

Highlights
-
•
Amplification of RAD54B promotes HCC progression by wnt/β-catenin pathway.
Keywords: HCC, Wnt/β-catenin signaling, Gene amplification, RAD54B, Metastasis
Abbreviations: HCC, hepatocellular carcinoma; IHC, immunohistochemistry; LRP5/6, lipoprotein receptor-related protein-5 or -6; TCF, T-cell factor; ScRad54, Saccharomyces cerevisiae Rad54; aa, amino acid; HRR, homologous recombination repair; ATCC, American Type Culture Collection; FBS, fetal bovine; TCGA, the Cancer Genome Atlas; ANT, adjacent normal tissues; GSEA, gene set enrichment analysis
Abstract
Liver cancer was reported to be the sixth most frequently diagnosed cancer, and hepatocellular carcinoma (HCC) accounts for 75%-85% of primary liver cancer. Nevertheless, the concrete molecular mechanisms of HCC progression remain obscure, which is essential to elucidate. The expression profile of RAD54B in HCC was measured using qPCR and western blotting. Moreover, the levels of RAD54B in paraffin-embedded samples were evaluated using immunohistochemistry (IHC). The effect of RAD54B on HCC progression was testified by in vitro experiments, and in vivo orthotopic xenograft tumor experiments. The mechanisms of RAD54B promoting HCC progression were investigated through molecular and function experiments. Herein, RAD54B are dramatically upregulated in HCC tissues and cell lines both on mRNA and protein levels, and RAD54B can servers as an independent prognostic parameter of 5-year overall survival and 5-year disease-free survival for patients with HCC. Moreover, up-regulation of RAD54B dramatically increases the capacity for in vitro cell viability and motility, and in vivo intrahepatic metastasis of HCC cells. Mechanistically, RAD54B promotes the HCC progression through modulating the wnt/β-catenin signaling. Notably, blocking the wnt/β-catenin signaling axis can counteract the activating effects of RAD54B on motility of HCC cells. Besides, further analysis illustrates that DNA amplification is one of the mechanisms leading to mRNA overexpression of RAD54B in HCC. Our findings indicate that RAD54B might be a promising potential prognostic marker and a candidate therapeutic target to therapy HCC.
Introduction
Liver cancer was reported to be the sixth most frequently diagnosed cancer and the fourth leading cause of cancer death, with approximate 841,000 new cases and 782,000 deaths annually worldwide, of which, hepatocellular carcinoma (HCC) accounts for 75%−85% [1]. The mortality of HCC is high. Nevertheless, the concrete molecular mechanisms of HCC progression remain obscure in HCC.
Cancer progression is composed of a series of steps. Firstly, the cancer cells obtain the ability to sustain chronic proliferation, and migrates from the original site of growth, and invades into the neighbouring tissues through basement membranes, further permeates lymphatic systems or blood, then, colonize in distant organs, and metastatic tumors ultimately establish [2], [3]. During these processes, it is the indispensable step that capability for cell viability and motility elevates, and many alterations contributes to this process, including the activating of Wnt/β-catenin [4]. As to the canonical Wnt/β-catenin signaling, Wnt ligands interact with the receptor frizzled and co-receptor lipoprotein receptor-related protein-5 or −6 (LRP5/6). Their interaction subsequently leads to the cytoplasmic accumulation of β-catenin, which further translocate into nuclear. In nuclear, β-catenin can bind to T-cell factor (TCF) to activate transcription of target genes [5], [6], [7]. Multiple extracellular and intercellular factors can modulate the Wnt/β-catenin pathway. However, these factors and their concrete molecular mechanisms are still dismal.
Human RAD54B was firstly identified as a homolog of Saccharomyces cerevisiae Rad54 (ScRad54), and is located in 8p22.1, and harbors 15 exons and about 103 kb DNA [8]. RAD54B was transcribed into a 3.074 kb mRNA and a ~103 kDa protein with 910 amino acid (aa) [9]. Further studies clarified that RAD54B belongs to SWI2/SNF2 helicase superfamily and is involves in modulating the DNA damage checkpoint response [10] and homologous recombination repair (HRR) pathway [11]. Accordingly, sequencing assay uncovered that multiple human cancers are implicated in alterations of RAD54B such as gene amplification, homozygous deletion and non-synonymous single nucleotide polymorphism [12], [13], [14], [15], [16], [17]. Of note, wang et al. demonstrated that downregulation of RAD54 can inhibit proliferation, and enhance of hepatoma cell apoptosis [18]. However, their study is limited, and there are many unknowns. For example, this study just explores the in vitro function of RAD54B on cell proliferation and apoptosis, is not involved in molecular mechanisms, and the patient samples are small. Since the mortality of HCC is metastasis, we further investigate the function and mechanisms of RAD54B on metastasis in HCC, and we collected more sample of patient with HCC.
