

Abstract
Despite the emerging evidence on ferroptosis implicated in diverse pathologies, molecular linkage between oxidative inducers and chromatin as epigenetic memory carrier for its propagation remains elusive. Here, we report the identification of two WD40 proteins DCAF8 and WDR76 as substrate adapter and molecular inhibitor respectively of the Cullin-4 RING ubiquitin ligase (CRL4) system for stability control of chromatin remodeler LSH. Degradation analysis and CRL4-DCAF8 complex reconstitution demonstrate that CRL4DCAF8 is a bona fide E3 ligase for LSH. In contrast, WDR76 antagonizes DCAF8-targeted LSH proteolysis through competitive inhibition of the holo-CRL4DCAF8-LSH complex assembly. Importantly, this opposing regulatory strategy is utilized in lipid hydroperoxide induced ferroptosis, where we identify key redox homeostasis genes significantly regulated by the DCAF8/WDR76/LSH axis through transcriptomic epistasis analysis. This regulation is mechanistically attributed to DNA hydroxymethylation fostered WDR76 interaction with LSH and increased ratio of DCAF8 to WDR76 for antagonistic LSH association accompanying decreased DNA oxidation along with ROS overproduction. Evaluation of epigenetic dynamics at ferroptosis gene promoters reveals linker histone H1- and LSH-associated transcriptional repression is coordinately removed upon lipid peroxidation stress. Together with the phenotypes driven by WDR76 and DCAF8 manipulations, these data identify DCAF8- and WDR76-adapted oxidative damage sensing through DNA hydroxymethylation for LSH degradation control as a crucial nexus in epigenetic regulation of ferroptosis.
Subject terms: Cancer genetics, Ubiquitylation, Gene regulation, Epigenetics
Introduction
Epigenetic regulators are master players in the genome-environment interactions, and their dysfunctions and deregulations are causally linked with many human malignancies and chronological aging [1–3]. Lymphoid-specific helicase (LSH; also known as HELLS or PASG), a member of SNF2 family chromatin remodeling ATPases, is a such epigenetic factor, and the coordination between LSH and DNA/histone modifiers is an evolutionally conserved theme for the maintenance of DNA methylome, guarding of repetitive elements, and transcriptional silencing [4–11].
The conjugation of eukaryotic proteins with multimers of ubiquitin for proteasomal degradation is catalyzed by the E1 activating enzyme, E2 conjugating enzyme, and E3 ligase in three consecutive reactions [12]. The cullin-RING E3 ligases (CRLs), comprising the largest superfamily of multisubunit E3s in eukaryotic cells, are characterized by a common modular architecture composed of a scaffold cullin protein, a RING finger protein (ROC1 or ROC2) which together with ubiquitin-charged E2 forms the catalytic core, a substrate recruiting receptor and its cognate adapter [13]. The CRL4 subfamily CRLs, share a Cullin-4 type scaffold (CUL4A or CUL4B) and a large β-propeller protein, DNA damage-binding protein-1 (DDB1) as the adapter bridging substrate receptors and CUL4 [14, 15]. DCAF (DDB1 and CUL4-associated factor) proteins responsible for substrate recognition and recruitment are molecular matchmakers of CRL4 E3s, which contain an array of WD40 repeats packed into propeller-like structure and determine the substrate specificity [16, 17].
Accumulation of overproduced reactive oxygen species (ROS) is implicated in a range of pathologies, including cancer, degenerative disease, and premature aging [18, 19]. ROS caused macromolecular damages have been extensively studied on the topics of DNA base mutation and protein conformational aberrance [19], yet mounting evidences have pointed to the function of lipid peroxidation in attacking membrane polyunsaturated lipids and inducing iron-dependent, nonapoptotic, and regulated form of cell death, ferroptosis [20]. Indeed, pharmacological and genetic profiling suggest that ferroptosis inducers or inhibitors generally target the redox homeostasis pathways, especially the glutamate/cystine antiporter system Xc−, which mediates cellular uptake of extracellular cystine in exchange for intracellular glutamate [20]. Interestingly, hypoxia, as an oxidative stress, is involved in the regulation of ten-eleven translocation (TET) family dioxygenase catalyzed oxidation of cytosine methylation (5-mC) to hydroxymethylation (5-hmC) in cancers, thus linking DNA methylation based epigenetic regulation to global oxygenation state [21]. A potential connection between LSH and oxidative stress response could then be grasped from the proteomic identification of LSH as a binder of 5-hmC [22].
Expression changes of LSH have been broadly observed in cancers [23–28], and its deregulations are crucially implicated in carcinogenesis and premature aging [29–31]. However, the protein stability control mechanisms for LSH have not been investigated yet. In this study, we demonstrated a counteracting theme of CRL4DCAF8-mediated LSH polyubiquitination and proteasomal degradation and WDR76-mediated LSH stabilization to control the overall protein levels of LSH. Importantly, we found this stability regulation was tuned by TET-catalyzed cytosine hydroxymethylation and functionally involved in epigenetic control of ferroptosis.
Results
Identification of LSH-associated proteins
Epigenetic regulators at the interface between carcinogenesis and aging have been the primary research focus of this laboratory [32–34]. In an effort to understand the molecular function of LSH, we employed affinity purification to capture LSH-associated proteins (Fig. 1a). Subsequent mass spectrometry analysis revealed their identities: two WD40 repeat proteins, DCAF8 (DDB1 and CUL4-associated factor 8) and WDR76 (WD repeat-containing protein 76); DDB1 (DNA damage-binding protein 1), the adapter protein of substrate receptors in CRL4 E3 complex [17]; nucleotide excision repair factor ERCC5 (ERCC excision repair 5) [35]; XRCC6 (X-ray repair cross complementing 6), a subunit of Ku complex in non-homologous end joining [36]; and linker histone H1 (H1.0, H1.1, H1.2, H1.3, H1.4, H1.5, H1FX). The identification of ERCC5 and XRCC6 as LSH-interacting partners was consistent with the function of LSH in DNA damage response and double-strand break repair [37]. Among these, we are particularly interested in DCAF8, WDR76 and DDB1, given the molecular composition of CRL4 family E3 ligase [17] and the possible implication of DCAF8 and WDR76 in the regulation of LSH protein levels.
Fig. 1. Identification and validation of LSH-interacting proteins.

a Cellular extracts from HeLa cells transfected with FLAG-LSH or control vector were immunopurified with anti-FLAG affinity resin and eluted with FLAG peptide. The eluates were resolved on SDS-PAGE followed by silver staining. The protein bands were retrieved and analyzed by mass spectrometry. Detailed information for the top-scoring proteins was shown in the list. b FLAG-LSH and each of the HA-tagged DCAF8, WDR76, DDB1, CUL4A, CUL4B, and ROC1, were co-tranfected into HEK293T cells. Co-immunoprecipitation experiments were then performed with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. c Co-immunoprecipitation experiment was performed with protein extract from cells simultaneously transfected with the plasmids encoding FLAG-DCAF8 and each of the HA-tagged CUL4A, CUL4B, DDB1 or ROC1. d Four groups of HEK293T cells co-transfected with HA-WDR76/FLAG-DDB1, FLAG-WDR76/HA-DDB1, FLAG-WDR76/HA-CUL4A, or FLAG-WDR76/HA-CUL4B were subjected to co-immunoprecipitation analysis with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. e GST pull-down experiments were performed with bacterially purified GST-ROC1, GST-CUL4A, GST-CUL4B, GST-DCAF8, GST-WDR76, and in vitro transcribed/translated LSH. Coomassie blue staining of the purified proteins were shown. f GST pull-down experiments were performed with bacterially purified truncated deletions of DCAF8 or WDR76 and in vitro transcribed/translated LSH. Coomassie blue staining of the purified proteins were shown.
Molecular interface between LSH and DCAF8, WDR76, and other CRL4 core components
To validate these interactions, FLAG-LSH and each of the HA-tagged DCAF8, WDR76, DDB1, CUL4A, CUL4B, and ROC1, were co-transfected into HEK293T cells. Co-immunoprecipitation results indicated that DCAF8, WDR76, and all the tested CRL4 subunits were efficiently precipitated along with LSH (Fig. 1b). We next asked whether these two WD40 proteins could be incorporated into a functional CRL4 complex. Co-immunoprecipitation experiment in cells simultaneously transfected with plasmids encoding DCAF8 and each of the CUL4A, CUL4B, DDB1 or ROC1 subunit of CRL4 verified the interaction between DCAF8 and other CRL4 components, supporting a possible CRL4-DCAF8 composition in vivo (Fig. 1c). In sharp contrast, interaction between WDR76 and DDB1, which is the key adapter bridging WD40 proteins with the rest of CRL4 complex [14], could not be detected in the co-immunoprecipitation analysis with either WDR76 or DDB1 as the bait protein, arguing against the CRL4-WDR76 composition (Fig. 1d). Moreover, the interaction between WDR76 and the scaffold CUL4A or CUL4B could not be detected either (Fig. 1d).
