

Abstract
Exploiting a pure culture strategy to investigate the composition of the human gut microbiota, two novel anaerobes, designated strains AF52-21T and CM04-06T, were isolated from faeces of two healthy Chinese donors and characterized using a polyphasic approach. The two strains were observed to be gram-negative, non-motile, and rod-shaped. Both strains grew optimally at 37 °C and pH 7.0. Phylogenetic analysis based on 16S rRNA gene sequences revealed that the two strains clustered with species of the genus Faecalibacterium and were most closely related to Faecalibacterium prausnitzii ATCC 27768T with sequence similarity of 97.18% and 96.87%, respectively. The two isolates shared a 16S rRNA gene sequence identity of 98.69%. Draft genome sequencing was performed for strains AF52-21T and CM04-06T, generating genome sizes of 2.85 Mbp and 3.01 Mbp. The calculated average nucleotide identity values between the genomes of the strains AF52-21T and CM04-06T compared to Faecalibacterium prausnitzii ATCC 27768T were 83.20% and 82.54%, respectively, and 90.09% when comparing AF52-21T and CM04-06T. Both values were below the previously proposed species threshold (95–96%), supporting their recognition as novel species in the genus Faecalibacterium. The genomic DNA G + C contents of strains AF52-21T and CM04-06T calculated from genome sequences were 57.77 mol% and 57.51 mol%, respectively. Based on the phenotypic, chemotaxonomic and phylogenetic characteristics, we conclude that both strains represent two new Faecalibacterium species, for which the names Faecalibacterium butyricigenerans sp. nov. (type strain AF52-21T = CGMCC 1.5206T = DSM 103434T) and Faecalibacterium longum sp. nov. (type strain CM04-06T = CGMCC 1.5208T = DSM 103432T) are proposed.
Subject terms: Bacteria, Genome, Microbiology, Genome informatics
Introduction
The human gastrointestinal tract harbours complex microbial communities1,2, dominated by bacteria from the phyla Bacteroidetes and Firmicutes3. The composition and diversity of the gut microbiota are affected by numerous factors, including host genetics4, long-term diet5,6, drugs1,7,8, and several other environmental factors9. Evidence suggests that the composition of the microbiota is associated with the development of obesity3,10–12, diabetes13,14, inflammatory bowel disease15,16, colorectal cancer17,18, and non-alcoholic fatty liver disease19,20. Therefore, the composition and function of the microbial species living in our gut are of crucial importance for maintenance of health. Short-chain fatty acids (SCFAs) produced by fermentation of dietary fibre by several abundant genera of the intestinal microbiota, including Roseburia, Eubacterium, and Faecalibacterium21, have been reported to elicit beneficial effects on energy metabolism and prevent colonization of pathogens22. Bacteria of the genus Faecalibacterium, abundant butyric acid-producing bacteria colonizing the human gut, display anti-inflammatory effects and may be used as potential probiotics for treatment of gut inflammation23,24.
The genus Faecalibacterium, belonging to the family Ruminococcaceae within the order Clostridiales, comprises only one validated species, Faecalibacterium prausnitzii25, and two non-validated published species, ‘Faecalibacterium moorei’26 and ‘Faecalibacterium hominis’27, all originally isolated from human faeces. F. prausnitzii is a gram-negative, non-spore-forming, and strictly anaerobic rod-shaped bacterium. The genomic G + C content of the genus Faecalibacterium ranges from 47 to 57%28. The fermentation products from glucose are butyrate, D-lactate, and formate. In the present study, we describe two novel species of the genus Faecalibacterium by using a polyphasic taxonomy approach along with whole genome sequence analysis.
Results
Phenotypic and chemotaxonomic characterization
Strains AF52-21T and CM04-06T were isolated from the faeces of two healthy Chinese donors. Both strains were observed to be obligate anaerobic, gram-negative, non-spore-forming, non-motile, and rod-shaped bacteria (Fig. 1). After incubation on MPYG agar at 37 °C for 2 days, the colonies appeared 1.0–2.0 mm in diameter, round, creamy white to yellowish, convex, and opaque with entire margins for AF52-21T, and 2.0 mm in diameter, round, yellowish, slightly convex, and opaque with entire margins for CM04-06T. The growth temperature was 20–42 °C (optimum 37 °C) for AF52-21T and 30–45 °C (optimum 37 °C) for CM04-06T. Growth was observed at pH 6.0–7.5 (optimum 7.0–7.5) for AF52-21T and pH 5.0–8.0 (optimum 7.0–7.5) for CM04-06T. Strains AF52-21T and CM04-06T grew with 0–1% and 0–3% NaCl, respectively. Both strains were found to be catalase-negative. The major metabolic end products for strains AF52-21T and CM04-06T were acetic acid, formic acid, butyric acid, and lactic acid. Differential physiological and biochemical characteristics of strains AF52-21T and CM04-06T with the closest related species of genus Faecalibacterium are listed in the species description and in Table 1 (Fig. 2).
Figure 1.