Herein, RAD54B are considerably upregulated in HCC, which leads to poor prognosis for patients with HCC. Moreover, up-regulation of RAD54B dramatically increases in vitro proliferation and motility, and in vivo intrahepatic metastasis of HCC cells. Further investigation uncovers that RAD54B promotes the progression of HCC through mediating wnt/β-catenin pathway. Notably, blocking wnt/β-catenin axis abolishes the effects of RAD54B on HCC progression. Besides, we found that genomic DNA amplification is one of the mechanisms that lead to mRNA overexpression of RAD54B. Our findings indicate that RAD54B might be a promising potential prognostic marker and a candidate therapeutic target for therapy HCC.
Materials and methods
Cell line and cell culture
HCC cell line BLC-2, Hep3B and Huh7 were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). MHCC97L, MHCC97H and SUHC-1 were purchased from the Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China), and HCCLM3 were purchased from China Center for Type Culture Collection (Wuhan, China). All the cell lines were cultured using DMEM adding 10% fetal bovine (FBS; HyClone, UT, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich, St Louis, MO, USA).
Patient specimens
Total HCC paraffin-embedded specimens and fresh HCC tissues were collected from Guangdong Second Provincial General Hospital. Before collecting the specimens for research, we acquired informed consents of patients with HCC and approval of the Institutional Ethics Committee.
Plasmids and retroviral infection
In order to construct the RAD54B-overexpressig plasmid, the full-length fragments of human RAD54B was amplified using PCR. Subsequently, RAD54B cDNA were cloned into the pMSCV plasmid. To downregulate RAD54B, two human RAD54B-targeting shRNA fragments were inserted into pSuper-retro-puro plasmid, respectively. The shRNA sequences are as follows: Scramble: TTCTCCGAACGTGTTCACGT; shRNA#1: GCTTTATCGAAAGCTGTTAAA; shRNA#2: GCAGATTGTTGATGGCTTTAA. Transfections of all plasmids were completed using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to protocol. The cells were continuously maintained in 0.5 μg/ml puromycin for 10 days, and the stable cells were obtained.
Quantitative PCR (qPCR)
Total RNA was extracted using Trizol reagent (Invitrogen). The RNA was reversed transcribed to synthesize cDNA using the First Strand cDNA Kit (Invitrogen). qPCR was conducted on the 7500 Fast Real time PCR system (Applied Biosystems, Rockville, MD, USA) using SYBR Green PCR Kit (Invitrogen). α-Tubulin was served as reference. All of the primers were summarized in Supplemental Table 5.
Protein extraction and western blotting assay
The nuclear and cytoplasm protein were isolated using Nuclear and Cytoplasmic Protein Extraction Kit (Thermo Scientific, Waltham, Massachusetts, USA) following the protocol provided. Equal proteins were isolated on SDS-PAGE gel, subsequently transferred to the PVDF membranes (Millipore, Billerica, MA, USA). Then, the membranes were probed using primary antibody overnight at 4 °C, and incubated using HRP-conjugated secondary antibody for 1 h at room temperature. Finally, the signal was examined using Super Signal Chemi-luminescent Substrate (Pierce, Rockford, IL, USA). The information of antibodies is as follows: RAD54B (1:100; HPA007087, Sigma-Aldrich); α-Tubulin (1:5000; ab7291, abcam); β-catenin (1:10,000; ab16051, abcam); Ki67 (1:5000; ab92742, abcam); cleaved caspase-3 (1:500; ab2302, abcam).
Immunohistochemistry (IHC)
Paraffin-embedded slides were deparaffinized by xylene, and then, rehydrated using the following concentration of ethanol: 100, 95 and 75%. The antigen retrieval was implemented by maintaining the slides in 10 mM Tris-EDTA (PH 9.0) for 10 min in a pressure boiler. 3% hydrogen peroxide was used to eliminate the activity of endogenous peroxidase, and blocked using Endogenous Biotin Blocking Kit (Ventana) for 30 min at room temperature. Next, the slides were probed by primary antibody at 4 °C for 12 h, and incubated using biotinylated secondary antibody and subsequently treated using streptavidin-HRP conjugate for 8 min at 37 °C. Finally, the proteins were visualized with a copper-enhanced DAB reaction. Two independent pathologists were blinded to assess the staining results. Multiply proportion of positively-stained cells and the intensity of staining get the staining results. Cell proportions were evaluated according to the following rule: 0, no positive cells; 1, < 10% positive cells; 2, 10%−35% positive cells; 3, 35%−75% positive cells; 4, > 75% positive cells. Staining intensity was assessed as follows: 0, no staining; 1, weak staining (light yellow); 2, moderate staining (yellow brown); 3, strong staining (brown). Through the above method, we evaluated protein expression with the possible scores as follows: 0, 1, 3, 4, 6, 8, 9 and 12. ≥ 6 was defined as high level, and < 6 was defined as low expression.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cells were seeded into 96-well plates and cultured in a cell incubator. Then, the cells were harvested at the indicated time points, and stained using 50 μL of MTT reagent for 4 h. Subsequently, 100 μL of dimethyl sulfoxide (DMSO) was used to dissolve the MTT formazan. The absorbance of the different cells at 540 nm was examined using a Falcon microplate reader (BD Labware, San Jose, CA, USA).
