

Abstract
Clinical application of somatic genome editing requires therapeutics that are generalizable to a broad range of patients. Targeted insertion of promoterless transgenes can ensure that edits are permanent and broadly applicable while minimizing risks of off-target integration. In the liver, the Albumin (Alb) locus is currently the only well-characterized site for promoterless transgene insertion. Here, we target the Apoa1 locus with adeno-associated viral (AAV) delivery of CRISPR-Cas9 and achieve rates of 6% to 16% of targeted hepatocytes, with no evidence of toxicity. We further show that the endogenous Apoa1 promoter can drive robust and sustained expression of therapeutic proteins, such as apolipoprotein E (APOE), dramatically reducing plasma lipids in a model of hypercholesterolemia. Finally, we demonstrate that Apoa1-targeted fumarylacetoacetate hydrolase (FAH) can correct and rescue the severe metabolic liver disease hereditary tyrosinemia type I. In summary, we identify and validate Apoa1 as a novel integration site that supports durable transgene expression in the liver for gene therapy applications.
Keywords: AAV, CRISPR-Cas9, gene therapy, genome editing, gene targeting, Apoa1, liver, inherited metabolic disorders
Graphical abstract
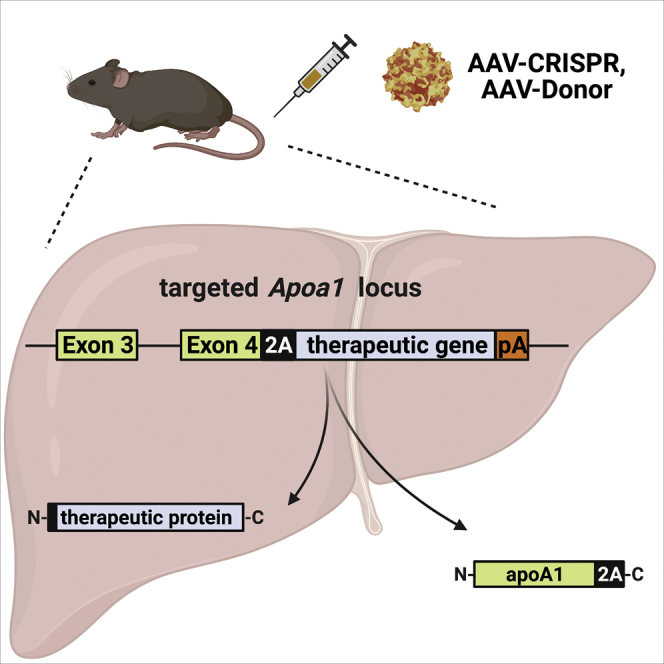
De Giorgi and colleagues identified the Apolipoprotein a1 (Apoa1) locus as a promising site for therapeutic genome engineering in the liver. The authors used adeno-associated viral (AAV) delivery of CRISPR-Cas9 and donor templates to target the Apoa1 locus to achieve sustained expression of both intracellular and secreted therapeutic proteins.
Introduction
There are at least 700 inherited metabolic disorders associated with the genetic loss of a specific liver function.1 Liver-directed genome editing is an exciting and potentially curative strategy for these diseases. However, a clinically relevant genome-editing approach should be generalizable to many patients rather than mutation specific. In addition, the transgene sequence must be integrated safely into a well-defined site to ensure permanent expression, while avoiding adverse events. Identification of safe and effective integration sites in the liver is important for the field, as it will enable more streamlined deployment of gene-editing therapies.
Adeno-associated viral (AAV) vectors are a leading platform for gene therapy and somatic genome editing.2,3 Recombinant AAVs are composed of a small, non-enveloped, protein capsid enclosing a single-stranded DNA genome flanked on either side by hairpin-like structures called inverted terminal repeats (ITRs). Following entry into the nucleus, the single-stranded AAV genome is converted to double-stranded episomes, circularized through the ITRs. AAV episomes provide strong and stable expression of transgenes.3 However, since episomes are generally not integrated into the host chromosome, they will be lost through cell death and division. Although hepatocyte turnover has not been well determined in humans, the average lifespan of a rodent hepatocyte is estimated to be between 200 and 400 days.4,5 Likewise, preclinical models of liver-directed neonatal AAV gene therapy show only short-term effects.6, 7, 8 Therefore, it is reasonable to predict that conventional AAV gene therapy may not provide lifelong correction of many liver disorders, especially in pediatric patients. Strategies are needed to capitalize on the high delivery efficiency of AAV, while ensuring that the therapeutic cargo is passed on to daughter cells.
Targeted integration of AAV transgenes is an attractive strategy that requires the identification of genomic sites, which can be modified safely and efficiently. Ideally, these sites should enable (1) access for the genomic modification machinery,9 (2) robust and sustained expression from an endogenous promoter rather than introduction of an external promoter that may be silenced,10, 11, 12, 13, 14 (3) no disruption or dysregulation of neighboring genes,15,16 and (4) no induction of any malignancies or toxicity.17 The Albumin (Alb) locus is currently the only site that has been characterized as a liver-specific platform for gene addition. Zinc finger nucleases (ZFN) have been used to insert several therapeutic transgenes at the Alb locus, including factor VIII (FVIII) and IX (FIX).18 Barzel et al.19 introduced the concept of “promoterless targeting” where an AAV vector was used to modify the 3′ end of the Alb gene, referred to as GeneRide. Alb targeting has been applied successfully to a mouse model of Crigler-Najjar syndrome type I,20 and the efficiency of this approach can be dramatically increased with CRISPR-Cas9.21 Clinical trials are currently underway using AAV and ZFN targeting of the ALB locus to treat Mucopolysaccharidosis I and II (MPSI and II) and Hemophilia B (NCT02702115, NCT03041324, NCT02695160). However, progress reports from the MPSI and II clinical trials show low targeted expression levels of therapeutic transgene from the ALB locus.22 Moreover, a decrease of the plasma Alb levels has been reported following Alb targeting in several preclinical models.22,23 Therefore, it remains to be determined whether ALB targeting will provide safe and durable correction, or if unexpected pathology will result. Recent data have identified ALB as one of the most commonly mutated genes in human hepatocellular carcinoma (HCC) biopsies24,25 with mutations observed in 13% of tumors. It is currently unclear whether ALB mutations are passengers in HCC tumor growth or potentially causative. Despite recent evidence showing no HCC development up to 15 months following Alb targeting in mice,26 the risk of unintended editing events in ALB in the setting of human gene therapy remains unknown. Therefore, there is a compelling need to explore additional targetable loci in the liver.
Apolipoprotein A1 (ApoA1) is the primary structural component of high-density lipoprotein (HDL) particles and is highly expressed by the liver. Here, we show that the locus is amenable to modification using AAV vectors for homology-directed repair (HDR), and that targeting efficiency is dramatically enhanced with CRISPR-Cas9 editing. Fusion of promoterless transgenes to the 3′ end of the Apoa1 gene supports sustained expression in the liver, without adverse consequences. Furthermore, Apoa1 targeting supports the expression of secreted therapeutic proteins FIX and Apolipoprotein E (APOE), and enables correction of inherited metabolic disorders, such as hypercholesterolemia and hereditary tyrosinemia type I. Taken together, our work identifies and validates Apoa1 as a therapeutically useful integration site for liver-directed genome editing.
Results
High chromatin accessibility and expression of Apoa1 in mouse and human liver
ApoA1 is the primary structural component of HDL particles and circulates in the bloodstream at concentrations > 1 mg/mL, making it one of the most abundant plasma proteins in humans.27 The Apoa1 gene is expressed in the liver and intestine, and the liver in particular is a major contributor to circulating apoA1 protein levels.28 To determine the feasibility of targeting the Apoa1 locus for expression of therapeutic transgenes from the liver, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) for histone H3K27 acetylation and RNA polymerase II (RNA Pol II) binding, as well as assay for transposase-accessible chromatin (ATAC)-sequencing (Figure S1). The data showed strong evidence of high rates of transcription, as well as accessible chromatin at the Apoa1 locus in both mouse and human liver tissue. The murine Apoa1 gene had a stronger H3K27 acetylation and RNA Pol II signature than Alb, which is a popular target for liver-directed transgene insertion18, 19, 20 (Figure S1A). ChIP-seq and ATAC-seq analysis on human liver also showed strong H3K27 acetylation and RNA Pol II binding, as well as chromatin accessibility for the human APOA1 locus in comparison to ALB (Figure S1B), strongly supporting the candidacy of this site.
