

Abstract
Estrogen-related receptor γ (ERRγ), a member of the orphan nuclear receptor family, is a key mediator in cellular metabolic processes and energy homeostasis. Therefore, ERRγ has become an attractive target for treating diverse metabolic disorders. We recently reported that ERRγ acts as a negative regulator of osteoclastogenesis induced by receptor activator of nuclear factor-κB ligand (RANKL). In the present study, we explored the effects of an ERRγ-specific modulator, GSK5182, on ERRγ-regulated osteoclast differentiation and survival. Interestingly, GSK5182 increased ERRγ protein levels much as does GSK4716, which is an ERRγ agonist. GSK5182 inhibited osteoclast generation from bone-marrow-derived macrophages without affecting cytotoxicity. GSK5182 also attenuated RANKL-mediated expression of c-Fos and nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), pivotal transcription factors for osteoclastogenesis. Arrested osteoclast differentiation was associated with reduced RANK expression, but not with the M-CSF receptor, c-Fms. GSK5182 strongly blocked the phosphorylation of IκBα, c-Jun N-terminal kinase, and extracellular signal-regulated kinase in response to RANKL. GSK5182 also suppressed NF-κB promoter activity in a dose-dependent manner. In addition to osteoclastogenesis, GSK5182 accelerated osteoclast apoptosis by caspase-3 activation. Together, these results suggest that GSK5182, a synthetic ERRγ modulator, may have potential in treating disorders related to bone resorption.
Keywords: Apoptosis, ERRγ, GSK5182, NFATc1, Osteoclast
INTRODUCTION
Bone is a very active tissue that experiences continual remodeling by dynamic communication between bone-degrading osteoclasts and bone-forming osteoblasts. The balance between their cellular functions is crucial for maintaining adult bone mass and integrity. Abnormally elevated bone resorption by osteoclasts breaks this balance and often causes pathologic bone diseases, such as osteoporosis and inflammatory arthritis (1-3). Therefore, regulating osteoclast differentiation, function, and survival is a promising strategy for treating erosive bone disorders.
Osteoclasts are giant polykaryons generated from hematopoietic progenitor cells under the control of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor (NF)-κB ligand (RANKL) (4-7). RANKL binding to its receptor, RANK, triggers osteoclastogenesis by activating downstream signaling pathways, including NF-κB and mitogen-activated protein kinases (MAPKs), c-Jun N-terminal kinase (JNK), extracellular sig-nal-regulated kinase (ERK), and p38 (8, 9). In addition, RANKL mediates c-Fos induction and activates calcium signaling by cooperating with immunoglobulin-like receptors. These signaling cascades ultimately induce nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) expression, which upregulates expression of osteoclast marker genes (10, 11).
Estrogen-related receptors (ERRs), termed ERRα, ERRβ, and ERRγ, are orphan receptors that belong to the nuclear receptor superfamily (12-15). ERRα and ERRγ play a role in regulating metabolic gene and energy metabolism, whereas ERRβ maintains embryonic stem-cell pluripotency. Although the endogenous ERR ligands have not been found to date, synthetic compounds that regulate their activity and expression have been developed. Among them, a small synthetic molecule, GSK5182 (4-[(Z)-1-[4-2-dimethylaminoethyloxy)phenyl]-hydroxy-2-phenylpent-1-enyl]phenol, a 4-hydroxy tamoxifen analog), has been identified as an ERRγ-specific inverse agonist that does not bind to other nuclear receptors (16). GSK5182 has an anti-diabetic effect by suppressing hepatic gluconeogenesis in a type 2 diabetic mouse model (17). GSK5182 also prevents vascular calcification by downregulating expression of osteogenic marker genes (18). In addition, GSK5182 reduces osteoarthritis (OA) in an experimental animal OA model (19). A recent study also showed that GSK 5182 has anti-tumor activity in human breast cancer, where reactive oxygen species (ROS) play an important role (20). These findings suggest that GSK5182 may help treat various types of human diseases.
The role of ERRγ in bone metabolism has been reported, with most studies demonstrating its function in osteoblast and cartilage formation (19, 21-23). Recently, we reported the role of ERRγ in osteoclast differentiation (24). We showed that ERRγ expression is downregulated during the osteoclastogenesis and that ERRγ plays a key role in osteoclastogenesis modulation. In the current study, we evaluated the in vitro pharmacological effect on ERRγ regulation in osteoclast differentiation and survival by using GSK5182. We show that GSK5182 inhibits osteoclasto-genesis by blocking RANKL signaling molecules, demonstrating that this compound may help treat osteoclast-related disorders. Interestingly, GSK5182 increased ERRγ protein levels, and ERRγ-mediated NF-κB promoter suppression was further accelerated by GSK5182. Therefore, in this context, GSK5182 acts as an agonist of ERRγ.
