

Abstract
Incorporation of microbiome data has recently become important for prevention, diagnosis, and treatment of colorectal cancer, and several species of bacteria were shown to be associated with carcinogenesis. However, the role of commensal fungi in colon cancer remains poorly understood. Here, we report that mice lacking the c‐type lectin Dectin‐3 (Dectin‐3 −/−) show increased tumorigenesis and Candida albicans burden upon chemical induction. Elevated C. albicans load triggered glycolysis in macrophages and interleukin‐7 (IL‐7) secretion. IL‐7 induced IL‐22 production in RORγt+ (group 3) innate lymphoid cells (ILC3s) via aryl hydrocarbon receptor and STAT3. Consistently, IL‐22 frequency in tumor tissues of colon cancer patients positively correlated with fungal burden, indicating the relevance of this regulatory axis in human disease. These results establish a C. albicans‐driven crosstalk between macrophages and innate lymphoid cells in the intestine and expand our understanding on how commensal mycobiota regulate host immunity and promote tumorigenesis.
Keywords: Candida albicans, colorectal cancer, dectin‐3, IL‐22, ILC3
Subject Categories: Cancer, Immunology, Metabolism
Lack of the Candida albicans recognition receptor Dectin‐3 in mice triggers aberrant immune cell metabolism and tumorigenic cytokine signaling.

Introduction
Colorectal cancer (CRC) is presently considered as world’s fourth most commonly diagnosed cancer and the second leading cause for mortality, resulting in death of more than 500,000 people every year (Bray et al, 2018; Siegel et al, 2020). Inflammatory bowel disease (IBD) has been reported to be associated with the development of colitis‐associated colon cancer (CAC) (Rutter et al, 2004; Gupta et al, 2007). Thus, exploring the relationship between intestinal inflammation and CRC progress may provide novel approaches for treatment of CAC.
More than 100 trillion microbes are colonized in mammalian gastrointestinal tract, mostly composed of gut commensal bacteria, fungi, and viruses. Gut microbiota participates in the modulation of both innate and adaptive immune responses and maintain host intestinal homeostasis (Round & Mazmanian, 2009; Belkaid & Hand, 2014). Microbial dysbiosis is associated with IBD and CRC (Willing et al, 2010; Lepage et al, 2011). By studying colitis and tumor formation in germ‐free (GF) animals and animals treated with antibiotics, scientists have proved the contribution of commensal bacteria to colitis and CAC, including tumor progression, chemotherapeutic drug resistance, and cancer immunotherapy (Arthur et al, 2012; Yu et al, 2017; Matson et al, 2018; Routy et al, 2018; Baruch et al, 2021; Pleguezuelos‐Manzano et al, 2020). Beside bacteria, mycobiota is also an important member in gut microbiota. Several kinds of fungi reside on the gastrointestinal tract of healthy individuals, among which Candida is the dominant (Iliev et al, 2012; Mukherjee et al, 2015). Fungi are conditionally pathogenic in certain environments, especially in an immune‐suppressive system. It is reported that Candida tropicalis, a kind of Candida, can translocate into lamina propria (LP) of intestine and aggravate colitis (Iliev et al, 2012). Our previous study also confirmed the function of C. tropicalis on the development of CAC (Wang et al, 2018). We found an increased C. tropicalis burden in the gut of caspase recruitment domain‐containing protein 9 (CARD9)‐deficient mice, resulting in increased myeloid‐derived suppressor cells (MDSCs) accumulation and increased inhibition of effector T cells. Interestingly, the cell wall components of C. guilliermondii, C. krusei, C. tropicalis, C. auris, and C. albicans are different, which may trigger different recognition by human innate immune cells (Navarro‐Arias et al, 2019). However, whether other fungi, besides C. tropicalis, can trigger colonic tumorigenesis remains unknown.
Mammalian C‐type lectin receptors which consist of Dectin‐1, Dectin‐2, and Dectin‐3 are pattern recognition receptors (PRR) on innate immune cell surface for sensing fungi infection and inducing activation of downstream signaling (Saijo et al, 2007; Saijo et al, 2010; Zhu et al, 2013). Dectin‐3 (also named as MCL/CLECSF8/Clec4d) is expressed by multiple myeloid cells like neutrophils, monocytes, and dendritic cells (Balch et al, 1998; Arce et al, 2004; Graham et al, 2012). Dectin‐3 was found to function as a PRR for recognizing multiple fungi including C. albicans, Paracoccidioides brasiliensis, and Cryptococcus through recognition of a‐mannans on their surface (Kerscher et al, 2016; Huang et al, 2018). Our previous study demonstrated that Dectin‐3 forms a heterodimeric complex with Dectin‐2 for sensing a‐mannans (Zhu et al, 2013). We also found the deficiency of Dectin‐3 impairs phagocytic and fungicidal abilities of C. tropicalis in macrophages and promotes colitis (Wang et al, 2016). Although functional interactions of Dectin‐3 with commensal fungi are crucial for colonic immune homeostasis, the impact of Dectin‐3 on CAC development is still unknown.
Innate lymphoid cells (ILCs) have been identified as a novel family of lymphoid effector cells (Serafini et al, 2015). ILCs can be classified into three major subgroups (ILC1, ILC2, and ILC3) based on different transcription factors (Spits et al, 2013). ILC3 contains two major subtypes, NCR+ ILC3 and LTi‐like ILC3, depending on the expression of natural cytotoxicity receptors (NCR, including NKp30, NKp44, and NKp46) on cell surface (von Burg et al, 2015). Generation and differentiation of ILC3 require the nuclear hormone receptor RORγt (Luci et al, 2009; Sanos et al, 2009; Takatori et al, 2009). The interactions among ILC3, T cells, and macrophages have a crucial impact on promoting intestinal homeostasis of gut bacteria and preventing inflammation‐related diseases (Hepworth et al, 2013; Mortha et al, 2014; Hepworth et al, 2015). Previous studies found that interleukin (IL)‐22 and IL‐23 production by ILCs resulted in tumor progression in colon cancer mouse models (Kirchberger et al, 2013; Chan et al, 2014). However, the relationship between ILC3 and commensal fungi remains unknown.
In this study, we evaluated the effect of Dectin‐3 on microbial dysbiosis and tumor progression. The direct tumor promotion role of C. albicans was confirmed by using fecal microbiota transplantation experiment, intragastrical fungal administration experiment, and anti‐fungal experiment. Furthermore, we confirmed that C. albicans triggered IL‐7 production of macrophage, led to IL‐22 production in ILC3, and eventually promoted CAC development.
Results
Dectin‐3 −/− mice display increased CAC than wild‐type (WT) mice
To explore the function of Dectin‐3 on CAC, we employed the CAC mouse model in mono‐housed WT and Dectin‐3 −/− mice. By using azoxymethane (AOM) and dextran sodium sulfate (DSS), colon tumors can be observed in all mice in distal colon and rectum. We found that tumor number, tumor size, and tumor burden in Dectin‐3 −/− mice were significantly higher than those in WT littermates (Fig 1A and B). During induction of tumor, the clinical colitis scores were also higher in Dectin‐3 −/− mice (Fig EV1A). Histologically, the lesions of Dectin‐3 −/− mice presented more adenocarcinomas with higher grade, which frequently invading into the submucosa and invading into the muscularis propria occasionally (Fig 1C and D). Immunohistochemical staining indicated the positive proportion of Ki‐67 and p‐STAT3 in tumors of Dectin‐3 −/− mice were markedly higher than that in WT mice (Fig 1E and F). However, the positive proportion of cleaved‐caspase 3 showed no difference between WT and Dectin‐3 −/− group (Fig EV1B). These data suggest that Dectin‐3 deficiency promotes the occurrence of CAC by inducing epithelial cell proliferation rather than inhibiting cell apoptosis.
Figure 1. Dectin‐3−/− mice have increased tumor load upon AOM‐DSS treatment than WT mice.

-
ARepresentative images of colon tumors were shown. Scale bars, 10 mm.
-
BTumor number, tumor size, and tumor load in each mouse were measured.
-
C, DHistological analysis of colon tumors was shown by hematoxylin and eosin (HE) staining. Tumors were microscopically analyzed and classified as low or high grade. Histological score was assessed by a pathologist. Scale bars, 25 µm.
-
E, FTumor tissues were stained for Ki67 and p‐STAT3. The percentage of Ki67‐positive and p‐STAT3‐positive cells was quantified. Scale bars, 25 µm.
-
GProportion of CD11b+F4/80+ and RORγt+/CD45+lin− cells was analyzed in colonic LP cells and mLNs among tumor‐bearing WT and Dectin‐3−/− mice by flow cytometry.
-
HRelative expression of Il‐6, Il‐22, Cxcl1, and Il‐17a in LP cells from tumor‐bearing WT and Dectin‐3−/− mice were detected using qPCR.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by unpaired Student’s t‐test. See also Fig EV1.
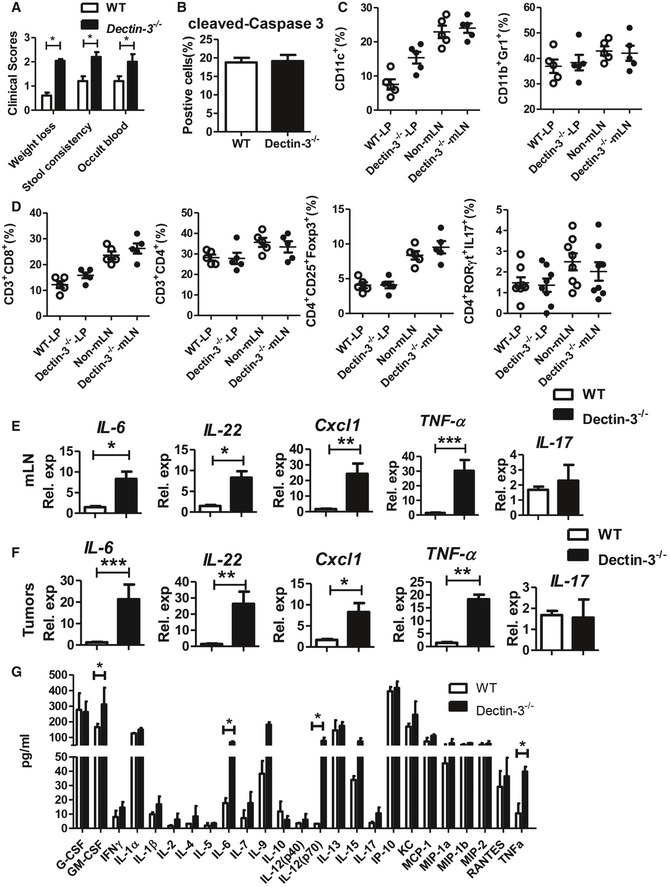
Figure EV1. Dectin‐3 −/− mice have increased tumor burden and impaired immune responses upon AOM‐DSS treatment than WT mice. Related to Fig 1 .
