

Abstract
Objectives
Long‐term adrenocorticotropic therapy (LT‐ACTH), which consisted of 2‐4 weeks of daily injections of adrenocorticotropic hormone (ACTH) and subsequent months of weekly injections, was tried for relapsed West syndrome (WS) or other intractable epilepsies in small case reports. Our aim was to explore the efficacy of LT‐ACTH for preventing WS relapse, as well as the prevalence of its adverse events.
Methods
This is a retrospective, nationwide, multicenter case series of patients with WS who underwent LT‐ACTH. Clinical information of the patients and protocol of LT‐ACTH were collected from participating institutes in this study. We defined clinical response to ACTH as achievement of hypsarrhythmia and epileptic spasms resolution. Patients who responded to daily ACTH injections were identified and assessed whether they experienced WS relapse during/after the weekly ACTH injection period. The outcome was measured by the nonrelapse rate at 24 months after daily ACTH injections using the Kaplan‐Meier method.
Results
Clinical information of 16 children with WS was analyzed. The median age at LT‐ACTH initiation was 14.5 months (range: 7‐68 months). Thirteen (81%) patients had previously undergone conventional ACTH treatment. The LT‐ACTH regimens comprised a median of 16 days of daily injections (range: 11‐28 days) and 10 months of weekly injections (range: 3‐22 months). Seven patients experienced WS relapse during/after subsequent weekly ACTH period, and the nonrelapse rate at 24 months after daily injections was estimated at 60.6% (95% confidence interval: 32.3%‐80.0%). Height stagnation, hypertension, and irritability were observed; lethal adverse events were not reported.
Significance
Our study firstly explored the efficacy of LT‐ACTH for preventing WS relapse. LT‐ACTH might be a treatment option for patients with relapsed or intractable WS; however, we note that our study is limited by its small sample size and the lack of an appropriate control group.
Keywords: adrenocorticotropic hormone, long‐term treatment, Nationwide survey, retrospective case series, West syndrome
Key Points.
First retrospective, nationwide, multicenter case series of long‐term ACTH therapy (LT‐ACTH) for patients with West syndrome (WS)
Daily and weekly ACTH injections were administered for a median duration of 16 days (range: 11‐28 days) and 10 months (range: 11‐28 months), respectively
The nonrelapse rate of WS symptoms observed at 24 months after the daily ACTH injections was estimated at 60.6% (95% confidence interval: 32.3%‐80.0%)
The adverse events reported during the weekly ACTH injections included height stagnation, hypertension, and irritability
1. INTRODUCTION
West syndrome (WS) is an age‐dependent, epileptic encephalopathy characterized by epileptic spasms (ES) in clusters, hypsarrhythmia on electroencephalogram (EEG), and developmental stagnation or regression. 1 , 2 , 3 , 4 WS mainly occurs in the first 2 years of life, and its incidence is estimated to be around 2‐5 per 10 000 live births. 5 , 6 WS is associated with further intellectual disability, 7 autism spectrum disorder, 8 and subsequent development of other types of epilepsy including refractory epileptic encephalopathy, such as Lennox‐Gastaut syndrome (LGS). 9 Early treatment administration is desired, as delays in diagnosis and treatment 10 or prolonged hypsarrhythmia 11 are associated with poor intellectual outcomes.
Treatment using adrenocorticotropic hormone (ACTH) is widely used for West syndrome. The response rates of ES and hypsarrhythmia to ACTH range from 38.8%‐86.6%. 12 , 13 The ideal treatment regimen remains controversial, as insufficient data exist to define the optimal preparation, dosage, and duration of treatment. 1 , 2 , 3 , 4 Low‐dose short‐term regimens could reduce the risk of serious adverse events and have comparable effectiveness to high‐dose long‐term regimens. 14 , 15 , 16 , 17 , 18 However, the effect of ACTH treatment duration on its effectiveness for WS is unclear.
Evidence is scarce regarding the management of patients with WS who lack a sufficient response to initial ACTH therapy or who relapse soon after. Secondary treatment regimen effectiveness is often inferior to the initial regimen since failed initial ACTH therapy is a poor prognostic factor for long‐term seizure control. 19 Vigabatrin is a good treatment option for patients who did not benefit from initial ACTH therapy, 20 but vigabatrin may not be suitable for long‐term treatment because cumulative vigabatrin doses increase the risk of permanent visual field loss. 21 , 22 Moreover, vigabatrin was not available until 2016 in Japan; therefore, clinicians in Japan had limited treatment options for managing relapsed or intractable WS.
A study in 2008 reported successful long‐term weekly ACTH therapy in three patients with WS who had relapsed after 2‐3 weeks of conventional ACTH treatment. 23 This regimen consisted of 2‐3 weeks of daily ACTH injections followed by 7 months to 1 year of weekly or biweekly injections. This regimen has been employed for various intractable epilepsies. 24 , 25 , 26 Long‐term, weekly ACTH therapy might be a good treatment option for pediatric patients with intractable epilepsy; however, as there has been no relevant large cohort study, and its effectiveness, ideal protocol, patients assumed to be good candidates, and incidence of adverse effects are not known.
In this article, we present a retrospective analysis of a multicenter series of patients with WS who received long‐term weekly ACTH therapy. The main goals of this study were to explore the efficacy of long‐term weekly ACTH therapy for WS patients for preventing relapse. We also investigated the incidence of adverse events related to the long‐term therapy. Finally, we hope to find an effective long‐term regimen and identify candidate factors that might be associated with the treatment response.
