

Abstract
The hepatic immune system is designed to tolerate diverse harmless foreign moieties to maintain homeostasis in the healthy liver. Constant priming and regulation ensure that appropriate immune activation occurs when challenged by pathogens and tissue damage. Failure to accurately discriminate, regulate, or effectively resolve inflammation offsets this balance, jeopardizing overall tissue health resulting from an either overly tolerant or an overactive inflammatory response. Compelling scientific and clinical evidence links dysregulated hepatic immune and inflammatory responses upon sterile injury to several pathological conditions in the liver, particularly nonalcoholic steatohepatitis and ischemia-reperfusion injury. Murine and human studies have described interactions between diverse immune repertoires and nonhematopoietic cell populations in both physiological and pathological activities in the liver, although the molecular mechanisms driving these associations are not clearly understood. Here, we review the dynamic roles of inflammatory mediators in responses to sterile injury in the context of homeostasis and disease, the clinical implications of dysregulated hepatic immune activity and therapeutic developments to regulate liver-specific immunity.
Keywords: homeostasis, immunology, inflammation, liver
Subject terms: Immunology, Inflammation
An introduction to the immunological structure of the liver
The liver is an immunologically complex organ that functions as a physiological bridge between gut-derived molecules and the systemic circulation. Approximately 80% of the hepatic vascular supply is provided by the portal venous system, which results in a low-pressure blood system with large amounts of bacterial products and other foreign molecules that are presented to the immune system. Consequently, the liver must tolerate these potentially immunogenic or inflammatory foreign molecules while maintaining constant immunosurveillance for infectious pathogens and liver malignancies. The hepatic microstructure is composed of an extensive repertoire of immune cells embedded in a meshwork of liver sinusoids through which hepatic venous blood passes and mixes with oxygen-rich arterial blood from the hepatic artery. These sinusoids are lined by specialized liver sinusoidal endothelial cells (LSECs) that contain numerous fenestrations, allowing molecules to freely diffuse through to the underlying hepatocytes, which perform the central metabolic functions of the liver1 (Fig. 1). This alignment facilitates the rapid exchange of blood-borne molecules and metabolic processing in the liver, as well as extensive interactions with local and systemic immune populations.
Fig. 1.

The cellular composition of the liver. Venous blood from the gut mixes with oxygenated arterial blood, drains through the sinusoids and passes through plates of hepatocytes to the central vein. Hepatocytes secrete bile into the canaliculi, and these secretions ultimately flow into the bile ducts. The sinusoids are lined by specialized, fenestrated liver sinusoidal endothelial cells that allow blood to pass through the space of Dissé to the underlying hepatocytes. Within the blood, Kupffer cells adhere to the endothelial wall, while resident lymphoid and myeloid immune cell populations are found around the portal tract and throughout the parenchyma
Hepatic homeostasis relies on effective regulation of the intimate interactions between resident and infiltrating immune cells and nonhematopoietic hepatic cells. Resident immune cells populate the liver sinusoids and the subendothelial compartment known as the space of Dissé, which is located between hepatocytes and LSECs; these immune cells are a diverse population, including professional antigen-presenting cells (APCs), myeloid cells, and specialized innate and adaptive lymphoid cell populations.1 Many hepatic immune populations play vital roles in homeostasis, tissue repair, maintaining immune tolerance, and modulating liver inflammation. Here, we review the dynamic roles of hepatic inflammatory immune responses in homeostasis and how inflammatory responses to sterile injury lead to pathology, specifically in the context of nonalcoholic steatohepatitis (NASH) and ischemia-reperfusion injury (IRI).
Hepatic immune cell repertoires
Hepatic immune cell populations play diverse roles in regulating inflammation and immune responses. Kupffer cells (KCs), which account for 80–90% of the total population of fixed tissue macrophages and almost one-third of the nonparenchymal cell population in the liver, are key.2 These liver-specific myeloid cells play critical roles in the recognition of blood-borne pathogens and the clearance of invading microbes.3,4 These cells have key roles in identifying, phagocytosing, and eliminating “foreign” or “dangerous” antigens via PPRs, complement receptors, and Fc receptors. Consistent with their macrophage-like functions, KCs exhibit increased phagocytic activity; however, in contrast to other myeloid populations, KCs produce proinflammatory and anti-inflammatory factors, illustrating their dichotomous role in tolerogenic defense and inflammation.5
Dendritic cells (DCs) are also important inducers of tolerance in the liver, although these cells are also capable of antigen presentation and type 1 interferon production.6–8 DCs are categorized into myeloid and plasmacytoid subtypes and are primarily located around the central veins and portal tracts.9 Under physiological conditions, DCs are phenotypically immature and tolerogenic and are thought to play an important role in tolerogenicity posttransplantation;10 however, proinflammatory DCs develop in response to liver injury, stimulating cytokine secretion and T cell responses.11 There is evidence of DC participation in anti-inflammatory activities, particularly in the context of IRI, indicating their dual immunosuppressive and proinflammatory roles.12
Classic neutrophils are rarely detected in the healthy liver. However, neutrophil subsets with immunoregulatory properties, also referred to as type 2 neutrophils (N2), have been found among tumor-associated neutrophils (TANs) in hepatocellular carcinoma.13 These neutrophil subsets share phenotypic and functional features with mature low-density neutrophils or granulocyte-like myeloid-derived suppressor cells (MDSCs),14 which are known to suppress T-cell activation and, in the context of cancer, promote cancer growth.15,16 MDSC expansion has also been described in nonalcoholic fatty liver disease (NAFLD).17 The immunosuppressive activity of N2/TANs/G-MDSCs has been associated with the upregulation of various factors that promote an immunosuppressive environment, such as arginase or indoleamine 2,3-dioxygenase.18
Classic T lymphocytes account for up to 50% of lymphocytes in the liver, and B cells make up only 5% of the hepatic lymphocyte population.19 While CD8+ T cells usually outnumber CD4+ T cells at a two-to-one ratio in the liver, both cell types have activated phenotypes. A large proportion of liver T lymphocytes have innate-like features.20 These T cells coexpress CD56 and/or CD161 and include NK T (NKT) cells, mucosal-associated invariant T cells, and γδ T cells, which are involved in hepatic immunity and tissue remodeling.21–23
Innate B cells that express high levels of CD5 are also found in the human liver and are likely to be immunoregulatory, although it is unknown whether these cells produce the same suppressive cytokine milieu as the systemic subtype.24 Regulatory T cell-induced Breg cell formation in a mouse model of hepatic autoimmunity requires IL-21 but neither IL-10 nor TGF-β.25 Human studies have consistently demonstrated B-cell expansion in hepatitis C-infected liver tissue, and these cells potentially contribute to the suppressive immune environment that is typically observed in chronic HCV-infected livers.24,26–28
In humans, NK cells account for 30–50% of the hepatic lymphocyte population,29 while in mice, only 5–10% of hepatic lymphocytes are NK cells.5,30 These cells have important antiviral and antitumor activities and can also interact directly or indirectly with liver APCs, KCs, and DCs to regulate hepatic immune responses.31
Several studies using murine and other animal models have defined the role of the liver in hematopoiesis.32–34 Mouse studies have demonstrated that in the absence of a spleen, extramedullary hematopoiesis can occur in the liver.35 In adult mice, the lymphopoietic capacity of adult liver cells was similar to that of bone marrow transplantation, suggesting a key role in liver transplantation tolerance and T-cell reconstitution in recipients.36 Most human hematopoietic stem cell (HSC) studies are centered around fetal liver development, although these stem cells are also present in the adult liver.34 Lymphoid progenitors (LPs) have also been detected in healthy liver tissue,33 and LPs that express CD56 are more common in the liver than in the bone marrow.37 The expansion of the HSC pool that expresses markers of myeloid differentiation has been described in liver metastasis38 and may contribute to the increased numbers of granulocytic cells or MDSCs observed in metastatic livers.