Colony formation assay
Cells were seeded into 12-well plates and maintained in incubator at 37 °C. Ten days later, the cells were fixed using 4% paraformaldehyde, and then stained with 0.1% crystal violet. The stained colonies were counted under a light microscope.
Flow cytometry
The cells were seeded into 10-cm plates and cultured for 72 h. Then, the cells were harvested and washed twice using phosphate-buffered saline (PBS), and re-suspended using binding buffer. Subsequently, fluorescein isothiocyanate (FITC) Annexin V (to detect apoptotic cells)/RNase A (to detect the cell cycle) and propidium iodide were supplemented into the cells. The mixture was incubated at room temperature in the dark for 30 min. Finally, the proportions of different cell features were detected on a flow cytometer (Beckman-Coulter, Indianapolis, IN, USA).
Cell wound healing assay
The cells were seeded into 6-well plates, and maintained in incubator until the cell confluence reached approximately 90%. A sterilized 10 μL pipette tip was used to generate wound across the cell monolayer. The cell debris was removed by means of two washing of PBS. 24 hour later, the wound healing was detected using Olympus camera.
Transwell invasion assay
To perform cell invasion assay, firstly, the transwell filter (BD Biosciences, Franklin Lake, New Jersey, USA) was coated with Matrigel. Secondly, 1 × 104 cells were added into the transwell plate that was filled with DMEM without FBS. Thirdly, the transwell plate was put into the 24-well plate that was added DMEM containing 0.5% FBS. Then, the plate was maintianed in incubator for 24 h. And the cells that didn't traverse the filter were softly removed by cotton swabs. Finally, the filter was fixed by 4% paraformaldehyde and stained with 1% crystal violet.
In vivo metastasis assay
All in vivo experiments were approved by the Medical Experimental Animal Care Commission of Guangdong Second Provincial General Hospital. The female BALB/c nude mice were randomly grouped into four groups (n = 6/group). 1 × 106 cells stably transfected with RAD54B or sh#1 were suspended in serum-free DMEM/Matrigel (1:1), subsequently, through a 1 cm transverse incision in the upper abdomen under anesthesia, the complex were injected into the left hepatic lobe of the mice using 28-gauge needle. The liver was returned to the peritoneal cavity and the abdominal wall was closed. After one month, all mice were sacrificed. And their livers were dissected, and the numbers of intrahepatic metastasis node were counted.
Luciferase reporter assay
1 × 103 cells were added into the 48-well plates, and maintained in humid incubator for 24 h. 100 ng luciferase reporter plasmids and 10 ng Renilla plasmids were transfected into cells using Lipofectamine 3000 (Invitrogen). The luciferase and Renilla signal was measured using dual luciferase reporter assay kit (Promega, Madison, Wisconsin, USA) following the protocol.
Genomic DNA quantitative PCR
Genomic DNA was collected with PureLink Genomic DNA Mini Kit (Invitrogen). qPCR of genomic DNA was finished using the KAPA SYBR FAST PCR polymerase with 20 ng template and 200 nM primers following the manufacturer's protocol (Kapa Biosystems, Woburn, MA, USA).
Public data processing and visualization
The levels of RAD54B (Fig. 1A and B) in HCC were downloaded from The Cancer Genome Atlas (TCGA) database (http://cancergenome.nih.gov/). Gene Set Enrichment Analysis (GSEA) was performed using GSEA 2.0.9 (http://www.broadinstitute.org/gsea/). The genetic alteration of RAD54B (Fig. 7A and B) in HCC were downloaded from cBioPortal (http://www.cbioportal.org/).
Fig. 1.

RAD54B is overexpressed in HCC. (A) RAD54B mRNA expression is significantly elevated in HCC tissues compared with normal tissues using the data from TCGA. (B) RAD54B mRNA levels were drastically increased in paired HCC tissues relative to matched adjacent normal tissues (ANT) using the data from TCGA. (C) High level of RAD54B is closely related with poor prognosis of HCC using data from TCGA. (D) The mRNA (left panel) and protein levels (right panel) of RAD54B is markedly upregulated in HCC tissues (T) compared with matched adjacent normal tissues (ANT) using paired fresh HCC tissues. (E) The mRNA (left panel) and protein levels (right panel) of RAD54B is markedly upregulated in HCC cell lines relative to 3 normal liver tissues. N: normal liver tissues; ANT: adjacent normal tissues.
Fig. 7.

Amplification of RAD54B gene in HCC tissues. (A) RAD54B was amplified in HCC tissues using data from public dataset cBioportal. (B) RAD54B mRNA expression was correlated with the copy-number alterations of RAD54B using data from public dataset cBioportal. (C) Genomic DNA expression in HCC tissues (left panel) and correlation analysis between genomic DNA levels and mRNA expression in HCC tissues (right panel). (D) The correlation analysis of β-catenin protein level in nuclear and DNA (left panel) or mRNA (right panel) of RAD54B.
