonstrate infiltration of the capillary proliferation into bronchi, intralobular fibrous septa, and walls of small pulmonary arteries and veins. Rare capillary proliferation into pleura, mediastinal lymph nodes and pericardium were described [11,17,18]. PCH can be distinguished from other conditions with prominent capillaries, including PVOD with capillary invasion of pulmonary veins, arteries, and other tissues [11,19,20].
Clinical features
The incidence of PCH remains uncertain. El-Gabaly et al, [21] reported a frequency of almost 4 cases per million individuals. The age of onset is poorly defined, the reported age in the literature is wide-ranging from 2 to 71 years with a mean age of 30 years. It usually affects adults aged 20 to 40 years [21] but it was also described in elderly or even in premature infants and newborns [8,22,23]. PCH has an equal frequency in both sexes in patients of any age [24-26]. It commonly occurs in a sporadic form, although familial association has been reported [22,27]. Progressive dyspnea and fatigue are the principal clinical manifestations of both PVOD and PCH [24,26]. Chronic cough (dry or productive), chest pain, syncope, digital clubbing, fever, respiratory tract infection, thrombocytopenia and hemorrhagic complications may occur [26]. However, hemoptysis (reported in 30% of PCH patients but absent in PVOD) and hemorrhagic pleural effusion (present in 25% of PCH patients but not reported in PVOD) is the only clinical manifestations which can differentiate PCH from PVOD [6,24-26]. With further progression of PCH, PH and right-sided heart failure develop with peripheral oedema, ascites, tender hepatomegaly, hepatojugular reflex and right parasternal leave [8].
Hemodynamics
PCH is classically present with an increased pulmonary arterial pressure with normal or low pulmonary capillary wedge pressures (PCWP) [8]. Increased pulmonary arterial pressures at right-sided heart catheterization can be explained by the reflection of the sustained back pressure from capillary beds [28]. PCWP is actually a misnomer since the pressure of the capillary bed is not measured. The actual measured pressure is the pressure distal to the wedged catheter tip beyond the venules and small veins to the largest pulmonary veins and left atrium which is unaffected in PCH and this feature can distinguish PCH from other cases of PAH causing elevated PCWP [8].
Diagnostic modalities
In PCH, Electrocardiography (ECG) typically displays right axis deviation and RVH [8]. Chest radiography shows PAH signs such as enlarged central pulmonary arteries and right-sided prominence of the heart which also occurs in PVOD. However diffuse or bibasilar reticulonodular or micronodular areas of opacity can only occur in PCH. In contrast to PVOD, septal lines or pleural effusions are unusual. Mediastinal lymphadenopathy is reported occasionally at chest radiography [8]. TTE is the first-line modality for diagnosis of PAH [23]. It can diagnose PH and define certain etiologies such as left-sided heart lesions as mitral stenosis, left ventricular dysfunction, or intracardiac shunts [29,30]. However, TTE has a limited ability to evaluate pulmonary arteries distal to the main pulmonary artery and branches and a far limited ability to evaluate right ventricular function [30]. PH probability can be classified into low, intermediate, or high based on echocardiographic findings, Further evaluation is required for intermediate or high probability of PH patients [14].
HRCT chest plays a crucial role in the diagnosis of PCH as it shows a group of characteristic cardiovascular and pulmonary parenchymal findings. The cardiovascular findings include enlarged central pulmonary arteries, dilated right-sided heart, IV contrast reflux into the IVC, normal caliber pulmonary veins and average-sized left atrium and pericardial effusion while pulmonary parenchymal findings are diffuse well-defined small centrilobular ground-glass pulmonary nodules with no zonal predominance with or without few and sparse smooth inter-lobular septal thickening. The associated findings, which may be encountered in PCH patients, include mediastinal lymphadenopathy and pleural effusions [30].
The predominance of the centrilobular ground-glass nodules over the septal lines help to distinguish PCH from PVOD, which shows predominant septal lines, and to a lesser extent centrilobular ground-glass nodules. Additionally, the lack of dilated pulmonary veins in PCH patients is particularly important to differentiate PCH/PVOD from other causes of post-capillary pulmonary hypertension, which similarly show smooth septal thickening and ground-glass opacities due to constriction/impedance of the pulmonary venous drainage [30].