-
AClinical colitis scores were evaluated on day 56.
-
BTumor tissues were stained for cleaved‐caspase 3. The percentages of positive cells were quantified.
-
C, DColonic LP cells and mLNs were isolated from each mouse. The proportion of immune cells was determined by flow cytometry.
-
E, FRelative expression of Il‐6, Il‐22, Cxcl1, TNF‐α and Il‐17 in mLNs cells and tumors from tumor‐bearing WT and Dectin‐3 −/− mice were detected using qPCR.
-
GCytokine and chemokine production in the serum of WT and Dectin‐3−/− tumor‐bearing mice was determined by a multiple cytokine detection assay.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by unpaired Student’s t‐test.
We then analyzed the innate and adaptive immune cells in colonic LP and mesenteric lymph nodes (mLN). Compared with WT tumor‐bearing mice, more macrophages (CD11b+F4/80+) and ILC3 were recruited to mLN and LP tissues in Dectin‐3 −/− tumor‐bearing mice (Fig 1G). Our previous study found an aberrant proportion of MDSCs in CAC models in Card9 −/− mice. However, we did not find significant difference in proportion of DCs (CD11c+) and MDSCs (CD11b+Gr1+) between WT and Dectin‐3 −/− tumor‐bearing mice (Fig EV1C). For adaptive immune cells, no significant difference of CD4+ T cells, CD8+ T cells, Tregs, and Th17 cells was found in mLN and LP between WT and Dectin‐3 −/− tumor‐bearing mice (Fig EV1D), suggesting that anti‐tumor immune responses mediated by T cells did not contribute to the increased tumor burden in Dectin‐3 −/− mice.
We also detected cytokine expressions in LP cells. Expressions of IL‐6, IL‐22, and Cxcl1, detected by qPCR, were increased in Dectin‐3 −/− tumor‐bearing mice compared with WT mice (Fig 1H). Similar results were found in mLN cells and tumor cells (Fig EV1E and F). The systemic production of cytokines and chemokines in serum was detected using multiplex cytokine assays. Production of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), IL‐6, IL‐12, and tumor necrosis factor‐alpha (TNF‐α) were higher in the serum of Dectin‐3 −/− tumor‐bearing group than those in WT group (Fig EV1G).
Gut mycobiota from Dectin‐3 −/− tumor‐bearing mice promote CAC development
To determine the influence of microbes on tumorigenesis, co‐housed WT and Dectin‐3 −/− mice were used to repeat the above CAC experiment. WT and Dectin‐3 −/− littermates were co‐housed for more than 4 weeks before and during the whole experiments. Tumor burden and clinical colitis scores did not display any difference between co‐housed Dectin‐3 −/− and WT mice (Fig EV2A and B), indicating that the endogenous microbes have significant impact on the increase of tumor burden in mono‐housed Dectin‐3 −/− mice. Next, germ‐free (GF) mice were transferred with feces from WT (WT → GF) or Dectin‐3 −/− (D3 → GF) tumor‐bearing mice during the treatment of AOM‐DSS. GF mice with microbiota from tumor‐bearing Dectin‐3 −/− mice displayed significantly more severe colitis and more tumor lesions, compared with WT‐microbiota‐receiving mice (Figs 2A and EV2C). Consistently, higher histological score and more proportion of Ki‐67‐positive cells were found in Dectin‐3 −/−‐microbiota‐receiving mice, compared with WT‐microbiota‐receiving mice (Fig 2B and C). These data indicated that the altered microbiota in Dectin‐3 −/− mice, rather than the lack of Dectin‐3 in recipient GF mice, resulted in the increased CAC progress.
Figure EV2. Feces from tumor‐bearing Dectin‐3 −/− mice promote CAC. Related to Fig 2 .

- WT and Dectin‐3 −/− mice were co‐housed for at least 4 weeks before and throughout the CAC experiments (n = 8, each group). After induction of tumorigenesis, mice were euthanized and colons were removed. Tumor number, tumor size, and tumor load in colons were measured.
- Mice were treated as described in Fig EV2A. Clinical colitis scores were evaluated on day 56.
- Mice were treated as described in Fig 2A. Clinical colitis scores were evaluated on day 56.
- Mice were treated as described in Fig 1A. Fungal ITS2 rDNA gene sequence was performed in each group. OTU numbers were counted.
- Shannon analysis related to alpha diversity was used. The central band presents the mean value. The box represents the 25th–75th percentile, and the whisker plots represent the minimum and maximum percentiles.
- Mice were treated as described in Fig 2A. Fungal ITS2 rDNA gene sequence was performed in each group. Fungal‐taxon‐based analysis at the phylum level in feces of mice.
- Specific fungal burden of C. tropicalis and C. glabrata in the feces were quantified using qPCR.
- Mice were treated as described in Fig EV2A. Total fungal burden, bacteria burden, and specific burden of C. albicans in feces were quantified using qPCR.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01 as determined by unpaired Student’s t‐test.
Figure 2. Feces from tumor‐bearing Dectin‐3−/− mice promote CAC.

-
ATumor number, tumor size, and tumor load in colons were measured.
-
BTumors were microscopically analyzed and classified as low or high grade. Histological score was assessed by a pathologist.
-
CTumor tissues were stained for Ki67. The percentages of Ki67‐positive tumor cells were quantified. Scale bars, 25 µm.
- D
-
EMice were treated as described in Fig 1A. Fungal ITS2 rDNA gene sequence was performed in each group. Fungal‐taxon‐based analysis at the phylum level in feces of mice. Color from blue to red indicates enrichment of fungus.
-
FFungal‐taxon‐based analysis at the genus level in feces of mice. 1 Candidan 2 Fusarium 3 Kazachstania 4 Thermomyces 5 Phaeococcomyces 6 Mycosphaerella 7 Penicillium 8 Aspergillus 9 Coprinellus 10 Cryptococcus 11 Mortierella 12 Guehomyces 13 Preussia 14 Chaetomium 15 others.
-
GThree‐dimensional principal component analysis (PCA) based on fungal ITS2 rDNA gene sequence abundance in the feces. WT: samples are from WT mice, De: samples are from Dectin‐3−/− mice.
-
HSpecific fungal burden of C. albicans in the feces were quantified using qPCR.
-
IGF mice were orally gavaged with C. albicans (twice a week, 1 × 107) during administration with AOM‐DSS (n = 8, each group). Mice were euthanized on Day 100, tumor load in each mouse were measured.
-
JELISA results for binding assays of Dectin‐3 with C. albicans. Plates were coated with C. albicans and then were added with 100 ml/well recombinant Dectin‐3 at indicated concentrations.
-
K, LGFP‐C. albicans (5 × 106) were added onto 1 × 106 BMDMs, and incubated at 37°C for 1 h. Cells were washed and fresh media containing fluconazole (300 μg/ml) was added. At 6 and 18 h, C. albicans CFU inside BMDMs were photographed and calculated by plating on YPD agar. Scale bars, 25 µm.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test. See also Fig EV2 and Appendix Fig S1.
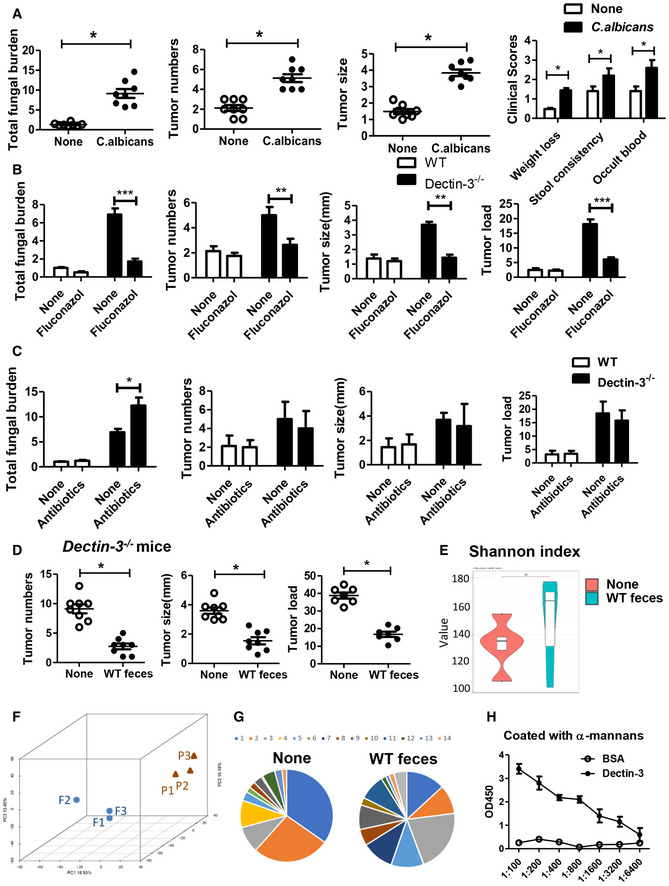
Based on our previous study, the basic level of total fungal burden in colons was almost the same between Dectin‐3 −/− and WT mice (Wang et al, 2016). We next explored microbiota composition in Dectin‐3 −/− and WT tumor‐bearing mice. The total fungal burden in feces was significantly higher in tumor‐bearing Dectin‐3 −/− group than in WT group (Fig 2D), while the bacteria burden did not display any difference (Fig EV2D). Next, we used high‐throughput internal transcribed spacer 2 (ITS2) sequencing (NCBI SRP database: PRJNA661172) to analyze the fungal composition in the feces. Although fungal biodiversity showed no difference between tumor‐bearing Dectin‐3 and WT group (Fig EV2E and F), the mycobiota was dominated by Ascomycota in tumor‐bearing Dectin‐3 −/− mice (Fig 2E), in which the percentages of Candida in tumor‐bearing Dectin‐3 −/− and WT group were significantly different (Fig 2F). The illustrative diagrams of fecal principal component analysis (PCA) showed that the principal fecal fungi component in Dectin‐3 −/− mice was changed, compared with WT mice (Fig 2G). Similar result was found in transplantation experiments in GF mice (NCBI SRP database: PRJNA 661186, Fig EV2G). To identify the specific species of Candida, we next detected the relative burden of C. albicans, C. tropicalis, and C. glabrata in LP using qPCR. We found that the fungal burden of C. albicans were obviously increased in Dectin‐3 −/− tumor‐bearing mice and Dectin‐3 −/−‐microbiota‐receiving mice (Figs 2H and EV2H). Moreover, this difference in fungal burden and specific fungal amount disappeared in co‐housing experiment (Fig EV2I). We also used 16S ribosomal DNA sequencing to analyze the bacterial composition in mouse feces. We found that the composition of bacterial genera did not display any difference between tumor‐bearing WT and Dectin‐3 −/− group by using bacteria‐taxon‐based analysis, alpha diversity analysis, and PCA analysis.