Efficient targeting of the Apoa1 locus for the expression of transgenic proteins in vivo
To target the Apoa1 locus, we used AAV vectors based on serotype 8, to deliver the Staphylococcus aureus Cas9 (SaCas9) nuclease and guide RNA (gRNA; AAV-CRISPR), along with a donor template for HDR (AAV-Donor). In order to avoid indels in the Apoa1 coding sequence, we identified an efficient gRNA that cuts the 3′ untranslated region (3′ UTR) of Apoa1 at 29 base pairs downstream of the termination codon (Figure S2). The AAV-Donor vector includes exon 4 of Apoa1 followed by a 2A skipping peptide and the therapeutic transgene, flanked on each side by homology arms (Figure 1). Correct in-frame integration by HDR should result in removal of the endogenous stop codon, and transcription of a bicistronic mRNA driven by the endogenous Apoa1 promoter. This mRNA should be translated into two proteins: apoA1 C-terminally tagged with 2A peptide and the therapeutic protein including an N-terminal proline (Figure 1).
Figure 1.

Strategy for therapeutic targeting of the Apoa1 locus for liver gene therapy
AAV-CRISPR encodes a gRNA for targeting the Apoa1 3′ UTR, and SaCas9 driven by a liver-specific promoter (HLP). The gRNA target site (red) and protospacer adjacent motif (PAM) sequence (blue) in the Apoa1 3′ UTR are shown. AAV-Donor contains the final coding exon of Apoa1 (exon 4) fused to a 2A ribosomal skipping sequence upstream of a therapeutic gene and synthetic polyadenylation signal (pA), flanked by homology arms (HR) to the Apoa1 locus. Following correct integration by HDR, the Apoa1 promoter drives the expression of a bicistronic mRNA consisting of Apoa1 and therapeutic transgene. Translation results in expression of apoA1 with a C-terminal 2A epitope tag, as well as the therapeutic protein including an N-terminal proline. Created with BioRender.
We injected adult mice with AAV-CRISPR and/or an AAV-Donor encoding the far-red fluorescent protein mKate2 and examined expression at 12 weeks post-injection (Figure 2A). Two major integration events of AAV-Donor were observed by PCR: (1) HDR integration and (2) non-homologous end joining (NHEJ)-insertion of the AAV genome, including ITRs (Figure 2B). In addition to integration, SaCas9-cutting of the Apoa1 3′ UTR generated indels with high frequencies of 54.5% ± 2.5% and 51.5% ± 3.2% in CRISPR and CRISPR + Donor livers, respectively (Figure 2C; Table S1). Editing appeared to be very specific, with no detectable off-target activity above background in any of the predicted sites examined (Figure S3; Table S1). We performed droplet digital PCR (ddPCR) to quantify the Apoa1 alleles with NHEJ-insertion, using a primer specific to the ITRs present in both AAV vectors. We observed 11.8% ± 5.5% and 11.8% ± 2.5% NHEJ-insertion events in livers from CRISPR and CRISPR + Donor mice, respectively (Figure 2D). We were also able to detect correct HDR-insertion events of AAV-Donor with a frequency of 1.8% ± 0.6% in livers from CRISPR + Donor mice and 0.3% ± 0.2% in livers from Donor mice (Figure 2E). Immunohistochemistry revealed that 0.35% ± 0.32% of hepatocytes in Donor mice expressed the mKate2-FLAG transgene (Figures 2F and 2G), which increased dramatically to 5.7% ± 2.9% with the addition of AAV-CRISPR (Figures 2F, 2G, and S4). By western blot, we were able to detect mKate2-FLAG and 2A-tagged apoA1 only in CRISPR + Donor mice (Figures 2H, 2I, 2K, and 2L). Targeting the Apoa1 locus did not adversely impact endogenous apoA1 protein levels (Figures 2J, 2M, and S5B) or expression of neighboring genes (Figure S5). Likewise, no liver toxicity or histopathological abnormalities were observed (Figures S6–S9).
Figure 2.

Targeted integration and expression from the Apoa1 locus in vivo
(A) Adult C57BL/6J mice were intraperitoneally injected with AAV-CRISPR (5 × 1011 GC) and/or an AAV-Donor template (5 × 1011 GC) encoding a far-red fluorescent protein (mKate2). Control mice were injected with AAV-GFP (5 × 1011 GC). Livers and plasma were harvested for analysis at 12 weeks post-injection. (B) PCR from liver showing integration of AAV-Donor into the Apoa1 locus. Two main products were observed: correct HDR (1,139 bp) and NHEJ insertion (2,031 bp). The forward primer binds to the Apoa1 locus upstream of the 5′ HR and reverse primer binds to the mKate2 coding sequence. Minus (–) indicates water-only control. (C) Frequency of indel formation in the Apoa1 3′ UTR by deep sequencing. (D) Frequency of Apoa1 alleles with NHEJ insertions of AAV genomes by ddPCR. (E) Frequency of correct HDR targeting of AAV-Donor by ddPCR. (F) Representative immunohistochemistry for mKate2-FLAG (brown cells) in Apoa1-targeted mice. Scale bar is 100 μm. (G) Quantification of FLAG positive hepatocytes relative to total nuclei per field. (H) Western blot of mKate2-FLAG in liver lysates with β-tubulin (β-tub) as a loading control. (I and J) Western blot analysis of 2A-tagged (I) and total apoA1 (J) in plasma with alpha-1 antitrypsin (aat) as loading control. Four representative mice per group are shown in western blots. (K) Densitometry analysis of mKate2-FLAG in liver lysates relative to β-tub loading control. Densitometry analysis of apoA1-2A (L) and apoA1 (M) in plasma relative to aat loading control. Data are shown as mean ± standard deviation (n = 5; n = 4 in densitometry analyses), with significance determined by one-way ANOVA followed by Tukey test. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (A) Created with BioRender.
Improved expression of transgenic proteins from the Apoa1 locus in mice injected at post-natal day 4
HDR occurs at very low rate in post-mitotic tissues such as the adult liver, where NHEJ is the preferred repair pathway.29 In an attempt to take advantage of hepatocyte division during liver growth, we injected neonatal mice at post-natal day 4 (P4) with the same vectors and examined expression and integration 20 weeks later (Figure 3A). Relative to adult mice, there was a marked enhancement of HDR versus NHEJ-insertion of the AAV-Donor (Figure 3B versus 2B). The frequency of NHEJ insertion was 17.4% ± 5% and 21.5% ± 8% in CRISPR and CRISPR + Donor mice, respectively (Figure 3C). The frequency of HDR events in CRISPR + Donor mice increased to 7.8% ± 1.7%, almost 8-fold compared with adult injected mice (Figure 3D). Donor mice showed a HDR frequency of 0.1% ± 0.06%. Indel formation was significantly lower in CRISPR + Donor than CRISPR mice (21.4% ± 3.6% versus 29.2% ± 0.4%, Figure 3E). By immunohistochemistry, we observed only rare targeted hepatocytes in Donor-only treated mice, 0.6% ± 0.1% (Figures 3F and 3G), in contrast to numerous positive hepatocytes arranged in colonies in the CRISPR + Donor livers at 16.4% ± 3.6% (Figures 3F, 3G, and S10). The mKate2 protein and 2A-tagged apoA1 were readily detectable in CRISPR + Donor mice by western blot (Figures 3H, 3I, 3K, and 3L). Interestingly, the increased targeting of Apoa1 did not affect total apoA1 levels in plasma (Figures 3J and 3M). Moreover, in spite of the high rate of integration at the Apoa1 locus we did not observe any histopathological abnormalities, toxicity, or tumor development (Figure S11). In order to investigate whether the greater proportion of targeted cells could be due to the higher AAV dose relative to body weight, we performed a dose-ranging experiment by injecting pups with several doses of AAV-CRISPR + AAV-Donor at a constant 1:1 molar ratio (Figure S12). We observed a dose-dependent decrease in the frequency of both HDR- (7.5% ± 4.2%, 1% ± 0.7% and 0.2% ± 0.1%) and NHEJ-integration events (18.5% ± 0.8%, 4.2% ± 1.3%, and 0.9% ± 0.5%) in pups injected with a total of 1012, 1011, and 5 × 1010 GC of AAV, respectively (Figures S12B and S12C). Similarly, the frequency of mKate2-FLAG-positive hepatocytes correlated well with descending AAV doses at 13.5% ± 6.3%, 2.5% ± 1.8%, and 0.6% ± 0.7% (Figures S12D and S12E). These data demonstrate a high efficiency of targeting and expression from the Apoa1 locus in pups and that such effects are dose dependent.