Glutathione S-transferase (GST) pull-down analysis using purified proteins demonstrated that LSH could only directly interact with DCAF8 but no other component from the CRL4-DCAF8 complex (Fig. 1e), consistent with a possible substrate receptor function of DCAF8 to target LSH [16, 17]. Interestingly, although WDR76 is not likely to be incorporated into a CRL4 complex, GST pull-down results suggested that WDR76 could directly associate with LSH showing even stronger affinity (Fig. 1e), in line with co-immunoprecipitation data (Fig. 1b). Detailed mapping using a series of truncated deletions of either DCAF8 or WDR76 indicated that the N-terminal portion of both proteins is indispensable for their association with LSH (Fig. 1f). These results together suggested that a molecular architecture of CRL4-DCAF8 complex could be formed in association with LSH, while WDR76 is not detectable within this protein assembly.
CRL4DCAF8 is a ubiquitin E3 complex targeting LSH for its proteasomal degradation
In line with the involvement of ubiquitin-proteasome system in regulation of LSH proteins, proteasome inhibitor MG132 treatment resulted in LSH accumulation (Fig. 2a). Importantly, treatment with MLN4924, an inhibitor of NEDD8-activating enzyme to prevent cullin neddylation and CRL E3 activation [38], also up-regulated LSH protein levels (Fig. 2a), indicating the specific implication of CRL system in LSH degradation.
Fig. 2. CRL4DCAF8 is a ubiquitin E3 complex targeting LSH for its proteasomal degradation.

a Western blotting analysis for the indicated proteins from HEK293T and HT-1080 cells treated with MG132 (20 μM, 5 h), MLN4924 (1 μM, 24 h), or DMSO (dimethyl sulfoxide, 1.1 mg/ml) as a control. The relative densities of LSH indicated below the blots were first normalized to that of TUBULIN and then calculated as ratio relative to the value in DMSO-treated cells. b U2OS cells stably transfected with DCAF8 or HEK293T and HT-1080 cells transiently transfected with DCAF8 together with the control cells were collected for western blotting analysis of indicated proteins. The relative densities of LSH indicated below the blots were first normalized to that of TUBULIN and then calculated as ratio relative to the value in control cells. c HEK293T and HT-1080 cells were transfected with control or siRNA against DCAF8. CRISPR-Cas9-mediated DCAF8 ablation was performed in HeLa cells. Protein extracts from these cells were subjected to western blotting analysis with indicated antibodies. siNS, nonspecific RNAi control. The relative densities of LSH indicated below the blots were first normalized to that of TUBULIN or GAPDH and then calculated as ratio relative to the value in control cells. d Individual or combinational transfection of the indicated CRL4DCAF8 complex components was performed in HEK293T cells. Protein extracts from these cells were subjected to western blotting analysis with indicated antibodies. The relative densities of LSH indicated below the blot were first normalized to that of TUBULIN and then calculated as ratio relative to the value in the absence of the four plasmids. e HEK293T cells transfected with DCAF8 were further incubated with MG132 (20 μM) for 5 h. Protein extracts from these cells together with nontreated control cells were subjected to western blotting analysis. The relative densities of LSH shown below the blot were first normalized to that of TUBULIN and then calculated as ratio relative to the value in the absence of MG132 and DCAF8. f HEK293T cells transfected with indicated siRNAs were treated with 40 μg/mL cycloheximide (CHX) for indicated times, and harvested for western blotting analysis. The relative densities of LSH shown were first normalized to that of GAPDH or TUBULIN and then calculated as ratio to the value in the absence of CHX. g HEK293T cells transfected with plasmids expressing indicated proteins were treated with 40 μg/mL cycloheximide for indicated times and harvested for western blotting analysis. The relative densities of LSH shown were first normalized to that of GAPDH and then calculated as ratio to the value in the absence of CHX. h HEK293T cells transfected with control, MYC-tagged wild-type, or R314A mutated DCAF8 were subjected to western blotting analysis with indicated antibodies. The relative densities of LSH indicated below the blot were first normalized to that of GAPDH and then calculated as ratio relative to the value in vector-transfected cells. i 48 h post-transfection with indicated plasmids, HEK293T cells were further treated with MG132 (20 μM) for 5 h. The cells were then collected for immunoprecipitation with anti-FLAG antibody to enrich LSH proteins. Ubiquitination of LSH was then examined by immunoblotting analysis by using of anti-HA antibody. j HEK293T cells transfected with control, wild-type, or mutated DCAF8 together with other indicated plasmids were subjected to LSH ubiquitination analysis as in i. k An in vitro ubiquitination system with purified E1, E2, and ubiquitin proteins and immunopurified core components of CRL4DCAF8 complex and FLAG-LSH from eukaryotic cells.
Gain-of-function experiments through either transient overexpression or stably integration of DCAF8 indicated significantly decreased LSH protein levels (Fig. 2b). Consistently, loss-of-function for DCAF8 through either RNAi-meditated DCAF8 depletion or CRISPR-Cas9-mediated DCAF8 ablation resulted in LSH accumulation (Fig. 2c). Analysis of the combinational effect of CRL4-DCAF8 complex components indicated that LSH protein levels were reduced in cells co-transfected with CUL4B/DDB1/ROC1 or DDB1/DCAF8, and were nearly undetectable when CUL4A, DDB1, ROC1 and DCAF8 co-transfected, suggesting a functional coordination of the whole CRL4DCAF8 complex (Fig. 2d). Once again, this DCAF8 overexpression resulted LSH degradation was nearly abolished by MG132 treatment (Fig. 2e). Concordantly, half-life measurement of LSH by cycloheximide (CHX) chase indicated that any depletion of DCAF8, DDB1 or CUL4 subunit substantially improved LSH protein stability (Fig. 2f), while DDB1 or DCAF8 overexpression decreased it (Fig. 2g). Interestingly, point mutations at the WDXR motifs of DCAF8 (DCAF8R314A) to disrupt its interaction with DDB1 [39], even showed a dominant negative effect to stabilize LSH proteins (Fig. 2h), implying the integrity of CRL4DCAF8 complex was essential for its functionality.
We next tested whether DCAF8-promoted LSH degradation was a consequence of CRL4DCAF8-catalyzed LSH ubiquitination. As expected, the in vivo ubiquitination result indicated that DCAF8 significantly promoted LSH polyubiquitination, compared to the control or unrelated DCAF factor DDB2 [16] (Fig. 2i). Furthermore, in line with the defect of DCAF8R314A to promote LSH degradation, LSH polyubiquitination level in these cells was lower (Fig. 2j). Importantly, using an in vitro ubiquitination system with immunopurified CRL4DCAF8 complex and LSH, we demonstrated that CRL4DCAF8 could directly induce the polyubiquitination of LSH in a molecularly defined reaction (Fig. 2k). Together, these data indicated that CRL4DCAF8 is a bona fide ubiquitin E3 ligase to target LSH for ubiquitination and proteasomal degradation.
WDR76 is a negative regulator of CRL4DCAF8-mediated ubiquitination of LSH
The similar domain architecture of WDR76 and DCAF8 as WD40 proteins and the lack of WDR76-DDB1 physical association (Fig. 1d) promoted us to examine the role of WDR76 in CRL4DCAF8-mediated LSH ubiquitination and degradation. A survey of LSH and WDR76 levels in a collection of 26 cell lines revealed a co-expressing pattern of these two proteins (Fig. 3a). Importantly, ectopic WDR76 expression increased LSH protein levels significantly (Fig. 3b), and the half-life of LSH could be greatly extended by transient or stable WDR76 overexpression (Fig. 3c). Consistently, WDR76 removal either by RNAi or by CRISPR-Cas9-mediated genetic ablation resulted in decreased LSH protein levels (Fig. 3d), and WDR76 depletion shortened LSH protein half-life (Fig. 3e).
Fig. 3. WDR76 is a negative regulator of CRL4DCAF8-mediated ubiquitination of LSH.