To confirm the function of C. albicans on CAC development, we colonized GF mice with C. albicans during AOM‐DSS administration. We found C. albicans colonization‐induced higher tumor load and more severe colitis in GF mice (Figs 2I and EV3A), with increased fungal burden and undetectable bacteria burden. Furthermore, the anti‐fungal treatment, fluconazole, inhibited CAC development in Dectin‐3 −/− tumor‐bearing mice (Fig EV3B). However, the antibiotics had no significant effect on the tumor load in Dectin‐3 −/− tumor‐bearing mice (Fig EV3C). Moreover, transferring feces from WT mice to Dectin‐3 −/− mice protected mice from tumor progression (Fig EV3D). Detailed mycobiota profiling data (NCBI SRP database: PRJNA 661617) showed fecal transferring changed the composition and principal component in Dectin‐3 −/− tumor‐bearing mice (Fig EV3, EV4, EV5). Together, these results suggested that the deficiency of Dectin‐3 gene led to the increasing of C. albicans, which subsequently promote CAC development in Dectin‐3−/− mice.
Figure EV3. Up‐regulation of IL‐22 in Dectin‐3 −/− mice contributes to CAC development. Related to Fig 3 .
-
AGF mice were orally gavaged with C. albicans (twice a week, 1 × 107) during administration with AOM‐DSS (n = 8, each group). After induction of tumorigenesis (100 days), mice were euthanized and colons were removed. Tumor number, tumor size, and tumor load in colons were measured. Total fungal burden of colon was detected by using qPCR. Clinical colitis scores were evaluated on day 56.
-
BWT and Dectin‐3 −/− mice were treated with fluconazole during administrated with AOM‐DSS (n = 8, each group). Tumor number, tumor size, and tumor load in colons were measured. Total fungal burden of colon was detected by using qPCR.
-
CWT and Dectin‐3 −/− mice were treated with antibiotics during administrated with AOM‐DSS (n = 8, each group). Tumor number, tumor size, and tumor load in colons were measured. Total fungal burden of colon was detected by using qPCR.
-
DDectin‐3 −/− mice were oral transferred with feces (400 ml each time, twice a week) from tumor‐bearing WT mice during administrated with AOM‐DSS (n = 8, each group). After induction of tumorigenesis, mice were euthanized and colons were removed. Tumor number, tumor size, and tumor load in colons were measured.
-
E, FMice were treated as described in Fig EV3D. Fungal ITS2 rDNA gene sequence was performed in each group. Principal component analyses (PCA) based on fungal composition and Shannon analysis related to alpha diversity were used. F: samples are from Dectin‐3−/− mice, P: samples are from Dectin‐3−/− mice treated with WT feces.
-
GFungal‐taxon‐based analysis at the genus level in feces of mice. 1 Candidan 2 Fusarium 3 Kazachstania 4 Thermomyces 5 Phaeococcomyces 6 Mycosphaerella 7 Penicillium 8 Aspergillus 9 Coprinellus 10 Cryptococcus 11 Mortierella 12 Guehomyces 13 Preussia 14 Chaetomium.
-
HELISA results for binding assays of Dectin‐3 with α‐mannans. Plates were coated with α‐mannans and then were added with 100 ml/well recombinant Dectin‐3 at indicated concentrations.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test.
Figure EV4. IL‐22 is mainly produced by RORγt+ ILC3. Related to Fig 4 .

-
AGating strategy for ILC3. Live, Lin‐, CD90hi lymphocytes expressing RORγt (ILC3) were gated to analyze ILC3 function.
-
BMice were treated as described in Fig 1A. mLNs were isolated from each mouse. The proportions of IL‐22+ cells in CD3−RORγt+, CD3−RORγt−, CD3+RORγt−, and CD3+RORγt+ and the absolute number of IL‐22+ cells were determined by flow cytometry.
-
C, DMice were treated as described in Fig 4E. Clinical colitis scores were evaluated on day 56. Colon tissues were stained for p‐STAT3. The percentage of p‐STAT3‐positive tumor cells was quantified.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01 as determined by Student’s t‐test.
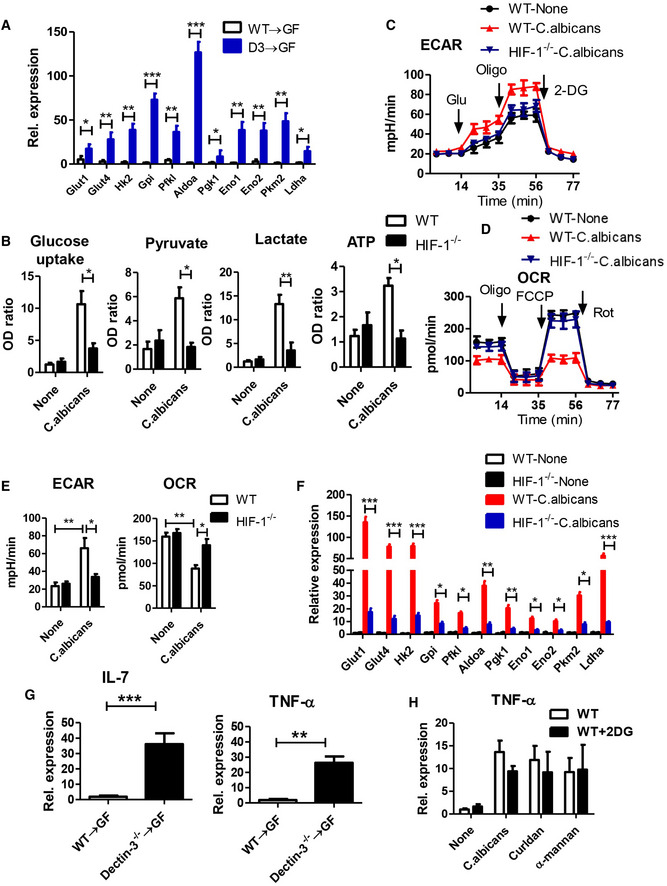
Figure EV5. Candida albicans induce glycolysis and IL‐7 production in macrophages. Related to Fig 5 .
-
AMice were treated as described in Fig 2A. Primary macrophage were isolated from LP in WT and Dectin‐3 −/− tumor‐bearing mice. mRNA expression of glycolysis‐relative genes was detected by using qPCR.
-
B–FBMDMs were acquired from WT and HIF‐1 −/− mice and were stimulated with C. albicans for 24 h. Glucose uptake, pyruvate level, lactate production, and ATP level were determined using assay kit. ECAR and OCR were also examined. mRNA expression of glycolysis‐relative genes was detected by using qPCR.
-
GPrimary macrophage were isolated from LP tissues in indicated mice. mRNA expressions of IL‐7 and TNF‐α were detected by qPCR.
-
HWT‐derived BMDMs were stimulated with C. albicans, curdlan, or α‐mannan in combination with or without 2‐DG (2.5 mM) for 24 h. mRNA expression of TNF‐α was detected by using qPCR.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test.
Since the burden of C. albicans is crucial in Dectin‐3 −/− tumor‐bearing mice, we next aimed to explore the impact of Dectin‐3 on recognition and fungicidal abilities of C. albicans in macrophages. By using ELISA methods, we found that recombinant Dectin‐3 displayed high affinity to bind to both C. albicans hyphae‐coated plates and α‐mannans (Figs 2J and EV3H), suggesting an effective binding of Dectin‐3 with C. albicans. We further examined fungicidal abilities of primary macrophages obtained from bone marrows (BMDMs) which were subsequently challenged with C. albicans. We found that BMDMs derived from WT mice limited the intracellular replication of C. albicans, while Dectin‐3 −/−‐derived BMDMs had a much higher fungal load (Fig 2K). Colony‐forming unit (CFU) assays indicated an increased number of viable yeasts recovered from Dectin‐3 −/−‐derived BMDMs (Fig 2L). Therefore, the increased burden of C. albicans in Dectin‐3 −/− tumor‐bearing mice may due to the impaired anti‐fungal ability of macrophage.
Up‐regulation of IL‐22 in Dectin‐3 −/− mice contributes to CAC development
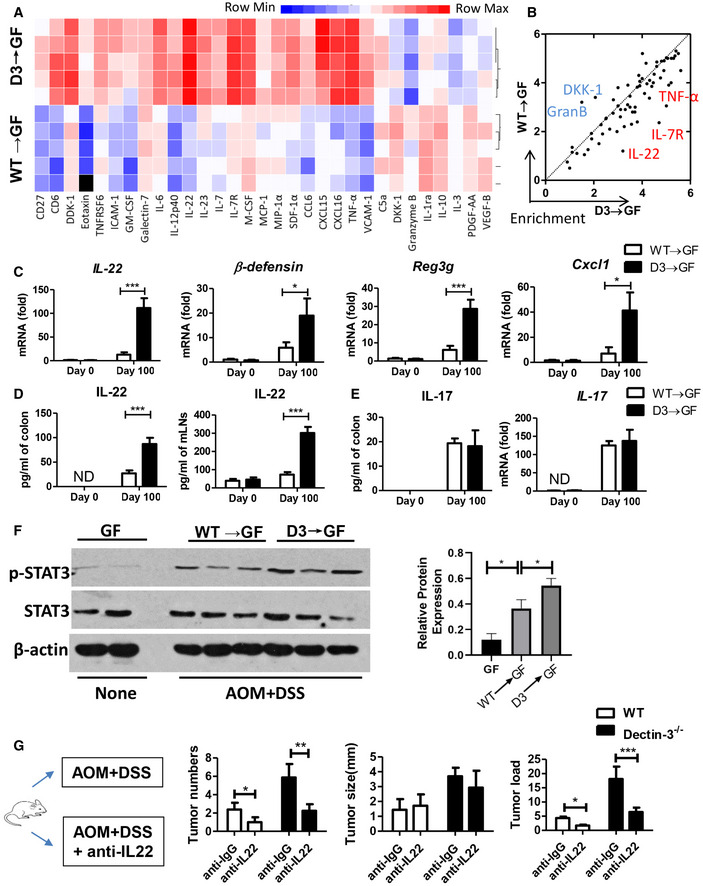
To explore the mechanism of C. albicans ‐induced CAC progression, we used a multiple cytokine profiling assay, which revealed 31 signal transduction pathways participating in different gene expressions (Fig 3A). We found the production of IL‐22, IL‐7R, and TNF‐α was markedly up‐regulated in Dectin‐3 −/−‐microbiota‐receiving GF mice, compared with WT‐microbiota‐receiving mice (Fig 3B). The increased expression of IL‐22 was confirmed by using qPCR (Fig 3C). Previous study has proved that the expressions of antimicrobial proteins, like β‐defensin and regenerating islet‐derived protein III (RegIII‐γ), by intestinal epithelial cells were triggered by IL‐22. We also found up‐regulated expression of β‐defensin, regenerating islet‐derived protein 3 gamma (Reg3g; encoding RegIII‐γ), and Cxcl1 in both LP and mLNs of GF mice transferred with feces from tumor‐bearing Dectin‐3 −/− mice (Fig 3C and D, and Appendix Fig S1A). Since IL‐17a also has a protective function in concert with IL‐22, we then detected IL‐17 expression and production. However, no difference was found in the expression and production of IL‐17 between two groups (Fig 3E).