Within the liver, nonhematopoietic cell populations also possess important immune functions.5 LSECs are involved in hepatic leukocyte recruitment via Toll-like receptor 4 (TLR4) activation and appear to exhibit both tolerogenic and proinflammatory functions through CD4+ and CD8+ interactions. Antigen-presenting capabilities have also been described in hepatocytes.39 In response to hepatocyte antigen presentation, an effector CD8+ T-cell response is initiated when the initial antigen load is low, in contrast to the functionally exhausted population of CD8+ cells with PD-1 expression that is observed during abundant hepatic antigen presentation.22,40 Furthermore, hepatic injury and inflammation can result in IL-10 induction by CD4+ T cells as a consequence of hepatocyte activation to restore immunological homeostasis and regulate a potentially overwhelming immune response.41
Identification and characterization of immune cell populations by single-cell RNA sequencing analyses
The spectrum of resident hepatic immune cell populations is currently expanding through the use of single-cell RNA sequencing approaches.42,43 This technology has allowed for the intimate analysis of a comprehensive immune cell population and has identified a number of novel intrahepatic immune subsets (Table 1). The challenge in applying these approaches to liver tissue relates to the spatially graded hepatic microenvironment and zonation patterns, although new technologies are beginning to include spatial localization data.44,45 The loss of fragile liver cells during sample processing and the consequent underrepresentation of non-parenchymal cell populations in subsequent single-cell RNA sequencing datasets is another limitation of these technologies.42
Table 1.
The expanding repertoire of intrahepatic immune cell populations identified by single-cell RNA sequencing
| Immune cell compartment | Subpopulations identified | Reference |
|---|---|---|
| Kupffer cells (CD163+VSIG4+) |
LIRB5+CD5L+MARCO+HMOX1high immunoregulatory subset CD1C+FCER1A+ antigen-presenting subset |
Aizarani et al.46 |
| Kupffer cells (CD68+) |
LYZ+CSTA+CD74+ inflammatory macrophages CD5L+MARCO+VSIG4+LILRB5+HMOX1+ tolerogenic macrophages |
MacParland et al.42 |
| Mononuclear phagocytes |
CD163+MARCO+TIMD4+/− Kupffer cells TREM2+CD9+ scar-associated macrophages |
Ramachandran et al.47,48 |
| Dendritic cells |
CLEC9A+XCR1+CADM1+ cDC1 CD1C+FCER1A+CLEC10A+ cDC2 LAMP3+CD80+CD83+CCR7+ activated migratory DCs |
Zhang et al.134 |
| Natural killer cells |
CD160+IL2RB+CXCR6+ tissue-resident NK cells CX3CR1+GNLY+GZMB+ classic NK cells HMGB2+MKI67+ cycling NK cells |
Zhao et al.49 |
A number of liver single-cell RNA sequencing studies have focused on heterogeneity within the KC population. These studies have identified two subpopulations in the human liver: a proinflammatory antigen-presenting KC subset and a tolerogenic KC subset (Table 1).42,46 There is also a distinct scar-associated macrophage subpopulation present in patients with liver cirrhosis,47,48 highlighting the importance of comparative analysis of healthy and diseased tissue.
In addition to KCs, unique populations of both DCs and NK cells have been identified in single-cell RNA sequencing datasets, in addition to previously identified populations of DCs and liver-resident NK cells (Table 1),49 and it is likely that further liver-specific immune cell subpopulations will be identified as this technology matures. Beyond immune cells, it is evident that zonal and disease-specific populations of hepatocytes, endothelial cells, and stellate cells are also present within the liver.44,47,50 The technological advances that have enabled the analysis of transcriptional differences across these zonal regions within the liver are now also being applied to resident immune cell populations to uncover further immune cell heterogeneity associated with different regions of the liver.
The homeostatic response to sterile injury in the liver
Inflammatory activity in the liver that is stimulated in the absence of foreign microbial antigens can occur in response to a wide range of stimuli and is commonly referred to as sterile inflammation.51 This inflammatory response can lead to the pathological production of reactive oxygen species (ROS), lipid-derived metabolites (retinoic acid and endocannabinoids), and damage-associated molecular patterns (DAMPs), which amplify inflammatory signals through TLRs, nuclear/neuronal receptors, and the inflammasome (Fig. 2). While maintaining an overall tolerogenic environment, the healthy liver is well equipped to restore homeostasis after disturbances. Large populations of innate immune cells clear senescent, injured, or apoptotic cells, as well as microbes and their associated products.24 Activated neutrophils and cytotoxic cells that are no longer responsive to inflammatory signals or that have already completed their immunologic functions and need to be removed from the circulation migrate to the liver to die via apoptosis. This detoxification mechanism or resolution is largely mediated by KCs, which are the first cells in the liver to encounter cells and molecules from the systemic circulation. High quantities of microbial DAMPs and MAMPs arriving via portal venous blood bind to PRRs expressed on KCs and some hepatocytes, facilitating microbial phagocytosis and degradation without initiating the overwhelming immune response that is usually stimulated by PRRs.52,53 In a cirrhotic mouse model, endotoxin accumulation appeared to result from decreased KC uptake and low levels of TNF alpha release.54 In this context, hepatic detoxification ensures that inflammatory antigens from the gut do not travel to the rest of the body, causing excessive immune activity.55 While it is unsurprising that failed detoxification mechanisms are pathognomonic of hepatic failure predicated on the assumption of immunoregulatory disturbances, few studies have defined the inflammatory interplay in hepatic detoxification and its clinical implications.
Fig. 2.

The acute response to sterile injury in the liver. During liver injury, hepatocyte death causes DAMP release, resulting in Kupffer cell activation, neutrophil recruitment to the site of injury and the activation of hepatic stellate cells, leading to cytokine secretion. Kupffer cells and neutrophils release inflammatory mediators and begin to phagocytose necrotic debris. T cells, dendritic cells, and Kupffer cells facilitate leukocyte infiltration from the systemic circulation by releasing chemoattractants such as CCL2 and IL-17. NK and NKT cells release IFNγ, stimulating the release of proinflammatory mediators. During resolution, monocytes acquire a reparative phenotype, and macrophage reprogramming occurs in a subset of macrophages that exhibit a resolution phenotype. Monocytes migrate across the liver sinusoidal endothelial cell layer, differentiating into macrophages to either replenish the Kupffer cell pool or restore homeostasis by secreting anti-inflammatory cytokines and promoting angiogenesis. Following injury, a restorative macrophage phenotype emerges, promoting neutrophil apoptosis. Hepatocyte proliferation occurs to replace the lost parenchyma131
Some NK cells respond rapidly to injury following direct lipid recognition or cytokine secretion by other immune cells, leading to the downstream accumulation of additional macrophages and neutrophils in the liver.56 In murine models, type 1 NKT cell-deficient mice do not exhibit neutrophil and macrophage infiltration in response to injury and display hepatic resistance to IRI or a high-fat diet, suggesting that one subset of NKT cells promotes injury.57–59 In contrast, sulfatide-reactive type 2 NKT cells play an opposing role in hepatic IRI by protecting against injury, and a novel anti-inflammatory mechanism has been proposed to exploit this process and ultimately result in NKT cell recruitment in response to liver injury.59
This response to acute sterile inflammation is physiological and self-limiting and results in the restoration of tissue homeostasis. However, the ongoing presence of the stimuli driving chronic sterile inflammation leads to pathology. These stimuli include alcohol metabolites that can induce hepatocyte cell death and excessive fat deposition within hepatocytes, resulting in lipotoxicity and oxygen depletion during ischemia and reperfusion that leads to ischemic damage.