a Western blotting analysis on the expression of WDR76 and LSH in 26 cell lines. The relative densities of WDR76 and LSH indicated below the blots were first normalized to that of TUBULIN and then calculated as ratio relative to the value in MCF-7 or Hela cells. b Protein extracts from H1299, A549 or HeLa cells stably expressing empty vector or lentiviral WDR76 were subjected to western blotting analysis with indicated antibodies. The relative densities of LSH indicated below the blots were first normalized to that of TUBULIN and then calculated as ratio relative to the value in control cells. c Cycloheximide chase analysis on the half-life of LSH proteins in HeLa cells transiently expressing control or WDR76 and A549 cells stably expressing control or lentiviral WDR76. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. d HEK293T cells were transfected with control or two independent siRNA against WDR76. CRISPR-Cas9-mediated WDR76 ablation was performed in HeLa cells. Protein extracts from these cells were subjected to western blotting analysis with indicated antibodies. The relative densities of LSH indicated below the blots were first normalized to that of GAPDH and then calculated as ratio relative to the value in control cells. e HEK293T cells were transfected with control or two independent siRNA against WDR76. These cells were then subjected to cycloheximide chase analysis on the half-life of LSH proteins. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. f HEK293T cells co-transfected with FLAG-LSH and indicated amount of HA-DCAF8 (upper panel) or HA-WDR76 (lower panel) were subjected to co-immunoprecipitation analysis with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. The relative densities of immunoprecipitated (IPed) HA-WDR76 or HA-DCAF8 indicated at the bottom were first normalized to that of FLAG-LSH, and then calculated as ratio relative to the value in the absence of HA-DCAF8 or HA-WDR76. g GST pull-down experiments were performed with bacterially purified truncated deletions of LSH and in vitro transcribed/translated DCAF8 or WDR76. Coomassie blue staining of the purified proteins were shown. NT, N-terminus. CT, C-terminus. h HEK293T cells transfected with indicated plasmids were collected for co-immunoprecipitation analysis with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. The relative densities of immunoprecipitated HA-DCAF8 indicated at the bottom were first normalized to that of Input, and then calculated as ratio relative to the value in vector-transfected cells. i HEK293T cells co-transfected with HA-WDR76 and FLAG-DCAF8 were collected for co-immunoprecipitation analysis with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. GST pull-down experiments were performed with bacterially purified GST-WDR76 and in vitro transcribed/translated DCAF8. j HEK293T cells co-transfected with HA-DDB1 and HA-WDR76 together with MYC-tagged wild-type or R314A mutated DCAF8 were collected for co-immunoprecipitation analysis with MYC antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. k Global 5-mC abundance in cells overexpressing control, WDR76, or DCAF8 was determined through dot blotting analysis. Methylene blue staining provided genomic DNA loading control. LSH protein levels were determined by western blotting analysis. The relative 5-mC levels shown were first normalized to that of methylene blue, and then calculated as the ratio relative to the value of 1 µg vector-transfected cells.
To understand the molecular basis for this WDR76-meidated LSH stabilization, we firstly asked whether WDR76 could compete with DCAF8 for LSH binding. By maintaining the amount of transfected DCAF8 and varying the co-transfected WDR76, we found overexpression of WDR76 had minimal effect on the interaction between DCAF8 and LSH (Fig. 3f). Similarly, increasingly transfected DCAF8 had little effect on the interaction between WDR76 and LSH (Fig. 3f). On the other hand, DCAF8 and WDR76 directly associate with different regions of LSH, as DCAF8 binds to the DEXD domain, and WDR76 binds to the N-terminal fragment (Fig. 3g), thus disfavoring the model of direct competition between DCAF8 and WDR76 in substrate binding for LSH. However, WDR76 overexpression greatly suppressed the interaction between DCAF8 and DDB1 (Fig. 3h), suggesting that WDR76 could interfere with the incorporation of DCAF8-LSH subcomplex into an intact CRL4DCAF8-LSH complex. In support of this notion, we captured the DCAF8-WDR76 interaction by ectopically expressing both DCAF factors (Fig. 3i). Interestingly, further analysis indicated that compared to wild-type DCAF8, the ratio of DDB1 to WDR76 proteins interacted with DCAF8 was reduced by R314A mutation, implying a competition between the DCAF8-DDB1 and DCAF8-WDR76 interactions (Fig. 3j). These data together suggested that WDR76 could suppress CRL4DCAF8-mediated LSH ubiquitination and proteasomal degradation through competitive inhibition of the holo-CUL4-DDB1-DCAF8-LSH complex formation.
WDR76 and DCAF8 modulate global DNA methylation levels correlating with their regulated LSH protein abundance
Given the critical implication of LSH in cytosine methylation [4], measurement of global 5-mC abundance indicated that, WDR76 overexpression led to an obvious increase of the total 5-mC levels, while DCAF8 overexpression elicited an inverse change, correlating with the increased and decreased LSH protein levels, respectively (Fig. 3k), thus functionally linking WDR76/DCAF8-regulated LSH degradation to global DNA methylation maintenance.
Differential regulation of LSH stability by the counteracting CRL4DCAF8 and WDR76 system corresponds to distinct ferroptosis sensitivities of cancer cells
We next sought to address whether the opposing stability control of LSH by CRL4DCAF8 and WDR76 could be recapitulated in vivo. Ferroptosis is a recently characterized iron-dependent form of cell death caused by the accumulation of lethal level lipid peroxidation which targets polyunsaturated fatty acids at cellular membranes and impairs lipid homeostasis [40]. In light of the implication of LSH in ferroptosis [41], we asked whether the changes of redox microenvironment triggered by ferroptosis inducers could affect LSH stability through the CRL4DCAF8 and WDR76 opponents. Surprisingly, in contrast to the nearly unchanged LSH protein levels in the tested cell lines poorly responding to erastin (Fig. 4a, Supplementary Figs. 1 and 2), an established ferroptosis inducer to target system Xc− and prevent cystine import [20], its addition elicited an obvious decline of LSH proteins in all of the tested ferroptosis sensitive cell lines (Fig. 4b). Meanwhile, the half-life of LSH proteins was concordantly shortened by erastin exposure (Fig. 4c).
Fig. 4. Differential regulation of LSH stability by the counteracting CRL4DCAF8 and WDR76 system corresponds to distinct ferroptosis sensitivities of cancer cells.

a HeLa, U2OS, HuH7, MDA-MB-231 and HEK293T cells treated with increasing concentration of erastin were collected for western blotting analysis on the expression of indicated proteins. b The fibrosarcoma cell line HT-1080, renal clear cell cancer cell line Caki-1, human proximal tubular epithelial cell line HK-2, and human pancreatic cancer cell line Panc-1 were treated with increasing concentration of erastin and collected for western blotting analysis on the expression of indicated proteins. c Cycloheximide chase analysis on the half-life of LSH proteins in HT-1080 cells treated with DMSO, erastin, or a combination of erastin and MLN4924. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. d HT-1080 cells treated with DMSO, erastin, or a combination of erastin and MLN4924 were collected for western blotting analysis on the expression of indicated proteins. The relative densities of LSH indicated below the blot were first normalized to that of TUBULIN and then calculated as ratio relative to the value in the absence of erastin and MLN4924. e HT-1080 cells transfected with control or RNAi molecules against DCAF8 were further incubated with erastin (10 μM) followed by western blotting analysis on the expression of indicated proteins. The relative densities of LSH indicated below the blot were first normalized to that of TUBULIN and then calculated as ratio relative to the value in the absence of erastin in siNS-transfected cells. f CRISPR-Cas9-mediated DCAF8 ablation was performed in HT-1080 cells. These together with control cells were further treated with erastin (10 μM) before cycloheximide chase analysis on the half-life of LSH proteins. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. g HT-1080 cells stably integrated control or lentiviral-WDR76 were further treated with erastin (10 μM) before cycloheximide chase analysis on the half-life of LSH proteins. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. h HEK293T cells transfected with indicated expression constructs were collected for co-immunoprecipitation analysis with HA antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. i Global 5-hmC abundance in HT-1080 cells and Caki-1 cells in the absence or presence of erastin was determined through dot blotting analysis. Methylene blue staining provided genomic DNA loading control. The relative densities of 5-hmC to that of methylene blue were shown. j HEK293T cells transfected with indicated expression constructs were further treated with DMOG (100 μM). The treated or nontreated cells were then collected for co-immunoprecipitation analysis with FLAG antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. k HT-1080 cells treated with DMSO, DMOG, or a combination of DMOG and MLN4924 were subjected to cycloheximide chase analysis on the half-life of LSH proteins. The relative densities of LSH indicated at the bottom were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of CHX. l HEK293, HEK293T, MDA-MB-231, SH-SY5Y, and SMMC-7721 cells in the absence or presence of DMOG (100 μM) were subjected to western blotting analysis on the expression of indicated proteins. m HT-1080 cells treated with DMSO, DMOG, or a combination of DMOG and MLN4924 were collected for western blotting analysis on the expression of indicated proteins. The relative densities of LSH indicated below the blot were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of DMOG and MLN4924. n HT-1080 cells transfected with control or siRNA against DCAF8 were further exposed to DMOG (100 μM). The treated or untreated cells were then collected for western blotting analysis on the expression of indicated proteins. The relative densities of LSH indicated below the blot were first normalized to that of TUBULIN and then calculated as ratio to the value in the absence of DMOG in siNS-transfected cells.