Figure 3. Up‐regulation of IL‐22 in Dectin‐3 −/− mice contributes to CAC development.
-
A, BMice were treated as described in Fig 2A. LP cells were isolated from each mouse and were culture for 48 h. Cytokine and chemokine production of LP cells were detected using multiplex cytokine assay. Color from blue to red indicates enrichment of gene expression.
-
CmRNA expressions of IL‐22, β‐defensin, Reg3g, and Cxcl1 in LP cells were detected by qPCR.
-
DLP cells and mLNs were isolated from each mouse and were culture for 48 h. Production of IL‐22 in LP cells and mLNs were detected by ELISA.
-
ELP cells were isolated from each mouse and were culture for 48 h. Expression of Il‐17 in LP cells were detected by qPCR. Production of IL‐17 in LP cells were detected by ELISA.
-
FProtein level of p‐STAT3 and STAT3 in colonic epithelial cells was detected by using Western blot (one mouse per lane). The relative expression of p‐STAT3 was calculated.
-
GWT and Dectin‐3−/− mice were intraperitoneally treated with anti‐IL22 antibody or anti‐IgG antibody as control during AOM‐DSS administration (n = 5, each group). Mice were euthanized on Day 100, tumor number, tumor size, and tumor load in colons were measured.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test. See also Fig EV3.
The IL‐22 and IL‐22R binding on intestinal epithelial cells could activate JAK/STAT signaling, mainly STAT3, through its phosphorylation process (Lindemans et al, 2015). Consistent with the increased production of IL‐22, we also found a higher protein level of p‐STAT3 in intestinal epithelial cells in Dectin‐3 −/− microbiota‐received GF mice compared with WT‐microbiota‐received GF mice (Fig 3F). To confirm the function of IL‐22 in increased tumor load in Dectin‐3 −/− mice, mono‐housed WT and Dectin‐3 −/− mice were treated with anti‐IL22 antibody during the treatment of AOM‐DSS (Fig 3G). We found anti‐IL22 treatment had no effect on fungal burden and clinical colitis scores (Appendix Fig S1B and C). But tumor loads and histological scores were downregulated in Dectin‐3 −/− mice after anti‐IL22 treatment (Fig 3G and Appendix Fig S1D). We also found the percentage of p‐STAT3‐positive cells was decreased after anti‐IL22 treatment (Appendix Fig S1E and F).
IL‐22 is mainly produced by RORγt+ ILC3
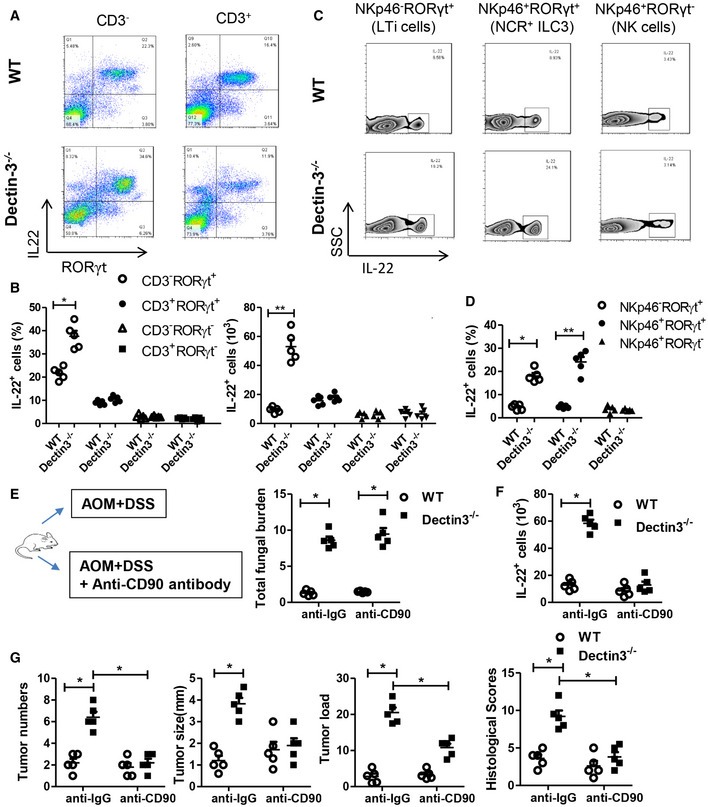
Considering the up‐regulation of IL‐22 could promote CAC development, we next aimed to detect the source of IL‐22 in Dectin‐3 −/− tumor‐bearing mice. Previous reports suggest that IL‐22 is produced by cells in both innate and adaptive immune systems. Both ILC3 and RORγt+ T helper 17 cells produce large number of IL‐22 in host intestine. In our Dectin‐3 −/− tumor‐bearing mice, we observed that most IL‐22‐producing cells in the intestine resided in the compartment with the CD3−CD45+RORγt+ phenotype, which was consistent with the RORγt+ ILC3 phenotype (Figs 4A and B, and EV4A). Moreover, IL‐22 could be produced by both two subsets of RORγt+ ILC3s, including LTi cells and NCR+ ILC3s, but not by NKp46+ RORγt− natural killer cells (NK cells) (Fig 4C and D). Similar results were found in mLN tissues (Fig EV4B).
Figure 4. IL‐22 is mainly produced by RORγt+ ILC3.
-
A, BMice were treated as described in Fig 1A. LP cells were isolated from each mouse. The proportions of IL‐22+ cells in CD3‐RORγt+, CD3−RORγt−, CD3+RORγt−, and CD3+RORγt+ were determined by flow cytometry.
-
C, DMice were treated as described in Fig 1A. LP cells were isolated from each mouse. The proportions of IL‐22+ cells in NKp46−RORγt+, NKp46+RORγt− and NKp46+RORγt+ were determined by flow cytometry.
-
EWT and Dectin‐3−/− mice were intraperitoneally treated with anti‐CD90 antibody or anti‐IgG antibody during AOM‐DSS administration (n = 5, each group). Mice were euthanized on Day 100, feces were collected in colon from each mouse. Total fungal burden in feces were quantified using 18S rDNA qPCR.
-
FMice were treated as described in Fig 4E and colonic LP cells were isolated. Numbers of IL‐22+ cells in LP cells were determined by flow cytometry.
-
GMice were treated as described in Fig 4E. Tumor number, tumor size, tumor load, and histological score in colons were measured.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01 as determined by Student’s t‐test. See also Fig EV4.
To confirm the critical role of ILC3 cells in Dectin‐3 −/− tumor‐bearing mice, CD90 antibodies were used to deplete ILC3 subpopulation. Although anti‐CD90 treatment had no effect on fungal burden and colitis scores (Figs 4E and EV4C), the absolute number of IL‐22+ cells was markedly decreased after anti‐CD90 treatment in Dectin‐3 −/− tumor‐bearing mice (Fig 4F). Tumor numbers, tumor size, tumor loads, and histological malignance scores were all decreased upon anti‐CD90 treatment (Fig 4G), suggesting the effect of ILC3 and its producing IL‐22 on increased tumor burden in Dectin‐3 −/− mice. We also found the percentage of p‐STAT3‐positive cells was decreased after anti‐CD90 treatment in Dectin‐3 −/− tumor‐bearing mice (Fig EV4D).
IL‐22 production depends on microbial signals
Since the effector function of RORγt+ ILC3s relies on commensal microbiota‐driven signals (Satoh‐Takayama et al, 2008; Sanos et al, 2009), we therefore detected whether IL‐22 production depends on gut microbial signals. We found that IL‐22 production could not be detected in newborn mice, slightly increased in day 7, and significantly increased from day 28 after birth (Appendix Fig S2A). This accumulating production of IL‐22 is in consistent with the increase in number and complexity of the intestinal microbe. To further confirm the impact of commensal fungi on IL‐22 production in the intestine, we analyzed IL‐22 production in our previously described mouse model. As shown in Fig 5A, both the proportion of IL‐22+ cells and the absolute numbers of IL‐22+ cells were up‐regulated in Dectin‐3 −/− microbiota‐received GF mice, comparison to WT‐microbiota‐received GF mice, and were increased in C. albicans colonization mice compared with control mice. Upon fluconazole treatment, both the proportion of IL‐22+ cells and the absolute numbers of IL‐22+ cells were decreased in Dectin‐3 −/− tumor‐bearing mice (Appendix Fig S2B). However, treatment of antibiotics did not affect IL‐22 production in Dectin‐3−/− tumor‐bearing mice (Appendix Fig S2C). These data together indicated that the increased production of IL‐22 in Dectin‐3 −/− tumor‐bearing mice was dependent on the intestinal microbial signals, especially the fungal signal.
Figure 5. Candida albicans induce glycolysis and IL‐7 production in macrophages.