2. METHODS
2.1. Definition of long‐term ACTH therapy
Based on previous case reports, 23 , 24 , 25 , 26 we defined long‐term ACTH therapy (LT‐ACTH) as the consecutive combination of daily and weekly injection periods (Figure 1). The daily injection period lasted the first 1‐4 weeks, while the weekly injection period involved weekly or biweekly injections for the subsequent 3 months or longer.
FIGURE 1.
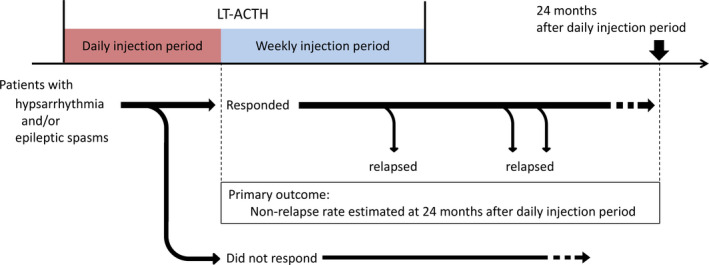
The LT‐ACTH procedure and assessment protocol. Patients with WS who responded to the daily ACTH injections were followed up and checked whether they experienced WS relapse during/after the subsequent weekly injection period. The nonrelapse rate of WS symptoms was estimated at 24 months after the daily injection period (primary outcome). Abbreviations: LT‐ACTH, long‐term adrenocorticotropic hormone therapy; WS, West syndrome
2.2. Retrospective patient identification
On February 2018, we recruited patients with pediatric‐onset intractable epilepsy scheduled to receive weekly or biweekly ACTH injections. To identify suitable patients for participation, we used the Zao Seminar Mailing list, which is comprised of >1000 pediatric neurologists in Japan (available from: http://square.umin.ac.jp/shiihara/), and questionnaires were sent to 484 institutional facilities of pediatric neurologists certified by the Japanese Society of Child Neurology. Consent for submitting data to the study was obtained from the physicians in charge of the patients or from the head of the relevant department.
2.3. Clinical information
In March 2018, we sent detailed questionnaires to pediatric neurologists who had administered LT‐ACTH to patients meeting the inclusion criteria. We retrospectively collected the following information: gender, age at onset of epilepsy, gestational age at birth, weight at birth, early childhood life‐threatening events, developmental assessment, physical anomalies, epilepsy syndrome, seizure types, etiology, EEG findings, magnetic resonance imaging findings, genetic and metabolic test findings, treatments attempted before LT‐ACTH, age at LT‐ACTH initiation, clinical response to LT‐ACTH, change of antiepileptic drugs (AEDs) during LT‐ACTH, and adverse events during LT‐ACTH. As there was no standardized protocol for LT‐ACTH, we also collected information about the protocol used in each institute, such as duration of daily or weekly injection period and dose of ACTH per an injection.
We conducted development assessment based on the model established by Knupp et al. 20 We obtained the clinician's perception of the overall development, motor, and cognitive status of each patient (normal, mild or equivocal delay, or definite abnormality) upon initiating the first ACTH and LT‐ACTH treatments. We used these three domains to categorize the overall development assessment as normal, mild delay, moderate delay, or severe delay.
We used the International League Against Epilepsy (ILAE) 2017 classification of seizure types to categorize them into ES, generalized onset, focal onset, and unknown onset seizure. 27 The pediatric neurologists were allowed to add detailed symptoms for each seizure. If a patient presented multiple seizure types, each seizure was assessed and classified into the four aforementioned groups. We did not collect ictal EEG information.
We classified the EEG findings into the following five groups: hypsarrhythmia (including modified hypsarrhythmia variants 2 ), diffuse slow spike and wave (DSSW), diffuse or multifocal epileptic discharges, focal epileptic discharges, and no epileptic discharges.
Based on the ILAE guidelines, we classified the etiology into the following six categories: structural cerebral abnormality, genetic, infectious, metabolic, immune, and unknown. 28
Each collaborating physician was asked whether he or she had observed specific adverse events during the daily/weekly injection period. The participating physicians were asked to describe the administered treatments to address each adverse event, the necessity of terminating the daily/weekly injection, and any unlisted adverse events as required. We did not standardize the definitions of events or treatment protocols (eg, definition of hypertension or timing of any treatment for irritability) performed at each hospital.
We excluded those patients who exhibited epilepsy syndromes other than WS. Among patients with WS, we excluded patients who did not present both hypsarrhythmia and ES at LT‐ACTH administration, since we aimed to assess the efficacy of LT‐ACTH for these conditions.
2.4. Assessment of clinical response to LT‐ACTH
Figure 1 presents the assessment scheme. Firstly, we identified the patients who responded to daily ACTH injections—that is, who achieved the disappearance of both hypsarrhythmia and ES, as well as the lack of DSSW or generalized tonic seizure. Clinical information of these patients was extracted. These patients were followed up to determine whether they experienced WS relapse during/after the subsequent weekly injection period. WS relapse was defined as the reappearance of either hypsarrhythmia or ES. We set the primary outcomes as the estimated nonrelapse rate of WS at 24 months after daily injections.