Statistical analysis
All statistical analysis was finished using the SPSS 22.0 software package. The statistical significance of two groups was performed by 2-tailed paired student's t-test. The statistical analysis between sh-1/2 and scramble were performed using analysis of variance (ANOVA) followed by Dunnett's test. The correlation of RAD54B level and clinicopathologic features were evaluated by χ2 test. Survival curves were plotted by Kaplan–Meier method and compared using log-rank test. P less than 0.05 is considered as statistically significance, and all experiments were performed more than three times.
Results
RAD54B is overexpressed in HCC
To explore the expression and roles of RAD54B in HCC, we firstly analyzed its expression using availably public dataset the Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/). As illustrated in Fig. 1A, RAD54B mRNA levels is dramatically increased in 351 HCC tissues relative to 50 normal liver tissues. Besides, we checked its level using 50 paired HCC tissues using data also from TCGA. In accordance with the above results, RAD54B mRNA expression dramatically is upregulated in HCC tissues relative to corresponding adjacent normal tissues (ANT) (Fig. 1B). Moreover, Kaplan–Meier survival curves and log-rank tests were accomplished using the data from TCGA. Firstly, the patients with HCC were grouped into high level group and low level group using medium mRNA level as cutoff. The results revealed that HCC patients with high RAD54B levels have shorter 5-year overall survival and relapse-free survival relative to that with low RAD54B levels (Fig. 1C). Next, we checked RAD54B levels using paired fresh HCC specimens and HCC cell lines using qPCR and western blotting assay, respectively, which illustrates that RAD54B significantly upregulates in fresh HCC specimens compared with ANT both on mRNA and protein levels (Fig. 1D). Similarly, the expressions of RAD54B are remarkably increased in HCC cell lines relative to 3 normal liver specimens (Nor-1, Nor-2 and Nor-3; Fig. 1E).
Altogether, our analysis shows that RAD54B level is dramatically upregulated in HCC tissues and cell lines.
High level of RAD54B leads to poor prognosis for patients with HCC
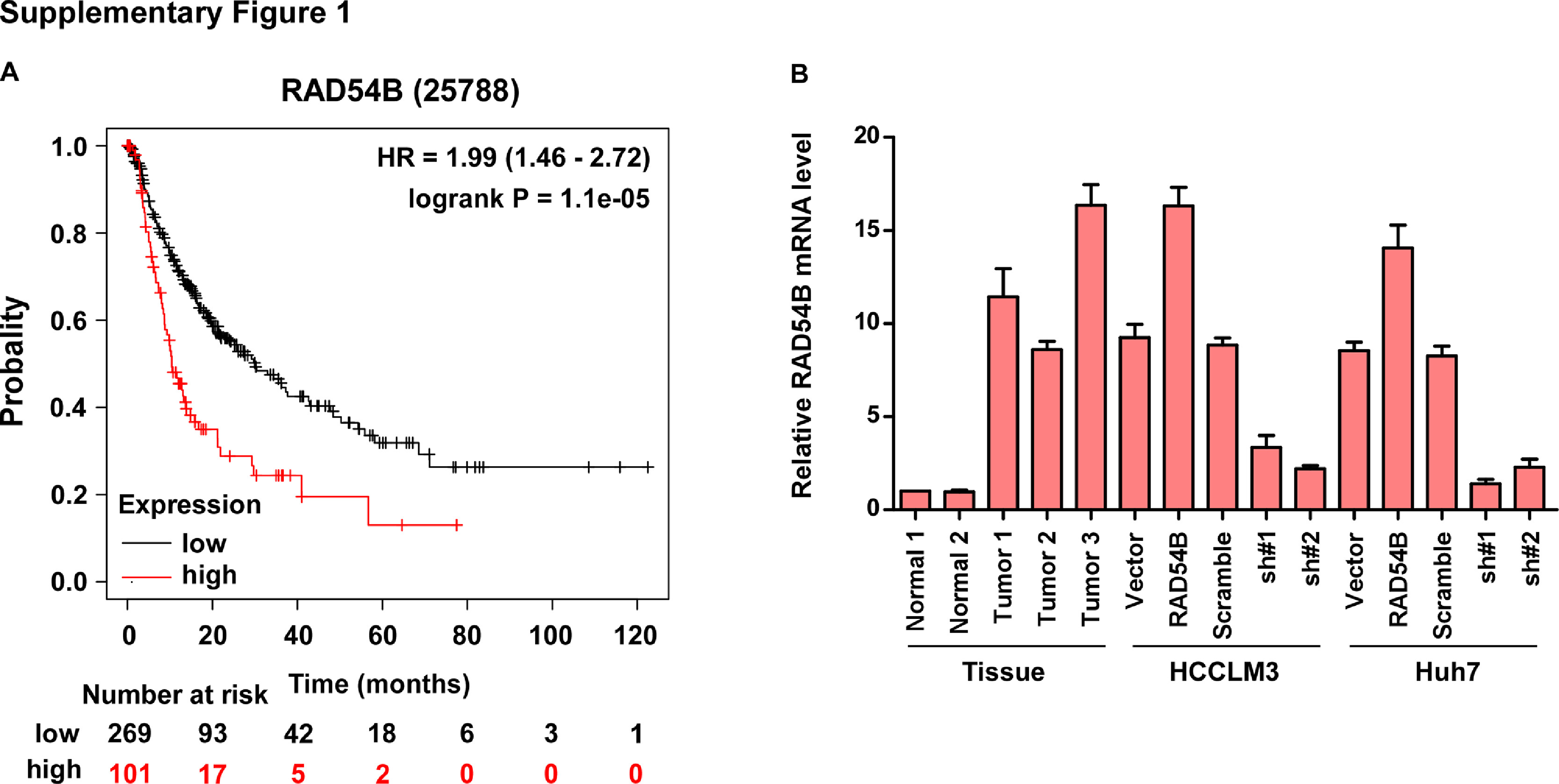
Subsequently, the correlation of RAD54B level and clinical characteristics was analyzed. Firstly, we examined the RAD54B expression in 257 paraffin-embedded, archived HCC tissues by IHC assay (Supplemental file: Table S1). IHC assay showed that RAD54B levels are upregulated in HCC specimens relative to normal liver specimens (Fig. 2A). Furthermore, the 5-year overall survival and disease-free survival time of HCC patients with high RAD54B levels are both significantly shorter relative to that with low RAD54B levels (Fig. 2B). The correlation analysis of RAD54B expression and the clinicopathologic characters reveals that RAD54B expression is closely related with clinical stage (p = 0.011), T stage (p = 0.027), M stage (p = 0.011), alpha fetal protein (AFP; p = 0.015) and histologic grade (p = 0.024) (Supplemental file: Table S2). Additionally, multivariate analysis indicates that RAD54B might be an independent prognostic factor for patients with HCC as to 5-year overall survival (Supplemental file Table S3, Fig. 2C) and disease-free survival time (Supplemental file Table S4, Fig. 2C). In addition, we used the available public dataset Kaplan–Meier Plotter (http://kmplot.com/analysis/index.php?p=background). The patients with HCC were divided into high expression and low expression group using medium mRNA expression as cutoff. The results showed that high mRNA level of RAD54B results in short disease progression free survival compared with low level for patients with HCC (Supplemental Figure 1A).