Interestingly, this erastin-induced LSH destabilization was blocked by CRL inhibitor MLN4924 (Fig. 4c, d). More importantly, DCAF8 depletion greatly suppressed erastin-induced LSH degradation (Fig. 4e), and the half-life of LSH proteins in the presence of erastin was extended by genetic ablation of DCAF8 (Fig. 4f), suggesting a specific implication of the CRL4DCAF8 E3 ligase in ferroptosis associated LSH degradation. Consistently, the cells with stably integrated lentiviral-WDR76 showed both higher basal levels of LSH protein and improved LSH stability during erastin exposure (Fig. 4g). These data together supported that the counteracting CRL4DCAF8 and WDR76 system controlled LSH degradation in erastin-induced ferroptosis.
The opposing regulation of LSH stability by WDR76 and DCAF8 is tuned by TET-mediated DNA oxidation
We next sought to determine the molecular connection between erastin inflicted oxidative stress and the DCAF8-WDR76-LSH axis. In this regard, it is worthy of noting that both LSH and WDR76 have been identified as reader proteins of 5-hmC or its oxidative derivatives [22]. Since the iron and 2-oxoglutarate (2OG)-dependent TET family dioxygenases, responsible for enzymatic catalysis of 5-mC oxidation [42], have been reported to show decreased activities in hypoxia associated ROS production [21], we firstly asked whether ectopic TET expression could affect the association between LSH and DCAF8/WDR76. Co-immunoprecipitation analysis indicated that overexpression of the catalytic domain of TET2 (TET2-CD) greatly compromised the interaction between DCAF8 and LSH, whereas the interaction between WDR76 and LSH was nearly unchanged, thus resulting in a net decreased ratio of DCAF8 over WDR76 for their antagonistic association with LSH (Fig. 4h). Meanwhile, dot blotting analysis revealed a global decrease of 5-hmC abundance in erastin-treated cells, suggesting a similarly decreased TET activity in lipid peroxidation provoked oxidative response (Fig. 4i). Accordingly, pharmacological blockade of TET enzymes through dimethyloxalylglycine (DMOG) [43], potentiated the interaction between LSH and DCAF8 or DDB1, and weakly decreased the association between LSH and WDR76, implying increased LSH incorporation into the CRL4DCAF8 E3 ligase complex along with decreased 5-mC oxidation (Fig. 4j).
In line with these observations, DMOG treatment was associated with shortened half-life of LSH (Fig. 4k) and enhanced LSH degradation (Fig. 4l). Importantly, this DMOG-induced LSH destabilization was largely abolished by further addition of MLN4924 (Fig. 4k and m), and could be rescued, at least partially, by DCAF8 depletion (Fig. 4n). Given the oxidative stress nature of ferroptosis and the decreased TET activities within excessive ROS production context [19, 21], these data together suggested that the declined oxidative modification of DNA could regulate LSH stability by shifting the association preference of LSH towards CRL4DCAF8 versus WDR76 in ferroptosis.
Transcriptomic profiling revealed key ferroptosis genes regulated by the DCAF8/WDR76/LSH axis
Since LSH is functionally implicated in epigenetic regulation of gene transcription [11, 30, 44–47], and ferroptosis has been recently linked to epigenetic mark associated transcriptional activation [48, 49], it is intriguing to determine the influence of DCAF8/WDR76/LSH axis on erastin-induced transcriptomic changes. We thus performed RNA sequencing (RNA-seq) in the HT-1080 ferroptosis sensitive cells. By use of a stringent cutoff (absolute fold change > 2, adjusted p value < 1e−3), we firstly identified a total of 63 genes significantly up-regulated, and 207 genes significantly down-regulated by erastin treatment (Fig. 5a). Interestingly, HMOX1, FTH1, GCLM, SAT1 and ALOX5AP, with critical roles in ferroptosis [40, 50–52] were transcriptionally activated by erastin; while the negative regulator SLC7A11, as core component of glutamate/cystine antiporter system Xc- pharmacologically targeted by erastin [53], and CTH [54], were repressed, indicating a nuclear transcriptional response accompanying redox/metabolic changes in ferroptosis (Fig. 5a). We then profiled erastin-induced transcriptomic changes in DCAF8-depleted or WDR76-overexpressed HT-1080 cells. Set-wise comparisons on the trends of whole groups of genes showing either upward or downward mRNA expression changes in control HT-1080 cells revealed that DCAF8 silencing led to loss of erastin responsiveness in both directions (Fig. 5b, c). Similarly, WDR76 overexpression blunted these erastin-induced transcriptional alterations (Fig. 5b and c). Interestingly, SLC7A11 and CTH, two genes transcriptionally inhibited by erastin, were reverted to an inducible state when DCAF8 was depleted (Fig. 5c and e).
Fig. 5. Transcriptomic profiling revealed key ferroptosis genes regulated by the DCAF8/WDR76/LSH axis.

a Volcano plot shows the differentially regulated genes in HT-1080 cells treated with erastin or vehicle. The significantly altered genes (absolute fold change > 2, adjusted p value < 1e−3) were highlighted in red or blue. Ferroptosis related genes with significant expression changes were labeled. b Boxplot shows average mRNA expression levels of the differentially regulated sets of genes from a in the indicated cells. oe overexpression, rep replicate. c RNA sequencing coverage at the HMOX1 and SLC7A11 loci in the indicated cells. All the coverage tracks in the same locus have same data ranges. d Erastin responsiveness was measured as absolute value of the log2 transformed fold changes of the mRNA expression levels in cells treated with erastin relative to the mRNA levels in cells treated with vehicle. The significantly induced or suppressed genes in control cells were then partitioned into nine subgroups according to their differential responses to erastin in DCAF8 knockdown cells compared to the control cells (adjusted p value cutoff: 5e−2), and their differential responses in DCAF8/LSH co-depleted cells compared to the single DCAF8-depleted cells both in the presence of erastin (adjusted p value cutoff: 5e−2). e Gene expression matrix of the significantly induced or inhibited genes by erastin was scaled within the expression vector for the same gene and shown as a heatmap with each column representing a gene and each row representing a cellular state with indicated gene manipulations and treatment. The genes are clustered together according to their partitions in d. Ferroptosis related genes are highlighted below.
We next sought to determine the LSH dependency in these transcriptomic deviations. With the additional RNA-seq data in HT-1080 cells with DCAF8 and LSH co-depleted, we partitioned the significantly induced or suppressed genes in control cells into nine subgroups. Among them, DCAF8-regulated genes were defined as the ones showing significantly differential responses to erastin in DCAF8 knockdown cells versus control cells (adjusted p value < 5e−2), and LSH-dependent/DCAF8-regulated genes were defined as the ones showing restored responsiveness when comparing mRNA levels in DCAF8/LSH co-depleted cells to the levels in single DCAF8-depleted cells both treated by erastin (Fig. 5d, e). This analysis revealed a total of 17 (Up/Cluster1, ~27%) out of the 63 erastin-induced genes lost their erastin-mediated transcriptional activation upon DCAF8 depletion, but showed rescued erastin responsiveness by LSH co-depletion (Fig. 5d, e). Concordantly, a subset of 21 genes (Down/cluster1, ~10%) in the downward direction showed corresponding changes (Fig. 5d and e). Importantly, 4 (FTH1, HMOX1, GCLM and ALOX5AP) out of the 5 key up-regulated ferroptosis genes were consistently subjected to DCAF8/LSH-mediated transcriptional regulation, and the two down-regulated ones (SLC7A11 and CTH) showed blunted activation upon LSH co-depletion (Fig. 5c–e). These data together suggested that the DCAF8-WDR76-LSH axis was critically implicated in erastin-induced ferroptosis through transcriptional regulation of metabolic and redox homeostasis genes.
Transcriptional suppression of ferroptosis genes by LSH is linked to its physical association with linker histone H1
With the use of H3 lysine 27 acetylation (H3K27ac) antibody to probe active chromatin context [55, 56], we found that erastin treatment fostered the enrichment of H3K27ac at promoters of HMOX1, ALOX5AP, GCLM, and FTH1, four Up/Cluster1 ferroptosis genes with regulatory dependence on the DCAF8-WDR76-LSH axis (Fig. 5), indicating an erastin-induced epigenetic activation correlating with the stimulated transcription (Fig. 6a). We then depleted LSH in the absence of erastin to investigate whether only LSH removal was sufficient to unleash these promoters from repressive chromatin microenvironment. Indeed, ChIP results suggested that LSH knockdown led to a substantial increase of H3K27ac occupancy on the four tested promoters correlating with their elevated mRNA levels at basal state (Fig. 6b).