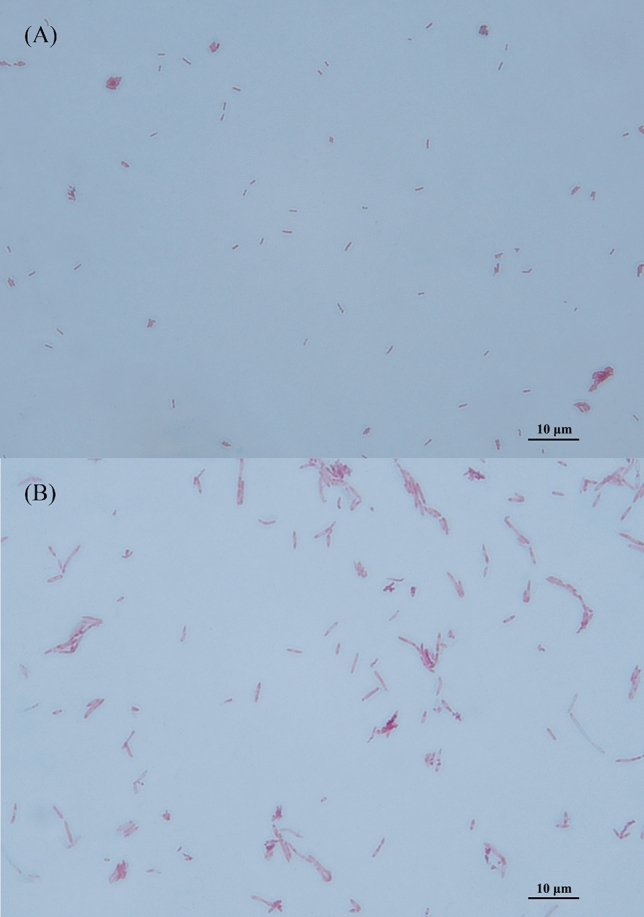
Micrographs of strains AF52-21T and CM04-06T after Gram staining. (A) AF52-21T; (B) CM04-06T.
Table 1.
Differential phenotypic characteristics of strains AF52-21T, CM04-06T, and the related species F. prausnitzii ATCC 27768T.
| Phenotypic features | 1 | 2 | 3 |
|---|---|---|---|
| Growth | |||
| Temperature range (optimum) (°C) | 20–42 (37) | 30–45 (37) | 20–42 (37) |
| pH range | 6.0–7.5 | 5.0–8.0 | 6.0–7.5 |
| Salt tolerance (%) | 1 | 3 | 3 |
| Fermentation products | |||
| formic acid (mM) | 4.86 | 7.62 | 18.20 |
| acetic acid (mM) | 69.70 | 44.8 | 29.67 |
| butyric acid (mM) | 15.08 | 40.03 | 39.10 |
| lactic acid (mM) | 29.25 | 30.53 | 5.70 |
| Hydrolysis of | |||
| Aesculin | + | − | + |
| Gelatin | − | + | − |
| Acid from (API 20A and API 50CHL) | |||
| Cellobiose | + | − | w |
| D-Fructose | w | − | + |
| D-Fucose | w | − | w |
| D-Galactose | w | − | − |
| D-Glucose | w | − | + |
| D-Lactose | + | − | − |
| D-Maltose | + | + | w |
| D-Mannitol | + | − | − |
| D-Mannose | + | + | − |
| D-Raffinose | − | w | − |
| D-Trehalose | + | w | w |
| Gluconate | − | − | + |
| Glycogen | + | − | − |
| Inositol | w | − | − |
| Inulin | + | − | + |
| Methyl-β-D-Xylopyranoside | w | − | − |
| Enzyme activity (API ZYM) | |||
| N-acetyl-β-Glucosaminidase | − | w | − |
| Naphthol-AS-BI-Phosphohydrolase | + | − | + |
| α-Glucosidase | − | − | + |
| β-Galactosidase | − | − | w |
| β-Glucosidase | + | − | − |
| β-Glucuronidase | + | w | + |
| DNA G + C (mol %) | 57.77 | 57.51 | 52 − 57 |
Strains: 1, F. butyricigenerans AF52-21T; 2, F. longum CM04-06T; 3, F. prausnitzii ATCC 27768T. + , positive; w, weakly positive; −, negative. All data are from this study.
Figure 2.

Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationships of strains AF52-21T, CM04-06T and the representatives of related taxa within the family Ruminococcaceae. Clostridium butyricum DSM 10702T (AQQF01000149) was used as an out-group. Bootstrap values based on 1000 replications higher than 70% are shown at the branching points. Bar, substitutions per nucleotide position.
The result of cellular fatty acid profiles of strains AF52-21T and CM04-06T and related species are shown in Table 2. The major components of fatty acids (constituting > 5% of the total) present in strain AF52-21T were found to be C18:1 ω9c (39.0%), C16:0 (16.3%), iso-C19:0 (12.9%), C18:1 ω7c (8.1%), and C14:0 (5.9%). The profiles including C18:1 ω9c (32.5%), C16:0 (25.5%), iso-C17:1 I/anteiso B (9.7%), C18:1 ω7c (7.5%), and iso-C19:0 (5.9%) were detected as the predominant fatty acids for strain CM04-06T. The highest levels of fatty acids, including C16:0 and C18:1 ω9c, were found to be similar, but not identical comparing strains AF52-21T, CM04-06T, and ATCC 27768T. Furthermore, strains AF52-21T, CM04-06T, and ATCC 27768T could be differentiated by less abundant fatty acids, such as C18:1 2OH, anteiso-C15:0, anteiso-C17:0, C13:0 3OH/Iso-C15:1 I, C16:1 ω7c/C16:1 ω6c, and antei-C18:0 /C18:2 ω6, 9c (Table 2). Strains AF52-21T and CM04-06T were found to contain meso-diaminopimelic acid as the diamino acid of the peptidoglycan. The polar lipid profiles of strains AF52-21T, CM04-06T, and F. prausnitzii ATCC 27768T are shown in Supplementary Fig. S1. The polar lipid profiles of AF52-21T and CM04-06T were observed to be similar to that of the most closely related strain F. prausnitzii ATCC 27768T, with diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), and several unidentified glycolipids (GL1, GL3) being present in both strains. However, the presence/absence of three unidentified lipid (L, L1, L2), unidentified phospholipid (PL), unidentified phosphoglycolipids (PGL) and an unidentified glycolipid (GL2) can be used to distinguish strains AF52-21T and CM04-06T from the closest relative. Quinones were not detected in either strain (Table 3).
Table 2.