Figure 3.

Improved targeting of the Apoa1 locus in neonatal mice injected at P4
(A) Experimental design and AAV-Donor scheme. C57BL/6J mice were subcutaneously injected with AAV-CRISPR (5 × 1011 GC) and/or an AAV-Donor template (5 × 1011 GC) encoding a far-red fluorescent protein (mKate2) at P4 and liver and plasma were evaluated for the expression of the mKate2 protein at 20 weeks post-injection. Control mice were injected with AAV-GFP (5 × 1011 GC). (B) Integration PCR on liver showed two main products corresponding to correct HDR (1,139 bp) and NHEJ insertion (2,031 bp) of AAV-Donor at the Apoa1 locus. Minus (–) indicates water-only control. (C) Frequency of Apoa1 alleles with NHEJ insertions of AAV genomes by ddPCR. (D) Frequency of correct HDR targeting of AAV-Donor by ddPCR. (E) Indel formation in the 3′ UTR of Apoa1 by ICE analysis. (F) Representative immunohistochemistry for mKate2-FLAG (brown cells) in Apoa1-targeted mice. Scale bar is 100 μm. (G) Quantification of FLAG-positive hepatocytes relative to total nuclei per field. (H) Western blot of mKate2-FLAG in liver lysates with β-tub as a loading control. (I and J) Western blot analysis of 2A-tagged (I) and total apoA1 (J) in plasma with aat as loading control. Aat shows a slight difference in protein expression based on sex: lanes 1, 4, 7, 8, 9, 10, 13, 14, and 15 are male mice; lanes 2, 3, 5, 6, 11, 12, and 16 are female mice. Four representative mice per group are shown in western blots. (K) Densitometry analysis of mKate2-FLAG in liver lysates relative to β-tub loading control. Densitometry analysis of apoA1-2A (L) and apoA1 (M) in plasma relative to aat loading control. Data are shown as mean ± standard deviation (n = 5; n = 4 in densitometry analyses), with significance determined by one-way ANOVA followed by Tukey test. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (A) Created with BioRender.
Sustained expression of secreted therapeutic proteins
The liver is of particular interest for gene therapy, both for correcting inherited metabolic disorders and as a biofactory for secretion of therapeutic proteins. To test whether our Apoa1 targeting strategy could support the secretion of a therapeutic protein, we constructed an AAV-Donor vector encoding human FIX, the clotting factor deficient in patients with hemophilia B (Figure 4A). We injected adult mice with AAV-Donor alone or in combination with AAV-CRISPR and collected plasma every 2 to 4 weeks up to 24 weeks post-injection (Figure 4B). As expected from previous experiments, we detected the 2A-tagged apoA1 only in plasma from CRISPR + Donor mice (Figures 4C and 4E), which showed editing and integration in the Apoa1 locus (Figures S13A and S13B). Similar to the adult mice injected with the mKate2 Donor construct (Figure 2), CRISPR + Donor mice showed a frequency of 1% ± 0.2% and 14.9% ± 4.2% for HDR- and NHEJ-integration events at the Apoa1 locus (Figures S13C and S13D). In Donor mice, the HDR frequency was around 0.1% ± 0.05% (Figure S13C). Despite this, we were able to detect the bicistronic mRNA using a primer binding the 2A sequence only in CRISPR + Donor mice (Figure S13E). No changes in apoA1 levels were observed following Apoa1 targeting (Figures 4D and 4F). The production of FIX was constant and significantly higher in CRISPR + Donor than in Donor mice—228 ± 65.6 versus 139 ± 22.8 ng/mL at experimental endpoint (Figure 4G). Similarly, the expression of total FIX transgene mRNA was ∼3 fold higher in CRISPR + Donor as compared to Donor livers (Figure S13F). Together, these data show that the Apoa1 locus can drive stable and sustained expression of therapeutically relevant secreted proteins.
Figure 4.

Sustained expression of factor IX over time in Apoa1-targeted mice
(A) AAV-Donor scheme. (B) Adult C57BL/6J mice were intraperitoneally injected with AAV-CRISPR (5 × 1011 GC) and/or an AAV-Donor (5 × 1011 GC) encoding human FIX. Control mice were injected with AAV-GFP (5 × 1011 GC). Plasma was collected every 2 to 4 weeks up to 24 weeks post-injection for analysis of human FIX protein levels. (C and D) Western blot analysis of 2A-tagged (C) and total apoA1 (D) in plasma isolated at 24 weeks post-injection, with aat as loading control. Five representative mice per group are shown in western blots. (E and F) Densitometry analysis of apoA1-2A (E) and apoA1 (F) in plasma relative to aat loading control. (G) Quantitative measurement of plasma FIX over time by ELISA (green line, control; blue line, Donor; red line, CRISPR + Donor mice). Data are shown as mean ± standard deviation (n = 5 in densitometry analyses; n = 6 in FIX ELISA). A one-way and two-way ANOVA followed by Tukey test were used respectively for densitometry analyses (E and F) and FIX ELISA (G). ∗p < 0.05 CRISPR + Donor versus Donor; ∗∗p < 0.01 CRISPR + Donor versus Donor; ∗∗∗p < 0.001 CRISPR + Donor versus Donor, # p < 0.0001 Control versus Donor and CRISPR + Donor, ∗∗∗∗p < 0.0001. (A and B) Created with BioRender.
Amelioration of hypercholesterolemia through Apoa1 targeting
Next, we asked whether the Apoa1-targeted expression of a secreted protein could correct an inherited metabolic disorder. ApoE is a constituent of several lipoprotein classes and serves as a high-affinity ligand for the low-density lipoprotein receptor (LDLR). Mutations at the APOE gene have been associated with familial type III hyperlipoproteinemia. Similar to the phenotype observed in patients, Apoe−/− mice are deficient in clearing circulating apoB-containing lipoproteins, resulting in development of hypercholesterolemia and atherosclerosis upon high fat diet feeding.30 First, we demonstrated the feasibility of targeting Apoa1 with an AAV-Donor encoding human APOE in a small group of adult Apoe−/− mice (Figures 5A and S14). Then, we injected P4 Apoe−/− pups with AAV-CRISPR plus the AAV-Donor encoding human APOE (Figure 5A) or saline as control, and we fed mice with western diet starting at weaning for 20 weeks. We collected plasma samples every 2 to 4 weeks up to 23 weeks of age (Figure 5B). We detected a high proportion of correct HDR integration of the AAV-Donor in the Apoa1 locus (Figure 5C), which resulted in secretion of apoA1-2A and APOE in plasma of CRISPR + Donor mice (Figures 5D, 5E, 5H, and 5I). While the endogenous levels of apoA1 did not vary between mice (Figures 5F and 5J), CRISPR + Donor mice showed less apoB-48 in plasma as compared to control mice (Figures 5G and 5K), consistent with enhanced clearance of apoB-containing lipoprotein particles via APOE. Control mice showed gradual and continuing accumulation of total cholesterol in plasma up to 1,325 ± 131 mg/dL (Figure 5L). On the contrary, CRISPR + Donor mice showed significantly lower levels of total cholesterol over time, which were reduced to about half relative to control mice (638 ± 58.7 mg/dL at endpoint, Figure 5L). Similarly, CRISPR + Donor mice showed significantly lower endpoint plasma triglycerides as compared to control mice (83.4 ± 7.8 versus 155.7 ± 20.5 mg/dL; Figure 5M). These data show that Apoa1-targeted expression of APOE can dramatically reduce hypercholesterolemia.