RESULTS
The ERRγ modulator, GSK5182, increases ERRγ content
We first investigated the effect of the synthetic ERRγ modulators, GSK5182 and GSK4716, on ERRγ protein expression. We isolated and cultured bone-marrow-derived macrophages (BMMs) with two different concentrations of ERRγ modulators. Immunoblotting analysis using an ERRγ-specific antibody showed that GSK5182 treatment significantly increased ERRγ protein levels in a dose-dependent manner (Fig. 1A). We also observed that GSK4716, a known ERRγ agonist compound, elevated ERRγ protein levels at both concentrations (Fig. 1B). We then examined whether GSK5182 affects ERRγ expression at the RNA level. As determined by real-time PCR, GSK5182 dose-dependently increased mRNA expression of ERRγ (Fig. 1C). These results demonstrate that GSK5182 regulates ERRγ expression at the transcriptional level, but the precise mechanism of this modulation remains to be further investigated.
Fig. 1.
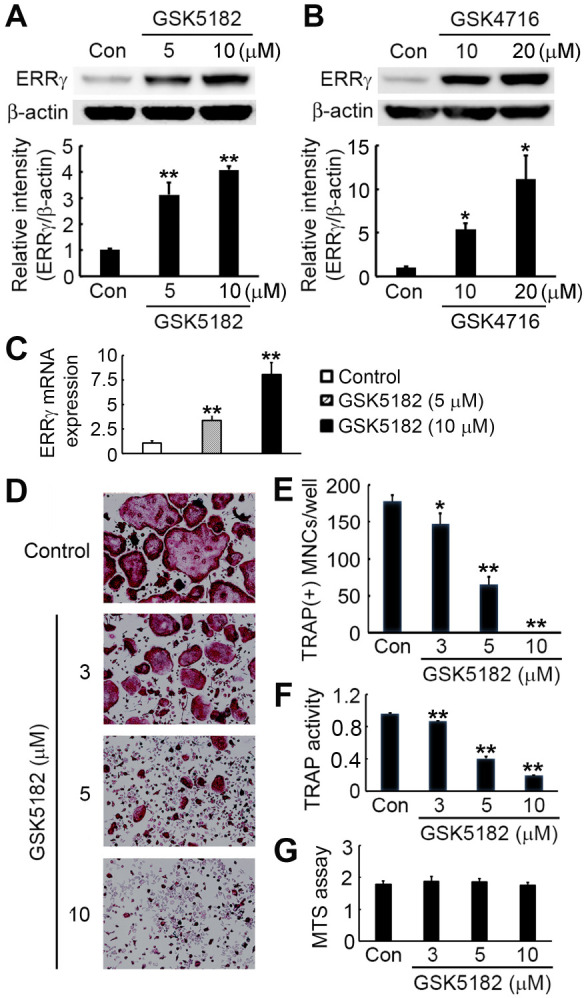
GSK5182 inhibits RANKL-induced osteoclast formation. We cultured BMMs with the indicated concentrations of ERRγ modulators GSK5182 (A) or GSK4716 (B) for 24 h. We used immunoblotting to detect the ERRγ protein contents. Graphs represent the results of quantitative analysis of ERRγ protein levels from three independent experiments. (C) We did real-time PCR to assess ERRγ mRNA levels. (D-F) BMMs were cultured at the indicated GSK5182 concentrations in the presence of RANKL (20 ng/ml) and M-CSF (20 ng/ml) for four days. (D) The cells were fixed and stained for TRAP. (E) TRAP-positive multinucleated cells (MNCs) were counted. (F) TRAP activity in the culture medium generated by cells in (D) was quantified at 405 nm. (G) We cultured BMMs with the indicated concentrations of GSK5182 in the presence of M-CSF (30 ng/ml) for three days. We evaluated cell viability by MTS assay. The data presented in (A-C, E, and F) are expressed as the mean ± SD. *P < 0.05, **P < 0.001 vs. the vehicle-treated control (Con).