Fatty acid profiles of strains AF52-21T, CM04-06T, and the closest related species F. prausnitzii ATCC 27768T.
| Fatty acids composition | F. butyricigenerans AF52-21T | F. longum CM04-06T | F. prausnitzii ATCC 27768T |
|---|---|---|---|
| C12:0 | 1.5 | 1.8 | 1.9 |
| C13:1 | – | – | 1.25 |
| C14:0 | 5.9 | 4.6 | 11.8 |
| C16:0 | 16.3 | 25.5 | 21.1 |
| C17:1 ω8c | 1.3 | – | 1.1 |
| C18:1 ω7c | 8.1 | 7.5 | 5.7 |
| C18:1 ω9c | 39.0 | 32.5 | 31.4 |
| C18:0 | 4.5 | 3.5 | 4.1 |
| C18:1 2OH | 2.9 | – | 2.0 |
| Iso-C19:1 I | 1.2 | 1.1 | 2.1 |
| Iso-C19:0 | 12.9 | 5.9 | – |
| Anteiso-C15:0 | – | 2.6 | – |
| Anteiso-C17:0 | – | 2.1 | – |
| C13:0 3OH/ Iso-C15:1 I | – | – | 2.1 |
| C16:1 ω7c/ C16:1 ω6c | 1.5 | 1.9 | 4.0 |
| Iso-C17:1 I/anteiso B | 4.7 | 9.7 | 7.6 |
| Antei-C18:0 /C18:2 ω6, 9c | – | 1.9 | 1.3 |
Numbers represent percentages of the total fatty acids. −, not detected (< 1%). All data are from this study.
Table 3.
Levels of 16S rRNA gene sequence similarity and ANI values (in percentages) based on BLAST for strains AF52-21T, CM04-06T, and the phylogenetically related species F. prausnitzii ATCC 27768T and the unrecognized species ‘Faecalibacterium hominis’ 4P-15.
| Strain | Accession no. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| 16S rRNA gene sequence similarity (%) | |||||
| AF52-21T | N_000011117 | 100 | 98.53 | 97.27 | 98.65 |
| CM04-06T | N_000011118 | 98.53 | 100 | 96.51 | 97.68 |
| ATCC 27768T | AJ413954 | 97.27 | 96.51 | 100 | 98.35 |
| 4P-15 | NMDCN000012L | 98.65 | 97.68 | 98.35 | 100 |
| ANI values (%) | |||||
| AF52-21T | CNA0017730 | 100 | 90.01 | 83.16 | 85.72 |
| CM04-06T | CNA0017731 | 90.19 | 100 | 82.53 | 85.40 |
| ATCC 27768T | CNA0017732 | 83.32 | 82.58 | 100 | 85.79 |
| 4P-15 | NMDC60014083 | 85.72 | 85.40 | 85.79 | 100 |
Taxa: 1, F. butyricigenerans AF52-21T; 2, F. longum CM04-06T; 3, F. prausnitzii ATCC 27768T; 4, ‘Faecalibacterium hominis’ 4P-15.
Genome analysis
The assembled draft genomes of strains AF52-21T and CM04-06T comprised total lengths of 2,851,918 bp and 3,011,178 bp with 73 and 47 scaffolds, respectively (Table 4). The G + C contents calculated from the genome sequences were 57.77% and 57.51%, which are slightly higher than the range reported previously for the genus Faecalibacterium (47–57 mol%)25. CheckM analysis of the genomes showed high completeness (> 90%) and low contamination (< 5%) (Table 4), indicating these are high-quality genomes sequences. The genome comparison between strains AF52-21T, CM04-06T, ATCC 27768T, and ‘Faecalibacterium hominis’ 4P-15 showed ANI values ranging from 82.53% to 90.19% (Table 3), which are significantly below the proposed cutoff value of 95–96% for delineating bacterial species, indicating that strains AF52-21T and CM04-06T represent novel species in the genus Faecalibacterium. Circular maps of the two strains AF52-21T and CM04-06T are shown in Fig. 3.
Table 4.
Genome properties of F. butyricigenerans AF52-21T and F. longum CM04-06T.
| Feature | AF52-21T | CM04-06T |
|---|---|---|
| Accession no. | CNA0017730 | CNA0017731 |
| Approximate genome Size (bp) | 2,851,918 | 3,011,178 |
| G + C content (mol%) | 57.77 | 57.51 |
| DNA scaffolds | 73 | 47 |
| N50 Length | 191,233 | 119,299 |
| Completeness | 100 | 99.32 |
| Contamination | 0 | 0 |
| Genes total number | 2291 | 2506 |
| Gene average length (bp) | 939 | 920 |
| rRNAs (5S, 16S, 23S) | 4 | 5 |
| tRNAs | 60 | 61 |
| sRNA | 0 | 0 |
| Genes assigned to COGs | 2029 | 2164 |
Figure 3.

Circular map of AF52-21T and CM04-06T. Innermost circle, GC skew; circle 2, G + C content; circle 3, contigs; circles 4, predicted prophage remnants; circle 5, tmRNA, tRNA and rRNA genes; circles 6, CDS; circles 7–9, (A) homologous genomic segments from CM04-06T, F. prausnitzii ATCC 27768T and ‘F. hominis’ 4P-15, (B) homologous genomic segments from AF52-21T, F. prausnitzii ATCC 27768T and ‘F. hominis’ 4P-15.