Figure 5.

Reduction of plasma lipids in a mouse model of hypercholesterolemia through Apoa1-targeting
(A) AAV-Donor scheme. (B) P4 Apoe−/− male pups were subcutaneously injected with AAV-CRISPR (5 × 1011 GC) and an AAV-Donor (5 × 1011 GC) encoding human APOE or saline (control). Mice were fed a western diet starting at weaning for 20 weeks. Plasma was collected every 2 to 4 weeks up to 23 weeks of age. (C) Integration PCR on liver DNA showed two main products corresponding to HDR (1,289 bp) and NHEJ (2,213 bp) insertion of AAV-Donor in the Apoa1 cut site. Minus (–) indicates a water-only PCR control. (D–G) Western blot analysis of 2A-tagged apoA1 (D), APOE (E), total apoA1 (F), and apoB-48 and apoB-100 (G) in plasma isolated at endpoint, with aat as loading control. Eight representative mice per group are shown in western blots. (H–K) Densitometry analysis of apoA1-2A (H), APOE (I), apoA1 (J), and apoB-48 (K) in plasma relative to aat loading control. (L and M) Plasma total cholesterol (L) and triglycerides (M) measurement over time (green line, control; red line, CRISPR + Donor mice). Data are shown as mean ± standard deviation (n = 8 for densitometry analyses; n = 11 control and 9 CRISPR + Donor mice for lipid analyses). Significance was determined by a two-tailed Student’s t test in densitometry analyses (H–K). A two-way ANOVA followed by Bonferroni test was used for plasma lipid analyses in (L) and (M). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001. (A and B) Created with BioRender.
Correction of a lethal metabolic liver disorder through Apoa1 targeting
We next sought to determine whether targeted integration at the Apoa1 locus could correct a lethal metabolic liver disease. Fumarylacetoacetate hydrolase (FAH) catalyzes the final step of tyrosine catabolism and its deficiency results in the accumulation of toxic catabolites, leading to liver and kidney failure in hereditary tyrosinemia type I (HT-I). Liver and kidney pathology can be averted by supplying 2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione (NTBC), which blocks the pathway upstream.31 Fah−/− mice were maintained on NTBC and then injected with either saline (control) or AAV-CRISPR plus an AAV-Donor encoding a human FAH transgene. NTBC was removed from both groups, which were then followed for 40 days (Figure 6A). Control mice showed a rapid loss of body weight reflecting the dehydration associated with kidney disease starting at days 12–15 and met the humane endpoint for euthanasia of >20% body weight loss between 23 and 29 days (Figures 6B and 6C). In contrast, the CRISPR + Donor mice showed only a transient decrease in body weight around day 20, after which time the animals fully recovered (Figures 6B and 6C). Integration analysis by PCR showed a strong enrichment for HDR-modified Apoa1 alleles versus NHEJ insertion (Figure S15A). FAH protein levels were restored to ∼50% (Figure 6D), along with high levels of 2A-tagged apoA1 in the plasma (Figures S15B and S15C). Control livers showed no FAH staining and a histopathology typical of HT-I including enlarged hepatocytes, vacuolation, and immune cell infiltration (Figure 6E). Liver function was restored in the CRISPR + Donor mice based on improvements in blood chemistry (Figure S16) and liver morphology (Figure 6F). As a sign of integration, expression and function of the Apoa1-targeted FAH, the corrected hepatocytes can clonally expand in this disease model (Figure 6F). Despite the high rate of Apoa1 targeting and expansion of targeted cells, CRISPR + Donor mice showed unaffected levels of plasma apoA1 (Figure S17). These data show that Apoa1-targeting enables correction of a severe metabolic liver disorder.
Figure 6.

Correction of HT-I through targeted integration at the Apoa1 locus
(A) Adult Fah−/− mice were intraperitoneally injected with either saline (control) or AAV-CRISPR plus an AAV-Donor encoding a human FAH transgene (5 × 1011 GC each; CRISPR + Donor). NTBC was withdrawn at 7 days post-injection (time 0). Body weight was monitored over time up to 40 days and liver and plasma samples were collected for analyses. (B) Body weight ratios normalized to time zero (red line, CRISPR + Donor; green line, control mice). (C) Kaplan-Meier curve showing disease-free survival. Mice were euthanized upon loss of >20% body weight (red line, CRISPR + Donor; green line, control mice). One control mouse was found dead at day 30. (D) Western blot analysis of FAH in liver lysates with β-tub as a loading control. Plus (+) indicates a wild-type mouse liver as a positive control for endogenous Fah levels. (E) Representative FAH and hematoxylin and eosin staining in liver from control mice. (F) Representative FAH and hematoxylin and eosin staining in liver from CRISPR + Donor mice. Red squares are amplified in the images on the right. Scale bar is 100 μm. (A) Created with BioRender.
Discussion
Here, we identify the Apoa1 locus as a useful targeted integration site for liver-directed genome engineering. Apoa1 is highly transcriptionally active with accessible chromatin in both mouse and human livers. We show that the 3′ UTR of Apoa1 can be efficiently targeted with AAV-CRISPR, achieving correct HDR in up to ∼8% of the Apoa1 alleles. Furthermore, the endogenous Apoa1 promoter drives robust and sustained expression of several therapeutic proteins. Most importantly, we demonstrate functionality and correction of metabolic liver diseases using the Apoa1 locus.
Promoterless transgenes have been efficiently targeted to the Alb locus by using ZFN, CRISPR-Cas9, or without nucleases.18, 19, 20, 21, 22, 23 However, mutations in the ALB gene have been found in two independent cohorts of human HCC with a frequency of 13%.24,25 The role of these mutations in HCC susceptibility and pathology is not clear but raises questions about the long-term safety of this approach. Moreover, a slight decrease of plasma Alb levels has been recently reported following Alb targeting in preclinical models.22,23 To the best of our knowledge, mutations in the Apoa1 locus are not enriched in human HCC, nor have they been associated with cancer risk. Furthermore, there was no evidence of the following adverse outcomes: (1) Apoa1 3′ UTR targeting did not disrupt the gene or alter its expression level (Figures 2, 3, 4, 5, and S5), even when a high rate of targeting was observed (Figure S17); (2) expression of neighboring genes was not affected (Figure S5); (3) plasma cholesterol levels were unchanged suggesting preservation of the apoA1 function (Figure S6); and (4) we did not observe liver toxicity or tumors, at least within the time frame of these experiments (<6 months; Figures S6–S9 and S11). Finally, we recently demonstrated that our AAV-CRISPR system is liver restricted, resulting in no Cas9 activity in extra-hepatic tissues.32
Targeted integration in post-mitotic tissues, such as the adult liver, is limited by the very low rate of HDR-mediated repair. We determined the targeting frequency with three approaches: (1) ddPCR, (2) immunohistochemistry, and (3) co-expression of apoA1-2A and the transgenic proteins. In mice injected as adults with CRISPR + Donor, we observed the correct HDR integration in ∼1% of Apoa1 alleles, which resulted in ∼6% of mKate2-expressing hepatocytes (Figures 2E–2G). In mice injected at P4 with the same AAVs, we observed ∼8% of HDR-modified Apoa1 alleles and ∼16% positive hepatocytes (Figures 3D, 3F, and 3G). The greater relative ratio of HDR versus NHEJ editing events in the pups (Figures 3B versus 2B) could be partially due to a larger proportion of dividing cells at time of injection in the neonates. However, it should be noted that the dose used in pups was significantly higher than that used in adults relative to body weight. In a dose-ranging experiment in pups, we found that total editing events and positive hepatocytes were both reduced in a dose-dependent manner, but similar to adults when comparing equivalent doses (Figure S12). Therefore, the greater percentage of positive hepatocytes we observed in pups is mostly attributable to a higher AAV dose, rather than an increased propensity for HDR in neonatal liver. This rate is higher than what has been described previously, although a direct comparison is complicated by different viral dosages, routes of injection, transgenes, and time points.21 Importantly, all the transgenic proteins achieved their expected localization (cytoplasmic for mKate2 and FAH and secreted for APOE and FIX).