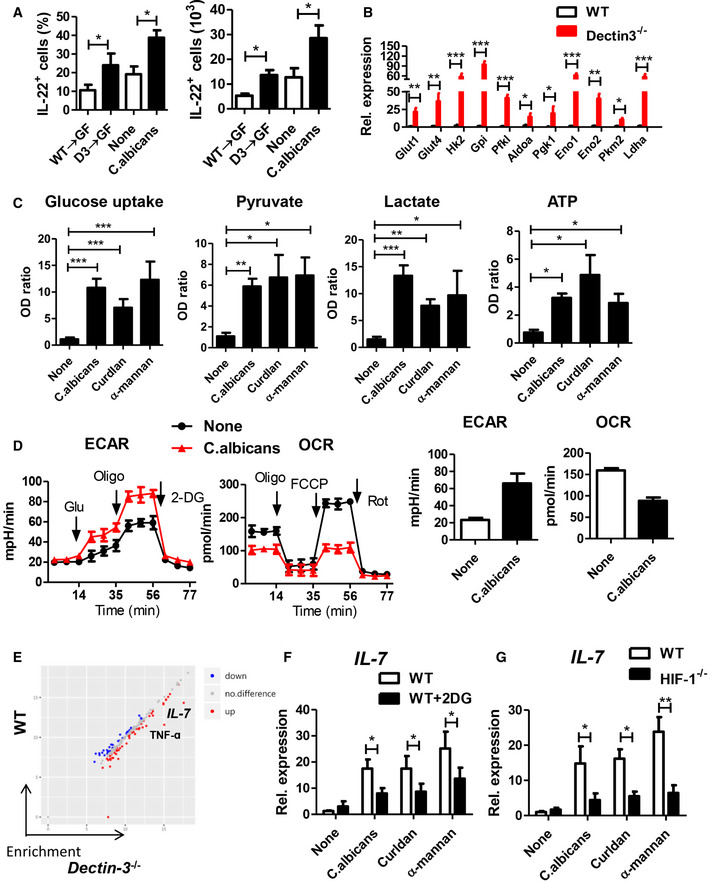
- Mice were treated as described in Fig 2A and I. Colonic LP cells were isolated. Proportion and absolute number of IL‐22+ cells in LP cells were detected by flow cytometry.
- Primary macrophages were isolated from LP cells in WT and Dectin‐3−/− tumor‐bearing mice. mRNA expression of glycolysis‐relative genes was detected by using qPCR.
- BMDM cells were stimulated with C. albicans (5 × 106), Curdlan, and α‐mannan for 24 h. Glucose uptake, pyruvate level, lactate production, and ATP level were determined using assay kit.
- BMDM cells were stimulated with C. albicans (5 × 106) for 24 h, and ECAR and OCR were examined.
- BMDMs acquired from WT and Dectin‐3−/− mice were stimulated with C. albicans for 24 h. Cytokine and chemokine production of BMDMs were detected using multiplex cytokine assay.
- WT‐derived BMDMs were stimulated with C. albicans, curldan, or α‐mannan in combination with or without 2‐DG (2.5 mM) for 24 h. mRNA expression of Il‐7 was detected by using qPCR.
- BMDMs were acquired from WT and HIF‐1−/− mice and were stimulated with C. albicans, curldan, or α‐mannan for 24 h. mRNA expression of Il‐7 was detected by using qPCR.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test. See also Fig EV5 and Appendix Fig S2.
Candida albicans induce glycolysis and IL‐7 production in macrophages
We next try to find out how intestinal fungi promote IL‐22 production by ILC3 cells. Murine RORγt+ ILC3s, lacking toll‐like receptors (TLRs) and C‐type lectin receptors (Appendix Fig S2D), cannot directly recognize gut microbial signals. Therefore, these cells are dependent on other cellular sensors to translate signals from gut microbiota into effector function. Macrophages, which have several different PRRs, are of great significance for innate immune defense system against pathogenic bacterial and fungal infections. Activated macrophages go through profound reprogramming of their cellular metabolism. We isolated primary macrophages in LP from WT and Dectin‐3−/− tumor‐bearing mice. The expressions of glycolytic genes, including Glut1, Glut4, Gpi, Pfkl, Hk2, Aldoa, Pkm2, Pgk1, Eno1, Eno2, and Ldha were significantly increased in primary macrophage from Dectin‐3 −/− tumor‐bearing mice, compared with those in WT mice (Fig 5B). These glycolytic genes were also significantly increased in primary macrophages isolated from Dectin‐3 −/− microbiota‐received GF mice, compared with those from WT‐microbiota‐received GF mice (Fig EV5A). To test whether C. albicans can induce glycolytic phenotype in macrophages, BMDMs were then co‐cultured with C. albicans, Curdlan, and α‐mannans. Curdlan and α‐mannans are exposed on the cell wall of C. albicans. Fungal stimulation significantly up‐regulated activity of the glycolytic pathway in macrophages, presenting as increased glucose uptake level, pyruvate level, ATP level, and lactate production in BMDMs (Fig 5C). We also found BMDMs treated with C. albicans showed decreased oxygen consumption rate (OCR) indicating the mitochondrial oxidative respiration and increased extracellular acidification rate (ECAR) reflecting overall glycolytic flux (Fig 5D).
Hypoxia inducible factor‐1 (HIF‐1) is a heterodimeric transcription complex that has been proved to work as a significant transcriptional factor in modulating functions of innate immune and glycolysis. Previous study has demonstrated that HIF‐1‐dependent glycolysis is crucial for functional differentiation of macrophages in preventing from bacterial infection. We tested whether C. albicans‐induced glycolysis is also dependent on HIF‐1. BMDMs were acquired from HIF‐1α‐deficient mice (HIF‐1−/−) and WT mice. We found HIF‐1 deficiency led to increased OCR and decreased glucose uptake level, pyruvate level, ATP level, lactate production, and ECAR, upon C. albicans stimulation (Fig EV5B–E). Glycolytic relative genes were also decreased in HIF‐1−/−‐derived BMDMs, compared with WT‐derived BMDMs (Fig EV5F).
The cytokine production function of macrophages was detected using multiple cytokine detection assay. Among 200 kinds of cytokines, 81 cytokines were differentially expressed, with fold changes > 2, in primary macrophages between WT‐microbiota‐received GF mice and Dectin‐3−/− microbiota‐received GF mice (Fig 5E). Of note, the production of TNF‐ɑ and IL‐7 were the most markedly differential expression proteins, with fold changes 15.6 and 30.5, respectively (Fig 5E). This increased IL‐7 and TNF‐ɑ mRNA expressions were also confirmed by using qPCR (Fig EV5G). To explore the association between macrophage glycolysis and cytokine production, 2‐deoxy‐D‐glucose (2‐DG) was added for glycolysis inhibition. We found the production and expressions of IL‐7 were decreased after 2‐DG treatment (Fig 5F), while the expression of TNF‐ɑ did not change after 2‐DG administration (Fig EV5H). We also found a decreased expression of IL‐7 in HIF‐1−/−‐derived BMDMs upon fungal stimulation, compared with WT‐derived BMDMs (Fig 5G). Together, these data suggest C. albicans regulate IL‐7 production through HIF‐1‐dependent glycolysis in macrophage.
IL‐7 induces IL‐22 production in ILC3
Primary ILC3 cells were sorted from LPs in large intestinal and were stimulated with IL‐7. Previous studies have proved that IL‐1β and IL‐23 induce IL‐22 production in ILC3s, which can be used as positive control in our study (Longman et al, 2014; Bergmann et al, 2017). Here, we found IL‐7 alone induced IL‐22 production in ILC3, while in combination with IL‐1β and IL‐23, we found a significant synergistic effect on IL‐22 expression and production (Fig 6A). To confirm the role of macrophage‐derived IL‐7 on IL‐22 production, BMDMs were stimulated with or without C. albicans, and the supernatant was isolated and was added into ILC3. We found supernatant from C.albican‐stimulated macrophages significantly induced IL‐22 production from ILC3s, while IL‐7 blockade by neutralizing antibody reduced IL‐22 production (Fig 6B). Furthermore, we found ILC3s activated by IL‐7 had increased expression of il1r and il23r (Fig 6C). Taken together, these results indicate that IL‐7, alone or combined with IL‐1β or IL‐23, induces IL‐22 production in ILC3.
Figure 6. IL‐7 induces IL‐22 production in ILC3s through AhR and STAT3.

-
APrimary ILC3 cells were sorted from colonic LP cells and were stimulated with indicated cytokines. Productions of IL‐22 were detected by ELISA. mRNA expressions of Il‐22 were detected by qPCR.
-
BBMDMs were stimulated with C. albicans for 24 h and the supernatant was isolated and added to primary ILC3 cells. (1) Supernatant from BMDMs without stimulation. (2) Supernatant from BMDMs with stimulation of C. albicans. (3) Supernatant from BMDMs with stimulation of C. albicans and IL‐7 antibody. Productions of IL‐22 by ILC3 cells were detected by ELISA. mRNA expressions of Il‐22 in ILC3 cells were detected by qPCR.
-
C, DPrimary ILC3 cells were sorted from LPs and were stimulated with (IL‐7 group) or without IL‐7 (None group). mRNA expressions of relative genes were detected by qPCR.
-
EPrimary ILC3 cells were sorted in LPs from STAT3fl/fl and STAT3fl/fl VillinCre mice and were stimulated with IL‐7. Productions of IL‐22 were detected by ELISA. mRNA expressions of Il‐22 in ILC3 cells were detected by qPCR.
-
FPrimary ILC3 cells were sorted from LPs and were stimulated with IL‐7 in combined with FIZC and AhR Inh. Cells without stimulation were used as control (None). Production of IL‐22 was detected by ELISA.
-
GAhR, STAT3, ROR binding sites in the il‐22 promoter.
-
HHEK293T cells were transiently transfected with the indicated expression constructs (Ctl or AhR) and were stimulated with (IL‐7 group) or without IL‐7 (none group). ChIP analysis of the interaction of AhR and STAT3 to binding sites in the il22 promoter with or without IL‐7, respectively.
-
IHEK293T cells were transiently transfected with the indicated expression constructs. Whole cell extracts were subjected to anti‐HA immunoprecipitation, and were then immunoblotted with HA antibody and anti‐STAT3 antibody.
-
JEL4 cells were transduced with retroviruses encoding AhR and STAT3. IL‐22 mRNA expression was analyzed by qPCR.
Data information: Error bars represented SD of triplicate samples. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by unpaired Student’s t‐test.
Both Stat3 and AhR modulate IL‐22 expression triggered by IL‐7
We further explore the molecular mechanism mediating IL‐22 production by ILCs stimulating with IL‐7. mRNA expressions of genes downstream the IL‐7 signaling and genes coding for transcription factors were detected, including AhR, Tbx21, Rora, Rorc, Socs3, Jak1, Jak3, Akt1, Gsk3b, Stat3, Stat5a, and Stat5b (Fig 6D). Among these genes, we observed an increased expression of Stat3 and AhR in IL‐7 stimulated ILCs. Although ROR has a significant role in the control of IL‐22 production in Th17 cells, the expression of Rora and Rorc did not change upon IL‐7 stimulation (Fig 6D).