16S rRNA gene sequence extraction and phylogenetic analysis
The almost complete 16S rRNA gene sequences of strains AF52-21T and CM04-06T were extracted from the genomes, in which the locations are Scaf2_220520-222018 and Scaf13_51882-53380, respectively. The length of 16S rRNA gene sequences was found to be 1499 bp for both strains. BLAST analysis of the 16S rRNA gene sequences against the EzBioCloud server showed that the two strains are most closely related to F. prausnitzii ATCC 27768T, which is the sole valid species of the genus Faecalibacterium, with similarity values of 97.27% and 96.51%, respectively. Strains AF52-21T and CM04-06T share a 16S rRNA gene sequence similarity of 98.65% and 97.68% with ‘Faecalibacterium hominis’ 4P-15. The 16S rRNA gene sequence similarity between strains AF52-21T and CM04-06T is 98.53% (Table 3). All these values are lower than the recommended threshold (98.7%) for classification of human-associated bacterial isolates at the species level29. Phylogenetic analysis based on the maximum-likelihood, neighbour-joining, and minimum-evolution (Fig. 2, Supplementary Figs. S2 and S3, respectively) confirmed the affiliation of the novel isolates with the genus Faecalibacterium, revealing that the two isolates form a distinct cluster with F. prausnitzii ATCC 27768T, supported independently of the treeing method by a high bootstrap value.
Function annotation
For genome annotation, the distributions of the genes into clusters of orthologous groups (COGs) functional categories are depicted in Supplementary Fig. S4 and Table S1. Both strains AF52-21T and CM04-06T share identical COGs functional categories, but different functional genes numbers. Annotated genes associated with synthesis of diaminopimelic acid (DAP), teichoic and lipoteichoic acids, lipopolysaccharides, and metabolism of polar lipids and polyamines by RAST annotation, comparing strains AF52-21T and CM04-06T with ATCC 27768T are shown in Table S2. For strain AF52-21T, 11 genes/proteins were observed to be associated with biosynthesis of DAP, 18 genes/proteins with biosynthesis of polar lipids, 12 genes/proteins with biosynthesis of polyamines, 3 genes/proteins with biosynthesis of teichoic and lipoteichoic acids, and 14 genes/proteins with biosynthesis of quinones. For strain CM04-06T, 12 genes/proteins were found to be associated with biosynthesis of DAP, 19 genes/proteins with biosynthesis of polar lipids, 13 genes/proteins with biosynthesis of polyamines, 2 genes/proteins with biosynthesis of teichoic and lipoteichoic acids, and 16 genes/proteins with biosynthesis of quinones. We detected no genes involved in the biosynthesis of lipopolysaccharides or mycolic acids in strains AF52-21T and CM04-06T.
The functional annotation showed that AF52-21T, CM04-06T, and ATCC 27768T contain a complete acetyl-CoA to butyrate synthesis pathway, but possess butyryl-CoA:acetate CoA-transferase activity only in the final step (Fig. 4), as discussed previously30,31. The antiSMASH analysis of biosynthetic gene clusters (BGCs) showed that strains AF52-21T and CM04-06T both contain two potential BGCs, which encode bacteriocin and sactipeptide, respectively, while ATCC 27768T contains BGCs encoding microcin and sactipeptide, respectively (Supplementary Fig. S5). Prophages were identified using the PHAST software, and the results are shown in Supplementary Fig. S6. Two incomplete phage sequences were detected in the AF52-21T genome, one of which encodes the Phd_YefM protein, an antitoxin component. Three incomplete phage sequences and two intact prophages were detected in the CM04-06T genome, encoding the Phd_YefM protein, relaxase/mobilisation nuclease domain, bacterial mobilisation protein (MobC) /ribbon-helix-helix protein, helix-turn-helix, and predicted transcriptional regulators. Moreover, the antibiotic resistance analysis indicated that strain AF52-21T contains macrolide antibiotic, lincosamide antibiotic, and streptogramin antibiotic genes, while strains CM04-06T and ATCC 27768T contain aminoglycoside antibiotic genes (Fig. 5). To better understand the biosynthetic pathway contributing to the in vitro characteristics of strains AF52-21T and CM04-06T, we explored genes related to important pathways involved in carbohydrate metabolism. The comparison of in vitro and in silico characteristics is presented in Table 5.
Figure 4.
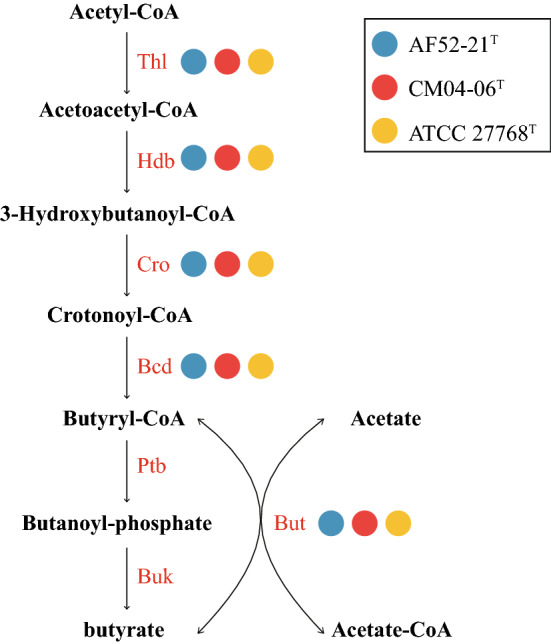
The synthesis pathways from acetyl-CoA to butyrate. Strains AF52-21T, CM04-06T and ATCC 27768T are presented as blue, red, and yellow, respectively. Thl, thiolase; Hdb, β-hydroxybutyryl-CoA dehydrogenase; Cro, crotonase; Bcd, butyryl-CoA dehydrogenase; But, butyryl-CoA:acetate CoA transferase; Ptb, phosphate butyryltransferase; Buk, butyrate kinase.
Figure 5.

Comparison of antibiotics genes in strains AF52-21T, CM04-06T, and F. prausnitzii ATCC 27768T.
Table 5.