We assessed the EEG findings and seizure condition for each patient at 3 points: before initiation of daily injections, before weekly injection period, and after the weekly injection period. At each point, we checked whether hypsarrhythmia or ES disappeared or relapsed. If seizures other than ES were newly observed during the LT‐ACTH, they were classified as generalized onset, focal onset, or unknown onset seizure. We did not regard resolution of hypsarrhythmia with presentation of DSSW or resolution of ES with newly developed generalized tonic seizures as a favorable outcome. This determination was made because we could not rule out the possibility that the epilepsy transformed to LGS. Therefore, patients who developed DSSW or generalized tonic seizure at the end of the daily injection period were excluded from the time course analysis of WS relapse.
Regarding patients who did not respond to daily ACTH injections, we did not include them in further analysis; instead, we described each course of treatment anecdotally focusing on whether they experienced EEG/seizure remission or exacerbation during/after the weekly injection period.
We additionally tried to seek candidate risk factors of WS relapse by comparing their background information, regimen of LT‐ACTH, and EEG/seizure condition between patients who experienced WS relapse during/after the weekly injection period and those who did not. We did not conduct statistical analysis for comparing these two groups, as our cohort size was too small to derive statistically reliable results.
2.5. Statistical analysis
The time course for occurrence of WS relapse from the weekly injection period was analyzed by the Kaplan‐Meier method. We regarded a P‐value <.05 as statistically significant. All statistical analyses were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), 29 a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria, version 3.6.0).
2.6. Ethical approvals
This study was approved by the institutional ethics committee of Seirei Hamamatsu General Hospital on January 17, 2018 (examination number 2649).
3. RESULTS
3.1. Patients and clinical information
Figure 2 presents the schemes of patients' identification. We collected clinical information regarding 29 patients from 20 hospitals. Among these patients, we excluded seven patients: two patients with LGS, one with ES without hypsarrhythmia, one with atypical benign partial epilepsy, two with epilepsy with continuous spike and wave during sleep, and one with WS who did not present both hypsarrhythmia and ES at LT‐ACTH initiation. Twenty‐one patients received daily injections and subsequent weekly injections. One patient who only received weekly injections was excluded because of the difference in the treatment regimen. Among the 21 patients from 15 hospitals, 16 patients responded to the daily ACTH injections and were included in the further analysis. Table S1 presents detailed clinical information for each patient included. The remaining five patients were excluded from the further analysis due to the following reasons: two experienced EEG transformation into DSSW after the daily injection period; three had persistent hypsarrhythmia and/or ES after the daily injection period. Table S2 presents detailed clinical information for these five patients.
FIGURE 2.
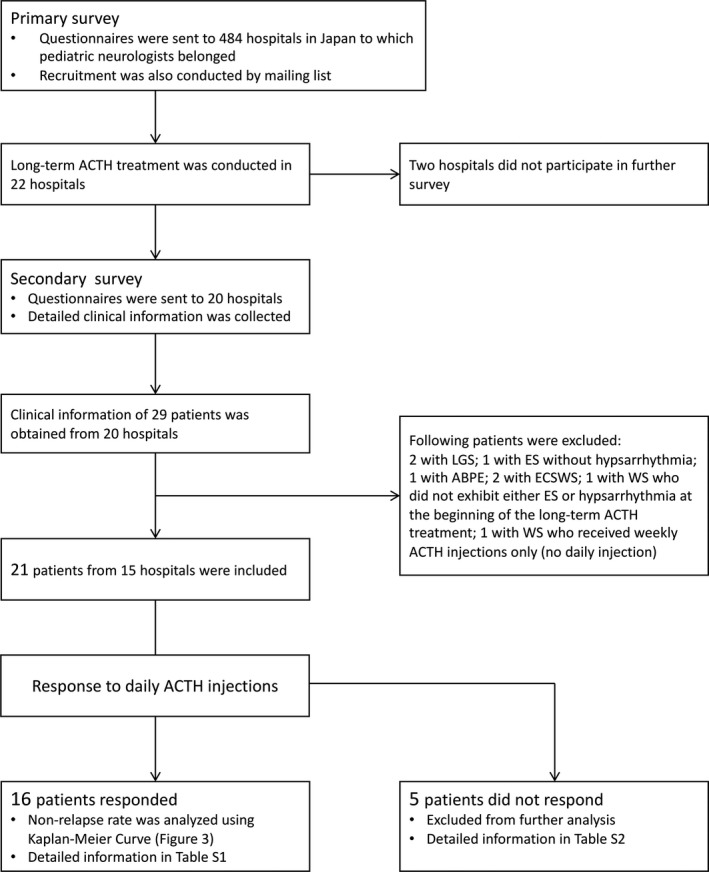
The scheme of patient identification. Abbreviations: ABPE, atypical benign partial epilepsy; ACTH, adrenocorticotropic hormone; ECSWS, epileptic encephalopathy with continuous spike and wave during sleep; ES, epileptic spasms; LGS, Lennox‐Gastaut syndrome; WS, West syndrome
All patients received intramuscular injection of synthetic, truncated, and depot ACTH, which is the only ACTH product available in Japan (Cortrosyn‐Z, Daiichi Seiyaku, Tokyo; zinc hydroxide suspension of tetracosactrin). Although there is no standardized ACTH protocol in Japan, ACTH is most often administered at a dose of 0.01‐0.014 mg/kg/day. Treatment schedules are usually comprised of daily injections for 2 weeks and thereafter reduced frequency of injection over a maximum of 8 weeks. 30
Table 1 presents the baseline characteristics for the 16 patients. We previously reported the clinical course of patient no. 6. 23 Of all the patients, eight patients each (50%) were boy and girl. The median age at epilepsy onset was 5 months (range: 2‐11 months), whereas the median age at LT‐ACTH initiation and the median duration from epilepsy onset to LT‐ACTH were 14.5 months (range: 7‐68 months) and 10.5 months (range: 2‐60 months), respectively. All patients were initially diagnosed with WS. At the beginning of LT‐ACTH treatment, five patients (31%) did not exhibit hypsarrhythmia or ES because they responded poorly or partially relapsed after their initial treatments. Regarding etiology, nine (56%), three (19%), and four (25%) patients had structural cerebral abnormality, genetic etiology, and unknown etiology, respectively. We found that two (13%) and 13 (81%) patients presented moderate and severe developmental delay at LT‐ACTH initiation, respectively. Autistic symptoms and aberrant behavior were reported in five (31%) and two (13%) patients, respectively. Thirteen patients (81%) had experienced conventional, short‐term ACTH treatment before LT‐ACTH. One patient (6%) had tried vigabatrin before LT‐ACTH, which resulted in no benefit.