GSK5182 blocks osteoclast formation induced by RANKL
To explore GSK5182’s effect on osteoclast formation, we cultured BMMs in the presence of RANKL, M-CSF, and various GSK 5182 doses or vehicles. Compared with the vehicle-treated control, GSK5182 exposure inhibited tartrate-resistant acid phosphatase (TRAP)-expressing multinucleated osteoclast generation from BMMs in a dose-dependent manner (Fig. 1D, E). GSK5182 completely blocked osteoclast formation at 10 μM (Fig. 1E). Consis-tent with a decrease in osteoclast number, TRAP activity from culture supernatants was also dose-dependently reduced by GSK5182 (Fig. 1F). Employing an MTS assay, we also found that the GSK5182 concentrations used in this study did not influence BMM cytotoxicity or proliferation (Fig. 1G).
GSK5182 attenuates osteoclastogenic marker induction
Because GSK5182 inhibited osteoclast formation, we next evaluated the GSK5182 effect on expression of osteoclastogenesis-associated marker genes. We cultured BMMs with RANKL and M-CSF in the absence or presence of GSK5182 (10 μM). RANKL stimulation in control BMMs increased c-Fos and NFATc1 mRNA levels, two key proteins involved in RANKL-mediated osteoclast differentiation. However, GSK5182 significantly attenuated the mRNA induction of these two transcription factors (Fig. 2A). These suppressive effects were further revealed by Western blot analysis. GSK5182 strongly reduced c-Fos and NFATc1 protein expression in a time- and dose-dependent manner (Fig. 2B, C). Reflecting the decrease in NFATc1 protein level, the expression of NFATc1 target genes, TRAP and cathepsin K (CTSK), was also attenuated in response to GSK5182 (Fig. 2A, B). We also found that GSK5182 suppressed the mRNA expression of the RANKL receptor, RANK, but not of the M-CSF receptor, c-Fms (Fig. 2A). We obtained similar results in the Western blot, showing that RANK protein levels were significantly reduced by GSK5182 treatment (Fig. 2D).
Fig. 2.
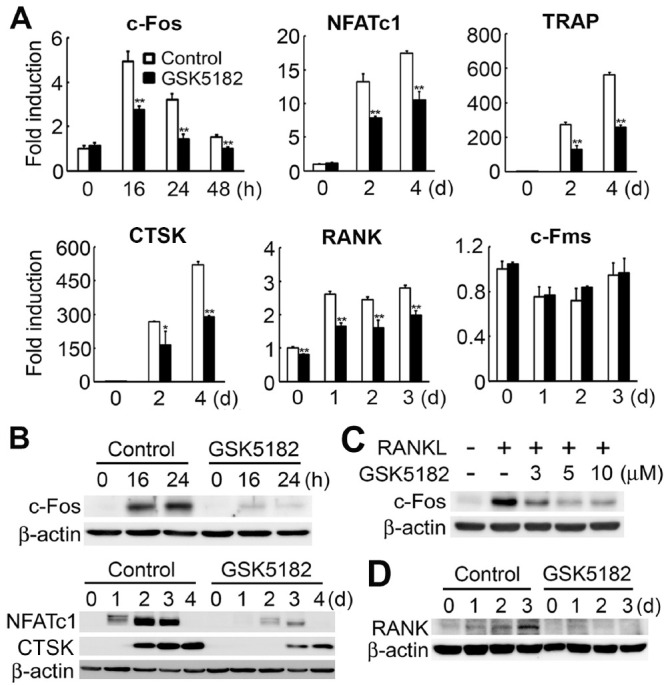
GSK5182 suppresses osteoclastogenic marker expression. (A and B) We cultured BMMs with RANKL and M-CSF in the absence or presence of GSK5182 (10 μM) for the indicated times. We did real-time PCR (A) or immunoblotting (B) to assess the expression of the indicated genes. *P < 0.05, **P < 0.001 vs. control. (C) We cultured BMMs with M-CSF in the absence (-) or presence (+) of RANKL with various GSK5182 concentrations for 24 h. Whole-cell lysates were subjected to immunoblotting to detect c-Fos. (D) RANK protein expression in cells treated with GSK5182 (10 μM) on the indicated days.
GSK5182 inhibits RANKL-stimulated NF-κB, JNK, and ERK activation
To further understand the mechanism for GSK5182’s inhibition of RANKL-induced osteoclastogenesis, we examined the GSK5182 effect on RANKL signaling cascades. We pretreated BMMs with GSK5182 or vehicle and assessed them for MAPK activation in response to RANKL. As expected, RANKL exposure in control cells activated MAPKs, including JNK, ERK, and p38 (Fig. 3A). In contrast, BMMs pretreated with GSK5182 abrogated RANKL-stimulated JNK and ERK phosphorylation (Fig. 3A). However, p38 activation by RANKL was not altered in the presence of GSK5182 (Fig. 3A).