Comparison of in vitro and in silico characteristics.
| Metabolic substrate or product | AF52-21T | CM04-06T | ATCC 27768T | |||
|---|---|---|---|---|---|---|
| In silico | In vitro | In silico | In vitro | In silico | In vitro | |
| Cellobiose | GH2, GH3 | + | GH1, GH2, GH3, GH88 | – | GH1, GH2, GH3, GH4, GH88, GH94 | w |
| Galactose | GH2 | w | GH2 | – | GH2, GH36 | – |
| Glucose | GH31 | w | GH1, GH31 | – | GH1, GH31, GH33, GH43 | + |
| Lactose | GH2, GH31 | + | GH1, GH2, GH31 | – | GH1, GH2, GH31, GH36, GH43 | – |
| Maltose | GH13, GH13_20, GH13_39, GH77, GT35 | + | GH1, GH13, GH13_20, GH13_39, GH77, GT35 | + | GH1, GH13, GH13_20, GH13_39, GH77, GT35 | w |
| Mannose | GH2, GH3 | + | GH2, GH3 | + | GH2, GH3 | – |
| Raffinose | GH2, GH13, GH32 | – | GH2, GH13, GH32 | w | GH2, GH4, GH13, GH32, GH36 | – |
| Trehalose | GH13 | + | GH13 | w | GH13 | w |
| Glycogen | GH13, GH13_9, GH13_39, GT5, GT35 | + | GH13, GH13_9, GH13_39, GT5, GT35 | – | GH13, GH13_9, GH13_39, GT5, GT35 | – |
| Inulin | GH32 | + | GH32 | – | GH32 | + |
| butyric acid | butyryl-CoA:acetate CoA transferase | + | butyryl-CoA:acetate CoA transferase | + | butyryl-CoA:acetate CoA transferase | + |
All data are from this study.
GH glycoside hydrolases, GT glycosyl transferases, + , positive, w weakly positive; − , negative.
Discussion
16S rRNA gene phylogeny, genome sequence comparison, and physiological results showed that the two new isolates AF52-21T and CM04-06T represent two novel species. The ANI values between AF52-21T, CM04-06T and the closest related species ATCC 27768T were found to be 82.54% and 90.09%, respectively, which support the delineation of new species. The result of biochemical and genomic functional analyses showed that both strains AF52-21T and CM04-06T are butyric acid-producing bacteria.
Most strains in the genus Faecalibacterium exhibit a common ability to produce butyric acid, bioactive peptides, and other anti-inflammatory substances with immunomodulatory effects23,24,32. Several studies have confirmed that a decreased abundance of this genus is related to the occurrence and development of inflammatory bowel diseases33–35. Accordingly, bacteria of the genus Faecalibacterium are receiving much attention as possible candidate next-generation probiotics (NGPs), which may be used for disease treatment36,37.
Previous studies based on comparative genomics of isolates suggested a wide diversity of this genus, with the presence of at least two phylotypes in F. prausnitzii26. A recent study analysing the Faecalibacterium-like MAGs, proposed that Faecalibacterium from the human gut can be divided into 12 clades37. These studies have expanded the diversity of the genus Faecalibacterium and proposed that different phylotypes have different functions with potentially different contributions in relation to health or diseases.
Moreover, as a candidate taxa for the NGPs, the bacteria of the genus Faecalibacterium can be used for in vitro functional verification and animal model experiments to further explore possible probiotic functions, and ultimately, used in clinical disease intervention trials.
Emended description the genus of Faecalibacterium
The genus description is as given by Duncan et al25 with the following changes. Cells are able to produce formic acid, acetic acid, and butyric acid. The major polar lipids are diphosphatidylglycerol, phosphatidylglycerol and several unidentified glycolipids. Genomic DNA G + C content is 47–63 mol%. Genome size is 2.68–3.32 Mb.
Emended description of Faecalibacterium prausnitzii
Cells are able to produce formic acid, acetic acid, butyric acid, and lactic acid. The major fatty acids (constituting > 5% of the total) are C16:0, C18:1 ω7c, and C18:1 ω9c. The rest of the species characteristics are as described by Cato et al38, Duncan et al25, and Fitzgerald et al26. The type strain is Faecalibacterium prausnitzii ATCC 27768T (= NCIMB 13872T).
Description of Faecalibacterium butyricigenerans sp. nov.
Faecalibacterium butyricigenerans (bu.ty.ri.ci.ge′ne.rans. N.L. n. acidum butyricum butyric acid; L. part. adj. generans, producing; N.L. adj. butyricigenerans, butyric acid-producing; referring to its production of butyric acid).
Cells are gram-negative, non-motile, non-spore-forming and rod-shaped. Strictly anaerobic and catalase negative. Colonies on PYG agar are round, creamy white to yellowish, convex, and opaque with entire margins, and colony size is approximately 1.0–2.0 mm in diameter after incubation at 37 °C for 2 days. Cells are able to grow at 20–42 °C with optimum temperature at 37 °C. The pH range for growth is 6.0–7.5 (optimum at 7.0–7.5). Growth occurs at NaCl concentrations 0–1%. Indole is not produced. Positive for hydrolysis of esculin and negative for gelatin. Formic acid, acetic acid, butyric acid, and lactic acid are the fermentation products. The major fatty acids are C14:0, C16:0, C18:1 ω7c, C18:1 ω9c, and iso-C19:0.
The type strain, AF52-21T (= CGMCC 1.5206T = DSM 103434T), was isolated from human faeces. The G + C content of the genomic DNA is 57.77 mol% as calculated from whole genome sequencing.
Description of Faecalibacterium longum sp. nov.
Faecalibacterium longum (lon′gum. L. neut. adj. longum long, the shape of the cells).