Nonalcoholic steatohepatitis (NASH)
Changes to the hepatic immune environment characterize the development of NASH, and murine and animal studies have been used to define the specific immunological events that occur throughout the clinical course of NASH (Fig. 3). Innate immune activation and dysregulated inflammation have both been implicated in the loss of tissue homeostasis and are a subject of considerable focus. Lipotoxicity, which results from aberrant lipid accumulation during prolonged nutrient excess or obesity exceeding adipose tissue capacity, underlies the generation of several harmful signaling intermediates.60 When storage is overwhelmed, lipid deposition occurs in ectopic sites, such as the liver, interfering with local immune regulation and resulting in a cycle of persistent metabolic dysregulation or “metainflammation”60,61 Inflammatory immune activation within the liver induces the infiltration of several immune cell subsets and drives the progression of hepatic injury.
Fig. 3.

Pathogenic immune mechanisms in NASH. NASH involves immune cell populations that functionally interact with each other. Increased fatty acid levels and lipotoxicity directly activate inflammation and result in hepatocyte injury and the release of DAMPs, triggering an amplified inflammatory response. Kupffer cell activation stimulates the subsequent release of several proinflammatory cytokines that activate monocytes and neutrophils. These cells exacerbate liver injury by secreting reactive oxygen species and profibrotic signals that promote fibrosis. Cytotoxic T cell activation contributes to perpetuating inflammation by the production of large amounts of IFNγ and TNFα, and natural killer cell activation directly stimulates hepatic stellate cells and hepatocytes, causing further inflammation and inducing profibrotic signals
KCs appear to play a pivotal role in NASH initiation and pathogenesis.62 Concurrent activation of KCs is seen following the introduction of a high-fat diet or methionine/choline-deficient diets in several murine models, indicating the early involvement of KCs in NASH.63,64 Human studies have also corroborated these findings, and KC activation and accumulation were observed in NASH samples, supporting the direct role of KCs in lipid processing and metainflammation.65 Thus, KC activation could be linked to direct lipid uptake or lipid metabolism and the resultant lipotoxicity that presents as foreign particles.66 In human studies, KC expansion was a key step in hepatic inflammatory initiation that, through a complex inflammatory network, resulted in the downstream activation of other components of the NASH complex.67 Furthermore, there is additional evidence to suggest the pathological role of KCs in hepatic insulin resistance upon activation, further suggesting the intricate interplay of immunological events and metabolic pathways in liver disease.63
The process by which circulating monocytes differentiate into macrophages upon arrival in the liver environment is poorly understood; however, monocyte infiltration may impact some of the inflammatory responses observed in NASH. In response to hepatic injury, Ly-6Chi monocytes differentiate into Ly-6C+ macrophages, stimulating proinflammatory and profibrogenic processes.62 In murine models, diet-induced hepatic steatosis induced Ly-6Chi monocyte recruitment via CC-chemokine receptor 2 (CCR2), which is largely expressed by KCs and HSCs.68 Activated monocytes were recruited following hepatic injury and guided by the expression of CC-chemokine ligand 2 (CCL2), and reduced inflammatory macrophage levels were observed in CCL2-knockout mice with MCD diet-induced NASH.69 In contrast, an anti-inflammatory, tissue-protective Ly-6Clow phenotype can emerge when apoptotic hepatocytes are phagocytosed, and these cells act to restore tissue homeostasis.70
Moreover, KC polarization into M1 and M2 phenotypes further reveals the cyclical and overlapping proinflammatory and immunoregulatory events in NASH, reflecting the tolerogenic and immunogenic nature of hepatic physiology.71,72 For example, M1 phenotype activation results in the production of several proinflammatory cytokines and chemokines, such as TNF-α, IL-1β, IL-12, CCL2, and CCL5, inducing additional hepatocyte injury and DAMP release, which further exacerbates KC activation and results in a vast influx of monocytes.11,62 In contrast, the role of M2 phenotype cells, although largely undefined in NASH, involves inflammatory resolution and healing processes to counteract the initial recruitment of various cellular populations.62,72 To date, few studies have examined M2 phenotypic changes in NASH compared to non-NASH in humans, although there is evidence to suggest PPAR-δ involvement in M2 activation, which may offer a therapeutic strategy in the clinical management of the entire NAFLD disease spectrum.73 In addition, mice fed a 30-week NASH diet exhibited substantial KC loss driven by genetic changes in KC enhancers and subsequent cell death.74 Consequently, increased expression of TREM2 and CD9 in several KC phenotypes was observed and could indicate the severity of steatosis and inflammatory injury in NASH.75,76 Recent single-cell analysis has further performed and identified the presence of scar-associated macrophages in murine NASH models and cirrhotic human livers.47,75 This TREM2+CD9+ macrophage subset induced collagen expression in HSCs, and their frequency correlated with fibrosis scoring and disease severity in NASH.47,48
Collagen deposition resulting from hepatic stellate cell activation has been linked to the effects of profibrogenic factors through KC activation, and in vivo research has indicated that KC depletion could attenuate fibrosis development in the late stage of the disease.77 Small animal and patient studies have reported increased levels of activin-A, a multifactorial cytokine belonging to the TGFβ superfamily, in liver disease, leading to the downstream activation of KCs.78,79 In a mouse model, the expression of proinflammatory molecules after activin-A KC activation further induced HSC-dependent fibrogenesis, which was observed in the later stages of NASH cirrhosis.80 KC activation promotes HSC transdifferentiation into myofibroblastic HSCs, which leads to the production of extracellular matrix elements, cytokine secretion, and alpha smooth muscle actin expression, which are necessary for the progression of fibrosis and hepatic scarring.9
In addition to in situ macrophage activation and monocyte recruitment, hepatic infiltration by additional cellular subtypes, such as lymphocytes and neutrophils, contributes to the underlying inflammatory disturbances that occur in NASH (Fig. 3).61 Following highly orchestrated immune processes in NASH, large amounts of proinflammatory cytokines and chemokines mediate the influx of additional populations of CD4+/CD8+ T and B lymphocytes, which further enhance macrophage activation and inflammatory activity.61 Increased levels of both CD4+ and CD8+ populations are found in the blood of NASH patients, and the degree of portal inflammation due to CD8+ cells correlates with disease severity.81,82 In high fructose murine models, CD8+ deficiency was associated with steatosis resistance compared to that of controls.83 Recent evidence has suggested a role of B lymphocytes in regulating T-cell activation in NASH, although some of the evidence has been conflicting.61 It is unclear whether B-cell lymphopoiesis is enhanced or compromised in obesity states; however, B cell-derived IL-10 has been shown to inhibit proinflammatory cytokines in metabolic syndromes.84
Ischemic liver damage
The dual blood supply of the liver consists of oxygenated blood delivered via the hepatic artery, which accounts for 20% of hepatic inflow, with the remaining 80% delivered by the portal venous system. The healthy liver, therefore, has characteristic zones of high and low oxygen levels, forming an oxygen gradient that is necessary for hepatic function, which is described as liver “zonation” and is required to maintain the structural integrity of hepatocytes85,86 (Fig. 4). The concentration of oxygen decreases along the sinusoids as blood passes from Zone 1 (periportal zone) through to Zones 2 (transitional zone) and 3 (perivenous zone). This understanding of zonation has important implications for homeostasis, and various metabolic capacities can be preferentially found in specific zones.87 Although the underlying regulatory pathways of oxygen zonation within the liver remain to be fully elucidated, complementary metabolic activities and spatially separated opposing pathways associated with carbohydrate, amino acid, and lipid metabolism have been shown to occur within distinct oxygen zones.88 In ischemic injury, the absence of oxygen disrupts this finely tuned gradient, resulting in several functional changes at the cellular level in response to oxidative injury that counteract and control the damage (Fig. 5). Hypoxia inducible factors (HIFs) play critical roles in maintaining homeostasis. However, despite the liver being exposed to variable and often low oxygen tensions, hypoxic responses are not induced in normal conditions.55,86 During prolonged periods of severe ischemia, as seen during transplantation and hepatic resections, HIF activation supports oxygen-independent ATP generation to mediate cellular processes and upregulates cell preservation mechanisms through antioxidant and antiapoptotic systems that facilitate cell survival. Unsurprisingly, hypoxia results in a proinflammatory response by increasing the expression of IFN‐γ, MHC II, and costimulatory molecules in hypoxic macrophages to induce T cell-driven cytokine production.89,90 In addition, both human and murine models have been used to study adaptive immune responses and T-cell recruitment in inflammatory hypoxia, and selective FoxP3 induction and upregulation plays a key role in homeostasis by eliciting potent anti-inflammatory responses to limit overwhelming hepatic injury via Treg signaling.91,92 These data suggest the complementary role of HIFs in hepatic immunomodulation and tolerance, but the details of these mechanisms require further clarification.