Fig. 6. Transcriptional suppression of ferroptosis genes by LSH is linked to its physical association with linker histone H1.

a HT-1080 cells in the absence or presence of erastin (10 μM, 24 h) were subjected to ChIP analysis on the promoters of indicated genes by use of normal IgG and H3K27ac antibodies. Results were represented as relative DNA content compared to the input DNA. Error bars represent mean ± SD for at least two independent experiments. *P < 0.05 (student’s t test). Normalized mRNA expression levels of indicated genes following erastin treatment were measured by quantitative RT-PCR analysis. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test). b HT-1080 cells transfected with control (siNS) or siRNA against LSH (siLSH) in the absence of erastin treatment were subjected to ChIP analysis as in a. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test). Normalized mRNA expression levels of indicated genes in HT-1080 cells transfected with control (siNS) or siRNA against LSH (siLSH) were measured by quantitative RT-PCR analysis. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test). c HeLa cells were transfected with plasmids encoding MYC-tagged H1 variants (H1.0~H1.5) respectively. The cell lysates were then subjected to co-immunoprecipitation analysis with MYC antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. d HeLa cells were collected for co-immunoprecipitation analysis on the interaction between endogenous LSH and H1 with LSH (left) or H1 (right) antibodies used for immunoprecipitation and indicated antibodies used for immunoblotting. e Recombinant HIS-H1 and GST-LSH proteins were bacterially purified and analyzed by Coomassie blue staining (left). These proteins were incubated with immobilized LSH followed by extensive washes. The bound proteins were then resolved on SDS-PAGE and detected with anti-HIS antibodies (right). f Mapping the interface of H1 for its interaction with LSH by pull-down experiments. Schematic diagram of mutated H1 constructs was shown on top (FL full-length, NG N-terminus and globular domain, GC globular domain and C-terminus, G globular domain). HIS-tagged wild-type and truncated H1 proteins were bacterially purified (bottom left). These proteins were incubated with immobilized LSH followed by extensive washes. The bound proteins were then resolved on SDS-PAGE and detected with anti-HIS antibody (bottom right). g Mapping the interface of LSH for its interaction with H1 by pull-down experiments. These GST-tagged LSH truncations were bacterially purified and immobilized, followed by incubation with purified HIS-H1 proteins. After extensive washes, the bound proteins were then resolved on SDS-PAGE and detected with anti-HIS antibody (right). FL full-length, NT N-terminus, DEXD DEXD or ATPase domain, CT C-terminus. h The N-terminal portion of LSH was further truncated into three fragments as illustrated on the top. These proteins were bacterially purified and subjected to GST pull-down analysis with the use of HIS-H1 proteins. i HT-1080 cells in the absence of erastin treatment were subjected to ChIP analysis on the promoters of indicated genes by use of normal IgG and H1 antibodies. Results were represented as relative DNA content compared to the input DNA. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test). j HT-1080 cells were transfected with plasmids encoding wild-type (LSH-WT) or Q90A mutated LSH (LSH-MT). These together with control HT-1080 cells were further treated with erastin. Normalized mRNA expression levels of indicated genes were measured by quantitative RT-PCR. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test).
Invoked by the presence of linker histone H1s in LSH’s interactome (Fig. 1a), we performed co-immunoprecipitation experiment and the results indicated that LSH could interact with four H1 subtypes, namely H1.2, H1.3, H1.4 and H1.5 (Fig. 6c), and endogenous LSH and H1 could be mutually immunoprecipitated (Fig. 6d). GST pull-down results demonstrated that LSH could directly associate with H1 (Fig. 6e), through an interface composed of N-terminal portion of LSH and C-terminal domain of H1 (Fig. 6f, g). Detailed analysis suggested that the 51-150 amino acid of the N-terminal portion of LSH were responsible for its association with H1 (Fig. 6h). Interestingly, further analysis with alanine scanning mutagenesis of conserved residuals within the 51-150 amino acid region revealed that the Q90A mutant showed mostly compromised interaction with H1 (Supplementary Figs. 3A, B.
In light of the contribution of Arabidopsis thaliana LSH (DDM1) to repressive chromatin configuration within H1-enriched epigenetic domains [57, 58], and also of the significant enrichment of H1 at the four Up/Cluster1 gene promoters at basal state (Fig. 6i), we tested whether human LSH possessed a similar H1-associated epigenetic silencing function for ferroptosis genes by employing wild-type and Q90A mutated LSH constructs. As expected, overexpression of wild-type LSH reduced erastin-stimulated transcription of HMOX1, FTH1, GCLM and ALOX5AP (Fig. 6j). However, this effect was attenuated by the Q90A mutation (Fig. 6j), indicating that H1 binding is essential for the full repression effect exerted by LSH in transcriptional regulation of ferroptosis genes.
Tumorigenic and senescent phenotypes driven by WDR76 and DCAF8 manipulations
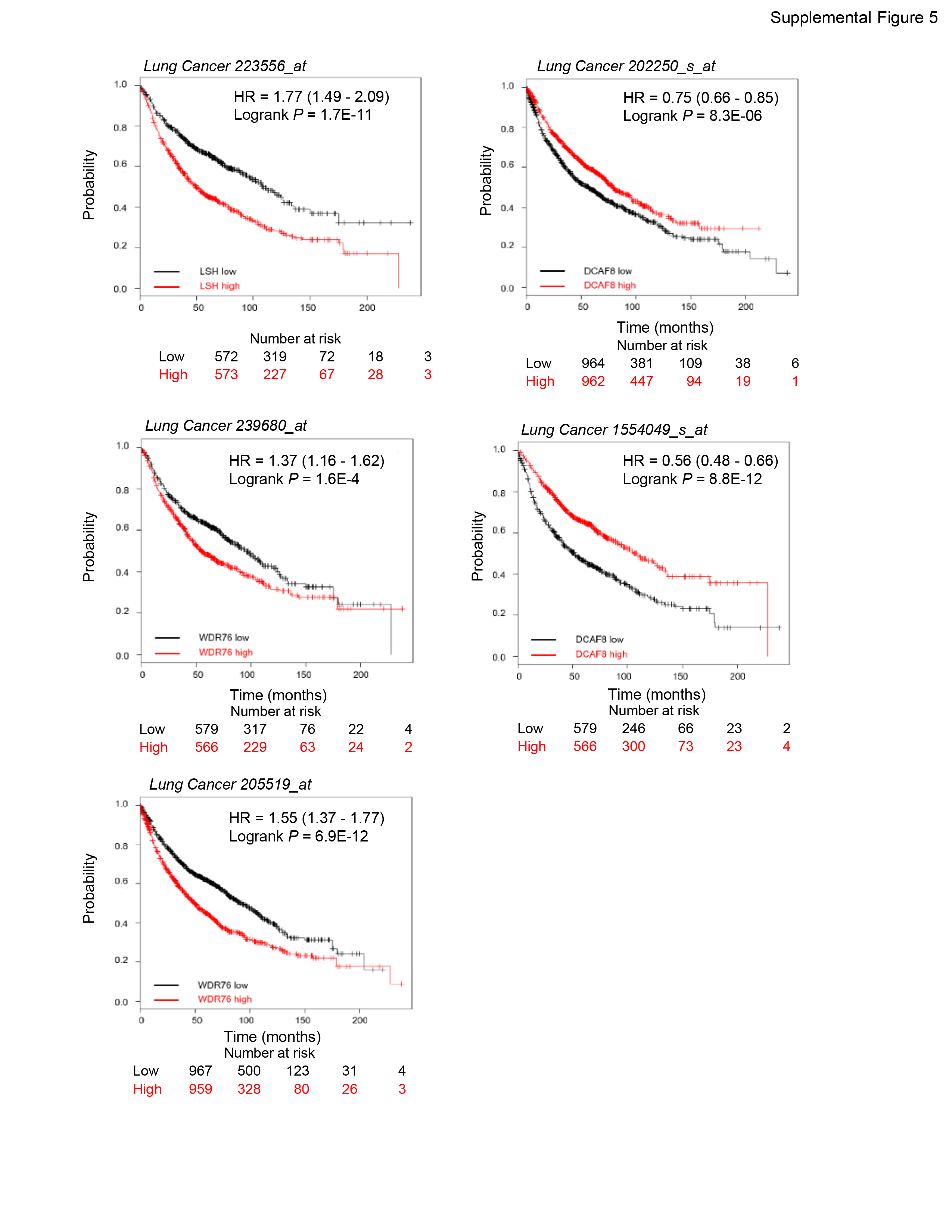
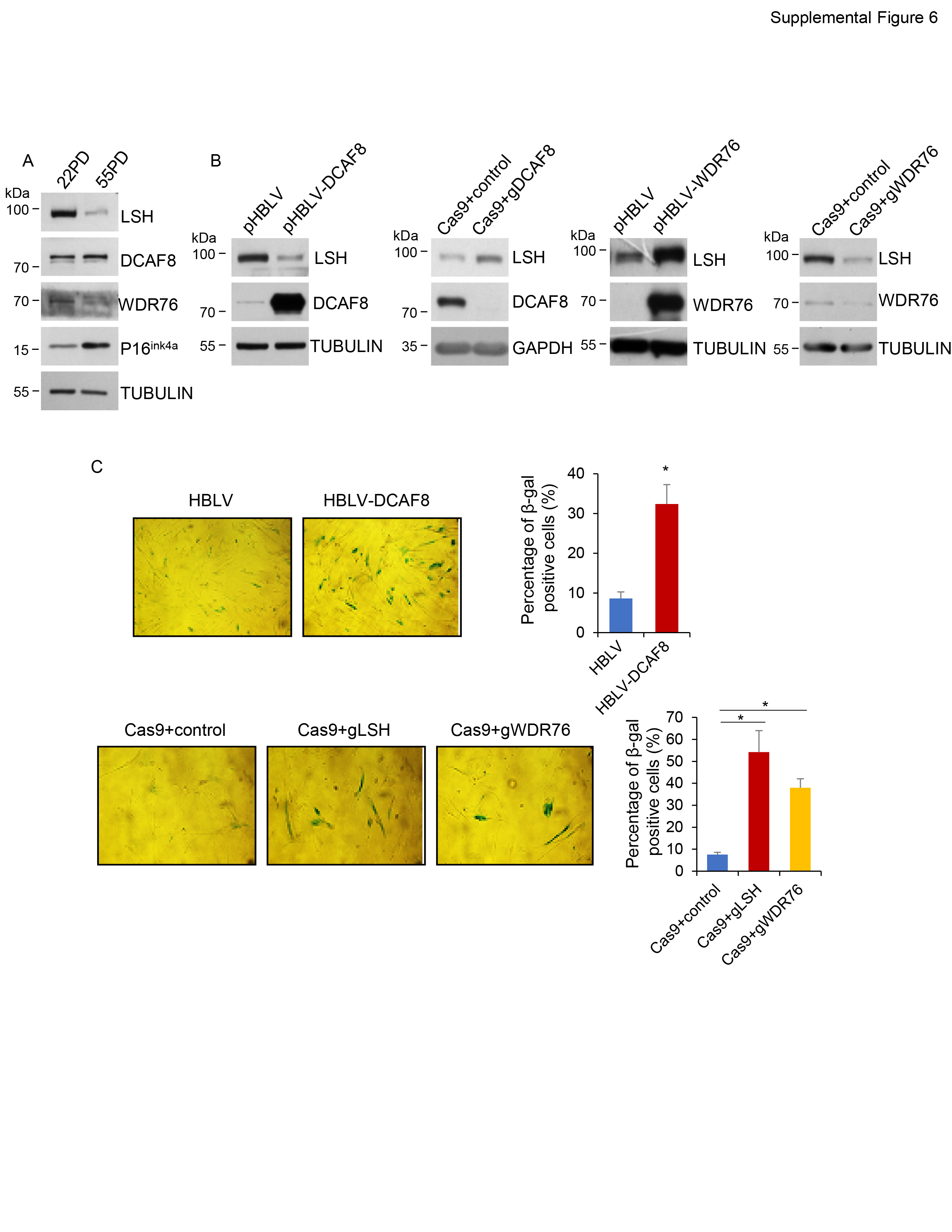