TABLE 1.
Baseline characteristics of the patients
| N = 16 | |
|---|---|
| Gender (boy: girl) | 8:8 (50%:50%) |
|
Age at epilepsy onset [months; median (range)] |
5 (2‐11) |
|
Duration from epilepsy onset to initial ACTH [months; median (range)] |
1.0 (0.3‐13) |
|
Age at LT‐ACTH initiation [months; median (range)] |
14.5 (7‐68) |
|
Duration from epilepsy onset to LT‐ACTH [months; median (range)] |
10.5 (2‐60) |
| Epilepsy syndrome | |
| West syndrome (N) | 16 (100%) |
| Etiology | |
| Structural cerebral abnormality (N) | 9 (56%) |
| Genetic (N) | 3 (19%) |
| Unknown (N) | 4 (25%) |
| Development before epilepsy onset | |
| Normal (N) | 7 (13%) |
| Mild delay (N) | — |
| Moderate delay (N) | 2 (13%) |
| Severe delay (N) | 6 (38%) |
| Not answered (N) | 1 (6%) |
| Development at LT‐ACTH initiation | |
| Moderate delay (N) | 2 (13%) |
| Severe delay (N) | 13 (81%) |
| Not answered (N) | 1 (6%) |
| Autistic symptom (N) | 5 (31%) |
| Aberrant behavior (N) | 2 (13%) |
| Treatment before LT‐ACTH | |
| Number of AED [N; mean (SD)] | 4.2 (1.5) |
| Previous attempts of conventional ACTH | |
| 0 (N) | 3 (19%) |
| 1 (N) | 8 (50%) |
| 2 (N) | 5 (13%) |
| Vigabatrin use (N) | 1 (6%) |
| Ketogenic diet (N) | 1 (6%) |
| Corpus callosotomy (N) | 1 (6%) |
| VNS (N) | 1 (6%) |
Abbreviations: ACTH, adrenocorticotropic hormone; AED, antiepileptic drugs; LT‐ACTH, long‐term ACTH therapy; VNS, vagus nerve stimulation.
3.2. LT‐ACTH protocol
Table 2 presents the LT‐ACTH protocol. The mean total ACTH dose during the daily injection period was 0.35 mg (SD: 0.15 mg), while the median duration of the daily injection period was 16 days (range: 11‐28 days). The mean ACTH dosage during the weekly injection period was 0.013 mg/kg (SD: 0.0026 mg/kg). The median duration of the weekly injection was 10 months (range: 3‐22 months) with two (13%) patients remaining under LT‐ACTH. The mean total ACTH dose during the LT‐ACTH period was 0.95 mg (SD: 0.31 mg).
TABLE 2.
LT‐ACTH protocol
| N = 16 | |
|---|---|
| LT‐ACTH procedure | 16 (100%) |
| Daily injection + weekly injection (N) | |
|
Total dose of ACTH during daily injection [mg/kg; mean (SD)] |
0.35 (0.15) |
|
Duration of daily injection [days; median (range)] |
16 (11‐28) |
| Dosage of ACTH during weekly injection [mg/kg; mean (SD)] | 0.013 (0.0026) |
|
Duration of weekly injection [months; median (range)] |
10 (3‐22) |
| Continuing LT‐ACTH therapy (N) | 2 (13%) |
| Total dose of ACTH (daily + weekly) [mg/kg; mean (SD)] | 0.95 (0.31) |
Abbreviations: ACTH, adrenocorticotropic hormone; LT‐ACTH, long‐term ACTH therapy.
3.3. Primary outcome: The nonrelapse rate at 24 months after daily injection period
The time course of relapses of 16 patients who responded to daily ACTH is shown in Figure 3. Patients who showed resolution of both hypsarrhythmia and ES during the daily injection period were subsequently followed up. Among them, WS relapsed in five and two patients during and after the weekly injection period, respectively. The nonrelapse rate of WS symptoms at 24 months after daily injection was estimated at 60.6% (95% CI: 32.3%‐80.0%). We estimated the nonrelapse rates of hypsarrhythmia alone and ES alone at 24 months after daily injection to be 65.9% (95% CI: 29.5%‐86.7%) and 67.0% (95% CI: 37.9%‐84.7%), respectively (Figure S1). Two patients (patients no. 9 who did not experience relapse and no. 15 who relapsed) continued undergoing LT‐ACTH.