We next study the function of Stat3 and AhR on IL‐22 production induced by IL‐7. Primary ILCs were sorted from large intestinal of Stat3cKO mice and were stimulated with IL‐7. IL‐22 production responsive to IL‐7, both protein and mRNA level, was markedly downregulated in STAT3‐deficient ILC3 cells (Fig 6E). It has been reported that AhR participates in IL‐22 production in Th17 cells (Veldhoen et al, 2008). In our study, the administration of the AhR agonist 6‐formylindolo [3,2‐b] carbazole (FICZ) caused an obvious increase in IL‐22 production induced by IL‐7 (Fig 6F). On the contrary, the addition of AhR‐specific antagonist (CH‐223191) led to an obvious decrease in IL‐22 production induced by IL‐7 (Fig 6F). These findings suggest that both Stat3 and AhR have significant impacts on the modulation of IL‐22 production activated by IL‐7.
Results of bioinformatic analysis showed that IL‐22 promoter displayed three putative AhR binding sites (xenobiotic response elements, AhRE‐1, AhRE‐2, and AhRE‐3), two putative STAT3 responsive elements (SRE‐1 and SRE‐2), and one putative binding site for RORγt (RORE) upstream of the transcription start site (Fig 6G). Therefore, chromatin immunoprecipitation (ChIP) assays were performed to detect whether STAT3 and AhR have interactions with these potential binding sites in the il22 locus. We found a significant interaction of AhR and STAT3 with the il22 promoter in EL4 cells with IL‐7 presence (Fig 6H). Using a co‐immunoprecipitation assay, we detected the interaction between AhR and STAT3 (Fig 6I). Next, we assessed the functional effects of STAT3 and AhR interaction with its DNA responsive elements by using a reporter plasmid which carries the firefly luciferase controlled by the il22 promoter. Transfection with vectors coding for AhR or constitutively activated STAT3 significantly induced luciferase activity (Fig 6J). Co‐transfection with constructs coding for constitutively activated AhR and STAT3 resulted in higher activity of luciferase, indicating a synergistic effect of AhR and STAT3 on IL‐22 production (Fig 6J).
IL‐22 is correlated with fungal burden in patients with CRC
Tumor tissues were collected from 172 patients with CRC. The mRNA expression level of Dectin‐3 in tumor tissues was detected using qPCR. Tumors in stage III and stage IV patients had significantly lower expressions of Dectin‐3 than those in stage I and II patients (Fig 7A). Fungal burden in fecal samples were determined by detecting 18sDNA. Patients with colon cancer were divided into two groups based on fungal burden. Dectin‐3 expression was markedly increased in group with low fungal burden than that with high fungal burden (Fig 7B), suggesting the function of Dectin‐3 on the host anti‐fungal immunity. Moreover, colon cancer patients with high fungal burden displayed poorer disease‐free survival and overall survival (Fig 7C). In consistent, the protein level and mRNA level of IL‐22 and STAT3 were higher in high fungal burden group, compared with those in low fungal burden group (Fig 7D and E).
Figure 7. IL‐22 is correlated with fungal burden in patients with colon cancer.

-
APatients with colon cancer were divided into four groups based on tumor stage. mRNA expressions of Dectin‐3 in tumor tissues were detected by qPCR. The box represents the 25th–75th percentile, and the whisker plots represent the minimum and maximum percentiles.
-
BPatients with colon cancer were divided into two groups based on fungal burden in the feces. mRNA expressions of Dectin‐3 were compared between these two groups.
-
COverall survival rate was shown by K‐M survival curve.
-
D, ETumor tissues were stained for IL‐22 and p‐STAT3. The percentages of IL‐22‐positive and p‐STAT3‐positive cells were quantified. mRNA expressions of IL22 and STAT3 were detected by qPCR. Scale bars, 25 µm.
Data information: Data with error bars are represented as mean ± SD. Each panel is a representative experiment of at least three independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by Student’s t‐test.
Summarily, we put forward the following working model: Dectin‐3 deficient macrophages display impaired fungicidal abilities, which lead to the increase of gut fungi, especially C. albicans. C. albicans regulate IL‐7 production through HIF‐1‐dependent glycolysis in macrophage. IL‐7 induces IL‐22 production in ILC3, which is controlled by transcriptional factor Stat3 and AhR. IL‐22 production from ILC3 caused increased p‐STAT3 level of epithelial cells and tumor progression.
Discussion
This study has several important findings: First, by using fecal transferring experiment, fungal oral administration experiment, and anti‐fungal experiment, we provide direct evidence that C. albicans could promote tumorigenesis in colon cancer. Second, C. albicans activates glycolysis and IL‐7 production in macrophage, suggesting that commensal fungi might function to reverse the metabolic program related to the inflammatory response in macrophage. Third, IL‐7 induces IL‐22 production in ILC3. Together, our results revealed a fungal‐dependent crosstalk between macrophage and IL‐22‐producing ILCs in the development of CAC.
The function of gut commensal microbes has achieved great attention, which was related to multiple diseases including colitis (Wang et al, 2016) and colon cancers (Arthur et al, 2012; Arthur et al, 2014; Geis et al, 2015). Most recently, CRC‐associated microbiota was found to contribute to oncogenic epigenetic alterations and induce increased expression of hypermethylated genes in colonic mucosa (Sobhani et al, 2019). Despite that most researches of gut commensal microbiota have focused on bacteria, a CRC‐associated mycobiome dysbiosis was reported and was characterized by altered fungal composition and ecology (Coker et al, 2019). Based on our previous study and this study, we found both C. albicans and C. tropicalis could directly promote the development of CAC. The fungus Candida lives harmlessly within most people. However, under illness condition, fungi can multiply and cause disease. In our mouse model, the increased burden of C. albicans was due to Dectin‐3 deficiency. Dectin‐3 has long been considered as a fungal recognition receptor, and our previous work has found that Dectin‐3 could form a heterodimer with Dectin‐2 to recognize C. albicans (Zhu et al, 2013). We also did acute colitis experiment and found Dectin‐3 −/− mice were susceptible to DSS‐induced colitis, presenting by increased mucosal erosion, inflammatory cell infiltration, crypt destruction, and loss of goblet cells in the colon of Dectin‐3 −/− mice, compared with WT mice. Although CAC is initialed by AOM and driven by the inflammation from DSS colitis, the development of CAC is still different with acute or chronic colitis, due to the complicated tumor microenvironment. For example, the role of ILC3 in colitis and colon cancer is different. Here, by using AOM/DSS‐induced CAC mouse model, we found co‐housing abolished the difference between WT and Dectin‐3 −/− mice and transferring feces from Dectin‐3 −/− to germ‐free mice increased susceptibility to CAC. Therefore, our study provided convincing evidence that deficiency of Dectin‐3 allows expansion of gut commensal fungi, which in turn promotes CAC development.
The function of macrophages was considered as crucial in gastrointestinal innate immunity homeostasis against IBD and colon tumorigenesis (Mantovani et al, 2017; Koelink et al, 2019; Na et al, 2019). Accumulating evidences suggest that metabolic patterns have a deep impact on the differentiation and activation of gut macrophages (Kelly & O'Neill, 2015; Murphy, 2019). Previous studies have indicated that the gut bacteria, especially Salmonella enterica serovar typhimurium‐mediated regulation of glycolysis level, could affect functional abilities of macrophages and participate in various gastrointestinal disorders (Bowden et al, 2009; Li et al, 2018; Ding et al, 2019). Recently, it is reported that both bacteria and fungi infection could up‐regulate macrophage glycolysis level and trigger rapid macrophage death (Tucey et al, 2018). Here, we found that macrophages with C. albicans infection also had an enhanced glycolysis level through HIF‐1‐dependent pathway. Moreover, the importance of glycolysis in the production of IL‐7 was also demonstrated, where glycolysis inhibition by 2‐deoxyglucose reduced IL‐7 expression. Therefore, our findings suggest that commensal fungi might function to reverse the metabolic program related to the inflammatory response in macrophages.
IL‐22 is a significant effector molecule produced by both innate and adaptive immune cells and participates in gastrointestinal inflammation and cancer development (Harrison, 2013). Production of IL‐22 from CD4 + T cells could stimulate colorectal tumor growth via activation of STAT3 transcription factor and H3K79 methyltransferase in cancer cells (Kryczek et al, 2014). Also, ILC‐secreted IL‐22 was proved to cause colon cancer progress via tumor cell intrinsic STAT3 phosphorylation and activation in multiple mouse models (Kirchberger et al, 2013; Bergmann et al, 2017). In our study, by using anti‐CD90 antibody and anti‐IL22 antibody, we found specific determination of RORγt+ ILC3, as a main source of IL‐22, reduced tumor load especially in Dectin‐3−/− mice. Although anti‐CD90 antibody also deplete subsets of T cells, experiments with detection of immune cells showed that T cells are not responsible for the phenotype of Dectin‐3−/− mice in CAC.
Environmental factors and transcriptional factors are crucial for IL‐22 modulation. In adaptive immune cells like Th22, differentiation and production of Th22 cells is modulated by cytokines including IL‐1β, IL‐6 and IL‐23 (Akdis et al, 2012). In ILC3s, the most potent environmental signals to trigger IL‐22 production are mostly IL‐23 or IL‐1β (Longman et al, 2014; Bergmann et al, 2017). In the intestinal microenvironment, crosstalk between macrophages and ILC3s was important to maintain immune homeostasis. IL‐1β production from gut macrophages could promote Csf2 release from ILC3s to modulate colonic Treg function (Mortha et al, 2014). In turn, Treg also inhibit IL‐22 production of ILC3s by suppressing IL‐23 and IL‐1β release from macrophages and prevent ILC3‐dependent colitis (Bauche et al, 2018). Furthermore, the crosstalk between macrophages and ILC3s could be regulated by microbiota. For example, the gut microbiota‐mediated IL‐23 release from DCs could promote ILC3 production of IL‐17 for colonic inflammation (Bhatt et al, 2018). Also, CX3CR1+ mononuclear phagocytes played an important role in integrating microbial signals to modulate colonic ILC3 function in IBD (Castellanos et al, 2018). Most recently, in the small intestine, ILC3s were found to be the major source of IL‐22 for intestinal homeostasis, which was regulated by microbiota‐induced release of IL‐1β from macrophages. Here we identified another kind of cytokine, IL‐7, that was produced by macrophages upon fungal stimulation and induced IL‐22 production from gut ILC3s. Therefore, our results provide evidence that C. albicans ‐driven release of IL‐7 from macrophages promote bindings of Ahr and p‐STAT3 at the IL‐22 locus, which subsequently stimulated the transcription process of IL‐22 for CAC development. These results represent a significant advance in our understanding of how gut commensal microbiota modulate host intestinal immunity and may provide potential applications of immunotherapies for CRC patients.