LSH dysfunction and deregulation have been involved in many cancers and premature aging, we thus tested the function of this newly identified LSH stability control program in tumorigenesis and cellular senescence. Indeed, we observed higher proliferation ratio (Fig. 7a) and increased colony formation (Fig. 7b) in WDR76-overexpressed cancer cells and a reverse trend in WDR76-ablated cells (Fig. 7c), while DCAF8 manipulations caused a counteracting effect correspondingly (Fig. 7d). Interestingly, expression levels of LSH target gene HMOX1, which is an essential heme oxygenase for ferroptotic cell death [50, 59], were lower in LSH- or WDR76-overexpressed or DCAF8-ablated cancer cells, but higher in cells with reverse manipulations (Supplementary Fig. 4), thus linking the ferroptosis pathway to LSH/DCAF8/WDR76-regulated cancer cell survival. Accordingly, both xenograft mouse model (Fig. 7e) and Kaplan–Meier survival analysis of lung cancer patients (Supplementary Fig. 5) supported a consistent role of the LSH/DCAF8/WDR76 axis in clinicopathologically relevant settings. Meanwhile, DCAF8-depleted cancer cells were more resistant to erastin treatment, but further knockout of LSH restored their sensitivities (Fig. 7f), suggesting a potential therapeutic value of this axis in ferroptosis associated tumor regression [20, 40]. On the other hand, both expression analysis and SA-β-galactosidase staining results (Supplementary Fig. 6) supported a conserved regulatory role of DCAF8/WDR76-controlled LSH proteolysis in replicative exhaustion.
Fig. 7. Tumorigenic and senescent phenotypes driven by WDR76 and DCAF8 manipulations.

a Growth curves of H1299 cells transduced with control or lentiviral LSH or WDR76 for their stable overexpression were measured by CCK-8 assay. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test), LSH versus Vector. #P < 0.05 (student’s t test), WDR76 versus Vector. Western blotting analysis for indicated proteins was shown on the left. b Colony formation in H1299 cells stably expressing control, LSH or WDR76. The relative colony areas were shown as mean ± SD for three independent experiments. *P < 0.05 (student’s t test). c CRISPR-Cas9-mediated LSH and WDR76 ablation was performed in H1299 cells. Colony formation in control and these cells was demonstrated. The relative colony areas were shown as mean ± SD for three independent experiments. *P < 0.05 (student’s t test). Western blotting analysis for indicated proteins was shown on the left. d Growth curves and colony formation in H1299 cells with stable DCAF8 overexpression, or with CRISPR-Cas9-mediated DCAF8 ablation were demonstrated. The number of cells used in initial seeding was indicated. Error bars represent mean ± SD for three independent experiments. *P < 0.05 (student’s t test). Western blotting analysis for indicated proteins was shown on the left. e CRISPR-Cas9-mediated DCAF8 and LSH double knockout was performed in H1299 cells. Control, DCAF8 single knockout, or DCAF8 and LSH double knockout H1299 cells were subcutaneously injected into the right flank of athymic nude mice (n = 5 for each group). After 6 weeks, the tumors were harvested. Western blotting analysis for indicated proteins was shown on the right. f CRISPR-Cas9-mediated DCAF8 single knockout, or DCAF8 and LSH double knockout were performed in HT-1080 cells. These cells were further exposed to the indicated concentrations of erastin. Colony formation in these cells was then measured. The number of cells used in initial seeding was indicated. The relative colony areas were shown as mean ± SD for three independent experiments at the bottom. *P < 0.05 (One-way Anova).
Discussion
Aberrant degradation regulation of oncogenes and tumor suppressors has been implicated in many malignancies [60–63]. Despite the known roles of LSH to cooperate with other oncogenic cascades to promote carcinogenesis [25, 26, 46, 64] and its deregulations in various cancers at both mRNA and protein levels [28, 30, 31, 46, 65–67], the degradation machinery responsible for stability control of LSH proteins, to our best knowledge, remained unexplored until this study. Hinted by the proteomic identification of DCAF8 and WDR76 as two potential WD40 family substrate receptors of CRL4 family E3, we evidenced that DCAF8 could be incorporated into a functional CRL4DCAF8 E3 ligase complex to catalyze LSH polyubiquitination, which is optimized by the co-expression of all common CRL4 components in vivo and could be recapitulated with defined factors in vitro, and to target LSH for proteasomal degradation. Indeed, in agreement with the biochemically identified interacting surface composed of two WDXR motifs of DCAF8 and β propeller B (BPB) of DDB1 [39], we found WDXR mutant of DCAF8 failed to promote LSH polyubiquitination, supporting a typical CRL4 molecular architecture using DDB1 to accommodate substrate receptor in CRL4DCAF8 complex. Interestingly, through both gain-of-function and loss-of-function analysis, WDR76 was identified as a negative regulator of CRL4DCAF8 mediated LSH degradation, which showed negligible interaction with DDB1 compared to the LSH-WDR76 or DCAF8-DDB1 interactions. Further analysis suggested that overexpression of WDR76 or DCAF8 had no gross effect on the interaction between LSH and the other one. This result, together with the data that WDR76 and DCAF8 directly associated with different regions of LSH, disfavored the explanation that WDR76 suppressed CRL4DCAF8-mediated LSH ubiquitination by competing for substrate binding. However, enforced WDR76 expression promoted the disruption of DCAF8-DDB1 interaction, pointing to the molecular action of WDR76 in suppressing LSH incorporation into an intact and functional CRL4DCAF8 E3 complex.