To test the potential of the Apoa1 locus for protein replacement therapy, we designed AAV-Donors encoding different therapeutic transgenes. We showed that the Apoa1 locus enables sustained expression of FIX (Figure 4G), the clotting factor deficient in patients with hemophilia B. In mice treated with AAV-CRISPR + AAV-Donor, FIX levels increased up to ∼230 ng/mL, which fall within the 2%–5% range required for therapeutic benefit.19 Barzel et al.19 previously reported much higher values of FIX produced from the Alb locus in adult injected-mice. However, a 2-fold higher dose of AAV-Donor was used as compared to our studies along with a different ELISA kit (homemade versus commercial in this study). We also showed that the Apoa1 locus drove the expression of comparable levels of another secreted therapeutic protein, APOE (Figure S14), whose deficiency results in familial hyperlipoproteinemia type III. As a result, the Apoa1-targeted expression of APOE dramatically reduced plasma lipids in a mouse model of hypercholesterolemia (Figure 5). Finally, we showed that the Apoa1-targeting could correct a severe metabolic liver disorder- HT-I (Figure 6). The targeted expression of FAH rescued lethality and restored liver function. These two disease models require different levels of gene correction for reaching therapeutic effects. While the Fah−/− model requires minimal gene correction for rescuing the hepatic toxicity,33 a frequency of at least ∼7% of HDR editing of Ldlr and ∼20% of edited hepatocytes was required for ameliorating plasma cholesterol in a mouse model of familial hypercholesterolemia.34 In agreement with these findings, we observed ∼8% of HDR-modified Apoa1 alleles and ∼16% positive hepatocytes when mice are injected with a high dose as neonates (Figure 3) along with the amelioration of hypercholesterolemia in the Apoe−/− mouse model (Figure 5).
When we compared the Apoa1-targeted expression of secreted therapeutic proteins in CRISPR + Donor versus Donor mice, we surprisingly observed only a modest increase in FIX plasma levels between the two groups (Figure 4G). These mice showed rates of HDR and NHEJ insertion events at the Apoa1 locus comparable with what was seen in the mKate2-targeted mice (Figures S13A–S13D and 2), suggesting that different AAV-Donors show similar targeted integration frequencies. We observed a ∼3-fold increase of total FIX transgene mRNA in CRISPR + Donor mice as compared to Donor mice (Figure S13F). This result is comparable to the relative difference in FIX levels detected by ELISA (Figure 4G). Despite this, we were not able to detect either the bicistronic mRNA by endpoint PCR or the 2A-tagged apoA1 in plasma from Donor mice. These data suggest that a significant amount of FIX in Donor-only mice may arise from either leaky expression or off-target integration events. In contrast to mKate2, both the FIX and APOE coding sequences retain the start codon in their respective Donor cassettes, potentially making them more amenable to translation. It is possible that expression of these secreted proteins could originate from either (1) cryptic promoter activity in the Donor cassette (i.e., ITR-driven expression), (2) from off-target integration events, or (3) secretion from non-hepatic tissues. The relative contributions of HDR targeting versus off-target integration with and without nucleases is a worthwhile area for future investigation.
Two major concerns with genome editing are the risk of off-target cutting, as well as unwanted editing events at the target locus. We observed very efficient on-target editing of ∼51%–54% (Figure 2C) in alleles that were not subject to HDR or NHEJ insertion. The Apoa1-targeting gRNA appears to be very specific, since no off-target activity was detected above background at the top 13 predicted sites (Figure S3). However, when we analyzed the integration events at the Apoa1 locus, we observed on-target NHEJ insertion of the AAV genome (Figures 2B, 3B, 5C, S13, and S14). By using a primer specific to the ITR sequence, we were able to quantify rates of ∼12% and 21% of Apoa1 alleles with NHEJ insertion in adult- and neonatal-injected mice, respectively (Figures 2D and 3C). It is possible that our analysis underestimates the total NHEJ insertion rate by not detecting the integration events that do not contain intact ITRs. Recently it has been reported that the majority of NHEJ-integrated AAV genomes contain ITR elements (up to 83.5%) and that all the ITR regions are detectable at relatively similar read counts.35 Despite the fact that most of the NHEJ insertion events were shown to not contain the full-length AAV genome,35 it is still possible that the integration of AAV-CRISPR at the Apoa1 locus could lead to an unwanted permanent expression of the Cas9. Overall, our data confirm previous reports on partial or full-length integration of AAV vectors at nuclease-induced double-strand breaks, observed not only in the liver but also in muscle, brain, heart, and cochlea.8,35, 36, 37, 38, 39 Therefore, NHEJ insertion of AAV genomes at the on-target site is a recurrent event and needs to be carefully evaluated in each context. This could be minimized by controlling the expression of Cas9 with inducible or self-deleting systems,36,40 or with alternative Cas9 delivery methods (i.e., nanoparticles, mRNA, or protein). Interestingly, in the experiments to correct HT-I, we found evidence of robust positive selection of correctly HDR-mediated edited cells over cells with NHEJ insertion. Combining Apoa1-targeting with synthetic selection strategies could be an effective way to selectively expand hepatocytes with HDR integration and merits further exploration.
In conclusion, we demonstrate that Apoa1 is a viable and useful integration site for liver-directed genome editing. Theoretically, any therapeutic transgene that does not exceed the cargo capacity of AAV could be targeted to the Apoa1 locus, making our method applicable to numerous genetic disorders. This platform is particularly valuable because it allows for permanent integration of entire transgenes, making therapies generalizable to larger patient populations and not mutation specific.
Materials and methods
ChIP-seq and ATAC-seq
Human and mouse liver ChIP-seq (H3K27 acetylation and RNA Pol II) and ATAC-seq datasets used in this study are listed in Table S2. With the exception of the mouse liver H3K27 acetylation ChIP-seq dataset, all other datasets were obtained from ENCODE project database (https://www.encodeproject.org/) as these datasets are highly validated. Relevant experiment, biosample and dataset descriptions, and accession numbers are provided in Table S2. Quality control reports for all ENCODE datasets can be found at https://www.encodeproject.org/ associated with the experiment summary accession numbers.
Mouse liver H3K27 acetylation ChIP-seq was performed for this study using the ChIPmentation protocol described at https://www.medical-epigenomics.org/papers/schmidl2015/.41 Briefly, hepatocytes were extracted from the livers of 8-week-old C57BL/6 mice using the protocol described in Foretz et al.42 1 million hepatocytes were cross-linked and sonicated for 6 cycles in order to shear chromatin in ∼200–400 bp length fragments. ChIPmentation was performed according to protocol and post amplification libraries were cleaned using AMPure bead size selection yielding a library of DNA fragments between 200–500 bp. Library was sequenced on an Illumina Next-Seq 500 to generate raw reads.
Both in-house and ENCODE ChIP-seq datasets were processed from raw read FASTQ files using the ENCODE (phase-3) transcription factor and histone ChIP-seq pipeline specification created by the Kundaje lab (https://github.com/NHLBI-BCB/TF_chipseq_pipeline). Alignments were performed on the NCBI GRCh38 (hg38) and GRCm38 (mm10) reference genomes.
ATAC-seq datasets were processed from raw read FASTQ files using the RIESLING pipeline43 developed by the Lin and Gordon laboratories (v2.8.1; https://github.com/GordonLab/riesling-pipeline).
ChIP-seq and ATAC-seq signal was plotted for specified loci from aligned read files (BAMs) that were filtered for duplicate reads using the Bamplot tool created from the Lin lab (https://github.com/linlabbcm/bamplot). This representation plots binned aligned read density in units of reads per million mapped reads per base pair (rpm/bp).