Fig. 3.
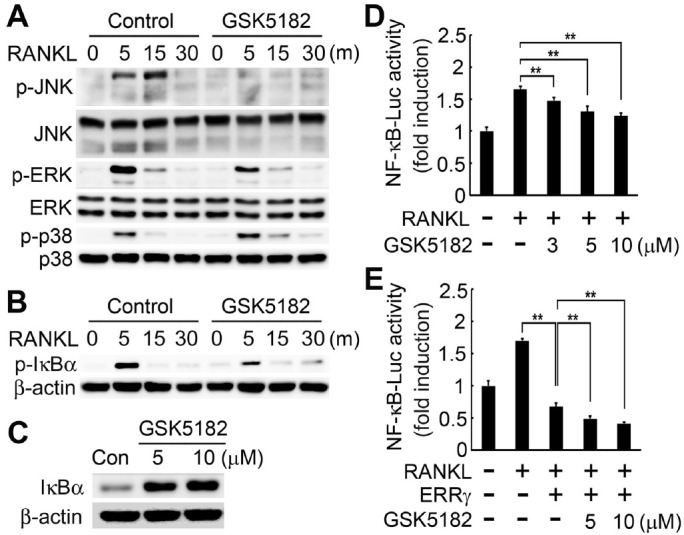
GSK5182 blocks RANKL-stimulated NF-κB, JNK, and ERK activation. (A and B) BMMs were serum-starved for 6 h, pretreated with GSK5182 (10 μM) or vehicle (control) for 1 h, and then stimulated with RANKL (30 ng/ml). (A) We assayed total protein extracts by immunoblotting to detect phosphorylated and total MAPK (JNK, ERK, p38) proteins. (B) Cell lysates were immunoblotted with a phospho-IκBα antibody. (C) IκBα protein levels in GSK5182-treated cells. (D, E) RAW264.7 cells were transfected with NF-κB luciferase reporter alone (D) or co-transfected with NF-κB luciferase together with the ERRγ expression plasmid (E). After transfection, we treated cells with the indicated concentrations of GSK5182 or vehicle for 24 h and then stimulated them with RANKL (100 ng/ml) for 24 h. We measured NF-κB luciferase activity using a dual-luciferase reporter assay system. **P < 0.001.
Another RANKL signaling cascade important for osteoclasto-genesis is the NF-κB signaling pathway. RANKL stimulation elevated IκBα phosphorylation levels in vehicle-treated control BMMs, whereas GSK5182 strongly inhibited RANKL-induced IκBα phosphorylation (Fig. 3B). Accordingly, IκBα protein amounts were increased in GSK5182-exposed cells at both concentrations (Fig. 3C). RANKL also triggers the induction of the NF-κB transcriptional activity. Hence, we next assessed the effect of GSK5182 on RANKL-mediated NF-κB promoter activity in RAW 264.7 macrophages. RANKL exposure increased NF-κB luciferase activity, whereas GSK5182 led to a dose-dependent reduction in NF-κB activity following RANKL stimulation (Fig. 3D). As previously reported (24), the induction of NF-κB promoter activity was significantly attenuated by ERRγ overexpression (Fig. 3E). Notably, GSK5182 treatment accelerated the ERRγ-reducing effect on the NF-κB promoter in a dose-dependent manner (Fig. 3E). These results reveal that GSK5182 inhibits the transactivation of NF-κB, much as ERRγ does, indicating that GSK5182 can act on ERRγ in an agonistic manner.
GSK5182 accelerates osteoclast apoptosis
Because osteoclastic bone resorption is also governed by the cell-survival rate, we next investigated the GSK5182 effect on osteoclast survival. We incubated mature osteoclasts with or without GSK5182 for 24 h and then stained them for TRAP (Fig. 4A). GSK5182 strongly decreased the survival rate of TRAP-positive osteoclasts (Fig. 4B). To examine if the reduced cell-survival rate was accompanied by apoptosis in GSK5182-treated cells, we carried out an apoptotic DNA fragmentation assay by ELISA. GSK5182 markedly increased apoptosis of mature osteoclasts (Fig. 4C). In agreement with its pro-apoptotic effect, GSK5182 accelerated both caspase-3 and caspase-9 activity (Fig. 4D).
Fig. 4.