FIGURE 3.
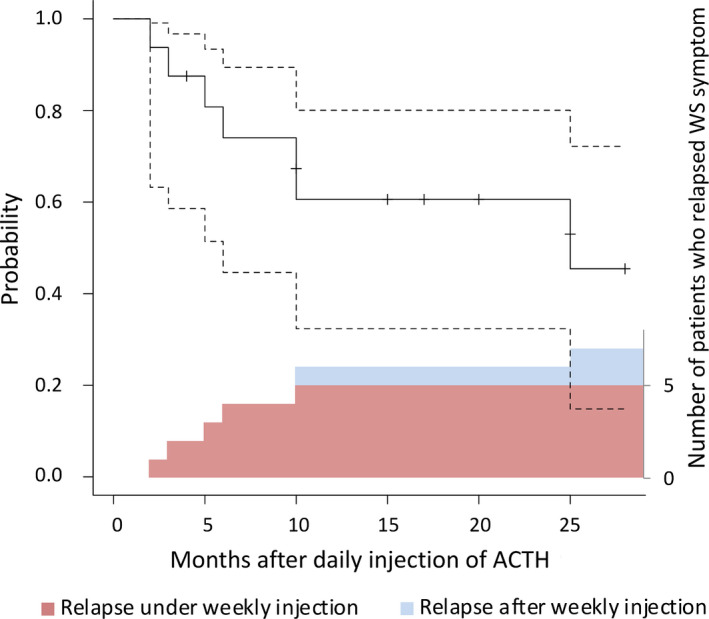
Kaplan‐Meier curve of nonrelapse rate after the daily injection period. The nonrelapse rate of WS symptom at 24 months after the daily injection period was 60.6%. Broken lines indicate 95% confidence intervals. We layered the column graph of the number of patients who relapsed to make it clear whether the patients relapsed during weekly injection period (red) or after weekly injection period (blue). In both plots, patients who developed DSSW or generalized tonic seizure were included in relapsed population. Patients whose EEG indicated transformation into DSSW at the end of the daily injection period were excluded from the Kaplan‐Meier analysis. Abbreviations: ACTH, adrenocorticotropic hormone; DSSW, diffuse slow spike and wave; EEG, electroencephalogram; WS, West syndrome
3.4. Response to weekly ACTH injections on patients who did not respond to daily ACTH injections
A total of five patients received weekly ACTH injections after insufficient response to the daily injection period (Table S2). Two patients underwent transformation to DSSW during the daily ACTH injection period even though their hypsarrhythmia and ES resolved. One experienced resolution of DSSW during the weekly injection period and no relapse thereafter; the other experienced hypsarrhythmia relapse during the weekly injection period. In the remaining three patients, hypsarrhythmia and/or ES did not cease during the daily injection period. These persisting symptoms did not resolve throughout the weekly injection period.
3.5. Adverse events of LT‐ACTH
Table 3 shows the observed adverse events during the LT‐ACTH period. During the daily injection period, irritability (15/20 patients), weight gain (15/20), electrolyte imbalance (6/20), and brain shrinkage (10/18) were frequently observed. Daily injections were stopped for one patient due to adverse events. During the weekly injection period, five patients presented with brain shrinkage, which had developed during the daily injection period. Height stagnation (4/15), hypertension that necessitated medical interventions (2/18), and irritability (2/21) were observed. The injection intervals for one patient were changed from weekly to every 10‐14 days due to adverse events. No case of early termination of weekly injections due to adverse events occurred. No patient required treatment for adrenal insufficiency during or after LT‐ACTH.
TABLE 3.
Adverse events during the LT‐ACTH period
| Daily injection period | |
|---|---|
| Irritability | 15/20 |
| Weight gain | 15/20 |
| Hypertension | 2/20 |
| Electrolyte imbalance | 6/20 |
| Urinary sugar | 1/20 |
| Infection | 1/20 |
| Gastrointestinal hemorrhage | 0/19 |
| Brain shrinkage | 10/18 |
| Others | |
| Hypertonia oculi | 1 |
| Transaminase elevation | 1 |
| Hypothyroidism | 1 |
| Chromatosis | 1 |
| Weekly injection period | |
|---|---|
| Infection | 1/21 |
| Hypertension | 2/18 |
| Cardiac hypertrophy | 0/12 |
| Gastrointestinal hemorrhage | 0/18 |
| Electrolyte imbalance | 0/19 |
| Hypertonia oculi | 0/10 |
| Cataract | 0/12 |
| Fracture | 0/19 |
| Skin trouble | 1/20 |
| Brain shrinkage | 5/10 |
| Subdural effusion/subdural hematoma | 0/12 |
| Obesity | 0/20 |
| Height stagnation | 4/15 |
| Depression | 0/21 |
| Irritability | 2/21 |
Patients who lacked information whether they had experienced each event or not were excluded from the number of corresponding denominators. Events classified as “Others” were obtained by free description in the questionnaire.
Abbreviations: LT‐ACTH, long‐term ACTH therapy.
3.6. Factors associating with WS relapse
For the aim of exploring candidate factors that associated with WS relapse, we classified 16 patients into nonrelapse group (9 patients) and relapsed group (7 patients). Then, we compared their clinical background, LT‐ACTH protocols, and EEG/seizure condition before the daily and weekly injection period between the two groups. Duration from epilepsy onset to LT‐ACTH was numerically longer in patients with WS relapse (9 months [range: 2‐28 months] vs 15 months [range: 7‐60 months]). Proportion of patients with etiology of structural cerebral abnormality was numerically larger in patients with WS relapse (3/9 patients [33%] vs 6/7 patients [86%]). Other clinical background, LT‐ACTH protocols, and EEG/seizure condition before the daily and weekly injection period were seemingly similar between the two groups (Table S3).