Cells are gram-negative, non-motile, non-spore forming, long rod in shape. Strictly anaerobic. Catalase and urease are negative. Colonies are round, yellowish, slightly convex, and opaque with entire margins with 2.0 mm in diameter on PYG agar for incubation at 37 °C for 48 h under anaerobic condition. The strain shows growth at 30–45 °C (optimum temperature is 37 °C). Growth is observed at pH 5.0–8.0 (optimum pH is 7.0–7.5). NaCl is tolerated with concentrations up to 3%. Indole is not produced. Gelatin is hydrolysed, but aesculin is not. Major end products are acetic acid, formic acid, butyric acid, and lactic acid. The major fatty acids (constituting > 5% of the total) are C16:0, C18:1 ω7c, C18:1 ω9c, iso-C19:0, and iso-C17:1 I/anteiso B.
The type strain, CM04-06T (= CGMCC 1.5208T = DSM 103432T), was isolated from human faeces. The G + C content of the genomic DNA is 57.51 mol% as calculated from whole genome sequencing.
Methods
Origin of bacterial strains
Faeces samples were collected from two healthy donors living in Shenzhen, Guangdong province, China, one donor is an adult female (AF), and the other is a male child (CM). The samples were stored refrigerated and kept anaerobically until processed. The collection of the samples was approved by the Institutional Review Board on Bioethics and Biosafety of BGI under number BGI-IRB17005-T2. All protocols were in compliance with the Declaration of Helsinki and explicit informed consent was obtained from the participant and the parents of the male child. 1 g of faecal sample was diluted with 0.1 M PBS (pH 7, supplemented with 0.5% cysteine) and spread onto modified peptone-yeast extract-glucose (MPYG, supplemented with 5 g/L sodium acetate in DSMZ 104 medium) agar plates in an anaerobic box (Bactron Anaerobic Chamber, BactronIV-2, shellab, USA). The plates were incubated at 37 °C under anaerobic conditions (90% N2, 5% CO2, and 5% H2, v/v) for 3–5 days. Single colonies were randomly picked and purified by repetitive subculturing on the new plates containing the same medium and incubated under the same conditions as described above. Among the pure cultures, two isolates, designated as AF52-21T and CM04-06T, respectively, were obtained and subsequently maintained in 20% (v/v) glycerol and frozen at -80 °C.
Phenotypic characterization
The morphological characteristics of strains AF52-21T and CM04-06T were performed on cultures grown on MPYG medium at 37 °C. Bacterial cell shape was examined by phase contrast microscopy (Olympus BX51, Japan) during the exponential phase of growth. Cell motility was examined using semi-solid MPYG medium containing 0.5% agar39. The Gram reaction was carried out using a Gram-staining kit (Solarbio, China). Spore formation and presence of flagella were determined by staining using spore stain kit and flagella stain kit supplied by Solarbio (China) following the manufacturer’s instructions. Colony morphology was observed following growth of the cultures on PYG agar for 2 days at 37 °C. Optimal temperature for growth was determined using growth in MPYG medium at 4, 10, 20, 25, 30, 35, 37, 45, and 50 °C for 7 days. The pH range for growth was also measured in MPYG medium covering the range of pH 3.0–10.0 (at an interval of 0.5 pH units) at 37 °C for 7 days, and the pH test medium stabilized with the appropriate buffers as described by Sorokin40. Growth at various NaCl concentrations (0–6%, in increments of 1.0%) was performed for determining tolerance to NaCl. Catalase activity was assessed by gas formation after dropping the fresh cells in 3% H2O2 solution. Biochemical properties, including utilization of substrates, acid production from carbohydrates, enzyme activities, hydrolytic activities, were determined using the API 20A, API 50CHL, and API ZYM systems (bioMérieux Inc., Marcy-l’Étoile, France) according to the manufacturer’s instructions with modification by adding sodium acetate at concentration of 0.5% in all tests. The reference type strain was tested under the same condition as used for strains AF52-21T and CM04-06T. In all tests, the strains were incubated under anaerobic conditions.
Chemotaxonomic characteristics
Chemotaxonomic features were investigated by analysing of cellular fatty acids, cell wall composition, polar lipids, and quinones. Biomasses of strains AF52-21T, CM04-06T, and ATCC 27768T were harvested from cells growing in MPYG at 37 °C under anaerobic conditions for 2 days. Whole cell fatty acid methyl esters (FAMEs) were extracted, separated and identified according to the MIDI Microbial Identifications System and performed by CGMGG (China General Microbiological Culture Collection Center, Beijing, China) identification service. The diagnostic isomer of diaminopimelic acid in whole-cell hydrolysates was identified by TLC as described by Zou et al.41. The polar lipids of strain AF52-21T, CM04-06T, and ATCC 27768T were extracted from lyophilized bacterial cells and analysed using two-dimensional TLC as described42. Menaquinone components were extracted and identified by HPLC (LC-20AD; Shimadzu) coupled with a single quadrupole mass spectrometer (LCMS-2020; Shimadzu) as described42.
Fermentation products analysis
For analysis the metabolic end products from glucose fermentation, including SCFAs and organic acids, cells were cultured in MPYG broth at 37 °C under anaerobic conditions for 2 days. Supernatant harvested from the cultures centrifuged at 10,000 g for 10 min was used for determining SCFAs and organic acids. SCFAs detection was performed using a gas chromatograph (GC-7890B, Agilent) equipped with a flame ionization detector (FID) and capillary column packed with Agilent 19091 N-133HP-INNOWax porapak HP-INNOWax (30 m × 0.25 mm × 0.25 μm). Organic acids were analysed by equipping capillary column packed with Agilent 122-5532G DB-5 ms (40 m × 0.25 mm × 0.25 μm).