Fig. 4.

Oxygenation within the liver. The oxygen gradient decreases in blood as it travels through the hepatic sinusoids (Zone 1) toward the pericentral region (Zone 3), significantly altering the local microenvironment and influencing immune cell localization and activity. Kupffer cells are larger and more numerous in periportal zones than in perivenous zones132
Fig. 5.

Pathological immune mechanisms in ischemia-reperfusion injury (IRI). The mechanisms of hepatic IRI are complex and result in a series of cellular responses influencing inflammatory, metabolic, and antioxidant pathways.133 Tissue hypoxia, oxidative stress, and the resulting anaerobic cell metabolism lead to structural cellular injury and a large amount of chemokine and cytokine release to enhance downstream immune cell accumulation. ATP adenosine triphosphate, IL interleukin, ROS reactive oxygen species, IRI ischemia-reperfusion injury, IFN-γ interferon-gamma, ICAM intercellular adhesion molecule, VCAM vascular cell adhesion molecule, TNF tumor necrosis factor, HGF hepatocyte growth factor, VEGF vascular endothelial growth factor
KCs that are activated during ischemia produce ROS, TNFα, and IL-1β, resulting in subsequent leukocyte recruitment, hepatocyte death, and endothelial damage.62,93 In addition, ROS and cytokine secretion activate CD4+ T cells and NKT cells, which produce IFN‐γ, potentiating KC activation.94 Widespread cytokine activation also upregulates the expression of T cell-associated cell surface adhesion molecules on SECs and ROS production by hepatocytes, triggering a multicellular immune process that contributes to an overwhelming inflammatory response.95,96 The end result is a complex communication network of immune cells that propagate IRI. Hepatic immunity during ischemia is mainly associated with immune restoration and regeneration without generating detrimental immune responses in normal contexts. As mentioned previously, liver zonation and the maintenance of an oxygen gradient are required to support the structural integrity of healthy hepatocytes.85 Thus, the pathological consequences of ischemia are particularly apparent during ischemia/reperfusion, which is a widely accepted example of sterile inflammation, and the rapid restoration of oxygen to ischemic tissue triggers apoptosis and oncotic necrosis (Fig. 5).97,98 Ischemia can be categorized as warm or cold, depending on the temperature at which the ischemia occurs. While warm ischemia is known to cause major disruptions in molecular signaling and cellular pathways, cold ischemia has been considered protective due to reduced ATP demands for regular molecular processes. Thus, cold ischemia is being utilized in liver surgery, especially transplantation, to minimize the injuries encountered during warm ischemia. In both contexts, the physiological repercussions are proportional to the extent of ischemia and the duration of oxygen blockade. Therefore, ischemic injury and IRI, although oftentimes used synonymously, are distinct entities. During ischemia, hepatocytes are immediately disrupted, and metabolic processes shift from oxidative phosphorylation to glycolysis.98 The subsequent ATP depletion and calcium accumulation lead to necrosis in the absence of ROS, which distinguishes this process from the pathological processes of IRI.98,99 In routine hepatic surgery, the ischemic time is limited, and severe ischemic injury can be avoided. The main inflammatory response occurs during reperfusion and tissue reoxygenation, whereby hepatocyte leakage results in DAMP release and subsequent KC activation, which is the main driving factor of ROS production.98,100 In addition, activation of the complement system and its components, of which several are potent neutrophil activators, contribute to the widespread inflammatory cascade during reperfusion.101,102
The liver parenchymal condition strongly determines the extent of ischemic injury, and several animal models have been used to establish reduced hepatic regeneration and extensive IRI injury in the background of chronic liver disease. It has been demonstrated in murine models that steatotic and cirrhotic livers exhibit devastating inflammation and hepatic injury following IRI with a reduced abilities to recover normal cellular function and healing.93,103,104 Molecular aberrations in older livers are likely to weaken immunotolerogenic responses to ischemic injury. Older livers exhibit reduced tolerance to IRI and a consistent inability to regenerate, partly explaining why younger donors are considered optimal in the context of transplants.105,106 In a mouse model, mature adult mice displayed significantly reduced neutrophil accumulation, suppression of the inflammatory transcription factor NF-κB and almost complete absence of macrophage inflammatory protein-2, which is necessary for neutrophil recruitment.107 Tissue remodeling roles of neutrophils have also been described in IRI. In a model of sterile liver injury, neutrophils were shown to dismantle injured vessels and create channels for new vascular growth,108 while in a chronic liver injury model, neutrophils promoted fibrolysis and suppressed the development of fibrotic lesions.109
A significant proportion of harvested donor livers are rejected because of fat deposition and damage that is likely caused by fat-induced sterile inflammation. Fat-related damage may significantly enhance IRI during transplantation and the subsequent loss of the graft.110 A major aim in liver transplantation research is to provide protection to minimize ischemic reperfusion injury, particularly in marginal livers, allowing them to withstand surgery.111
New therapeutic targets and strategies for immune-mediated liver pathology
NASH
Accumulating evidence implicates the hepatic microenvironment and immune repertoire with chronic liver injury, particularly when the balance between inflammation and tolerance is disrupted, resulting in important clinical consequences. The increasing prevalence of metabolic syndrome and consequences such as NASH pose several challenges that demand novel therapies to target the inflammatory microenvironment.5 The altered immune environment in NASH has been a topic of considerable focus in recent times, and several therapeutic strategies have been proposed to decrease local hepatic responses without compromising overall immunity. Inflammatory genes are overexpressed in NASH, and an array of proinflammatory factors are secreted by KCs, NKT cells, HSCs, DCs, monocytes, and lymphocytes, perpetuating a cycle of injury and DAMP release, which intensifies the innate immune response.112 To dampen T-cell responses during NASH, the induction of oral tolerance by the administration of anti‐CD3 mAbs is currently being explored in murine models.61 In a leptin-deficient model of fatty liver, oral anti‐CD3 mAbs alleviate hepatic fat accumulation, improve liver enzymes, and normalize serum glucose levels.113 Thus, a randomized, controlled clinical trial was conducted to determine the efficacy of oral anti‐CD3 mAbs on human NASH subjects over a 30-day period (Table 2).114 Treated patients demonstrated significant reductions in triglyceride levels and liver enzymes and elevations in TGF-β, indicating anti-inflammatory augmentation. Overall, this work highlights the important role T-cell responses play in NASH and suggest a therapeutic strategy for immune modulation. Further research is warranted to determine the utility of anti-CD3 mAbs in all stages of disease and potential interactions with the overlapping pathways seen in metabolic syndrome.