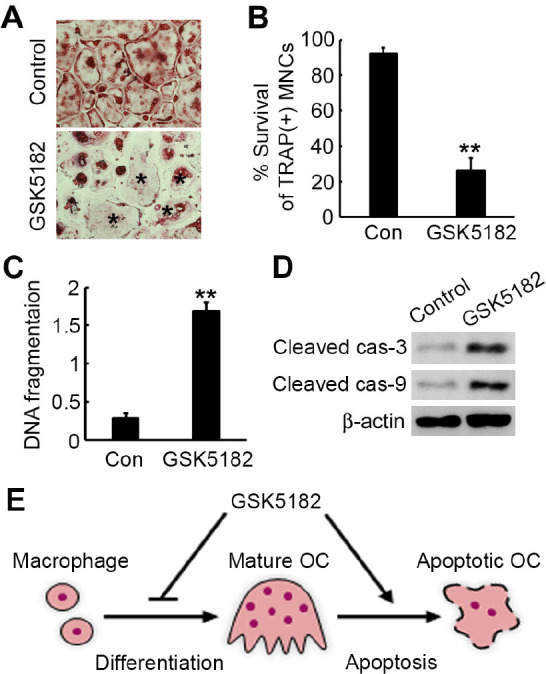
GSK5182 promotes osteoclast apoptosis. We incubated mature osteoclasts with GSK5182 (10 μM) or vehicle for 24 h. (A) The cells were fixed and subjected to TRAP staining. Apoptotic osteoclasts are marked by asterisks. (B) We measured osteoclast survival as the TRAP-positive MNC percentage. **P < 0.001. (C) We measured the osteoclast apoptosis rate by ELISA. **P < 0.001. (D) Total cell lysates were immunoblotted for cleaved caspase (cas)-3 and caspase-9. β-Actin served as a loading control. (E) A diagram showing the GSK5182 effect on osteoclast differentiation and apoptosis.
DISCUSSION
Because ERRγ is implicated in the pathogenesis of various meta-bolic events, regulating ERRγ using small synthetic compounds provides a useful clue for a novel therapeutic strategy. ERRγ is an orphan nuclear receptor because of the absence of its natural ligand. In this regard, a small synthetic compound, GSK5182, has attracted attention as a highly specific ERRγ modulator that does not associate with other nuclear receptors. To date, several studies have highlighted the therapeutic GSK5182-mediated effects on regulating ERRγ in a variety of metabolic diseases (17-20, 25-27). Along with these previous studies, our recent study also demonstrates that regulating ERRγ may be a way to treat bone disorders in an excessive bone-resorption pathological state. Employing loss- and gain-of-function systems, we reported that ERRγ negatively controls RANKL-induced osteoclast differentiation (24). In the current study, we show that an ERRγ modulator, GSK5182, inhibits osteoclastogenesis and promotes mature osteoclast apoptosis, suggesting that GSK5182 might work well to treat bone-resorption-associated diseases (Fig. 4E).
In an attempt to explore GSK5182’s effect on osteoclast formation, we first examined changes in ERRγ protein levels. We found that GSK4716, an ERRγ agonist, increased ERRγ protein levels. Our recent study demonstrated that both ERRγ overexpression and GSK4716 treatment inhibited RANKL-mediated osteoclastogenesis (24), indicating that GSK4716 is indeed an ERRγ agonist. Interestingly, GSK5182, a compound identified as an ERRγ inverse agonist, also dose-dependently increased ERRγ protein levels. Furthermore, like ERRγ and GSK4716, GSK 5182 blocked osteoclast formation and attenuated osteoclastogenic marker gene expression. These findings suggest that GSK 5182-mediated ERRγ protein increase may impair osteoclast differentiation. Therefore, we conclude that GSK5182 functions as an agonist and expression modulator of ERRγ, which serves as a strong negative regulator of osteoclast differentiation. The GSK5182 behavior is similar to that of the ERRα modulator, XCT790. A previous study showed that XCT790 is an ERRα antagonist that is also an ERRα agonist that suppresses cell proliferation and tumorigenicity (28). Thus, like XCT790, GSK5182 may act in a cell- and/or context-specific manner as a receptor agonist or antagonist.
c-Fos and NFATc1 are critical transcription factors for the RANKL-induced osteoclastogenesis. c-Fos targeted disruption results in severe osteopetrosis because it fails to generate osteoclasts from their precursors (29, 30). NFATc1-deficient mouse embryonic stem cells could not form osteoclasts following RANKL stimulation (31). Also, c-Fos is recruited to the NFATc1 promoter after 24 h of RANKL exposure (32), indicating that NFATc1 is a c-Fos target gene during the early stages of osteoclastogenesis. In this study, we observed that GSK5182 attenuated c-Fos and NFATc1 mRNA and protein expression. Consequently, GSK5182 suppressed NFATc1 target genes, such as TRAP and cathepsin K. These data reveal that GSK5182 inhibits osteoclast differentiation by suppressing the two key transcription factors, c-Fos and NFATc1.