Genome sequencing, assembly, and annotation of isolates
For genome sequences of strains AF52-21T and CM04-06T, genomic DNA was extracted following the method described above. The draft genome was sequenced on an Ion Proton Technology (Life Technologies) platform at BGI-Shenzhen (Shenzhen, China) after constructing a paired-end DNA library with insert size of 500 bp. The resulting reads were assembled using the SOAPdenovo 2 package43. CheckM (v1.1.2) was used to estimate genome completeness and contamination44. Genome assemblies were visualized using CGView Server45 (http://stothard.afns.ualberta.ca/cgview_server/index.html). Annotation of the assembled genome was performed using the Rapid Annotation Using Subsystem Technology (RAST) server46 and COG database47. The G + C content in genomic DNA was calculated from the whole genome sequence. The genes in known pathways from acetyl-CoA to butyrate were annotated by BLAST (evalue = 1e−5, identity ≥ 60%, coverage ≥ 90%)30. AntiSMASH 5.0 was used to predict BGCs. A search for prophages was performed by PHAST (http://phast.wishartlab.com/)48. Antibiotic resistance was analysed using the CARD database49. The carbohydrate active enzymes genes were annotated by dbCAN250. The dbCAN-PUL51 database was used to determine genes related to important carbohydrate metabolism pathways.
Average nucleotide identities
Genome relatedness was investigated by calculating average nucleotide identity (ANI)52, with a value of 95–96% proposed for delineating bacterial species, corresponding to the traditional 70% DNA-DNA reassociation standard53,54. The ANI values between strains AF52-21T, CM04-06T, and closely related species were determined using the FastANI55.
Phylogenetic analysis based on 16S rRNA genes sequence
16S rRNA gene sequences were extracted from the genomes using RNAmmer56. The obtained 16S rRNA gene sequences of strains AF52-21T and CM04-06T were compared with the sequences of type strains retrieved from the EzBioCloud database (https://www.ezbiocloud.net/)57 and an unrecognized species ‘Faecalibacterium hominis’ 4P-1527 using the BLAST program to determine the nearest phylogenetic neighbours and 16S rRNA gene sequence similarity values. Phylogenetic trees were reconstructed by using the neighbour-joining method58 (K2 + G model of substitution), maximum-likelihood method59 (GTR + G + I model of substitution) and minimum-evolution method60 (K2 + G model of substitution) with the MEGA X program package61, after Clustal W multiple alignment of the sequences. 1548 nucleotide positions were finally used for tree constructions. Robustness of the phylogenetic trees was evaluated by using the bootstrap resampling method (1000 resamplings) of Felsenstein62.
Supplementary Information
Acknowledgements
This work was supported by grants from National Key Research and Development Program of China (No. 2018YFC1313801) and Natural Science Foundation of Guangdong Province, China (No. 2019B020230001). We also thank the colleagues at BGI-Shenzhen for sample collection, and discussions, and China National Genebank (CNGB) Shenzhen for DNA extraction, library construction, and sequencing.
Author contributions
Conceived and designed the experiments: Y.Z. and L.X. Performed the experiments: Y.Z., W.X., L.T, M.C, X.C, F.L, and Y.D. Analyzed the data: Y.Z., L.X., L.T, and X.L. Contributed reagents/materials/analysis tools: Y.Z., W.X., S.L., C.S., and Y.D. Wrote the paper: Y.Z. and X.L. Revised the paper: K.K.
Data availability
The China National GeneBank DataBase (CNGBdb)63 accession numbers for the 16S rRNA gene sequences determined in this study are: AF52-21T (N_000011117) and CM04-06T (N_000011118). The data of draft genome sequences have been deposited into CNGB Sequence Archive (CNSA)64 of CNGBdb with accession number CNA0017730 and CNA0017731 for strains AF52-21T and CM04-06T, respectively.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yuanqiang Zou and Xiaoqian Lin.
Contributor Information
Yuanqiang Zou, Email: zouyuanqiang@genomics.cn.
Liang Xiao, Email: xiaoliang@genomics.cn.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-90786-3.
References
- 1.Russell SL, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13:440–447. doi: 10.1038/embor.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tagliabue A, Elli M. The role of gut microbiota in human obesity: recent findings and future perspectives. Nutr. Metab. Cardiovasc. Dis. 2013;23:160–168. doi: 10.1016/j.numecd.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Benson AK, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claesson MJ, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 6.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antunes LC, Finlay BB. A comparative analysis of the effect of antibiotic treatment and enteric infection on intestinal homeostasis. Gut Microbes. 2011;2:105–108. doi: 10.4161/gmic.2.2.15610. [DOI] [PubMed] [Google Scholar]
- 8.Wilson ID, Nicholson JK. The role of gut microbiota in drug response. Curr. Pharm. Des. 2009;15:1519–1523. doi: 10.2174/138161209788168173. [DOI] [PubMed] [Google Scholar]
- 9.Adlerberth I. Factors influencing the establishment of the intestinal microbiota in infancy. Nestle Nutr. Worksh. Ser. Pediatr. Program. 2008;62:13–29. doi: 10.1159/000146245. [DOI] [PubMed] [Google Scholar]
- 10.DiBaise JK, et al. Gut microbiota and its possible relationship with obesity. Mayo Clin. Proc. 2008;83:460–469. doi: 10.4065/83.4.460. [DOI] [PubMed] [Google Scholar]
- 11.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 12.Parekh PJ, Arusi E, Vinik AI, Johnson DA. The role and influence of gut microbiota in pathogenesis and management of obesity and metabolic syndrome. Front. Endocrinol. (Lausanne) 2014;5:47. doi: 10.3389/fendo.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsen N, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 15.Conte MP, et al. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut. 2006;55:1760–1767. doi: 10.1136/gut.2005.078824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joossens M, et al. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 17.Feng Q, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015;6:6528. doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- 18.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 19.Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin. Microbiol. Infect. 2013;19:338–348. doi: 10.1111/1469-0691.12140. [DOI] [PubMed] [Google Scholar]