Table 2.
Immunotherapeutic strategies that are currently under investigation for the treatment of immunopathological liver disease
| Therapeutic strategy | Targeted disease | Potential mechanism | Refs |
|---|---|---|---|
| Oral anti‐CD3 mAb | NASH |
Induction of CD4+ latency-associated peptide (LAP)-positive Tregs. Increased levels of TGF-β |
92,93 |
|
Bovine colostrum (IMM-124E) |
NAFLD | IgG antibody against bacterial LPS to prevent TLR4 signaling and cytokine release | 116 |
|
Cenicriviroc |
NASH Liver fibrosis |
Blockade of C-C chemokine receptors type 2 and 5 | 94,95 |
|
Hypothermic ex vivo machine perfusion |
Ischemia-reperfusion injury in liver transplantation | Hypothermic oxygenated perfusion with cytokine filtration of TNFα, IL-6, IL-8, and endothelin-1 | 98 |
In addition, the role of endotoxin in the inflammatory pathophysiology of NASH has long been hypothesized, and in human studies, increased plasma IgG levels were found in patients with biopsy-associated disease and correlated with disease severity.115 Following this discovery, hyperimmune colostrum preparations against bacterial LPS alleviated several metabolic abnormalities associated with NASH, primarily lessening chronic inflammation and improving insulin resistance.116 This strategy is currently being tested in pediatric patients with NAFLD for which no approved therapeutic intervention exists (NCT03042767) and follows the promising results observed in adult experiments in which IMM-124E, a hyperimmune bovine colostrum enriched with IgG against Escherichia coli, resulted in improvements in clinical parameters for alcoholic hepatitis (NCT01968382).
In murine and human models, monocytes have emerged as key mediators of hepatic injury through chemokine regulation and appear to be attractive targets for therapeutic interventions. Pharmacological blockade of CCR2 blocked macrophage infiltration, steatohepatitis, and fibrosis in mice with chronic liver injury.117,118 These results await successful translation into clinical use in affected patients, and the Centaur Trial (NCT02217475) is currently studying the effects of antagonizing CCR2 and CCR5 in a phase 2b clinical trial of NASH patients and fibrosis.
Liver surgery and ischemic injury
In hepatic surgery, the proinflammatory cytokines produced following IRI, especially in the context of transplantation, are potential therapeutic targets. The complexity and interactions of inflammatory factors have made it difficult to design a therapeutic strategy that could interact with the overlapping signaling pathways involved. The challenge in liver surgery is to identify agents that could abrogate ischemic insults and IRI within a realistic timeline while also minimizing collateral deleterious effects to the liver and other organs. One of the most significant improvements in transplantation surgery has been in the area of allograft machine perfusion and preservation. In this context, ex vivo machine perfusion strategies to minimize ischemic and inflammatory injury in transplanted livers have gained attention and are slowly being integrated into clinical practice.119,120 Two main perfusion approaches exist for the liver: (a) normothermic perfusion, which uses blood or oxygenated perfusates at physiologic temperatures, and (b) hypothermic perfusion using cooled oxygenated fluids.121,122 In both approaches, livers are perfused ex vivo immediately following procurement rather than undergoing static cold storage, and at both temperatures, acceptable viability measures and patient survival outcomes have been achieved.123,124 Normothermic interventions aim to scavenge ROS, DAMPs, and proinflammatory cytokines using perfusate scavengers.121,125 During hypothermic perfusion, metabolic processes are slowed to reduce endothelial cell injury and KC activation.126 An adjunct to pre-existing machine technology could be cytokine filtration techniques, which have shown some success in experimental lung perfusion models.127 This technique allows for cytokine removal and the clearance of accumulated inflammatory mediators that are associated with worse outcomes in transplantation, and this approach has seen favorable outcomes in allograft function. A similar tool is currently under investigation using novel hypothermic oxygenated perfusion techniques in liver transplantation (NCT04203004) and could potentially expand the donor pool by optimizing high-risk allograft function.
Immunometabolism
More recently, an intriguing link has been made between itaconate and antioxidant expression, suggesting that the former may promote significant anti-inflammatory phenotypes via an Nrf2 mechanism and offering an exciting therapeutic target for sterile inflammation128 (Fig. 6). Itaconate has recently been shown to control pulmonary fibrosis in a murine model by decreasing fibroblast activity, and this antifibrotic effect may be a viable therapeutic strategy in hepatic fibrosis in which the inflammatory cascade results in concurrent fibroblast activation and wound healing.129 An important emerging observation is that the same stimulus does not necessarily illicit the same damage in every liver. Some livers seem to be better protected from damage than others. The wide spectrum of immune activity is key to the diverse pathologies seen in response to a given stimulus, and of particular importance is the wide range of pathological injuries that can be induced in different individuals, indicating the importance of genetic influences. In a recent study, one-third of obese individuals undergoing bariatric surgery did not exhibit liver fibrosis, and a high proportion of these individuals also had normal insulin resistance.130 This finding was in contrast to the group who were metabolically unhealthy and who also had liver fibrosis. It is likely that metabolic activity in hepatocytes, particularly the production of Nrf2, which mediates HIF-1α activation, the induction of protective mechanisms against oxidation, and the activation of the mTOR pathway, is key to protecting against sterile inflammation during IRI. In this context, changing the nutritional regimen of donors prior to organ collection may have a profound effect on the response of the liver to IRI after transplantation.
Fig. 6.
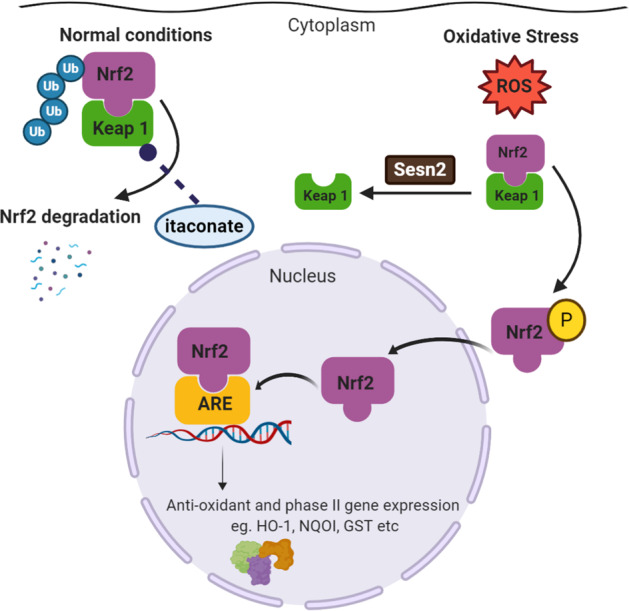
The Nrf2 activation pathway. Under basal conditions, Nrf2 is found in the cytoplasm bound to the inhibitory protein Keap1, and protein levels are kept low through a ubiquitin-dependent degradation mechanism. During oxidative stress and the release of reactive oxygen species (ROS), structural restriction of the Keap1 protein complex stimulates the release of Nrf2 and its subsequent translocation into the nucleus. Sestrin 2 (sesn2) is also induced by oxidative stress and facilitates the autophagic degradation of Keap1 to ensure Nrf2 activation. Within the nucleus, Nrf2 functions as a strong transcriptional activator of antioxidant response elements (AREs) in the promoter segments of corresponding genes. Itaconate can alkylate cysteine residues on Keap1, allowing Nrf2 to accumulate in the cytoplasm, translocate to the nucleus, and increase the expression of antioxidant and anti-inflammatory genes128
Concluding remarks
Hepatic inflammatory mechanisms maintain liver homeostasis and protect the body from injury and harmful pathogens while maintaining the liver parenchyma and other organs. Immunological tolerance is balanced by a proinflammatory and anti-inflammatory micromilieu generated by diverse immune cell populations that play both physiological and pathological roles. While there has been considerable progress in the field of hepatic immunology, several molecular mechanisms and the oftentimes contradictory interplay of immune factors require additional clarification. Although there are many branches of current research aiming to define the hepatic microenvironment, the myriad of local and systemic cellular interactions add to the complexity of immune biology and can obscure the responses to conventional and targeted immune therapies. The clinical implications of an altered hepatic immune environment that often results from global immune aberrations, rather than single isolated deviations from physiological cellular homeostasis, have been a clinical obstacle to date, but promising preliminary results from human studies in the experimental setting are currently awaiting translation to routine care. The challenge in this field is in identifying therapeutic strategies that can target the array of myeloid, lymphoid, and nonimmune cells without compromising the healthy parenchyma.