The majority of patients with PCH are misdiagnosed as primary PAH, PVOD, pulmonary fibrosis, sarcoidosis, pulmonary thromboembolism, or pulmonary hemosiderosis [30] so, HRCT chest is now considered the imaging modality of choice that is routinely used for the evaluation of patients with suspected PH [31].
Pathology (previously discussed in pathology section) remains the most definitive modality to diagnose PCH however there is a high risk of complications during the procedure such as pneumothorax, hemorrhage into the lung and infection that hider lung biopsy.
Treatment options
Pharmacologic management of PCH remains uncertain. Conventional medical therapies aiming to reduce pulmonary vascular resistance, increase cardiac output, and decrease volume overload are used such as diuretics, Angiotensin-converting-enzyme inhibitors (ACE) inhibitors, anticoagulants (warfarin), cardiac glycosides (digoxin), and oxygen supplementation [26]. Pulmonary vasodilator therapies such as calcium channel blockers and Prostacyclin have been proposed to improve hemodynamics and clinical course in PAH patients. Yet catastrophic pulmonary oedema has been reported following the initiation of pulmonary vasodilators in PCH patients [11,32]. This unfavorable outcome can be explained by the increased transcapillary hydrostatic pressure due to dilated pulmonary muscular arteries and arterioles and fixed pulmonary venous resistance leading to massive transudation of fluid into the lung parenchyma [26,32] so, High-resolution CT (HRCT) chest examination is now recommended for PAH patients to exclude unsuspected radiologic evidence of PVOD or PCH before initiation of vasodilator therapy [33]. Favorable responses have been reported to angiogenesis inhibitors such as doxycycline [34] and imatinib [35]. The only definite treatment of PCH is lung transplant or combined heart-lung transplantation in advanced disease [21].
Prognosis
The natural progression of PCH remains poorly determined but rarely reported survival beyond 5 years from the onset of symptoms [11,36]. The reported median survival is 3 years from the first clinical manifestation which can be explained with the slow progression of pulmonary hypertension to cause corpulmonale, right ventricular failure and eventually cardiovascular collapse and death [26].
Conclusion
PCH is a subclass of PAH group 1. It is characterized by uncontrolled capillary proliferation in the lung. Clinically, PCH can present with non-specific symptoms such as dyspnea and cough till irreversible severe PH with subsequent right-sided heart failure. Determining PCH as the underlying cause of PH is crucial as medications used for PAH are relatively ineffective in cases of PCH. Vasodilator therapy should be avoided due to an increased risk of pulmonary oedema, respiratory distress, and death. So far, no definitive treatment of PCH apart from lung transplantation.
Disclosure of conflict of interest
None.