Plasmid design and cloning
gRNAs targeting the Apoa1 3′ UTR (exon 4) were designed by manual inspection based on the presence of a canonical NNGRRT PAM for SaCas9. Cloning of gRNAs was accomplished by annealing oligonucleotides (Sigma-Aldrich) and ligating into the BbsI sites of 1313-pAAV-U6-SA-BbsI-MluI-gRNA-HLP-SaCas9-HA-OLLAS-spA (Addgene 109314)36 to obtain 1507-pAAV-U6-Apoa1-gRNA2-SA-HLP-SaCas9-HA-OLLAS-spA. This plasmid encodes both the gRNA under the control of a U6 promoter and SaCas9 under the control of the hybrid liver-specific promoter (HLP).44 The AAV donor plasmids were generated by gene synthesis (Integrated DNA Technologies) and standard molecular biology approaches. The donor plasmids include exon 4 of the murine Apoa1 gene (NCBI: NC_000075.7) but introduce the transgene coding sequences in place of the endogenous stop codon, flanked by homology arms to the Apoa1 locus on each side (5′ homology region, 700 bp; 3′ homology region, 480 bp). In place of the stop codon, each donor plasmid includes an XbaI cloning site (encoding serine, arginine), followed by a glycine-proline-glycine-P2A sequence (referred as 2A) in frame with the transgene coding sequence, followed by a small synthetic poly(A) signal. Plasmids 1729-pAAV-Apoa1-Target-2A-mKate-pA, 1730-pAAV-Apoa1-Target-2A-APOE-pA, 1731-pAAV-Apoa1-Target-2A-FIX-pA and 1771-pAAV-Apoa1-Target-2A-FAH-pA encode the mKate2-FLAG (gift of Dr. Joshua Wythe, Baylor College of Medicine), APOE, FIX, and FAH transgenes, respectively. Complete vector sequences are included in the Supplemental information, and plasmids will be made available through Addgene upon publication.
AAV production
Recombinant AAV8 vectors were generated as previously described45 with several modifications.46 Plasmids required for AAV packaging, adenoviral helper plasmid pAdDeltaF6 (PL-F-PVADF6), and AAV8 packaging plasmid pAAV2/8 (PL-T-PV0007) were obtained from the University of Pennsylvania Vector Core. Each AAV transgene construct was co-transfected with the packaging constructs into 293T cells (ATCC, CRL-3216) using polyethylenimine (PEI). Cell pellets were harvested and purified using a single cesium chloride density gradient centrifugation. Fractions containing AAV vector genomes were pooled and then dialyzed against PBS using a 100 kD Spectra-Por Float-A-Lyzer G2 dialysis device (Spectrum Labs, G235059) to remove the cesium chloride. Purified AAV were concentrated using a Sartorius Vivaspin Turbo 4 Ultrafiltration Unit (VS04T42) and stored at −80°C until use. AAV titers were calculated after DNase digestion using qPCR relative to a standard curve of the transgene plasmid. Primers used for titer are included in Table S3.
Animals
C57BL/6J (stock number: 000664) and Apoe−/− (B6.129P2-Apoetm1Unc/J; stock number: 002052) mice were obtained from The Jackson Laboratories. Fah−/− mice were generated by Dr. Karl-Dimiter Bissig and maintained as an in-house breeding colony at Baylor College of Medicine. Animals were allowed free access to food and water and maintained on a standard chow diet. All mice used were male, except for mixed sex mice used in experiment described in Figures 3 and S10–S12. Where indicated, Apoe−/− mice were fed a western diet (0.21% cholesterol and 21% fat, D12079B, Research Diets). Male Fah−/− mice were kept on 16 mg/L NTBC (Lab Network) in the drinking water before experiments. When NTBC was withdrawn, mice were monitored daily and euthanized upon loss of >20% body weight. AAV8 vectors (5 × 1011 genome copies [GC] each) were diluted in 300 μL of sterile saline and intraperitoneally injected to 8-week-old mice. For delivery at P4, AAV (5 × 1011, 5 × 1010, or 2.5 × 1010 GC each) were diluted in 50 μL sterile saline and delivered subcutaneously. Control mice were injected with AAV8-CB-EGFP (AAV-GFP), or just saline where indicated. All treatment conditions were randomly allocated within each cage of mice at the time of injection. Mice were fasted 5 h prior to injection and again before subsequent blood collection. Blood was collected via retro-orbital bleeding using heparinized Natelson collection tubes, and plasma was isolated by centrifugation at 10,000 × g for 20 min at 4°C. All experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC) and performed in accordance with institutional guidelines under protocol number AN-6243.
Integration PCR
Genomic DNA was extracted from livers using the DNeasy Blood and Tissue kit (QIAGEN) following the manufacturer’s protocol. The Apoa1 locus was PCR-amplified to detect integration of the AAV-Donor by using a forward primer within an endogenous genomic region upstream of the 5′ homology region and the reverse primer within the transgene cassette. APEX TaqRed Master Mix (Apex Bioresearch Products) was used following the manufacturer’s protocol and the PCR products were separated by agarose gel electrophoresis. Primers sequences are available in Table S3.
Targeted deep sequencing
Off-target sites for the Apoa1 gRNA were determined using the online bioinformatics tool COSMID at https://crispr.bme.gatech.edu/.47 Searches were completed on the least stringent settings (up to three indels, two one-base deletions, two one-base insertions). In addition, we allowed for some leniency in the PAM motif by searching both NNGRRT and NNGRR PAM sequences. Primers specific to the Apoa1 and to the 15 top-scored off-target loci were used to amplify these genomic sites with Herculase II Fusion DNA Polymerase (Agilent Technologies). PCR amplification failed for two off-target loci (OT9 and OT13). Secondary PCR was performed using 2 μL of the primary PCR product to add barcode sequences and the Illumina P5 and P7 adaptor sequences to each amplicon. The final barcoded amplicons were purified using magnetic beads, pooled in equimolar amounts, and sequenced using an Illumina MiSeq. Alignment of sequence reads to reference sequences and indel quantification was carried out as previously described.36 Indel data and deep sequencing primers are provided in Tables S1 and S3.
ICE analysis
Primers were designed to amplify the Apoa1 locus binding at least 350 bp away from the cut site at each side (Table S3). PCR was performed using Phusion DNA polymerase (Roche) and the 830 bp product was separated by agarose gel electrophoresis and gel extracted using the QIAquick gel extraction kit (QIAGEN). The PCR product was Sanger sequenced and the indel percentage was determined by Inference of CRISPR Edits (ICE) analysis (https://ice.synthego.com/#/) using a control chromatogram for comparison.
ddPCR
10 units of BamHI or MscI restriction enzymes (New England Biolabs) were used for fragmentation of 1 μg of genomic DNA, based on insensitivity to methylation, absence of restriction site in the targeted amplicon, and specific amplification of either the NHEJ- or HDR-integrated Apoa1 alleles. MscI cuts within the ITR regions enabling the specific amplification of the HDR-integrated alleles by using a forward primer binding the Apoa1 locus upstream of the 5′ homology region and a reverse primer in the 2A sequence of AAV-Donor. For the NHEJ-insertion amplification, genomic DNA was digested with BamHI and a reverse primer was designed to anneal within the ITR regions enabling the detection of NHEJ-insertion of either AAV-CRISPR or AAV-Donor, at either orientation. A genomic region spanning exon 1 and intron 3 of Apoa1 was PCR-amplified as reference for the total number of genome copies. Probes were designed to anneal within each specific amplicon by using Primer 3 plus (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and synthesized by Integrated DNA Technologies with FAM/Black Hole modifications. Each 20 μL PCR reaction contained 50 ng of appropriate digested DNA, 0.9 μM of each primer, 0.25 μM of FAM-probe, and 1× ddPCR Supermix for Probes (Bio-Rad Laboratories), which was combined with 70 μL of Droplet Generation Oil for Probes in a QX100 Droplet Generator (Bio-Rad Laboratories). Droplets were transferred into a 96-well plate and PCR-amplified using a three-step thermal cycling protocol with 2 min of extension time for 40 cycles. Droplet fluorescence was read using a QX100 Droplet Reader and analyzed with QuantaSoft Software (Bio-Rad Laboratories). ddPCR data are expressed as percentage of specific integration events relative to total genome copies. Primer and probe sequences used for ddPCR are listed in Table S3.