Author contributions
O.A. drafted the manuscript, designed the figures, and contributed to the main conceptual ideas. M. W. R. contributed to the writing, provided critical feedback, and helped shape the manuscript. C.O.F. designed and directed the review, supervised the work, and contributed to the writing of the manuscript.
Funding
This research was supported by an SFI Frontiers Grant.
Competing interests
The authors declare no competing interests.
References
- 1.Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell. Mol. Immunol. 2016;13:347–353. doi: 10.1038/cmi.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kubes P, Jenne C. Immune responses in the liver. Annu. Rev. Immunol. 2018;36:247–277. doi: 10.1146/annurev-immunol-051116-052415. [DOI] [PubMed] [Google Scholar]
- 3.Surewaard BGJ, Kubes P. Measurement of bacterial capture and phagosome maturation of Kupffer cells by intravital microscopy. Methods. 2017;128:12–19. doi: 10.1016/j.ymeth.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Zeng Z, et al. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe. 2016;20:99–106. doi: 10.1016/j.chom.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Horst AK, et al. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016;13:277–292. doi: 10.1038/cmi.2015.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomson AW, et al. Immunobiology of liver dendritic cells. Immunol. Cell Biol. 2002;80:65–73. doi: 10.1046/j.0818-9641.2001.01058.x. [DOI] [PubMed] [Google Scholar]
- 7.Webb LM, et al. Type I interferon is required for T helper (Th) 2 induction by dendritic cells. EMBO J. 2017;36:2404–2418. doi: 10.15252/embj.201695345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly A, et al. CD141+ myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 2014;60:135–142. doi: 10.1016/j.jhep.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Nati M, et al. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH) Rev. Endocr. Metab. Disord. 2016;17:29–39. doi: 10.1007/s11154-016-9339-2. [DOI] [PubMed] [Google Scholar]
- 10.Ochando J, et al. Tolerogenic dendritic cells in organ transplantation. Transpl. Int. 2020;33:113–127. doi: 10.1111/tri.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arrese M, et al. Innate immunity and inflammation in NAFLD/NASH. Digestive Dis. Sci. 2016;61:1294–1303. doi: 10.1007/s10620-016-4049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dou L, et al. Hepatic dendritic cells, the tolerogenic liver environment, and liver disease. Semin Liver Dis. 2018;38:170–180. doi: 10.1055/s-0038-1646949. [DOI] [PubMed] [Google Scholar]
- 13.Tsuda Y, et al. An immunosuppressive subtype of neutrophils identified in patients with hepatocellular carcinoma. J. Clin. Biochem. Nutr. 2012;51:12–32. doi: 10.3164/jcbn.12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pillay J, et al. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell. Mol. Life Sci. 2013;70:3813–3827. doi: 10.1007/s00018-013-1286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coffelt SB, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao L, et al. Characterization of liver monocytic myeloid-derived suppressor cells and their role in a murine model of non-alcoholic fatty liver disease. PLoS One. 2016;11:e0149948. doi: 10.1371/journal.pone.0149948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenne CN, Kubes P. Immune surveillance by the liver. Nat. Immunol. 2013;14:996–1006. doi: 10.1038/ni.2691. [DOI] [PubMed] [Google Scholar]
- 20.Doherty DG, O’Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunological Rev. 2000;174:5–20. doi: 10.1034/j.1600-0528.2002.017416.x. [DOI] [PubMed] [Google Scholar]
- 21.Shen Y, et al. Ambiguous roles of innate lymphoid cells in chronic development of liver diseases. World J. Gastroenterol. 2018;24:1962–1977. doi: 10.3748/wjg.v24.i18.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenna T, et al. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J. Immunol. 2003;171:1775–1779. doi: 10.4049/jimmunol.171.4.1775. [DOI] [PubMed] [Google Scholar]
- 23.Dusseaux M, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. 2011;117:1250–1259. doi: 10.1182/blood-2010-08-303339. [DOI] [PubMed] [Google Scholar]
- 24.Nemeth E, Baird AW, O’Farrelly C. Microanatomy of the liver immune system. Semin Immunopathol. 2009;31:333–343. doi: 10.1007/s00281-009-0173-4. [DOI] [PubMed] [Google Scholar]
- 25.Clemente-Casares X, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530:434–440. doi: 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- 26.Curry MP, et al. Expansion of peripheral blood CD5+ B cells is associated with mild disease in chronic hepatitis C virus infection. J. Hepatol. 2000;32:121–125. doi: 10.1016/s0168-8278(00)80198-1. [DOI] [PubMed] [Google Scholar]
- 27.Curry MP, et al. Expansion of innate CD5pos B cells expressing high levels of CD81 in hepatitis C virus infected liver. J. Hepatol. 2003;38:642–650. doi: 10.1016/s0168-8278(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 28.Monteverde A, Ballarè M, Pileri S. Hepatic lymphoid aggregates in chronic hepatitis C and mixed cryoglobulinemia. Springe. Semin. Immunopathol. 1997;19:99–110. doi: 10.1007/BF00945028. [DOI] [PubMed] [Google Scholar]
- 29.Doherty DG. Immunity, tolerance and autoimmunity in the liver: a comprehensive review. J. Autoimmun. 2016;66:60–75. doi: 10.1016/j.jaut.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 30.Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 31.Krueger DP, et al. Regulation of NK cell repertoire and function in the liver. Crit. Rev. Immunol. 2011;31:43–52. doi: 10.1615/critrevimmunol.v31.i1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taniguchi H, et al. Presence of hematopoietic stem cells in the adult liver. Nat. Med. 1996;2:198–203. doi: 10.1038/nm0296-198. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe H, et al. c-kit+ stem cells and thymocyte precursors in the livers of adult mice. J. Exp. Med. 1996;184:687–693. doi: 10.1084/jem.184.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crosbie OM, et al. In vitro evidence for the presence of hematopoietic stem cells in the adult human liver. Hepatology. 1999;29:1193–1198. doi: 10.1002/hep.510290402. [DOI] [PubMed] [Google Scholar]
- 35.Wolber FM, et al. Roles of spleen and liver in development of the murine hematopoietic system. Exp. Hematol. 2002;30:1010–1019. doi: 10.1016/s0301-472x(02)00881-0. [DOI] [PubMed] [Google Scholar]
- 36.Jiang X, et al. Characterizing the lymphopoietic kinetics and features of hematopoietic progenitors contained in the adult murine liver in vivo. PLoS One. 2013;8:e76762. doi: 10.1371/journal.pone.0076762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lysakova‐Devine T, O’Farrelly C. Tissue‐specific NK cell populations and their origin. J. Leukoc. Biol. 2014;96:981–990. doi: 10.1189/jlb.1RU0514-241R. [DOI] [PubMed] [Google Scholar]
- 38.Golden‐Mason L, et al. Differential expression of lymphoid and myeloid markers on differentiating hematopoietic stem cells in normal and tumor‐bearing adult human liver. Hepatology. 2000;31:1251–1256. doi: 10.1053/jhep.2000.7713. [DOI] [PubMed] [Google Scholar]
- 39.Arnold B. Parenchymal cells in immune and tolerance induction. Immunol. Lett. 2003;89:225–228. doi: 10.1016/s0165-2478(03)00150-0. [DOI] [PubMed] [Google Scholar]
- 40.Wong YC, et al. Immune outcomes in the liver: Is CD8 T cell fate determined by the environment? J. Hepatol. 2015;63:1005–1014. doi: 10.1016/j.jhep.2015.05.033. [DOI] [PubMed] [Google Scholar]
- 41.Burghardt S, et al. Hepatocytes contribute to immune regulation in the liver by activation of the Notch signaling pathway in T cells. J. Immunol. 2013;191:5574–5582. doi: 10.4049/jimmunol.1300826. [DOI] [PubMed] [Google Scholar]