NF-κB activation by RANKL is a critical signaling event for osteoclast differentiation. Deleting the NF-κB subunits (p50 and p52) leads to severe osteopetrosis because of defective osteoclastogenesis (33, 34). NF-κB is also recruited to the NFATc1 promoter in response to RANKL (32). We found that GSK5182 strongly blocked RANKL-stimulated phosphorylation of the NF-κB inhibitor, IκBα. In agreement with this observation, ERRγ overexpression and DY131 treatment, another ERRγ agonist, inhibited IκBα phosphorylation induced by RANKL (24). Furthermore, GSK5182 attenuated RANKL-mediated NF-κB transcriptional activity. As NF-κB promoter activity was also suppressed by ERRγ, and this ERRγ inhibitory effect was further acce-lerated by GSK5182, in this circumstance, GSK5182 functions as a receptor agonist. RANKL stimulation also activates the MAPK signaling pathways, including JNK, ERK, and p38. GSK 5182 inhibited JNK and ERK activation by RANKL, whereas this compound did not influence p38 activation. Collectively, our data reveal that blunting the RANKL-stimulated NF-κB, JNK, and ERK signaling pathways contributes to the suppressive effect of GSK5182 on NFATc1 induction.
Apoptosis of mature osteoclasts and/or osteoclast precursor cells is another important factor that governs osteoclast number and bone-resorptive capacity. In addition to its anti-osteoclastogenic effect, GSK5182 increased fully differentiated osteoclast apoptosis. GSK5182 increased mature osteoclast cell death by inducing caspase-3 and caspase-9 activation. However, this compound did not affect osteoclast precursor (BMM) cell viability (Fig. 1F).
In conclusion, our data showed that GSK5182 efficiently blocks osteoclast differentiation. The preventive effect of GSK5182 is associated with inhibiting c-Fos, NF-κB, JNK, and ERK signaling that leads to NFATc1 downregulation. Additionally, GSK5182 promotes osteoclast apoptosis by inducing caspase activation. Therefore, our findings suggest that the ERRγ modulator, GSK5182, might help treat skeletal diseases accompanied by excessive bone erosion, such as osteoporosis, periodontitis, and rheumatoid arthritis.
MATERIALS AND METHODS
Reagents and antibodies
GSK5182 was synthesized as described previously (16). We purchased:
Recombinant murine RANKL and human M-CSF from R&D Systems (Minneapolis, MN, USA).
Primary antibodies specific for IκBα, JNK, p38, ERK, phospho-IκBα, phospho-JNK, phospho-p38, and phospho-ERK from Cell Signaling Technology (Beverly, MA, USA).
ERRγ from R&D Systems.
NFATc1 from BD Pharmingen (San Diego, CA, USA).
c-Fos from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
RANK from Abcam (Cambridge, UK).
Osteoclast differentiation
We prepared osteoclasts from murine bone-marrow cells as previously described (35, 36). Whole bone marrow from C57/BL6 male mice femurs and tibiae was flushed out with α-minimum essential media (α-MEM). After removing the red blood cells, we incubated bone-marrow cells for three days in Petri dishes with α-MEM containing 10% FBS and 10% CMG 14-12 culture media (37) as a source of M-CSF. We selected the adherent cells as bone-marrow-derived macrophages (BMMs). To induce osteoclasts, we cultured precursor cells (BMMs) in α-MEM with RANKL (20 ng/ml) and M-CSF (20 ng/ml) for four or five days. Cells were fixed in 4% paraformaldehyde and stained with TRAP.
Cell viability assay
We seeded BMMs at 5 × 103 cells per well in 96-well plates and cultured them with different GSK5182 doses in the presence of M-CSF (30 ng/ml). After three days, the cells were incubated with 20 μl per well of CellTiter 96 AQueous One Solution Reagent containing MTS (Promega, Madison, WI, USA) at 37°C for 4 h. We quantified cell viability by measuring absorbance at 490 nm.