Immunohistochemistry
Livers were formalin-fixed for 24 h and then gradually dehydrated with ethanol and paraffin-embedded. Immunohistochemistry and hematoxylin and eosin staining were performed by the Texas Digestive Diseases Morphology Core at Baylor College of Medicine. Briefly, liver sections were deparaffinized and subjected to antigen retrieval with Target Retrieval Solution (S1699, DAKO). The sections were then incubated with 3% hydrogen peroxide, followed by incubation in normal serum to block nonspecific protein binding. Sections were incubated 1 h at room temperature with the following primary antibodies: anti-Ki67 (1:60, CRM325, Biocare), anti-FLAG (1:5000, 600-401-383, Rockland Antibodies), and anti-FAH (1:200, SAB2108553, Sigma-Aldrich). The Ki67 and FLAG antibodies were then detected respectively with a Rabbit-on-Rodent HRP-Polymer (RMR622H, Biocare) and ImmPRESS Horse Anti-Rabbit IgG Polymer Kit (MP-7401, Vector Laboratories) and visualized with DAB chromogen (DB801, Biocare). The FAH antibody was detected with a polymer and visualized with DAB chromogen (Leica Bond Polymer Detection kit, DS9800). TUNEL stainings were performed and detected using ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore, S7100). All slides were counterstained with hematoxylin, dehydrated, and mounted with a permanent mounting medium. A Nikon Ci-L bright field microscope was used for imaging at the Integrated Microscopy core (Baylor College of Medicine). Direct mKate2 fluorescence on liver slices was imaged by Leica DMIL LED Inverted Fluorescence Microscope. FLAG-positive cells quantification was performed by manual count of positive hepatocytes in ten 200× magnification images per liver taken across the whole section. Nuclei were quantified using ImageJ.48
Western blot
Liver tissue was homogenized in ∼10 volumes of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 150 mM sodium chloride, and protease inhibitors [Roche 11836153001]) using a Bead Blaster 24 (Benchmark D2400). Protein concentrations were determined using BCA assay (Thermo-Pierce #23227). Liver lysates (50 μg) or plasma (1 μL of 1:2 dilution) were diluted in 4× LDS buffer (Life Technologies, Ref. NP0007) supplemented with 5% beta-mercaptoethanol and separated by SDS-PAGE using 4%–12% gradient gels (Life Technologies NP0322BOX). Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore IPFL00010) followed by blocking for 2 h at room temperature in a 2:1 solution of Odyssey Blocking Buffer (Li-Cor 927-40000) and PBS with 0.05% Tween-20 (PBS-T). Primary antibodies to the FLAG tag (1:5,000, rabbit, 600-401-383, Rockland), 2A peptide (1:5,000, rabbit, ABS31, Sigma-Aldrich), APOE (1:1,000, rabbit, ab52607, Abcam), FAH (1:1,000, rabbit, SAB2100745, Sigma-Aldrich), apoB (1:5,000, rabbit, K23300R, Meridian), apoA1 (1:5,000, rabbit, K23500R, Meridian), alpha-1 antitrypsin (Aat; 1:1,000, rabbit, 16382-1-AP, Proteintech), and beta tubulin (1:500, mouse, University of Iowa Developmental Studies Hybridoma Bank E7) were diluted in 1% BSA in PBS-T and membranes were incubated overnight at 4 degrees. Goat anti-rabbit 680 nm and anti-mouse 800 nm antibodies (1:15,000, 611-144-002-0.5, and 610-145-002-0.5, Rockland) were incubated at room temperature for 1 h and imaged using an Odyssey Classic (Li-Cor). Densitometry analyses were performed using Image Studio Lite Version 5.2.5 and data are presented as normalized to loading control.
Plasma analysis
Alanine aminotransferase (ALT) was measured using the Teco ALT (SGPT) Kinetic Liquid Kit (A524-150). Total cholesterol was measured using the Wako Cholesterol E kit (999–02 601). Total triglycerides were measured using Infinity Triglycerides Reagent (TR22421 Thermo Fisher). Human APOE was measured in 1:12.5 diluted plasma by Human APOE ELISA Kit (ab108813, Abcam), following the manufacturer’s protocol. Human FIX measurement was measured in 1:50 diluted plasma by Factor IX Human SimpleStep ELISA Kit (ab188393, Abcam), following the manufacturer’s protocol. Plasma from Fah−/− mice was analyzed for ALT, aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), and total bilirubin by the pathology core (Center for Comparative Medicine at Baylor College of Medicine).
RNA isolation and analysis
RNA was isolated from liver using RNeasy Mini Kit (74106, QIAGEN), following the manufacturer’s protocol. 1 μg of RNA was used for generating cDNA by the iScript cDNA synthesis kit (170-8891, Bio-Rad). cDNA was diluted 1:25 and used as template for qPCR analysis using iTaq Universal SYBR Green Supermix (1725124, Bio-Rad) and ViiA 7 Real-Time PCR System (Thermo Fisher Scientific). In FIX qPCR, RNA was treated with DNase I and qPCR was performed also on RT (–) reactions as control for any potential contamination with AAV genome DNA. A dissociation curve was carried out at the end of qPCR for assessing the homogeneity of the PCR products. Relative gene expression was calculated using the ΔΔCt method and graphed as fold change relative to β-Actin. For detecting the bicistronic Apoa1-2A-FIX mRNA, 50 ng of cDNA were amplified using a forward primer binding the exon 3 of Apoa1 and the reverse primer binding the 2A sequence. APEX TaqRed Master Mix (Apex Bioresearch Products) was used following the manufacturer’s protocol and the PCR products were separated by agarose gel electrophoresis. All primers for qPCR and end point PCR are listed in Table S3.
Statistics
Graphpad Prism 7 was used for statistical analyses. All data are shown as the mean ± standard deviation. Comparisons involving two groups were evaluated by a two-tailed Student’s t test. For comparisons involving three or more groups, a one-way or two-way ANOVA was applied, with Tukey’s or Bonferroni’s post-test used to test for significant differences among groups. In all cases, significance was assigned at p < 0.05.
Acknowledgments
This work was supported by The National Institutes of Health (HL132840 and U42OD026645 to W.R.L., DK115461 and HL134510 to K.-D.B., and UG3HL151545 to G.B. and W.R.L.); The Cancer Prevention and Research Institute of Texas (RR140081 to G.B.); and The American Heart Association (19PRE34380467 to A.M.D. and 19POST34430092 to M.D.G.); This work was also supported by the Texas Digestive Diseases Morphology Core (P30DK56338) and the Integrated Microscopy Core (DK56338 and CA125123).
Author contributions
M.D.G., K.-D.B., G.B., and W.R.L. conceived the project and designed the studies; M.D.G. performed and analyzed most of the experiments; A.H. produced the viral vectors; M.D.G., A.L., A.H., M.B., and A.M.D. performed the in vivo experiments; N.A.C., H.E.S., C.Y.L., and J.D.B. performed ChIP-seq and ATAC-seq analysis; A.L. and M.D.G. performed analysis of genome editing; M.D.G. and W.R.L. wrote the manuscript, which was revised and approved by all authors.
Declaration of interests
C.Y.L. is an executive and shareholder of Kronos Bio. A provisional patent application has been filed by Baylor College of Medicine entitled “Selective expansion of gene targeted cells” on November 25, 2019. The inventors on this patent are W.R.L., A.H., Kelsey Jarrett, K.-D.B., M.D.G., and Mia Furgurson. The strategy of targeting the Apoa1 locus for docking therapeutic transgenes described in this manuscript is a component of the patent application.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.04.011.