Materials and Methods
Mice
Dectin‐3+/− mice were kindly provided by Dr. Xin Lin (Tsinghua University, Beijing, China). Dectin‐3 −/− mice and WT controls (Dectin‐3+/+) used in this study are from heterozygous Dectin‐3+/− breeding set‐ups, which generates both KO and WT littermates. Except co‐housing experiment, WT mice and Dectin‐3 −/− mice were separated for more than 4 weeks before use and during the whole experiments. Villin‐Cre mice, STAT3flox/flox mice, and HIF‐1α−/− mice were purchased from Model Animal Research Center of Nanjing University. All STAT3‐cKO were obtained by crossing Villin‐Cre mice with STAT3flox/flox mice on a C57BL/6 background. All mice were kept in a conventional and pathogen‐free facility at Medical School of Nanjing University. Germ‐free (GF) mice were breed and housed in germ‐free isolators. Conventional mice were fed with a standard chow diet, and GF mice were fed with a yeast‐free diet. All mouse handling and in vivo experiments were performed according to the NIH “Guide for the Care and Use of the laboratory Animals”. All animal procedures and experiments were carried out under protocols and obtained approval from Institutional Animal Care and Use Committee in Medical School of Nanjing University.
Mouse models
In order to establish CAC model, WT and Dectin‐3 −/− mice (10 weeks old) were injected into the abdominal cavity with AOM (10 mg/kg) on day 1. After 5 days, the drinking water for mice was added with 2% DSS for the seven subsequent days. Three cycles of DSS addition were employed. Mice were euthanized on day 100. For transferring experiment, fresh stool samples from WT or Dectin‐3 −/− tumor‐bearing mice were washed, aliquoted, and stored in −80°C. Fecal suspension was gavaged to GF mice orally (400 μl each time, twice a week) for 9 weeks after AOM injection. For fungal supplemental experiment, mice were treated with C. albicans (1 × 107 yeast, twice a week) for 9 weeks after AOM injection. 1.5% DSS was added to the drinking water during DSS treatment. For ILC3 deletion experiment, anti‐CD90 antibody (200 μg) was injected intraperitoneally every 3 days during DSS administration. For IL‐22 deletion experiment, anti‐IL‐22 antibody (250 μg) was injected intraperitoneally every 3 days during DSS administration. For fungi ablation experiments, fluconazole (0.5 mg/ml) were used during DSS administration. For bacteria ablation experiments, ampicillin (1 g/l), streptomycin (1 g/l), metronidazole (0.5 g/ml), and vancomycin (1 g/l) were added via drinking water for three cycles during DSS administration.
Histology and immunohistochemical (IHC) analysis
Hematoxylin‐eosin staining was did in paraffin‐embedded colon tissue sections. Colitis scores and tumor grades were analyzed by a pathologist (Wirtz et al, 2007; Kargl et al, 2013). For IHC staining, the indicated antibodies were used to stain colon sections. Proliferation index were counted in 10 continuously areas. Following antibodies were used (all from Cell Signaling Technology): Ki‐67(#12202), p‐STAT3(#9145), IL‐22(#5224).
Isolation of intestinal epithelial cells and LP lymphocytes
Colons of mice were isolated, resected, opened longitudinally, washed, and cut into pieces. Intestinal pieces were incubated in EDTA supplemented Hank’s balanced salt solution with Ca2+ and Mg2+ free (Gibco) for 15–20 min at 37°C with mild agitation. The epithelial cell layer was collected by vortexing. The remaining sheets of LP were digested in a digestion medium containing RPMI 1640, FBS (5%), collagenase type IV (1.5 mg/ml), penicillin‐streptomycin (1%), and DNase (5 U/ml), for 30 min at 37°C. Cell suspensions were filtered and were resuspended in 5 ml of 40% Percoll (GE Healthcare) and overlaid onto 5 ml of 80% Percoll. Lymphocytes in LP were collected at the interphase of the Percoll gradient media. Primary ILC3 (CD45+CD90+Lineage−) and primary macrophages (CD11b+F4/80+) were sorted from LP cells by using a FACS Aria II (BD Biosciences).
Flow cytometry
For surface staining, cells were washed and stained with fluorescent‐conjugated antibodies for 20 min at 4°C. For nuclear staining, cells were fixed and permeabilized by using a Mouse Regulatory T Cell Staining Kit (eBioscience). The following anti‐mouse antibodies were used: anti‐mouse F4/80 (FITC, #123107), anti‐mouse CD11b (APC, #101211), anti‐mouse/human CD11b (FITC, #101205), anti‐mouse CD11c (FITC, #117305), anti‐mouse CD3 (APC, #100235), anti‐mouse CD4 (FITC, #100405), anti‐mouse CD8 (FITC, #140403), anti‐mouse/rat/human FOXP3 (PE, #320007), anti‐mouse CD45 (PerCP, #103129), anti‐mouse CD90.2 (APC/Cy7, #105327), anti‐mouse Lineage Cocktail (Brilliant Violet 421 #133311), anti‐mouse CD335 (NKp46) (FITC, #137605), anti‐mouse CD127 (IL‐7Rα) (Alexa Fluor® 647, #135019), anti‐mouse IL‐17RB (Alexa Fluor® 647, #146303), anti‐mouse IL‐22 (Alexa Fluor® 647, #516406), anti‐mouse Gr1(APC, #108412) from BioLegend, Rorγt (Pacific Blue #562894) from BD Biosciences. Cells were examined using a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Raw data were evaluated using FlowJo software.
16S and ITS rDNA gene sequencing and analysis
Stool sample were acquired from the gut. Total genomic DNA was extracted using DNA Extraction Kit following the manufacturer’s instructions. Extracted DNA was diluted to a concentration of 1 ng/μl and stored at −20°C until further processing. The diluted DNA was used as template for PCR amplification of bacterial 16S rRNA and ITS genes with the barcoded primers and Takara Ex Taq (Takara). The detailed mycobiota and microbiota profiling data were detected by using ITS sequencing and 16S sequencing, respectively. Sequencing was performed by an Illumina MiSeq PE300 system (OEbiotech Co, Ltd.). Paired‐end sequences were merged to give an optimal alignment (overlap length ≥ 10 bp, mismatch proportion ≤ 20%). As an added quality control measure, the software package MacQIIME (version 1.9.1) pipeline was used to filter out and discard poor‐quality reads using the default settings. OTUs were assigned to the closest taxonomic neighbors and relative bacterial species using Seqmatch and Blastall. Relative abundance of each OTUs and other taxonomic levels (from phylum to genus) was calculated for each sample to account for different levels of sampling across multiple individuals. PCA and Shannon analysis related to alpha diversity were used. All the sequencing data are submitted into NCBI SRA database.
cDNA synthesis and qPCR
Total RNAs were extracted with TRIzol Reagent (Invitrogen, Carlsbad, CA) and were reverse‐transcribed into cDNA by using oligo (dT) primer. SYBR Green PCR master mix solution was employed for qPCR assays. The primers used in this study are listed in Appendix Table S1. The 2−ΔΔ C t quantification method was used with mouse β‐actin as an endogenous control.
Fungal strains and fungal burden assay
The C. albicans strain and GFP‐C. albicans strain (5314) were kindly provided by Dr. Xin Lin (Tsinghua University, Beijing, China). For the fungal hyphal form preparation, C. albicans was washed, resuspended, and incubated for 3 h. Total and specific fungal burden were analyzed using qPCR as previously described.
BMDMs preparation
Bone marrow cells were collected from mouse femurs and tibias. Erythrocytes were removed. Cells were cultured for 5 days in DMEM containing 2‰ macrophage colony‐stimulating factor (M‐CSF). Adherent cells were collected for subsequent experiments.
Fungal killing assay and phagocytic assay
GFP‐C. albicans (5 × 106) was resuspended in DMEM supplemented with 5% fetal bovine serum (FBS) and added into 1 × 106 WT and Dectin‐3 −/− BMDMs, subsequently incubated at 37°C in a 5% CO2 incubator for 1 h. Wells were washed and fresh media containing fluconazole (300 μg/ml) was added. At 6 and 18 h, BMDMs were washed with PBS for three times, lysed in water. Candida albicans CFU were photographed and calculated by plating on YPD agar.
Detection of cytokines
For multiple cytokine detection assay in serum, multiple cytokine and chemokine biomarkers with bead‐based multiplex assays with the Luminex technology (#MCYTMAG70PMX25BK) were used. For multiple cytokine detection assay in cell supernatant, Mouse Cytokine Antibody Array (RayBiotech, Inc, AAM‐CYT‐2000) were used following the manufacturer’s protocol. For single cytokine detection, Ready‐SET‐Go ELISA kits for IL‐22 (BioLegend, #436307) and IL‐17 (BioLegend, #436207) were used.
Western blot analysis
Protein concentration was measured after cell lysis. Blocking was conducted with TBST. Following antibodies were used (all from Cell Signaling Technology): p‐STAT3 (#9139), STAT3 (#9145) and β‐actin (#4970).
Glucose uptake, lactate, pyruvate, and ATP assays
Glucose uptake, levels of lactate, pyruvate, and ATP were analyzed with Glucose Uptake Colorimetric Assay Kit (Biovision, #K676), Lactate Assay Kit II (Biovision, #K627), Pyruvate Colorimetric Assay kit (Biovision, #K609), and ATP Colorimetric Assay Kit (Biovision, #K354) following protocols of the manufacturer. All results were normalized to cell number.
Oxygen consumption rate and extracellular acidification rate assays
Oxygen consumption rate (OCR) and extracellular acidification rate (EACR) of cells were analyzed with Seahorse XF Cell Mito Stress Test Kit and Seahorse XF Glycolysis Stress Test Kit (Agilent Technologies), respectively. Experiments were performed following instructions of the manufacturer using the Seahorse XFe 96 Extracellular Flux Analyzer (Seahorse Bioscience).
ChIP assay
ChIP assay was conducted with the EZ‐Magna ChIP™ G Chromatin Immunoprecipitation Kit (#17‐409, Millipore) in accordance to instructions of the manufacturer.
Human samples
172 colorectal cancer patients who received treatment in Department of Colorectal Surgery, The Affiliated Hospital of Nanjing University of Chinese Medicine and Nanjing University Affiliated Jinling Hospital from March 2016 to Feb 2018 were included in this research. Tumor tissues were obtained when patients received surgical operations. The feces from each patient were collected before surgery. All studies involved in human participants have been obtained approval from the ethics committee of “Medical School of Nanjing University”, and written informed consent was acquired from all subjects.
Statistical analysis
Statistical analysis was conducted in GraphPad Prism 7. A two‐tailed Student’s t‐test was employed to analyze statistical significance between two groups. Correlation significance was determined using linear regression. Data are shown as means ± SD. P < 0.05 was considered to be statistically significant.