- 42.MacParland SA, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018;9:1–21. doi: 10.1038/s41467-018-06318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol. Commun. 2019;3:730–743. doi: 10.1002/hep4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halpern KB, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017;542:352–356. doi: 10.1038/nature21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gebhardt R, Matz-Soja M. Liver zonation: novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 2014;20:8491. doi: 10.3748/wjg.v20.i26.8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aizarani N, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204. doi: 10.1038/s41586-019-1373-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stamataki Z, Swadling L. The liver as an immunological barrier redefined by single-cell analysis. Immunology. 2020;160:157–170. doi: 10.1111/imm.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramachandran P, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–518. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao J, et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020;6:22. doi: 10.1038/s41421-020-0157-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobie R, et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–1847.e8. doi: 10.1016/j.celrep.2019.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hossain M, Kubes P. Innate immune cells orchestrate the repair of sterile injury in the liver and beyond. Eur. J. Immunol. 2019;49:831–841. doi: 10.1002/eji.201847485. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 53.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Nakatani Y, et al. Endotoxin clearance and its relation to hepatic and renal disturbances in rats with liver cirrhosis. Liver. 2001;21:64–70. doi: 10.1034/j.1600-0676.2001.210110.x. [DOI] [PubMed] [Google Scholar]
- 55.Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016;13:267–276. doi: 10.1038/cmi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar V. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J. Hepatol. 2013;59:618–620. doi: 10.1016/j.jhep.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng ZB, et al. Immature myeloid cells induced by a high‐fat diet contribute to liver inflammation. Hepatology. 2009;50:1412–1420. doi: 10.1002/hep.23148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arrenberg P, Maricic I, Kumar V. Sulfatide-mediated activation of type II natural killer T cells prevents hepatic ischemic reperfusion injury in mice. Gastroenterology. 2011;140:646–655. doi: 10.1053/j.gastro.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Halder RC, et al. Type II NKT cell–mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J. Clin. Investig. 2007;117:2302–2312. doi: 10.1172/JCI31602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016;57:2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilan Y, Shailubhai K, Sanyal A. Immunotherapy with oral administration of humanized anti-CD3 monoclonal antibody: a novel gut-immune system-based therapy for metaflammation and NASH. Clin. Exp. Immunol. 2018;193:275–283. doi: 10.1111/cei.13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016;13:316–327. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lanthier N, et al. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;298:G107–G116. doi: 10.1152/ajpgi.00391.2009. [DOI] [PubMed] [Google Scholar]
- 64.Rivera CA, et al. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lefkowitch JH, Haythe JH, Regent N. Kupffer cell aggregation and perivenular distribution in steatohepatitis. Mod. Pathol. 2002;15:699–704. doi: 10.1097/01.MP.0000019579.30842.96. [DOI] [PubMed] [Google Scholar]
- 66.Bieghs V, Trautwein C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013;34:446–452. doi: 10.1016/j.it.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 67.Bertola A, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5:e13577. doi: 10.1371/journal.pone.0013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miura K, et al. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;302:G1310–G1321. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galastri S, et al. Lack of CC chemokine ligand 2 differentially affects inflammation and fibrosis according to the genetic background in a murine model of steatohepatitis. Clin. Sci. 2012;123:459–471. doi: 10.1042/CS20110515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H, et al. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021;18.1:73–91. doi: 10.1038/s41423-020-00579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 72.Dixon LJ, et al. Kupffer cells in the liver. Compr. Physiol. 2013;3:785–797. doi: 10.1002/cphy.c120026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fallowfield JA, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 74.Seidman JS, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057–1074.e7. doi: 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiong X, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell. 2019;75:644–660.e5. doi: 10.1016/j.molcel.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wen Y, et al. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2020;18:45–56. doi: 10.1038/s41423-020-00558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Hul N, et al. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol. 2011;179:1839–1850. doi: 10.1016/j.ajpath.2011.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang X, et al. Expression changes of activin A in the development of hepatic fibrosis. World J. Gastroenterol. 2001;7:37. doi: 10.3748/wjg.v7.i1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gressner OA, et al. Intracrine signalling of activin A in hepatocytes upregulates connective tissue growth factor (CTGF/CCN2) expression. Liver Int. 2008;28:1207–1216. doi: 10.1111/j.1478-3231.2008.01729.x. [DOI] [PubMed] [Google Scholar]
- 80.Kiagiadaki F, et al. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim Biophys. Acta Mol. Basis Dis. 2018;1864:891–899. doi: 10.1016/j.bbadis.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 81.Inzaugarat ME, et al. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J. Clin. Immunol. 2011;31:1120–1130. doi: 10.1007/s10875-011-9571-1. [DOI] [PubMed] [Google Scholar]
- 82.Gadd VL, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–1405. doi: 10.1002/hep.26937. [DOI] [PubMed] [Google Scholar]
- 83.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 84.Winer DA, et al. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell. Mol. Life Sci. 2014;71:1033–1043. doi: 10.1007/s00018-013-1486-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee-Montiel FT, et al. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. 2017;242:1617–1632. doi: 10.1177/1535370217703978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson GK, Tennant DA, McKeating JA. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J. Hepatol. 2014;61:1397–1406. doi: 10.1016/j.jhep.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 87.Katz N, Jungermann K. Autoregulatory shift from fructolysis to lactate gluconeogenesis in rat hepatocyte suspensions. The problem of metabolic zonation of liver parenchyma. Biol. Chem. 1976;357:359. doi: 10.1515/bchm2.1976.357.1.359. [DOI] [PubMed] [Google Scholar]
- 88.Kietzmann T. Metabolic zonation of the liver: the oxygen gradient revisited. Redox Biol. 2017;11:622–630. doi: 10.1016/j.redox.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bhandari T, et al. HIF-1α influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J. Mol. Med. 2013;91:1199–1205. doi: 10.1007/s00109-013-1052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Acosta-Iborra B, et al. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-γ production through the HIF-1α transcription factor. J. Immunol. 2009;182:3155–3164. doi: 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- 91.Clambey ET, et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl Acad. Sci. USA. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ben‐Shoshan J, et al. Hypoxia controls CD4+ CD25+ regulatory T‐cell homeostasis via hypoxia‐inducible factor‐1α. Eur. J. Immunol. 2008;38:2412–2418. doi: 10.1002/eji.200838318. [DOI] [PubMed] [Google Scholar]