Supplementary Videos
References
- 1.Lau EM, Humbert M. A critical appraisal of the updated 2014 nice pulmonary hypertension classification system. Can J Cardiol. 2015;31:367–374. doi: 10.1016/j.cjca.2014.09.033. [DOI] [PubMed] [Google Scholar]
- 2.Hatano S, Strasser T Organization WH. Primary pulmonary hypertension: report on a WHO meeting; 15-17 October 1973; Geneva. p. 1975. [Google Scholar]
- 3.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M. Clinical classification of pulmonary hypertension. JACC. 2004;43:S5–S12. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. JACC. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 5.O’Keefe MC, Post MD. Pulmonary capillary hemangiomatosis: a rare cause of pulmonary hypertension. Arch Pathol Lab Med. 2015;139:274–277. doi: 10.5858/arpa.2013-0500-RS. [DOI] [PubMed] [Google Scholar]
- 6.Wagenvoort C, Beetstra A, Spijker J. Capillary haemangiomatosis of the lungs. Histopathol. 1978;2:401–406. doi: 10.1111/j.1365-2559.1978.tb01734.x. [DOI] [PubMed] [Google Scholar]
- 7.Dhala A. Pulmonary arterial hypertension in systemic lupus erythematosus: current status and future direction. Clin Dev Immunol. 2012;2012:854941. doi: 10.1155/2012/854941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frazier AA, Franks TJ, Mohammed TL, Ozbudak IH, Galvin JR. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Radiographics. 2007;27:867–882. doi: 10.1148/rg.273065194. [DOI] [PubMed] [Google Scholar]
- 9.De Montpréville VT, Dulmet É, Fadel É, Dartevelle P. Lymph node pathology in pulmonary veno-occlusive disease and pulmonary capillary heamangiomatosis. Virchows Archiv. 2008;453:171–176. doi: 10.1007/s00428-008-0636-3. [DOI] [PubMed] [Google Scholar]
- 10.Chaisson NF, Dodson MW, Elliott CG. Pulmonary capillary hemangiomatosis and pulmonary veno-occlusive disease. Clin Chest Med. 2016;37:523–534. doi: 10.1016/j.ccm.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Langleben D, Heneghan JM, Batten AP, Wang NS, Fitch N, Schlesinger RD, Guerraty A, Rouleau JL. Familial pulmonary capillary hemangiomatosis resulting in primary pulmonary hypertension. Ann Intern Med. 1988;109:106–109. doi: 10.7326/0003-4819-109-2-106. [DOI] [PubMed] [Google Scholar]
- 12.Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, Rosenzweig EB, Bayrak-Toydemir P, Mao R, Cahill BC. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest. 2014;145:231–236. doi: 10.1378/chest.13-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, Coulet F, Bonnet D, Dorfmüller P. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. 2014;46:65. doi: 10.1038/ng.2844. [DOI] [PubMed] [Google Scholar]
- 14.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) EHJ. 2015;37:67–119. [Google Scholar]
- 15.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, Stenmark KR, Steinhorn R, Thébaud B, Fineman JR, Kuehne T, Feinstein JA, Friedberg MK, Earing M, Barst RJ, Keller RL, Kinsella JP, Mullen M, Deterding R, Kulik T, Mallory G, Humpl T, Wessel DL American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; and the American Thoracic Society. Pediatric pulmonary hypertension: guidelines from the american heart association and american thoracic society. Circulation. 2015;132:2037–99. doi: 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 16.Pulmonary capillary hemangiomatosis. PathologyOutlines.com website. http//www.pathologyoutlines.com/topic/lungtumorhemangiomatosis.html. Accessed July 13th.
- 17.Domingo C, Encabo B, Roig J, López D, Morera J. Pulmonary capillary hemangiomatosis: report of a case and review of the literature. Respiration. 1992;59:178–180. doi: 10.1159/000196053. [DOI] [PubMed] [Google Scholar]
- 18.Whittaker J, Pickering C, Heath D, Smith P. Pulmonary capillary haemangiomatosis. Diagn Histopathol. 1983;6:77–84. [PubMed] [Google Scholar]
- 19.Havlik DM, Massie LW, Williams WL, Crooks LA. Pulmonary capillary hemangiomatosis-like foci: an autopsy study of 8 cases. Am J Clin Pathol. 2000;113:655–662. doi: 10.1309/9R7N-19BP-P5QJ-U8E7. [DOI] [PubMed] [Google Scholar]