Fig. 2.

High expression of RAD54B is related with HCC progression and poor prognosis of patients with HCC through analyzing paraffin-embedded archived HCC tissues. (A) Representative images of RAD54B by IHC assay in paraffin-embedded archived HCC tissues. (B) The 5-year overall survival (left panel) and disease-free survival (right panel) of patients with HCC by Kaplan–Meier analysis. (C) Multivariate Cox regression analysis of various prognostic parameters for 5-year overall survival and disease-free survival in patients with HCC.
Altogether, aberrant overexpression of RAD54B might represent an independent prognostic factor for HCC and contributes to cancer progression.
Overexpression of RAD54B promotes HCC progression in vitro and in vivo
Since that RAD54B is correlated with overall and disease-free survival of patients with HCC, we further hypothesized that RAD54B is associated with cell viability and motility. Firstly, we established the stable cell lines-upregulated or -silenced RAD54B using two HCC cells HCCLM3 and Huh7, in which the levels of RAD54B are moderate (Fig. 3A). Then, we detected the roles of RAD54B on cell viability and apoptosis. As shown in Fig. 3B and C, MTT, colony formation and flow cytometry assay showed that overexpression of RAD54B significantly increases cell viability, while inhibits cell apoptosis. But, downregulation of RAD54B induces the opposite results.
Fig. 3.

Overexpression of RAD54B promotes cell viability, but inhibits cell apoptosis. (A) Western blotting assay of RAD54B levels in indicated stable HCC cell lines. (B) MTT assay showed that overexpression of RAD54B promotes, while downregulation inhibits cell viability. (C) Colony formation assay showed that overexpression of RAD54B promotes, while downregulation inhibits cell viability. (D) Flow cytometry showed that overexpression of RAD54B inhibits, while downregulation promotes cell apoptosis. * P < 0.05.
Subsequently, we detected the roles of RAD54B on cell motility. Firstly, we detected the enzyme activity of MMP2 and MMP9 by ELISA. The results show their activity significantly increases in RAD54B-overexpressed cells, while decreases in RAD54B-silenced cells (Fig. 4A). Subsequently, the wound healing assay shows that overexpression of RAD54B can dramatically promote, whereas, down-regulation of RAD54B remarkably inhibits migration ability of HCC cells (Fig. 4B). Accordingly, transwell assay illustrated that the capability for invasion increases in RAD54B-overexpressed cells, while inhibits in RAD54B-silenced cells (Fig. 4C). The above mentioned results suggests that aberrant overexpression of RAD54B contributes to cell viability and motility.
Fig. 4.

Aberrant overexpression of RAF54B promotes in vitro migration and invasion of HCC cells. (A) The enzyme activity of MMP2 and MMP9 in indicated stable cells by ELISA. (D) Wound healing assay showed that overexpression of RAD54B elevated the capability for migration of HCC cells. (E) Transwell assay illustrated that overexpression of RAD54B promote invasion of HCC cells. * p < 0.05.