The contribution of LSH/DDM1’s chromatin remodeling function in regard to linker histone H1 has come into light only recently [57, 58]. Our results suggested a similar H1-associated LSH targeting model for epigenetic suppression of ferroptosis genes in the absence of lipid peroxides inducers. In line with the established H1 functions in chromatin compaction and gene silencing [68, 69], we found that LSH depletion or degradation correlated with transcriptional activation of Up/Cluster1 genes and with increased H3K27ac enrichment at promoters, which also supported a gene specific regulatory role of H1 in controlling redox homeostasis genes [68, 70, 71]. Considering the variety of collaborating co-repressive complexes for LSH [6, 7, 10, 11], we could envision the involvement of LSH’s chromatin remodeling activities in facilitating cooperative histone and/or DNA modifiers to establish repressive chromatin microenvironment for ferroptosis genes.
Increasing evidence suggest that ferroptosis, as an iron-dependent regulated form of cell death, is tightly associated with metabolic and redox homeostasis, and is implicated in many degenerative diseases and cancers [40]. In this study, we found ferroptosis inducer could suppress DNA oxidation coordinated actions on basis of the opposing CRL4DCAF8/WDR76 system to control LSH turnover and downstream epigenetic remodeling at the promoter of redox homeostasis genes, thus functionally linking the DCAF8/WDR76/LSH axis as a critical epigenetic modulator to ferroptosis. According to the recent findings on DNA hydroxymethylation changes associated with hypoxia in cancers [21] and the connection between hypoxia and ROS production [72], it is reasonable to postulate that the overall redox state of cells could be sensed in part by dioxygenases, transmitted in the form of 5-hmC and other oxidative modification changes, and finally affect hydroxymethylation-tuned LSH stability through regulation of WDR76 and/or DCAF8 chromatin loading based on their differential affinity towards DNA hydroxymethylation [22].
Materials and methods
Cell culture and treatment
HT-1080, SH-SY5Y, HK-2, Caki-1, 786-0 and Panc-1 cell lines were purchased from China Infrastructure of Cell Line Resources (Beijing, China). HEK293T, HeLa, A549, U2OS, MCF-7, T47D, HCC1937, MDA-MB-231, HepG2, SMMC-7721, HuH-7, Caco-2, Lovo, SW620, HT-1080, SH-SY5Y, HK-2 and Panc-1 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO BRL, USA) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. Caki-1, HT-29 and HCT116 cells were cultured in McCoy’s 5A modified medium. MCF10A and MCF10F were grown in Dulbecco’s modified Eagle’s medium: nutrient Mixture F-12 (DMEM/F12). 2BS, 786-0 and H1299 cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. DMOG (HY-15893) was purchased from MedChemExpress. Erastin (S7242), MG132 (S2619), MLN4924 (S7109) and Ferrostatin-1 (S7243) were purchased from Selleck. Cycloheximide (C104450) was purchased from Sigma. The concentration of drug used was indicated in the relevant figure legends.
In vivo and in vitro ubiquitination assays
In vivo ubiquitination assays were performed as described previously with slight modification [73]. Briefly, HEK293T cells cultured in each 10-cm dish were transfected with different combinations of FLAG-LSH (8 µg), HA-ubiquitin (6 µg), and MYC-DCAF8 (6 µg), MYC-DCAF8R314A (6 µg), or MYC-DDB2 (6 µg). 48 h after transfection, the cells were treated with MG132 (20 μM) for 5 h and then lysed with denaturing lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 1 mM DTT, 1% SDS) by vortexing and boiling for 10 min. The supernatants were collected by centrifugation at 13,000 rpm for 10 min and diluted 10-fold with an NP-40 buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.3% Nonidet P-40). Further, the lysates were immunoprecipitated with anti-FLAG antibody and protein G agarose beads. Western blotting was conducted to probe ubiquitylated proteins with anti-FLAG, anti-MYC or anti-HA antibodies. For LSH ubiquitination analysis in vitro, we reconstituted and purified the CRL4DCAF8 complex as previously described with little modifications [74]. Briefly, HA-CUL4B, HA-DDB1, HA-ROC1, and FLAG-DCAF8 were transiently co-transfected into HEK293T cells. 48–72 h after transfection, the CRL4DCAF8 complex was purified by immunoprecipitation following the established protocol [74]. The purity and integrity of reconstituted CRL4DCAF8 complex were examined by western blotting analysis. The reconstituted CRL4DCAF8 complex together with eukaryotically purified FLAG-LSH were mixed with UBE1 (E1, Boston Biochem), UbcH5c (E2, Boston Biochem), and Ubiquitin (Ub, Boston Biochem) in a 20 μl system (1×Energy Regeneration Solution, Boston Biochem) for ubiquitination reaction for 30 min at 37 °C.
Cycloheximide chase assay
For protein half-life analysis, cycloheximide (CHX) (40 μg/ml) was added to cells, and aliquots of these cells were collected at specific time points. Protein levels were then determined by western blotting analysis. The relative intensities of LSH protein bands were measured by Image J software.
Measurement of lipid ROS by flow cytometry
Lipid peroxidation levels were measured as previously described [53]. Briefly, DMSO, erastin (10 μM) or a combination of erastin (10 μM) and ferrostatin-1 (2 μM) treated cells were harvested and resuspended in PBS containing 5 μM C11-BODIPY 581/591 (Invitrogen). Cells were incubated at 37 °C for 30 min, then washed, filtered through a 0.4 µm cell strainer and analyzed using the flow cytometer BD FACS Calibur. A minimum of 10,000 cells were analyzed per tube.
RNA sequencing (RNA-seq), quantitative reverse transcription PCR (qRT-PCR)
In-depth whole transcriptomic sequencing was performed by Beijing Genome Institute (BGI). Briefly, total RNA was processed for mRNA enrichment or rRNA depletion. The mRNA fragments were then subjected to reverse transcription by use of random N6 primer. After end processing and A tailing, the double stranded DNAs were further ligated with bubble adapter. These DNAs were then amplified by PCR, heat separated into single strand DNAs, and circularized into library ready for high-throughput sequencing. Raw reads were filtered with SOAPnuk and the resulting clean reads were aligned to human genome by Bowtie 2 [75]. We used R (http://www.R-project.org) and ggplot2 packages (http://ggplot2.org) to produce heatmaps, barplots, and other statistical plots [34]. For qRT-PCR, RNAs were isolated with Trizol (Invitrogen) and the cDNAs were synthesized by use of RevertAid First Strand cDNA synthesis kit (Thermo Scientific). Quantitative real-time PCR following reverse transcription was conducted on Applied Biosystems 7500 Real-Time PCR System. The primers used for qRT-PCR were as follows. GAPDH forward: 5’-AATTCCATGGCACCGTCAAGG-3’; GAPDH reverse: 5’-CCCCACTTGATTTTGGAGGGA-3’; HMOX1 forward: 5’-AGTCTTCGCCCCTGTCTACT-3’; HMOX1 reverse: 5’-CTTCACATAGCGCTGCATGG-3’; FTH1 forward: 5’-ACTTTGACCGCGATGATGTG-3’; FTH1 reverse: 5’-GAAGGAAGATTCGGCCACCT-3’; GCLM forward: 5’-AGACGGGGAACCTGCTGAA-3’; GCLM reverse: 5’-ATGAAGCTCCTCGCTGTGC-3’; ALOX5AP forward: 5’-TCATCAGCGTGGTCCAGAAT-3’; ALOX5AP reverse: 5’-GTAGACCCGCTCAAAGGCAA-3’; LSH forward: 5’-CATGGAACCCAGGAGGAACG-3’; LSH reverse: 5’-TGCTGTAACGCATTTCGGTC-3’.
Statistical analysis
Statistical analysis was performed with SPSS 13.0. Data in Figs. 6 and 7 were presented as mean ± SD from 3 independent experiments and assessed by independent 2-tailed Student’s t test or 1-way analysis of variance. Statistical significance was considered at P < 0.05.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 32070638 and 81771495 to YZ), the National Basic Research Programs of China (No. 2013CB530801 to TJT), and the Beijing Municipal Natural Science Foundation (No. 7192229 to QL).