Supplemental information
References
- 1.Pampols T. Inherited metabolic rare disease. Adv. Exp. Med. Biol. 2010;686:397–431. doi: 10.1007/978-90-481-9485-8_23. [DOI] [PubMed] [Google Scholar]
- 2.De Giorgi M., Lagor W.R. Gene Delivery in Lipid Research and Therapies. Methodist DeBakey Cardiovasc. J. 2019;15:62–69. doi: 10.14797/mdcj-15-1-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers A.E., Shaka J.A., Pechet G., MacDonald R.A. Regeneration of the liver. Absence of a “humoral factor” affecting hepatic regeneration in parabiotic rats. Am. J. Pathol. 1961;39:561–578. [PMC free article] [PubMed] [Google Scholar]
- 5.Magami Y., Azuma T., Inokuchi H., Kokuno S., Moriyasu F., Kawai K., Hattori T. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver. 2002;22:419–425. doi: 10.1034/j.1600-0676.2002.01702.x. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham S.C., Spinoulas A., Carpenter K.H., Wilcken B., Kuchel P.W., Alexander I.E. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol. Ther. 2009;17:1340–1346. doi: 10.1038/mt.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L., Bell P., Lin J., Calcedo R., Tarantal A.F., Wilson J.M. AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta) Mol. Ther. 2011;19:2012–2020. doi: 10.1038/mt.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L., Yang Y., Breton C., Bell P., Li M., Zhang J., Che Y., Saveliev A., He Z., White J. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020;6:eaax5701. doi: 10.1126/sciadv.aax5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen K.T., Fløe L., Petersen T.S., Huang J., Xu F., Bolund L., Luo Y., Lin L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. doi: 10.1002/1873-3468.12707. [DOI] [PubMed] [Google Scholar]
- 10.Lombardo A., Cesana D., Genovese P., Di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., Lo Riso P. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 11.Smith J.R., Maguire S., Davis L.A., Alexander M., Yang F., Chandran S., ffrench-Constant C., Pedersen R.A. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells. 2008;26:496–504. doi: 10.1634/stemcells.2007-0039. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ordovás L., Boon R., Pistoni M., Chen Y., Wolfs E., Guo W., Sambathkumar R., Bobis-Wozowicz S., Helsen N., Vanhove J. Efficient Recombinase-Mediated Cassette Exchange in hPSCs to Study the Hepatocyte Lineage Reveals AAVS1 Locus-Mediated Transgene Inhibition. Stem Cell Reports. 2015;5:918–931. doi: 10.1016/j.stemcr.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klatt D., Cheng E., Hoffmann D., Santilli G., Thrasher A.J., Brendel C., Schambach A. Differential Transgene Silencing of Myeloid-Specific Promoters in the AAVS1 Safe Harbor Locus of Induced Pluripotent Stem Cell-Derived Myeloid Cells. Hum. Gene Ther. 2020;31:199–210. doi: 10.1089/hum.2019.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papapetrou E.P., Schambach A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016;24:678–684. doi: 10.1038/mt.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura Y., Shofuda T., Higuchi Y., Nagamori I., Oda M., Nakamori M., Onodera M., Kanematsu D., Yamamoto A., Katsuma A. Human Genomic Safe Harbors and the Suicide Gene-Based Safeguard System for iPSC-Based Cell Therapy. Stem Cells Transl. Med. 2019;8:627–638. doi: 10.1002/sctm.18-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadelain M., Papapetrou E.P., Bushman F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2011;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 18.Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porro F., Bortolussi G., Barzel A., De Caneva A., Iaconcig A., Vodret S., Zentilin L., Kay M.A., Muro A.F. Promoterless gene targeting without nucleases rescues lethality of a Crigler-Najjar syndrome mouse model. EMBO Mol. Med. 2017;9:1346–1355. doi: 10.15252/emmm.201707601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Caneva A., Porro F., Bortolussi G., Sola R., Lisjak M., Barzel A., Giacca M., Kay M.A., Vlahoviček K., Zentilin L., Muro A.F. Coupling AAV-mediated promoterless gene targeting to SaCas9 nuclease to efficiently correct liver metabolic diseases. JCI Insight. 2019;5:e128863. doi: 10.1172/jci.insight.128863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou L., Przybilla M.J., Ahlat O., Kim S., Overn P., Jarnes J., O’Sullivan M.G., Whitley C.B. A Highly Efficacious PS Gene Editing System Corrects Metabolic and Neurological Complications of Mucopolysaccharidosis Type I. Mol Ther. 2020;28:1442–1454. doi: 10.1016/j.ymthe.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J.P., Cheng X.X., Zhao M., Li G.H., Xu J., Zhang F., Yin M.D., Meng F.Y., Dai X.Y., Fu Y.W. Curing hemophilia A by NHEJ-mediated ectopic F8 insertion in the mouse. Genome Biol. 2019;20:276. doi: 10.1186/s13059-019-1907-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulze K., Imbeaud S., Letouzé E., Alexandrov L.B., Calderaro J., Rebouissou S., Couchy G., Meiller C., Shinde J., Soysouvanh F. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandler R.J., Venturoni L.E., Liao J., Hubbard B.T., Schneller J.L., Hoffmann V., Gordo S., Zang S., Ko C.W., Chau N. Promoterless, nuclease-free genome editing confers a growth advantage for corrected hepatocytes in mice with methylmalonic acidemia. Hepatology. 2020 doi: 10.1002/hep.31570. Published online September 25, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson N.L., Anderson N.G. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 28.Eggerman T.L., Hoeg J.M., Meng M.S., Tombragel A., Bojanovski D., Brewer H.B., Jr. Differential tissue-specific expression of human apoA-I and apoA-II. J. Lipid Res. 1991;32:821–828. [PubMed] [Google Scholar]
- 29.Pankowicz F.P., Jarrett K.E., Lagor W.R., Bissig K.D. CRISPR/Cas9: at the cutting edge of hepatology. Gut. 2017;66:1329–1340. doi: 10.1136/gutjnl-2016-313565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meir K.S., Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- 31.Lindstedt S., Holme E., Lock E.A., Hjalmarson O., Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 32.De Giorgi M., Jarrett K.E., Burton J.C., Doerfler A.M., Hurley A., Li A., Hsu R.H., Furgurson M., Patel K.R., Han J. Depletion of essential isoprenoids and ER stress induction following acute liver-specific deletion of HMG-CoA reductase. J. Lipid Res. 2020;61:1675–1686. doi: 10.1194/jlr.RA120001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H., Li Y., He L., Pu W., Yu W., Li Y., Wu Y.T., Xu C., Wei Y., Ding Q. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation. 2020;141:67–79. doi: 10.1161/CIRCULATIONAHA.119.042476. [DOI] [PubMed] [Google Scholar]
- 35.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li A., Lee C.M., Hurley A.E., Jarrett K.E., De Giorgi M., Lu W., Balderrama K.S., Doerfler A.M., Deshmukh H., Ray A. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2018;12:111–122. doi: 10.1016/j.omtm.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L., Yang Y., Breton C.A., White J., Zhang J., Che Y., Saveliev A., McMenamin D., He Z., Latshaw C. CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood. 2019;133:2745–2752. doi: 10.1182/blood.2019000790. [DOI] [PubMed] [Google Scholar]
- 39.Pan X., Philippen L., Lahiri S.K., Lee C., Park S.H., Word T.A., Li N., Jarrett K.E., Gupta R., Reynolds J.O. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018;123:953–963. doi: 10.1161/CIRCRESAHA.118.313369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin H., Song C.Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015;12:963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Semenkovich N.P., Planer J.D., Ahern P.P., Griffin N.W., Lin C.Y., Gordon J.I. Impact of the gut microbiota on enhancer accessibility in gut intraepithelial lymphocytes. Proc. Natl. Acad. Sci. USA. 2016;113:14805–14810. doi: 10.1073/pnas.1617793113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McIntosh J., Lenting P.J., Rosales C., Lee D., Rabbanian S., Raj D., Patel N., Tuddenham E.G., Christophe O.D., McVey J.H. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121:3335–3344. doi: 10.1182/blood-2012-10-462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lagor W.R., Johnston J.C., Lock M., Vandenberghe L.H., Rader D.J. Adeno-associated viruses as liver-directed gene delivery vehicles: focus on lipoprotein metabolism. Methods Mol. Biol. 2013;1027:273–307. doi: 10.1007/978-1-60327-369-5_13. [DOI] [PubMed] [Google Scholar]
- 46.Jarrett K.E., Lee C., De Giorgi M., Hurley A., Gillard B.K., Doerfler A.M., Li A., Pownall H.J., Bao G., Lagor W.R. Somatic Editing of Ldlr With Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arterioscler. Thromb. Vasc. Biol. 2018;38:1997–2006. doi: 10.1161/ATVBAHA.118.311221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.