Real-time PCR
We did quantitative real-time PCR using SYBR Green dye and an ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). The following primer sequences were used:
ERRγ, 5’-GCTAAGTGCCAGAATTCAAACCA-3’ and 5’-ATCGACTCCTATGGATCAGGACTTT-3’;
c-Fos, 5’-AGGCCCAGTGGCTCAGAGA-3’ and 5’-GCTCCCAGTCTGCTGCATAGA-3’;
NFATc1, 5’-ACCACCTTTCCGCAACCA-3’ and 5’-TTCCGTTTCCCGTTGCA-3’;
TRAP, 5’-TCCCCAATGCCCCATTC-3’ and 5’-CGGTTCTGGCGATCTCTTTG-3’;
Cathepsin K, 5’-GGCTGTGGAGGCGGCTAT-3’ and 5’-AGAGTCAATGCCTCCGTTCTG-3’;
RANK, 5’-TCTGCAGCTCTTCCATGACACT-3’ and 5’-GAAGAGGAGCAGAACGATGAGACT-3’;
c-Fms, 5’-CCTGAATCTCCCGGAAGCA-3’ and 5’-CAAGCTCGGTACAACGGTAGGT-3’.
Western blotting
We harvested vehicle or compound-treated cells and lysed them in RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) supplemented with protease-phosphatase inhibitors. We measured the protein concentration using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Equivalent protein amounts were subjected to SDS-PAGE and Western blotting analysis. The immunoreactive proteins were detected using an ECL Plus kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and imaged with a LAS4000 luminescent image analyzer (GE Healthcare, Piscataway, NJ, USA).
Luciferase assay
We cultured RAW264.7 macrophages in Dulbecco’s modified Eagle’s medium containing 10% FBS at 1 × 105 cells per well in 48-well plates. On the following day, the cells were transfected with a NF-κB reporter plasmid (250 ng) using FuGENE HD transfection reagent (Promega, Madison, WI, USA). In another assay, we co-transfected RAW264.7 cells with the NF-κB luciferase reporter (250 ng) and an ERRγ expression plasmid (250 ng) provided by Dr. Hueng-Sik Choi (Chonnam National University, Gwangju, Korea). After 24 h, we incubated the cells with various GSK5182 concentrations for 24 h and then stimulated them with RANKL (100 ng/ml) for 24 h. We measured NF-κB luciferase activity using a dual-luciferase reporter assay system (Promega) according to the manufacturer’s protocols.
Apoptosis assay
We cultured mature osteoclasts in the presence of a vehicle or GSK5182 for one day. Osteoclast apoptosis was analyzed using a Cell Death Detection ELISAPLUS kit (Roche, Mannheim, Germany), which quantitatively detects cytoplasmic histone-associated DNA fragments.
Statistics
We did statistical analysis using Microsoft Excel 2016 (Microsoft, USA). Statistical significance was found by Student’s t-test, considering P < 0.05 as significant. The results are represented as the mean ± standard deviation (SD).
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (No. 2018R1A2B6001298); the Basic Science Research Program of NRF grant funded by the Ministry of Education, Science, and Technology (NRF-2015R1D1A1A01056666); and the Bio & Medi-cal Technology Development Program (NRF-2017R1A5A2015391).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 2.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 3.Tsukasaki M, Takayanagi H. Osteoimmunology: evolving concepts in bone-immune interactions in health and disease. Nat Rev Immunol. 2019;19:626–642. doi: 10.1038/s41577-019-0178-8. [DOI] [PubMed] [Google Scholar]
- 4.Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 5.Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- 6.Wong BR, Rho J, Arron J, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- 7.Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee ZH, Kim HH. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun. 2003;305:211–214. doi: 10.1016/S0006-291X(03)00695-8. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Lee NK, Lee SY. Current understanding of RANK signaling in osteoclast differentiation and matu-ration. Mol Cells. 2017;40:706–713. doi: 10.14348/molcells.2017.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JH, Kim N. Regulation of NFATc1 in osteoclast differentiation. J Bone Metab. 2014;21:233–241. doi: 10.11005/jbm.2014.21.4.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23:582–590. doi: 10.1016/j.tem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- 13.Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 14.Hong H, Yang L, Stallcup MR. Hormone-independent transcriptional activation and coactivator binding by novel orphan nuclear receptor ERR3. J Biol Chem. 1999;274:22618–22626. doi: 10.1074/jbc.274.32.22618. [DOI] [PubMed] [Google Scholar]