Since overexpression of RAD54B promotes the in vitro viability and motility of HCC cells, we further investigated the roles of RAD54B on HCC progression in vivo. We established an orthotopic animal transplant model using HCCLM3 and Huh7 cells. The results illustrates that overexpression of RAD54B can induce high, while downregulation inhibits intrahepatic metastasis node compared with control (Fig. 5A and B). In addition, western blotting assay showed that Ki67 levels were increased in liver tissues injected with cells-upregulated RAD54B compared with control. But, its levels were reduced in liver tissues injected with cells-downregulated compared with control (Fig. 5C). We also detected cleaved caspase-3 levels in liver tissues, the western blotting showed the opposite trend relative with Ki67 (Fig. 5C). Finally, ELISA assay demonstrates that the activity of MMP2 and MMP9 both markedly elevates in liver tissues injected with RAD54B-overexpressed cells compared with that injected with vector cells (Fig. 5D).
Fig. 5.

Upregulation of RAD54 promoted intrahepatic metastasis of HCC cells. (A) Representative images of intrahepatic metastasis node formed by indicated cells. (B) Numbers of intrahepatic metastasis node in orthotopic xenograft tumor model. (C) The Ki67 and cleaved caspase 3 levels in liver tissues. (D) The enzyme activity of MMP2 and MMP9 in mice injected with HCC cells. * p < 0.05.
Altogether, the function experiments demonstrated that aberrant overexpression of RAD54B can promote HCC progression in vitro and in vivo.
Overexpression of RAD54B can activate the Wnt/β-catenin signaling
Further GSEA shows that RAD54B may be involved in Wnt/β-catenin pathway (Fig. 5A). Moreover, we checked the hypothesis using the luciferase reporter assay, TOPFLASH (with three repeats of the TCF-binding site) or FOPFLASH (with three repeats of a mutated TCF-binding site) plasmids were transfected into HCC cells. The results illustrate that overexpression of RAD54B increases, while upregulation decreases the transcription activity of Wnt/β-catenin pathway (Fig. 6B). Moreover, we evaluated the role of RAD54B on the subcellular localization of β-catenin. The western blotting assay confirms that nuclear β-catenin increases in RAD54B-upregulating HCC cells, while decreases in RAD54B-silenced HCC cells (Fig. 6C). Consistently, RAD54B overexpression upregulates, while downregulation inhibits the mRNA levels of downstream target genes of Wnt/β-catenin pathway such as c-myc, CCND1, MMP7, CD44, VEGF, c-jun (Fig. 6D). Finally, we used a Wnt/β-catenin signaling antagonist, PNU-74654, which can prevent β-catenin binding with TCF to break this pathway, in RAD54B-upregulated cells. The transwell assay shows that PNU-74654 can markedly inhibit the activating effect of RAD54B on cell viability, migration and invasion, while promote effect on cell apoptosis relative to the control (Fig. 6E). Besides, we inhibited β-catenin in RAD54B-overexpressing cells using shRNA. Sh-β-catenin also inhibits the activating effect of RAD54B on cell biability, migration, invasion and intrahepatic metastasis ability, while promote effect of cell apoptosis compared with scramble (Fig. 6F), indicating that Wnt/β-catenin axis is necessary for the activating function of RAD54B on HCC progression.
Fig. 6.

Overexpression of RAD54B activates the Wnt/β-catenin signaling pathway. (A) GSEA showed that RAD54B may be implicated in Wnt/β-catenin signaling pathway. (B) Luciferase reporter assay showed that overexpression of RAD54B activated the transcription activity of Wnt/β-catenin signaling. (C) Western blotting assay demonstrated that overexpression of RAD54B can promote the nuclear accumulation of β-catenin. (D) The mRNA expression of Wnt/β-catenin downstream genes. (E) Wnt/β-catenin signaling inhibitorcounteracted the promoting effect of RAD54B on HCC progression. (F) Sh-β-catenin inhibit the promoting effect of RAD54B on HCC progression. * p < 0.05.
Altogether, aberrant upregulation of RAD54B can notably activate the Wnt/β-catenin pathway, and promote HCC progression.
Amplification of the RAD54B gene in HCC
Lastly, we explored the underlying mechanisms of RAD54B overexpression in HCC. Interesting, genetic amplification of RAD54B reaches up to 20% in HCC tissues using public dataset cBioPortal (http://www.cbioportal.org/) data (Fig. 7A). In addition, the mRNA expression of RAD54B is significantly related with the copy-number of RAD54B (Fig. 7B). The above results were confirmed in fresh HCC tissues. As shown in Fig. 7C, there are positively correlated between copy-number of RAD54B and mRNA levels of RAD54 (R = 0.6788; P = 0.0309). In addition, DNA and mRNA levels of RAD54B are both significantly correlated with β-catenin protein in nuclear, respectively. These results suggest that gene amplification is one of the mechanisms leading to RAD54B overexpression in HCC.