- 93.Abu-Amara M, et al. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review. Liver Transpl. 2010;16:1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 94.Kuboki S, et al. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G1054–G1059. doi: 10.1152/ajpgi.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taniai H, et al. Susceptibility of murine periportal hepatocytes to hypoxia-reoxygenation: role for NO and Kupffer cell-derived oxidants. Hepatology. 2004;39:1544–1552. doi: 10.1002/hep.20217. [DOI] [PubMed] [Google Scholar]
- 96.Hanschen M, et al. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation. 2008;86:710–718. doi: 10.1097/TP.0b013e3181821aa7. [DOI] [PubMed] [Google Scholar]
- 97.Quesnelle KM, Bystrom PV, Toledo-Pereyra LH. Molecular responses to ischemia and reperfusion in the liver. Arch. Toxicol. 2015;89:651–657. doi: 10.1007/s00204-014-1437-x. [DOI] [PubMed] [Google Scholar]
- 98.Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J. Clin. Transl. Res. 2017;3(Suppl 1):170–188. doi: 10.18053/jctres.03.2017S1.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaeschke H, Smith CV, Mitchell JR. Hypoxic damage generates reactive oxygen species in isolated perfused rat liver. Biochem Biophys. Res Commun. 1988;150:568–574. doi: 10.1016/0006-291x(88)90431-7. [DOI] [PubMed] [Google Scholar]
- 100.Jaeschke H, et al. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic. Res Commun. 1991;15:277–284. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 101.Jaeschke H, et al. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am. J. Physiol. 1993;264:G801–G809. doi: 10.1152/ajpgi.1993.264.4.G801. [DOI] [PubMed] [Google Scholar]
- 102.Tsung A, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hasegawa T, et al. Reduced inflammatory response and increased microcirculatory disturbances during hepatic ischemia-reperfusion injury in steatotic livers of ob/ob mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G1385–G1395. doi: 10.1152/ajpgi.00246.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jang JH, et al. Ischemic preconditioning and intermittent clamping confer protection against ischemic injury in the cirrhotic mouse liver. Liver Transpl. 2008;14:980–988. doi: 10.1002/lt.21467. [DOI] [PubMed] [Google Scholar]
- 105.Macshut M, et al. Older donor age is a risk factor for negative outcomes after adult living donor liver transplantation using small‐for‐size grafts. Liver Transplant. 2019;25:1524–1532. doi: 10.1002/lt.25601. [DOI] [PubMed] [Google Scholar]
- 106.Tanemura A, et al. Donor age affects liver regeneration during early period in the graft liver and late period in the remnant liver after living donor liver transplantation. World J. Surg. 2012;36:1102–1111. doi: 10.1007/s00268-012-1496-1. [DOI] [PubMed] [Google Scholar]
- 107.Okaya T, et al. Age-dependent responses to hepatic ischemia/reperfusion injury. Shock. 2005;24:421–427. doi: 10.1097/01.shk.0000181282.14050.11. [DOI] [PubMed] [Google Scholar]
- 108.Wang J, et al. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358:111–116. doi: 10.1126/science.aam9690. [DOI] [PubMed] [Google Scholar]
- 109.Saijou E, et al. Neutrophils alleviate fibrosis in the CCl4‐induced mouse chronic liver injury model. Hepatol. Commun. 2018;2:703–717. doi: 10.1002/hep4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu L, et al. Innate immune regulations and liver ischemia-reperfusion injury. Transplantation. 2016;100:2601–2610. doi: 10.1097/TP.0000000000001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Elnaggar, A. S. & Guarrera, J. V. The marginal liver donor and organ preservation strategies, in Liver Anesthesiology and Critical Care Medicine. (ed Wagener, G.) 207–220 (Springer, 2018).
- 112.Ilan Y. Review article: novel methods for the treatment of non-alcoholic steatohepatitis—targeting the gut immune system to decrease the systemic inflammatory response without immune suppression. Alimentary Pharmacol. Therapeutics. 2016;44:1168–1182. doi: 10.1111/apt.13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ilan Y, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc. Natl Acad. Sci. USA. 2010;107:9765–9770. doi: 10.1073/pnas.0908771107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mizrahi, M. et al. Oral administration of anti-CD3 MAB to patients with NASH is safe, promotes regulatory T cells, decreases liver enzymes, and alleviates insulin resistance: results of a phase IIA blinded placebo-controlled trial. Hepatology54, 117 (2011). [DOI] [PubMed]
- 115.Verdam FJ, et al. Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2011;45:149–152. doi: 10.1097/MCG.0b013e3181e12c24. [DOI] [PubMed] [Google Scholar]
- 116.Adar T, et al. Oral administration of immunoglobulin G-enhanced colostrum alleviates insulin resistance and liver injury and is associated with alterations in natural killer T cells. Clin. Exp. Immunol. 2012;167:252–260. doi: 10.1111/j.1365-2249.2011.04511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baeck C, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology. 2014;59:1060–1072. doi: 10.1002/hep.26783. [DOI] [PubMed] [Google Scholar]
- 118.Ehling J, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 2014;63:1960–1971. doi: 10.1136/gutjnl-2013-306294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Laing, R. W. et al. Viability testing and transplantation of marginal livers (VITTAL) using normothermic machine perfusion: study protocol for an open-label, non-randomised, prospective, single-arm trial. BMJ Open7 (2017). [DOI] [PMC free article] [PubMed]
- 120.Mergental H, et al. Transplantation of discarded livers following viability testing with normothermic machine perfusion. Nat. Commun. 2020;11:1–12. doi: 10.1038/s41467-020-16251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dutkowski P, et al. Evolving trends in machine perfusion for liver transplantation. Gastroenterology. 2019;156:1542–1547. doi: 10.1053/j.gastro.2018.12.037. [DOI] [PubMed] [Google Scholar]
- 122.Dutkowski P, et al. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. 2015;148:307–323. doi: 10.1053/j.gastro.2014.08.045. [DOI] [PubMed] [Google Scholar]
- 123.Nasralla D, et al. A randomized trial of normothermic preservation in liver transplantation. Nature. 2018;557:50–56. doi: 10.1038/s41586-018-0047-9. [DOI] [PubMed] [Google Scholar]
- 124.Schlegel A, et al. Outcomes of DCD liver transplantation using organs treated by hypothermic oxygenated perfusion before implantation. J. Hepatol. 2019;70:50–57. doi: 10.1016/j.jhep.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 125.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schlegel A, et al. Protective mechanisms of end-ischemic cold machine perfusion in DCD liver grafts. J. Hepatol. 2013;58:278–286. doi: 10.1016/j.jhep.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 127.Iskender I, et al. Cytokine filtration modulates pulmonary metabolism and edema formation during ex vivo lung perfusion. J. Heart Lung Transplant. 2018;37:283–291. doi: 10.1016/j.healun.2017.05.021. [DOI] [PubMed] [Google Scholar]
- 128.Mills EL, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ogger PP, et al. Itaconate controls the severity of pulmonary fibrosis. Sci. Immunol. 2020;5:eabc1884. doi: 10.1126/sciimmunol.abc1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.O’Connell J, et al. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PloS One. 2010;5:e9997. doi: 10.1371/journal.pone.0009997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Markose D, et al. Immune cell regulation of liver regeneration and repair. J. Immunol. Regenerative Med. 2018;2:1–10. [Google Scholar]
- 132.Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev. Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 133.Fu, P. & W. Li. Chapter 8—nitric oxide in liver ischemia–reperfusion injury, in Liver Pathophysiology (ed. Muriel, P.) 125–127 (Academic Press: Boston, 2017).
- 134.Zhang Q, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179:829–845.e20. doi: 10.1016/j.cell.2019.10.003. [DOI] [PubMed] [Google Scholar]