- 15.Misra J, Kim DK, Choi HS. ERRgamma: a junior orphan with a senior role in metabolism. Trends Endocrinol Metab. 2017;28:261–272. doi: 10.1016/j.tem.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Chao EY, Collins JL, Gaillard S, et al. Structure-guided synthesis of tamoxifen analogs with improved selectivity for the orphan ERRgamma. Bioorg Med Chem Lett. 2006;16:821–824. doi: 10.1016/j.bmcl.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Kim DK, Gang GT, Ryu D, et al. Inverse agonist of nuclear receptor ERRgamma mediates antidiabetic effect through inhibition of hepatic gluconeogenesis. Diabetes. 2013;62:3093–3102. doi: 10.2337/db12-0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JH, Choi YK, Do JY, et al. Estrogen-related receptor gamma plays a key role in vascular calcification through the upregulation of BMP2 expression. Arterioscler Thromb Vasc Biol. 2015;35:2384–2390. doi: 10.1161/ATVBAHA.115.306102. [DOI] [PubMed] [Google Scholar]
- 19.Son YO, Park S, Kwak JS, et al. Estrogen-related receptor gamma causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat Commun. 2017;8:2133. doi: 10.1038/s41467-017-01868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vernier M, Dufour CR, McGuirk S, et al. Estrogen-related receptors are targetable ROS sensors. Genes Dev. 2020;34:544–559. doi: 10.1101/gad.330746.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardelli M, Aubin JE. ERRgamma is not required for skeletal development but is a RUNX2-dependent negative regulator of postnatal bone formation in male mice. PLoS One. 2014;9:e109592. doi: 10.1371/journal.pone.0109592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardelli M, Zirngibl RA, Boetto JF, et al. Cartilage-specific overexpression of ERRgamma results in Chondrodysplasia and reduced chondrocyte proliferation. PLoS One. 2013;8:e81511. doi: 10.1371/journal.pone.0081511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong BC, Lee YS, Park YY, et al. The orphan nuclear receptor estrogen receptor-related receptor gamma negatively regulates BMP2-induced osteoblast differentiation and bone formation. J Biol Chem. 2009;284:14211–14218. doi: 10.1074/jbc.M808345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HJ, Kim BK, Ohk B, et al. Estrogen-related receptor gamma negatively regulates osteoclastogenesis and protects against inflammatory bone loss. J Cell Physiol. 2019;234:1659–1670. doi: 10.1002/jcp.27035. [DOI] [PubMed] [Google Scholar]
- 25.Kim DK, Jeong JH, Lee JM, et al. Inverse agonist of estrogen-related receptor gamma controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat Med. 2014;20:419–424. doi: 10.1038/nm.3483. [DOI] [PubMed] [Google Scholar]
- 26.Kim DK, Kim JR, Koh M, et al. Estrogen-related receptor gamma (ERRgamma) is a novel transcriptional regulator of phosphatidic acid phosphatase, LIPIN1, and inhibits hepatic insulin signaling. J Biol Chem. 2011;286:38035–38042. doi: 10.1074/jbc.M111.250613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DK, Ryu D, Koh M, et al. Orphan nuclear receptor estrogen-related receptor gamma (ERRgamma) is key regulator of hepatic gluconeogenesis. J Biol Chem. 2012;287:21628–21639. doi: 10.1074/jbc.M111.315168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bianco S, Lanvin O, Tribollet V, Macari C, North S, Vanacker JM. Modulating estrogen receptor-related receptor-alpha activity inhibits cell proliferation. J Biol Chem. 2009;284:23286–23292. doi: 10.1074/jbc.M109.028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson RS, Spiegelman BM, Papaioannou V. Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell. 1992;71:577–586. doi: 10.1016/0092-8674(92)90592-Z. [DOI] [PubMed] [Google Scholar]
- 30.Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- 31.Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/S1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 32.Asagiri M, Sato K, Usami T, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franzoso G, Carlson L, Xing L, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Lee DK, Jin X, Che X, Choi JY. Oleoyle-thanolamide exhibits GPR119-dependent inhibition of osteoclast function and GPR119-independent promotion of osteoclast apoptosis. Mol Cells. 2020;43:340–349. doi: 10.14348/molcells.2020.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung YK, Han SW, Kim GW, Jeong JH, Kim HJ, Choi JY. DICAM inhibits osteoclast differentiation through attenuation of the integrin alphaVbeta3 pathway. J Bone Miner Res. 2012;27:2024–2034. doi: 10.1002/jbmr.1632. [DOI] [PubMed] [Google Scholar]
- 37.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]