Altogether, gene amplification of RAD54B is frequently observed in HCC, and overexpression of RAD54B is partly attributed to gene amplification in HCC.
Discussion
Under normal conditions, RAD54B is implicated in regulation of cell cycle after DNA damage and homologous recombination repair to ensure the precise repair of the deleterious DNA lesions or double-stranded breaks, which is indispensable for the genome stability [19], [20], [21], [22]. Allow for the important role of RAD54 in physiological process, multiple cancers have been discovered alterations expression of RAD54B, including amplification or deletion, missense or monsense [23], which suggested that RAD54B can be a oncogene or suppressor gene according to the different tissues. For example, Hiramoto et al. found homozygous mutations of RAD54B at highly conserved positions in human primary lymphoma and colon cancer [8]. It is reporter that RAD54B is overexpressed in lung adenocarcinoma and negatively correlated with prognosis of patients [24]. Wang et al. found that the expression of RAD54B is overexpression in hepatoma cells, and inhibition of RAD54B suppressed proliferation and promotes apoptosis of hepatoma cells [18]. In our study, our experiments uncovers RAD54 is dramatically upregulated in HCC. Since metastasis is the main reason leading HCC patient death, we evaluated the role of RAD54B on HCC metastasis. Both loss-of- and gain-of-function experiments are carried out. Our findings shows that upregulation of RAD54B can significantly promote, while its knockdown inhibits HCC progression in vitro and in vivo.
Yasuhara et al. showed that negative correlation between RAD54B and p53 was observed, suggesting that aberrant upregulation of RAD54 can result in p53 degradation, which further results in tumorigenesis. Whereas, inhibition of RAD54B can elevate the level of p53, which increases anti-apoptosis after treating with genotoxic drugs [10]. Therefore, the underlying mechanisms of RAD54B are complicated. Herein, we found that overexpression of RAD54B can dramatically activate the transcription activity of Wnt/β-catenin. But, the concrete mechanisms of RAD54B activating Wnt/β-catenin pathway are unclear. It has been documented that p53 and Wnt modules are cross-talk, and p53 network can inhibit the Wnt cascade [25], [26], [27]. Besides, other study showed that p53 can modulate dynamics of Axin2 and GSK3, two essential component of Wnt/β-catenin pathway [28]. However, the mechanisms how p53 interacts with Wnt are still an open question. Therefore, we speculate that overexpression of RAD54B enhance the degradation of p53, and subsequently activate the Wnt signaling, which need our further study. Importantly, the prominent role of the EMT is strongly induced by the hyper-activation of Wnt pathway [29]. Our results showed that RAD54B contributes to activation of Wnt pathway, which suggests that RAD54B might be involved in EMT. We will validate in our future study.
Cancer is a consequence of accumulation of alterations on genetics (gene mutations, gene amplification, and so on) and epigenetics (aberrant DNA methylation, chromatin modifications, and so on) [30], [31], [32]. In the present research, we revealed that gene amplification of RAD54B plays a considerable role in the mRNA upregulation of RAD54B in HCC. Nevertheless, whether other epigenetic alterations of RAD54B exist in HCC need to further elucidate.
Conclusions
In conclusion, in the present study, our experiments demonstrate that RAD54B is dramatically upregulated in HCC. Subsequent assays show that upregulation of RAD54B can promote HCC progression. Further analysis clarified that upregulation of RAD54B can significantly activate the Wnt/β-catenin pathway. Finally, our analysis demonstrated that overexpression of RAD54B is partly attributed to gene amplification. In summary, RAD54B might be a promising prognostic marker and a candidate target to therapy metastatic HCC.
Declaration of Competing Interest
The authors declare no competing interests of this study.
Acknowledgments
Funding
This work is supported by Guangdong Natural Science Foundation (Nos. 2018A0303130184, 2019A1515011678), Science and Technology Programme of Guangzhou Municipal Government (No. 201707010304), The science foundation of Guangdong Second Provincial General hospital (YY2016-007, 3D-A2020005).
Ethics approval and consent to participate
This study was approved by Institutional Ethics Committee of Guangdong Second Provincial General Hospital and informed consent was obtained from all patients.
Consent for publication
All authors read the final version of manuscript and agreed on the publication.
Availability of data and materials
All data generated or analyzed during the study are included in the published article.
Authors’ contributions
XCZ and PG conceived and supervised the project. SWF, JHL, HLL and JFW performed the experiments. YJZ collected the data, XFL and GKL performed the analysis. SWF and JHL contributed to manuscript writing and editing. All authors read and approved the final manuscript.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.tranon.2021.101124.
Contributor Information
Senwen Feng, Email: fsw9295@163.com.
Junhao Liu, Email: 929382800@qq.com.
Li Hailiang, Email: lihailiang5612@163.com.
Jianfan Wen, Email: wenjianfan927@163.com.
Yujun Zhao, Email: huan1352115@126.com.
Xiaofeng Li, Email: lxf812495@foxmail.com.
Guankun Lu, Email: 458012938@qq.com.
Peng Gao, Email: gp13609741873@163.com.