4. DISCUSSION
This is the first nationwide, multicenter case series on the efficacy of a long‐term weekly ACTH regimen for WS. We found that the nonrelapse rate of WS at 24 months after daily injection was 60.6%. For cases that did not respond to the daily injections, resolution of hypsarrhythmia and/or ES rarely occurred during the weekly injection period. Indeed, the effectiveness of a long‐term weekly ACTH regimen is still unclear and the regimen is not a generally recommendable one for patients with WS; however, it may be a good option for those who responded well to initial ACTH therapy, but relapsed, or who responded insufficiently to initial ACTH therapy.
To evaluate whether the weekly ACTH injection contributed to suppressing WS relapse, we searched previous studies that reported long‐term relapse rate after WS remission achieved by conventional ACTH therapy. However, we could not find a study that we could directly compare with our data, mainly due to the difference in the patients' background. The nonrelapse rate after initial ACTH therapy in overall WS population is reported around 59.0%‐63.3%. 31 , 32 The nonrelapse rate might be lower if the patients have some risk factors for relapse, that is, delayed development at onset of WS 18 and persistent epileptic discharges after daily ACTH injections 32 , 33 , 34 . In addition, the effectiveness of secondary ACTH trial seems to be inferior to that of first trial, even though the evidence is scarce. For example, Knupp et al 20 reported a 21% effectiveness of secondary ACTH or oral corticosteroids in patients with WS who did not respond to initial ACTH or oral corticosteroid treatment. The nonrelapse rate in our study is similar to that of the overall WS population; however, it can be speculated that LT‐ACTH might improve the relapse rate, considering our patients had delayed development at onset of WS (8 [50%] patients), persistent epileptic discharges after daily ACTH injections (13 [81%]), and history of WS relapse after previous, conventional ACTH therapy (13 [81%]). Since our daily injection protocol was roughly similar to that of the previous Japanese studies, 19 , 35 improvement in relapse rate may be attributed to the subsequent weekly injection period. However, we cannot affirm the efficacy of LT‐ACTH by comparing previous studies with ours because of the small size of our cohort and the differences in the patients' backgrounds. A large prospective study with appropriate control is desired to confirm its effectiveness.
Whether ACTH treatment duration is associated with the relapse rate improvement remains unclear. Previous studies on the optimal balance between the effectiveness and side effects of ACTH for WS management have considered long‐term treatment. For example, Singer et al employed the following ACTH regimen: 40 units for 14 days with subsequent 80 units every other day for a minimum of 3 months or for 1 month after achievement of complete control. They treated 55 patients, with the mean ACTH treatment period being 10 months (range: 6‐14 months). They reported that 67% of the patients remained spasm‐free at ACTH termination. 36 A prospective study by Hrachovy et al 16 reported no significant difference in the responder rate, EEG normalization, and relapse rate between the short‐term, low‐dose regimen and the long‐term, high‐dose regimen. Moreover, multiple studies have not found a positive relationship between the cumulative ACTH dose and seizure/cognitive outcome. 17 , 35 However, a majority of these studies considered the total ACTH dose and not solely the regimen duration. Whether sufficient suppression of epileptic discharges or seizures could reduce the relapse rate of WS, for instance by long‐term ACTH regimen, may be a target for further investigation.
Although the mechanisms underlying the effects of ACTH in patients with WS are not clarified, two hypotheses have been proposed: (a) ACTH may accelerate central nervous system maturation through several processes, such as accelerating enzyme induction in the brain and modulating neurotropic factors in the cerebrospinal fluid 37 ; or (b) ACTH may modulate neuronal excitability by suppressing the production of corticotropin‐releasing hormone. 38 Despite the controversy of these hypotheses, current evidence suggests that a long‐term ACTH regimen may maintain favorable effects on epileptic symptoms for a sustained period and thus help lessen the probability of WS relapse.
In our study, we did not observe lethal adverse events, including adrenal insufficiency, during the weekly injection period. The mechanisms underlying the adverse effects of ACTH remain unclear; however, they could involve activation of glucocorticoid secretion. Since commonly used ACTH products are designed as sustained‐release preparations, the ACTH plasma level is maintained for several postinjection hours. Consequently, serum cortisol levels sustain elevation for more than 24 hours. 39 Side effects related to glucocorticoid therapy are well known. 40 The side effects of conventional daily ACTH therapy could be related to its cumulative dose. 35 Notably, varied adverse events force termination of ACTH therapy. 41 Previous studies on long‐term therapy have not reported any severe adverse events. 23 , 24 , 25 , 26 , 35 However, we observed nonlethal adverse effects, including height stagnation, hypertension, and irritability. Clinicians who plan to initiate LT‐ACTH should pay sufficient attention to these events, as well as give appropriate information to patients' caregivers. Also, clinicians should refrain from regarding LT‐ACTH as truly safe, as cumulative cases that experienced LT‐ACTH are still too small to determine its safety.