- 20.Gkolfakis P, Dimitriadis G, Triantafyllou K. Gut microbiota and non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2015;14:572–581. doi: 10.1016/S1499-3872(15)60026-1. [DOI] [PubMed] [Google Scholar]
- 21.Ríos-Covián D, et al. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016 doi: 10.3389/fmicb.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puertollano E, Kolida S, Yaqoob P. Biological significance of short-chain fatty acid metabolism by the intestinal microbiome. Curr. Opin. Clin. Nutr. Metab. Care. 2014;17:139–144. doi: 10.1097/MCO.0000000000000025. [DOI] [PubMed] [Google Scholar]
- 23.Quevrain E, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut. 2016;65:415–425. doi: 10.1136/gutjnl-2014-307649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokol H, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan SH, Hold GL, Harmsen HJ, Stewart CS, Flint HJ. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002;52:2141–2146. doi: 10.1099/00207713-52-6-2141. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald CB, et al. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genom. 2018;19:931. doi: 10.1186/s12864-018-5313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, et al. Enlightening the taxonomy darkness of human gut microbiomes with cultured biobank. Res. Square. 2020 doi: 10.21203/rs.3.rs-74101/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miquel S, et al. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013;16:255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Rossi-Tamisier M, Fournier PE, Benamar S, Raoult D. Cautionary tale of using 16S rRNA gene sequence similarity values in identification of human-associated bacterial species. Int. J. Syst. Evol. Microbiol. 2015;65:1929–1934. doi: 10.1099/ijs.0.000161. [DOI] [PubMed] [Google Scholar]
- 30.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio. 2014;5:e00889. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou Y, et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019;37:179–185. doi: 10.1038/s41587-018-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou L, et al. Faecalibacterium prausnitzii produces butyrate to maintain Th17/Treg balance and to ameliorate colorectal colitis by inhibiting histone deacetylase 1. Inflamm. Bowel Dis. 2018;24:1926–1940. doi: 10.1093/ibd/izy182. [DOI] [PubMed] [Google Scholar]
- 33.Fujimoto T, et al. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn's disease. J. Gastroenterol. Hepatol. 2013;28:613–619. doi: 10.1111/jgh.12073. [DOI] [PubMed] [Google Scholar]
- 34.Machiels K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Siles M, et al. Alterations in the abundance and co-occurrence of Akkermansia muciniphila and Faecalibacterium prausnitzii in the colonic mucosa of inflammatory bowel disease subjects. Front. Cell Infect. Microbiol. 2018;8:281. doi: 10.3389/fcimb.2018.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CJ, et al. Next generation probiotics in disease amelioration. J. Food Drug. Anal. 2019;27:615–622. doi: 10.1016/j.jfda.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Filippis F, Pasolli E, Ercolini D. Newly explored Faecalibacterium diversity is connected to age, lifestyle, geography, and disease. Curr. Biol. 2020 doi: 10.1016/j.cub.2020.09.063. [DOI] [PubMed] [Google Scholar]
- 38.Cato EP, Salmon CW, Moore W. Fusobacterium prausnitzii (Hauduroy et al.) Moore and Holdeman: emended description and designation of neotype strain. Int. J. Syst. Bacteriol. 1974;24:225–229. doi: 10.1099/00207713-24-2-225. [DOI] [Google Scholar]
- 39.Tittsler RP, Sandholzer LA. The use of semi-solid agar for the detection of bacterial motility. J. Bacteriol. 1936;31:575–580. doi: 10.1128/JB.31.6.575-580.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sorokin DY. Is there a limit for high-pH life? Int. J. Syst. Evol. Microbiol. 2005;55:1405–1406. doi: 10.1099/ijs.0.63737-0. [DOI] [PubMed] [Google Scholar]
- 41.Zou Y, et al. Lactobacillus shenzhenensis sp. nov., isolated from a fermented dairy beverage. Int. J. Syst. Evol. Microbiol. 2013;63:1817–1823. doi: 10.1099/ijs.0.041111-0. [DOI] [PubMed] [Google Scholar]
- 42.Liu S, et al. Desertihabitans brevis sp. nov., an actinobacterium isolated from sand of the Taklamakan desert, and emended description of the genus Desertihabitans. Int. J. Syst. Evol. Microbiol. 2020;70:1166–1171. doi: 10.1099/ijsem.0.003896. [DOI] [PubMed] [Google Scholar]
- 43.Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucl. Acids Res. 2008;36:W181–184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucl. Acids Res. 2015;43:261–269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucl. Acids Res. 2011;39:347–352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucl. Acids Res. 2017;45:566–573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucl. Acids Res. 2018;46:95–101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ausland C, et al. dbCAN-PUL: a database of experimentally characterized CAZyme gene clusters and their substrates. Nucl. Acids Res. 2021;49:523–528. doi: 10.1093/nar/gkaa742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goris J, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 54.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 55.Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagesen K, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucl. Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoon SH, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017;67:1613–1617. doi: 10.1099/ijsem.0.001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 59.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 60.Rzhetsky A, Nei M. Theoretical foundation of the minimum-evolution method of phylogenetic inference. Mol. Biol. Evol. 1993;10:1073–1095. doi: 10.1093/oxfordjournals.molbev.a040056. [DOI] [PubMed] [Google Scholar]
- 61.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 63.Chen FZ, et al. CNGBdb: China National GeneBank DataBase. Yi Chuan. 2020;42:799–809. doi: 10.16288/j.yczz.20-080. [DOI] [PubMed] [Google Scholar]
- 64.Guo X, et al. CNSA: a data repository for archiving omics data. Database (Oxford) 2020 doi: 10.1093/database/baaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The China National GeneBank DataBase (CNGBdb)63 accession numbers for the 16S rRNA gene sequences determined in this study are: AF52-21T (N_000011117) and CM04-06T (N_000011118). The data of draft genome sequences have been deposited into CNGB Sequence Archive (CNSA)64 of CNGBdb with accession number CNA0017730 and CNA0017731 for strains AF52-21T and CM04-06T, respectively.