Xiancheng Zeng, Email: 13710707889@163.com.
Appendix. Supplementary materials
{kind=link}
Reference
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 3.Qi J., Yu Y., Akilli Ozturk O., Holland J.D., Besser D., Fritzmann J. New Wnt/beta-catenin target genes promote experimental metastasis and migration of colorectal cancer cells through different signals. Gut. 2016;65:1690–1701. doi: 10.1136/gutjnl-2014-307900. [DOI] [PubMed] [Google Scholar]
- 4.Zhan T., Rindtorff N., Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao J., Wang J., Liu B., Pan W., Farr G.H., 3rd, Flynn C. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol. Cell. 2001;7:801–809. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 6.Gordon M.D., Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 7.Cui Y., Ma W., Lei F., Li Q., Su Y., Lin X. Prostate tumour overexpressed-1 promotes tumourigenicity in human breast cancer via activation of Wnt/beta-catenin signalling. J. Pathol. 2016;239:297–308. doi: 10.1002/path.4725. [DOI] [PubMed] [Google Scholar]
- 8.Hiramoto T., Nakanishi T., Sumiyoshi T., Fukuda T., Matsuura S., Tauchi H. Mutations of a novel human RAD54 homologue, RAD54B, in primary cancer. Oncogene. 1999;18:3422–3426. doi: 10.1038/sj.onc.1202691. [DOI] [PubMed] [Google Scholar]
- 9.Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., Lu S., Chitsaz F., Geer L.Y. CDD: NCBI's conserved domain database. Nucl. Acids. Res. 2015;43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuhara T., Suzuki T., Katsura M., Miyagawa K. Rad54B serves as a scaffold in the DNA damage response that limits checkpoint strength. Nat. Commun. 2014;5:5426. doi: 10.1038/ncomms6426. [DOI] [PubMed] [Google Scholar]
- 11.Miyagawa K., Tsuruga T., Kinomura A., Usui K., Katsura M., Tashiro S. A role for RAD54B in homologous recombination in human cells. EMBO J. 2002;21:175–180. doi: 10.1093/emboj/21.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.N. Cancer Genome Atlas Research Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dulak A.M., Stojanov P., Peng S., Lawrence M.S., Fox C., Stewart C. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013;45:478–486. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eirew P., Steif A., Khattra J., Ha G., Yap D., Farahani H. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAndrew E.N., McManus K.J. The enigmatic oncogene and tumor suppressor-like properties of RAD54B: insights into genome instability and cancer. Genes Chromosomes Cancer. 2017;56:513–523. doi: 10.1002/gcc.22458. [DOI] [PubMed] [Google Scholar]
- 18.Wang R., Li Y., Chen Y., Wang L., Wu Q., Guo Y. Inhibition of RAD54B suppresses proliferation and promotes apoptosis in hepatoma cells. Oncol. Rep. 2018;40:1233–1242. doi: 10.3892/or.2018.6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukas J., Lukas C., Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 21.Roos W.P., Thomas A.D., Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 2016;16:20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- 22.Wolters S., Schumacher B. Genome maintenance and transcription integrity in aging and disease. Front. Genet. 2013;4:19. doi: 10.3389/fgene.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang J.C., Sung W.W., Tu H.P., Hsieh K.C., Yeh C.M., Chen C.J. The Overexpression of FEN1 and RAD54B May Act as Independent Prognostic Factors of Lung Adenocarcinoma. PLoS One. 2015;10 doi: 10.1371/journal.pone.0139435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng X., Yang L., Chang H., Dai G., Wang F., Duan X. Wnt/beta-catenin signaling regulates the proliferation and differentiation of mesenchymal progenitor cells through the p53 pathway. PLoS One. 2014;9:e97283. doi: 10.1371/journal.pone.0097283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim N.H., Kim H.S., Kim N.G., Lee I., Choi H.S., Li X.Y. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011;4:ra71. doi: 10.1126/scisignal.2001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cha Y.H., Kim N.H., Park C., Lee I., Kim H.S., Yook J.I. MiRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling. Cell Cycle. 2012;11:1273–1281. doi: 10.4161/cc.19618. [DOI] [PubMed] [Google Scholar]
- 28.Kim N.H., Cha Y.H., Kang S.E., Lee Y., Lee I., Cha S.Y. p53 regulates nuclear GSK-3 levels through miR-34-mediated Axin2 suppression in colorectal cancer cells. Cell Cycle. 2013;12:1578–1587. doi: 10.4161/cc.24739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dituri F., Mancarella S., Cigliano A., Chieti A., Giannelli G. TGF-beta as multifaceted orchestrator in HCC progression: signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin. Liver Dis. 2019;39:53–69. doi: 10.1055/s-0038-1676121. [DOI] [PubMed] [Google Scholar]
- 30.Chin L., Gray J.W. Translating insights from the cancer genome into clinical practice. Nature. 2008;452:553–563. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grady W.M., Carethers J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feinberg A.P., Tycko B. The history of cancer epigenetics. Nat. Rev. Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during the study are included in the published article.