In our study, duration from epilepsy onset to LT‐ACTH was longer and proportion of patients with structural cerebral abnormality was larger in patients with WS relapse. However, due to the lack of reliable statistical analysis, we cannot verify whether these are associated with WS relapse during/after weekly ACTH injection. We hope our results to be a clue for identifying prognostic factors in future research so that we can foresee good candidates for LT‐ACTH.
Our study has several limitations. First, we did not include an appropriate control group. Second, the efficacy of LT‐ACTH may be influenced by the change in AEDs during the weekly injection period, which happened in many of our cases. Third, the follow‐up period was short for some of our patients. Fourth, we used a small sample size; however, this study included the largest series of long‐duration ACTH therapy ever published. Fifth, we could not determine the optimal LT‐ACTH regimen and risk factors for WS relapse, presumably because of the small size of our cohort. Sixth, reports of adverse events during LT‐ACTH were based on the clinicians' discretion due to the lack of a unified screening protocol. Hence, the true incidence of each condition may be higher than the presently reported.
In conclusion, this is the first case series that documented the efficacy of LT‐ACTH for preventing WS relapse. We did not observe lethal adverse events during the weekly injection period, although nonlethal events, such as height stagnation, hypertension, and irritability, were firstly noted. Our findings highlight the influence of treatment duration on relapse prevention, which has not been well studied. Large prospective cohort studies are needed to confirm the effectiveness of LT‐ACTH.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues regarding ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
We are grateful to Dr Tatsuya Yamamoto (Department of Pediatrics, Hirosaki University School of Medicine and Graduate School of Medicine), Dr Shinsuke Maruyama (Department of Pediatrics, Graduate School of Medical and Dental Sciences, Kagoshima University), Dr Ryosuke Urabe (Division of Neurology, National Center for Child Health and Development), and Dr Takafumi Sakakibara (Department of Pediatrics, Nara Medical University) for sharing their valuable experience on LT‐ACTH.
Baba S, Okanishi T, Homma Y, et al. Efficacy of long‐term adrenocorticotropic hormone therapy for West syndrome: A retrospective multicenter case series. Epilepsia Open. 2021;6:402–412. 10.1002/epi4.12497
REFERENCES
- 1. Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. Infantile spasms: A U.S. consensus report. Epilepsia. 2010;51:2175–89. [DOI] [PubMed] [Google Scholar]
- 2. Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi group. Epilepsia. 2004;45:1416–28. [DOI] [PubMed] [Google Scholar]
- 3. Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van Bogaert P, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia. 2015;56:1185–97. [DOI] [PubMed] [Google Scholar]
- 4. Go CY, Mackay MT, Weiss SK, Stephens D, Adams‐Webber T, Ashwal S, et al. Evidence‐based guideline update: Medical treatment of infantile spasms. Report of the guideline development subcommittee of the American Academy of Neurology and the practive committee of the Child Neurology Society. Neurology. 2012;78:1974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev. 2001;23:539–41. [DOI] [PubMed] [Google Scholar]
- 6. Cowan LD, Hudson LS. The epidemiology and natural history of infantile spasms. J Child Neurol. 1991;6:355–64. [DOI] [PubMed] [Google Scholar]
- 7. Trevathan E, Murphy CC, Yeargin‐Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40:748–51. [DOI] [PubMed] [Google Scholar]
- 8. Saemundsen E, Ludvigsson P, Rafnsson V. Risk of autism spectrum disorders after infantile spasms: a population‐based study nested in a cohort with seizures in the first year of life. Epilepsia. 2008;49:1865–70. [DOI] [PubMed] [Google Scholar]
- 9. Riikonen R. Long‐term outcome of patients with West syndrome. Brain Dev. 2001;23:683–7. [DOI] [PubMed] [Google Scholar]
- 10. O'Callaghan FJ, Lux AL, Darke K, Edwards SW, Hancock E, Johnson AL, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia. 2011;52:1359–64. [DOI] [PubMed] [Google Scholar]
- 11. Rener‐Primec Z, Stare J, Neubauer D. The risk of lower mental outcome in infantile spasms increases after three weeks of hypsarrhythmia duration. Epilepsia. 2006;47:2202–5. [DOI] [PubMed] [Google Scholar]
- 12. Baram TZ, Mitchell WG, Tournay A, Snead OC III, Hanson RA, Horton EJ. High‐dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics. 1996;97:375–9. [PMC free article] [PubMed] [Google Scholar]
- 13. Wanigasinghe J, Arambepola C, Ranganathan SS, Sumanasena S. Randomized, single‐Blind, parallel clinical trial on efficacy of oral prednisolone versus intramuscular corticotropin: a 12‐month assessment of spasm control in West syndrome. Pediatr Neurol. 2017;76:14–9. [DOI] [PubMed] [Google Scholar]
- 14. Riikonen R, Donner M. ACTH therapy in infantile spasms: side effects. Arch Dis Child. 1980;55:664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hrachovy RA, Frost JD Jr, Kellaway P, Zion TE. Double‐blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103:641–5. [DOI] [PubMed] [Google Scholar]