Data availability
RNA sequencing data was deposited at the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE135361.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by V. D'Angiolella
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Daoyuan Huang and Qian Li
Supplementary information
The online version of this article (10.1038/s41418-020-00689-5) contains supplementary material, which is available to authorized users.
References
- 1.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166:822–39. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dennis K, Fan T, Geiman T, Yan Q, Muegge K. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001;15:2940–4. doi: 10.1101/gad.929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muegge K. Lsh, a guardian of heterochromatin at repeat elements. Biochem Cell Biol. 2005;83:548–54. doi: 10.1139/o05-119. [DOI] [PubMed] [Google Scholar]
- 6.Myant K, Stancheva I. LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol. 2008;28:215–26. doi: 10.1128/MCB.01073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myant K, Termanis A, Sundaram AY, Boe T, Li C, Merusi C, et al. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83–94. doi: 10.1101/gr.108498.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vongs A, Kakutani T, Martienssen RA, Richards EJ. Arabidopsis thaliana DNA methylation mutants. Science. 1993;260:1926–8. doi: 10.1126/science.8316832. [DOI] [PubMed] [Google Scholar]
- 9.Jeddeloh JA, Stokes TL, Richards EJ. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet. 1999;22:94–7. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Geiman TM, Xi S, Jiang Q, Schmidtmann A, Chen T, et al. Lsh is involved in de novo methylation of DNA. EMBO J. 2006;25:335–45. doi: 10.1038/sj.emboj.7600925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi S, Zhu H, Xu H, Schmidtmann A, Geiman TM, Muegge K. Lsh controls Hox gene silencing during development. Proc Natl Acad Sci USA. 2007;104:14366–71. doi: 10.1073/pnas.0703669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–55. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 13.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 14.Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–3. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 15.Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat cell Biol. 2006;8:1277–83. doi: 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol cell. 2007;26:775–80. doi: 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochemical Sci. 2009;34:562–70. doi: 10.1016/j.tibs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol cell Biol. 2014;15:411–21. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 19.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol cell. 2012;48:158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends cell Biol. 2016;26:165–76. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537:63–8. doi: 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–59. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Lee DW, Zhang K, Ning ZQ, Raabe EH, Tintner S, Wieland R, et al. Proliferation-associated SNF2-like gene (PASG): a SNF2 family member altered in leukemia. Cancer Res. 2000;60:3612–22. [PubMed] [Google Scholar]
- 24.Gemenetzidis E, Bose A, Riaz AM, Chaplin T, Young BD, Ali M, et al. FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PloS one. 2009;4:e4849. doi: 10.1371/journal.pone.0004849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164–76. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benavente CA, Finkelstein D, Johnson DA, Marine JC, Ashery-Padan R, Dyer MA. Chromatin remodelers HELLS and UHRF1 mediate the epigenetic deregulation of genes that drive retinoblastoma tumor progression. Oncotarget. 2014;5:9594–608. doi: 10.18632/oncotarget.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi YJ, Yoo NJ, Lee SH. Mutation of HELLS, a chromatin remodeling gene, gastric and colorectal cancers. Pathol Oncol Res. 2015;21:851–2. doi: 10.1007/s12253-014-9862-y. [DOI] [PubMed] [Google Scholar]
- 28.Chen D, Maruschke M, Hakenberg O, Zimmermann W, Stief CG, Buchner A. TOP2A, HELLS, ATAD2, and TET3 are novel prognostic markers in renal cell carcinoma. Urology. 2017;102:e261–265 e267. doi: 10.1016/j.urology.2016.12.050. [DOI] [PubMed] [Google Scholar]
- 29.Sun LQ, Lee DW, Zhang Q, Xiao W, Raabe EH, Meeker A, et al. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev. 2004;18:1035–46. doi: 10.1101/gad.1176104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He X, Yan B, Liu S, Jia J, Lai W, Xin X, et al. Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res. 2016;76:5743–55. doi: 10.1158/0008-5472.CAN-16-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yano M, Ouchida M, Shigematsu H, Tanaka N, Ichimura K, Kobayashi K, et al. Tumor-specific exon creation of the HELLS/SMARCA6 gene in non-small cell lung cancer. Int J Cancer. 2004;112:8–13. doi: 10.1002/ijc.20407. [DOI] [PubMed] [Google Scholar]
- 32.Fu J, Huang D, Yuan F, Xie N, Li Q, Sun X, et al. TRAF-interacting protein with forkhead-associated domain (TIFA) transduces DNA damage-induced activation of NF-kappaB. J Biol Chem. 2018;293:7268–80. doi: 10.1074/jbc.RA117.001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Zhang Y, Fu J, Han L, Xue L, Lv C, et al. FOXA1 mediates p16(INK4a) activation during cellular senescence. EMBO J. 2013;32:858–73. doi: 10.1038/emboj.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Zhang D, Li Q, Liang J, Sun L, Yi X, et al. Nucleation of DNA repair factors by FOXA1 links DNA demethylation to transcriptional pioneering. Nat Genet. 2016;48:1003–13. doi: 10.1038/ng.3635. [DOI] [PubMed] [Google Scholar]
- 35.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, et al. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–24. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 36.Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5:367–78. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- 37.Burrage J, Termanis A, Geissner A, Myant K, Gordon K, Stancheva I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J Cell Sci. 2012;125:5524–34. doi: 10.1242/jcs.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 39.Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol cell. 2006;23:709–21. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7:3293–305. doi: 10.7150/thno.19988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amouroux R, Nashun B, Shirane K, Nakagawa S, Hill PW, D’Souza Z, et al. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat Cell Biol. 2016;18:225–33. doi: 10.1038/ncb3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan T, Hagan JP, Kozlov SV, Stewart CL, Muegge K. Lsh controls silencing of the imprinted Cdkn1c gene. Development. 2005;132:635–44. doi: 10.1242/dev.01612. [DOI] [PubMed] [Google Scholar]
- 45.Ren J, Hathaway NA, Crabtree GR, Muegge K. Tethering of Lsh at the Oct4 locus promotes gene repression associated with epigenetic changes. Epigenetics. 2018;13:173–81. doi: 10.1080/15592294.2017.1338234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Eyss B, Maaskola J, Memczak S, Mollmann K, Schuetz A, Loddenkemper C, et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012;31:972–85. doi: 10.1038/emboj.2011.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi S, Geiman TM, Briones V, Guang Tao Y, Xu H, Muegge K. Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells. 2009;27:2691–702. doi: 10.1002/stem.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92. doi: 10.1038/s41556-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Reports. 2019. [DOI] [PMC free article] [PubMed]
- 50.Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochimica et biophysica acta. 2015;1851:308–30. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hao S, Liang B, Huang Q, Dong S, Wu Z, He W, et al. Metabolic networks in ferroptosis. Oncol Lett. 2018;15:5405–11.. doi: 10.3892/ol.2018.8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci USA. 2010;107:2926–31. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lyons DB, Zilberman D. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife. 2017;6:e30674. doi: 10.7554/eLife.30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. 2019;116:2672–80. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lallemand-Breitenbach V, Zhu J, Chen Z, de The H. Curing APL through PML/RARA degradation by As2O3. Trends Mol Med. 2012;18:36–42. doi: 10.1016/j.molmed.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 61.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26:455–64. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–34. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 64.Janus JR, Laborde RR, Greenberg AJ, Wang VW, Wei W, Trier A, et al. Linking expression of FOXM1, CEP55 and HELLS to tumorigenesis in oropharyngeal squamous cell carcinoma. Laryngoscope. 2011;121:2598–603. doi: 10.1002/lary.22379. [DOI] [PubMed] [Google Scholar]
- 65.Waseem A, Ali M, Odell EW, Fortune F, Teh MT. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral Oncol. 2010;46:536–42. doi: 10.1016/j.oraloncology.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 66.Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PloS One. 2007;2:e594. doi: 10.1371/journal.pone.0000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoogland AM, Bottcher R, Verhoef E, Jenster G, van Leenders GJ. Gene-expression analysis of gleason grade 3 tumor glands embedded in low- and high-risk prostate cancer. Oncotarget. 2016;7:37846–56. doi: 10.18632/oncotarget.9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 69.Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol cell Biol. 2018;19:192–206. doi: 10.1038/nrm.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen X, Gorovsky MA. Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell. 1996;86:475–83. doi: 10.1016/s0092-8674(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 71.Sancho M, Diani E, Beato M, Jordan A. Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 2008;4:e1000227. doi: 10.1371/journal.pgen.1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21:894–9. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lv L, Wang Q, Xu Y, Tsao LC, Nakagawa T, Guo H, et al. Vpr targets TET2 for degradation by CRL4(VprBP) E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol Cell. 2018;70:961–70 e965. doi: 10.1016/j.molcel.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data was deposited at the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE135361.