- 20.Tron V, Magee F, Wright J, Colby T, Churg A. Pulmonary capillary hemangiomatosis. Hum Pathol. 1986;17:1144–1150. doi: 10.1016/s0046-8177(86)80420-8. [DOI] [PubMed] [Google Scholar]
- 21.El-Gabaly M, Farver CF, Budev MA, Mohammed TL. Pulmonary capillary hemangiomatosis imaging findings and literature update. J Comput Assist Tomogr. 2007;31:608–610. doi: 10.1097/01.rct.0000284393.76073.87. [DOI] [PubMed] [Google Scholar]
- 22.Silva CJ, Massie J, Mandelstam SA. Pulmonary capillary haemangiomatosis in a premature infant. Pediatr Radiol. 2005;35:635–640. doi: 10.1007/s00247-004-1374-6. [DOI] [PubMed] [Google Scholar]
- 23.Wirbelauer J, Hebestreit H, Marx A, Mark EJ, Speer CP. Familial pulmonary capillary hemangiomatosis early in life. Case Rep Pulmonol. 2011;2011:827591. doi: 10.1155/2011/827591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito K, Ichiki T, Ohi K, Egashira K, Ohta M, Taguchi K, Takeshita A. Pulmonary capillary hemangiomatosis with severe pulmonary hypertension. Circ J. 2003;67:793–795. doi: 10.1253/circj.67.793. [DOI] [PubMed] [Google Scholar]
- 25.Masur Y, Remberger K, Hoefer M. Pulmonary capillary hemangiomatosis as a rare cause of pulmonary hypertension. Pathology-Research and Practice. 1996;192:290–295. doi: 10.1016/S0344-0338(96)80232-9. [DOI] [PubMed] [Google Scholar]
- 26.Almagro P, JuliÀ J, Sanjaume M, GonzÁlez G, Casalots J, Heredia JL, MartÍnez J, Garau J. Pulmonary capillary hemangiomatosis associated with primary pulmonary hypertension: report of 2 new cases and review of 35 cases from the literature. Medicine (Baltimore) 2002;81:417–424. doi: 10.1097/00005792-200211000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Umezu H, Naito M, Yagisawa K, Hattori A, Aizawa Y. An autopsy case of pulmonary capillary hemangiomatosis without evidence of pulmonary hypertension. Virchows Archiv. 2001;439:586–592. doi: 10.1007/s004280100465. [DOI] [PubMed] [Google Scholar]
- 28.Lucas JR. Congenital causes of pulmonary venous obstruction. Clin Cardiol. 1972;4:19–51. [PubMed] [Google Scholar]
- 29.Peña E, Dennie C, Veinot J, Muñiz SH. Pulmonary hypertension: how the radiologist can help. Radiographics. 2011;32:9–32. doi: 10.1148/rg.321105232. [DOI] [PubMed] [Google Scholar]
- 30.Aluja Jaramillo F, Gutierrez FR, Díaz Telli FG, Yevenes Aravena S, Javidan-Nejad C, Bhalla S. Approach to pulmonary hypertension: from CT to clinical diagnosis. RadioGraphics. 2018;38:357–373. doi: 10.1148/rg.2018170046. [DOI] [PubMed] [Google Scholar]
- 31.François CJ, Schiebler ML. Imaging of pulmonary hypertension. Radiol Clin North Am. 2016;54:1133–1149. doi: 10.1016/j.rcl.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Gugnani MK, Pierson C, Vanderheide R, Girgis RE. Pulmonary edema complicating prostacyclin therapy in pulmonary hypertension associated with scleroderma: a case of pulmonary capillary hemangiomatosis. Arthritis Rheum. 2000;43:699–703. doi: 10.1002/1529-0131(200003)43:3<699::AID-ANR28>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Resten A, Maître S, Humbert M, Sitbon O, Capron F, Simoneau G, Musset D. Pulmonary arterial hypertension: thin-section CT predictors of epoprostenol therapy failure. Radiology. 2002;222:782–788. doi: 10.1148/radiol.2223010668. [DOI] [PubMed] [Google Scholar]
- 34.Freeman SV, Graham BB. C70. Call my name: case reports in pulmonary vascular disease. ATS 2017. Late presentation of pulmonary capillary hemangiomatosis and successful treatment with doxycycline; pp. A6201–A6201. [Google Scholar]
- 35.Nayyar D, Muthiah K, Kumarasinghe G, Hettiarachchi R, Celermajer D, Kotlyar E, Keogh A. Imatinib for the treatment of pulmonary arterial hypertension and pulmonary capillary hemangiomatosis. Pulm Circ. 2014;4:342–345. doi: 10.1086/675996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langleben D. Pulmonary capillary hemangiomatosis: the puzzle takes shape. Chest. 2014;145:197–199. doi: 10.1378/chest.13-2513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.