- 16. Hrachovy RA, Frost JD, Glaze DG. High‐dose, long‐duration versus low‐dose, short duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124:803–6. [DOI] [PubMed] [Google Scholar]
- 17. Yanagaki S, Oguni H, Hayashi K, Imai K, Funatuka M, Tanaka T, et al. A comparative study of high‐dose and low‐dose ACTH therapy for West syndrome. Brain Dev. 1999;21:461–7. [DOI] [PubMed] [Google Scholar]
- 18. Hamano SI, Yamashita S, Tanaka M, Yoshinari S, Minamitani M, Eto Y. Therapeutic efficacy and adverse effects of adrenocorticotropic hormone therapy in west syndrome: differences in dosage of adrenocorticotropic hormone, onset of age, and cause. J Pediatr. 2006;148:485–8. [DOI] [PubMed] [Google Scholar]
- 19. Matsumoto A, Watanabe K, Negoro T, Yoshinari S, Minamitani M, Eto Y. Long‐term prognosis after infantile spasms: a statistical study of prognostic factors in 200 Cases. Dev Med Child Neurol. 1981;23:51–65. [DOI] [PubMed] [Google Scholar]
- 20. Knupp KG, Leister E, Coryell J, Nickels KC, Ryan N, Juarez‐Colunga E, et al. Response to second treatment after initial failed treatment in a multicenter prospective infantile spasms cohort. Epilepsia. 2016;57:1834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maguire MJ, Hemming K, Wild JM, Hutton JL, Marson AG. Prevalence of visual field loss following exposure to vigabatrin therapy: a systematic review. Epilepsia. 2010;51:2423–31. [DOI] [PubMed] [Google Scholar]
- 22. Conway M, Cubbidge RP, Hosking SL. Visual field severity indices demonstrate dose‐dependent visual loss from vigabatrin therapy. Epilepsia. 2008;49:108–16. [DOI] [PubMed] [Google Scholar]
- 23. Okanishi T, Sugiura C, Saito Y, Maegaki Y, Ohno K, Togari H. Long‐term weekly ACTH therapy for relapsed West syndrome. Pediatr Neurol. 2008;38:445–9. [DOI] [PubMed] [Google Scholar]
- 24. Inui T, Kobayashi T, Kobayashi S, Sato R, Endo W, Kikuchi A, et al. Efficacy of long term weekly ACTH therapy for intractable epilepsy. Brain Dev. 2015;37:449–54. [DOI] [PubMed] [Google Scholar]
- 25. Nakata M, Kato T, Ide M, Saito K, Yoshida T, Awaya T, et al. Long‐term weekly ACTH therapy for relapsed West syndrome in tuberous sclerosis complex: a case report. Brain Dev. 2016;38:431–4. [DOI] [PubMed] [Google Scholar]
- 26. Maizuru K, Kato T, Nakata M, Ide M, Saito K, Yoshida T, et al. Treatment of epileptic encephalopathy after human herpesvirus 6‐induced post‐transplantation acute limbic encephalitis with adrenocorticotropic hormone therapy: a case report. Epilepsy Seizure. 2017;9:18–24. [Google Scholar]
- 27. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:522–30. [DOI] [PubMed] [Google Scholar]
- 28. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanda Y. Investigation of freely‐available easy‐to‐use software “EZR” (Easy R) for medical statistics. Bone Marrow Transplant. 2013;48:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ito M, Seki T, Takuma Y. Current therapy for West syndrome in Japan. J Child Neurol. 2000;15:424–8. [DOI] [PubMed] [Google Scholar]
- 31. Riikonen R. A long‐term follow‐up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982;13:14–23. [DOI] [PubMed] [Google Scholar]
- 32. Hayashi Y, Yoshinaga H, Akiyama T, Endoh F, Ohtsuka Y, Kobayashi K. Predictive factors for relapse of epileptic spasms after adrenocorticotropic hormone therapy in West syndrome. Brain Dev. 2016;38:32–9. [DOI] [PubMed] [Google Scholar]
- 33. Koo B, Hwang PA, Logan WJ. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic group. Neurology. 1993;43:2322–7. [DOI] [PubMed] [Google Scholar]
- 34. Yamada K, Toribe Y, Kimizu T, Kimura S, Ikeda T, Mogami Y, et al. Predictive value of EEG findings at control of epileptic spasms for seizure relapse in patients with West syndrome. Seizure. 2014;23:703–7. [DOI] [PubMed] [Google Scholar]
- 35. Ito M, Aiba H, Hashimoto K, Kuroki S, Tomiwa K, Okuno T, et al. Low‐dose ACTH therapy for West syndrome: initial effects and long‐term outcome. Neurology. 2002;58:110–4. [DOI] [PubMed] [Google Scholar]
- 36. Singer WD, Rabe EF, Haller JS. The effect of ACTH therapy upon infantile spasms. J Pediatr. 1980;93:485–9. [DOI] [PubMed] [Google Scholar]
- 37. Riikonen R. Infantile spasms: some new theoretical aspects. Epilepsia. 1983;24:159–68. [DOI] [PubMed] [Google Scholar]
- 38. Brunson KL, Avishai‐Eliner S, Baram TZ. ACTH treatment of infantile spasms: mechanisms of its effects in modulation of neuronal excitability. Int Rev Neurobiol. 2002;49:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Snead OC, Benton JW, Hosey LC, Swann JW, Spink D, Martin D, et al. Treatment of infantile spasms with high‐dose ACTH: efficacy and plasma levels of ACTH and cortisol. Neurology. 1989;39:1027–31. [DOI] [PubMed] [Google Scholar]
- 40. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and management of glucocorticoid‐induced side effects: a comprehensive review. J Am Acad Dermatol. 2017;76:1–9. [DOI] [PubMed] [Google Scholar]
- 41. Partikian A, Mitchell WG. Major adverse events associated with treatment of infantile spasms. J Child Neurol. 2007;22:1360–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3