Author contributions
Study concept (TW); study design (TW, YZ); data acquisition (TW, XL, TS, YZ, ZX, JQ, ZZ, GS, SS, YH, YC); data analysis and interpretation (TW, YC); statistical analysis (YZ); preparing the manuscript (TW, YZ, TS).
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Dr. Xin Lin for mice and reagents. This work is supported by grants from National Natural Science Foundation of China (81772542 and 82072648), the Natural Science Foundation of Jiangsu Province (BK20190134), the Fundamental Research Funds for the Central Universities (021414380472), A project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PADD), The Open Projects of the Discipline of Chinese Medicine of Nanjing University of Chinese Medicine Supported by the Subject of Academic Priority discipline of Jiangsu Higher Education Institutions, and Nanjing Medical Science and Technique Development Foundation (ZKX17033 & YKK18127).
The EMBO Journal (2021) 40: e105320.
See also: N Papon et al (2021)
Footnotes
Correction added on June 1 2021, after first online publication: Since the initial publication the NCBI (SRP) database was corrected to (SRA).
Data availability
The microbiota sequencing data from this publication have been deposited to the NCBI SRA‡ ‡ database and assigned the identifier accession number (PRJNA661172, PRJNA661186, and PRJNA661617).
References
- Akdis M, Palomares O, van de Veen W, van Splunter M, Akdis CA (2012) TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J Allergy Clin Immunol 129: 1438–1449; quiz1450‐1 [DOI] [PubMed] [Google Scholar]
- Arce I, Martinez‐Munoz L, Roda‐Navarro P, Fernandez‐Ruiz E (2004) The human C‐type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur J Immunol 34: 210–220 [DOI] [PubMed] [Google Scholar]
- Arthur JC, Perez‐Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB et al (2012) Intestinal inflammation targets cancer‐inducing activity of the microbiota. Science 338: 120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Gharaibeh RZ, Muhlbauer M, Perez‐Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C (2014) Microbial genomic analysis reveals the essential role of inflammation in bacteria‐induced colorectal cancer. Nat Commun 5: 4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch SG, McKnight AJ, Seldin MF, Gordon S (1998) Cloning of a novel C‐type lectin expressed by murine macrophages. J Biol Chem 273: 18656–18664 [DOI] [PubMed] [Google Scholar]
- Baruch EN, Youngster I, Ben‐Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick‐Necula D, Raskin S, Bloch N et al (2021) Fecal microbiota transplant promotes response in immunotherapy‐refractory melanoma patients. Science 371: 602–609 [DOI] [PubMed] [Google Scholar]
- Bauche D, Joyce‐Shaikh B, Jain R, Grein J, Ku KS, Blumenschein WM, Ganal‐Vonarburg SC, Wilson DC, McClanahan TK, Malefyt RW et al (2018) LAG3(+) regulatory T cells restrain interleukin‐23‐producing CX3CR1(+) gut‐resident macrophages during Group 3 innate lymphoid cell‐driven colitis. Immunity 49: 342–352.e5 [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157: 121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, Diefenbach A, Greten FR, Ruland J (2017) Card9‐dependent IL‐1beta regulates IL‐22 production from group 3 innate lymphoid cells and promotes colitis‐associated cancer. Eur J Immunol 47: 1342–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt B, Zeng P, Zhu H, Sivaprakasam S, Li S, Xiao H, Dong L, Shiao P, Kolhe R, Patel N et al 2018) Gpr109a limits microbiota‐induced IL‐23 production to constrain ILC3‐mediated colonic inflammation. J Immunol 200: 2905–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden SD, Rowley G, Hinton JC, Thompson A (2009) Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar typhimurium. Infect Immun 77: 3117–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68: 394–424 [DOI] [PubMed] [Google Scholar]
- von Burg N, Turchinovich G, Finke D (2015) Maintenance of immune homeostasis through ILC/T cell interactions. Front Immunol 6: 416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos JG, Woo V, Viladomiu M, Putzel G, Lima S, Diehl GE, Marderstein AR, Gandara J, Perez AR, Withers DR et al (2018) Microbiota‐induced TNF‐like ligand 1A drives group 3 innate lymphoid cell‐mediated barrier protection and intestinal T cell activation during colitis. Immunity 49: 1077–1089.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, Vives F, Moon C, Penaflor E, Turner S et al (2014) Interleukin‐23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol 7: 842–856 [DOI] [PubMed] [Google Scholar]
- Coker OO, Nakatsu G, Dai RZ, Wu WKK, Wong SH, Ng SC, Chan FKL, Sung JJY, Yu J (2019) Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 68: 654–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K, Zhang C, Li J, Chen S, Liao C, Cheng X, Yu C, Yu Z, Jia Y (2019) cAMP receptor protein of Salmonella enterica serovar typhimurium modulate glycolysis in macrophages to induce cell apoptosis. Curr Microbiol 76: 1–6 [DOI] [PubMed] [Google Scholar]
- Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F (2015) Regulatory T‐cell response to enterotoxigenic Bacteroides fragilis colonization triggers IL17‐dependent colon carcinogenesis. Cancer Discov 5: 1098–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M, Dennehy KM, Hornsell WG, Guler R, Campanero‐Rhodes MA, Palma AS et al (2012) The C‐type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem 287: 25964–25974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, Bodian C, Ullman T (2007) Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology 133: 1099–1105; quiz 1340–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C (2013) Cancer: IL‐22: linking inflammation and cancer. Nat Rev Drug Discovery 12: 504 [DOI] [PubMed] [Google Scholar]
- Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM et al (2013) Innate lymphoid cells regulate CD4+ T‐cell responses to intestinal commensal bacteria. Nature 498: 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W et al (2015) Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria‐specific CD4(+) T cells. Science 348: 1031–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HR, Li F, Han H, Xu X, Li N, Wang S, Xu JF, Jia XM (2018) Dectin‐3 recognizes glucuronoxylomannan of cryptococcus neoformans serotype AD and Cryptococcus gattii serotype B to initiate host defense against cryptococcosis. Front Immunol 9: 1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M et al (2012) Interactions between commensal fungi and the C‐type lectin receptor Dectin‐1 influence colitis. Science 336: 1314–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargl J, Haybaeck J, Stancic A, Andersen L, Marsche G, Heinemann A, Schicho R (2013) O‐1602, an atypical cannabinoid, inhibits tumor growth in colitis‐associated colon cancer through multiple mechanisms. J Mol Med 91: 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, O'Neill LA (2015) Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25: 771–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher B, Wilson GJ, Reid DM, Mori D, Taylor JA, Besra GS, Yamasaki S, Willment JA, Brown GD (2016) Mycobacterial receptor, Clec4d (CLECSF8, MCL), is coregulated with Mincle and upregulated on mouse myeloid cells following microbial challenge. Eur J Immunol 46: 381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F (2013) Innate lymphoid cells sustain colon cancer through production of interleukin‐22 in a mouse model. J Exp Med 210: 917–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelink PJ, Bloemendaal FM, Li B, Westera L, Vogels EWM, van Roest M, Gloudemans AK, van 't Wout AB, Korf H, Vermeire S et al (2019) Anti‐TNF therapy in IBD exerts its therapeutic effect through macrophage IL‐10 signalling. Gut 69: 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S et al (2014) IL‐22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40: 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Dore J, Raedler A et al (2011) Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 141: 227–236 [DOI] [PubMed] [Google Scholar]
- Li C, Wang Y, Li Y, Yu Q, Jin X, Wang X, Jia A, Hu Y, Han L, Wang J et al (2018) HIF1alpha‐dependent glycolysis promotes macrophage functional activities in protecting against bacterial and fungal infection. Sci Rep 8: 3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G et al (2015) Interleukin‐22 promotes intestinal‐stem‐cell‐mediated epithelial regeneration. Nature 528: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR (2014) CX(3)CR1(+) mononuclear phagocytes support colitis‐associated innate lymphoid cell production of IL‐22. J Exp Med 211: 1571–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D et al (2009) Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol 10: 75–82 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P (2017) Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14: 399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF (2018) The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science 359: 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M (2014) Microbiota‐dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343: 1249288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee PK, Sendid B, Hoarau G, Colombel JF, Poulain D, Ghannoum MA (2015) Mycobiota in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 12: 77–87 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2019) Rerouting metabolism to activate macrophages. Nat Immunol 20: 1097–1099 [DOI] [PubMed] [Google Scholar]
- Na YR, Stakenborg M, Seok SH, Matteoli G (2019) Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol 16: 531–543 [DOI] [PubMed] [Google Scholar]
- Navarro‐Arias MJ, Hernandez‐Chavez MJ, Garcia‐Carnero LC, Amezcua‐Hernandez DG, Lozoya‐Perez NE, Estrada‐Mata E, Martinez‐Duncker I, Franco B, Mora‐Montes HM (2019) Differential recognition of Candida tropicalis, Candida guilliermondii, Candida krusei, and Candida auris by human innate immune cells. Infect Drug Resist 12: 783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleguezuelos‐Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB et al (2020) Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli . Nature 580: 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9: 313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP et al (2018) Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science 359: 91–97 [DOI] [PubMed] [Google Scholar]
- Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A (2004) Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 126: 451–459 [DOI] [PubMed] [Google Scholar]
- Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N et al (2007) Dectin‐1 is required for host defense against Pneumocystis carinii but not against Candida albicans . Nat Immunol 8: 39–46 [DOI] [PubMed] [Google Scholar]
- Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH et al (2010) Dectin‐2 recognition of alpha‐mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans . Immunity 32: 681–691 [DOI] [PubMed] [Google Scholar]
- Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A (2009) RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22‐producing NKp46+ cells. Nat Immunol 10: 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh‐Takayama N, Vosshenrich CA, Lesjean‐Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf‐Bensussan N, Mandelboim O et al (2008) Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29: 958–970 [DOI] [PubMed] [Google Scholar]
- Serafini N, Vosshenrich CA, Di Santo JP (2015) Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol 15: 415–428 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7–30 [DOI] [PubMed] [Google Scholar]
- Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, de’Angelis N, Rabot S, Canoui‐Poitrine F, Mestivier D et al (2019) Colorectal cancer‐associated microbiota contributes to oncogenic epigenetic signatures. Proc Natl Acad Sci USA 116: 24285–24295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE et al (2013) Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 13: 145–149 [DOI] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O'Shea JJ (2009) Lymphoid tissue inducer‐like cells are an innate source of IL‐17 and IL‐22. J Exp Med 206: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucey TM, Verma J, Harrison PF, Snelgrove SL, Lo TL, Scherer AK, Barugahare AA, Powell DR, Wheeler RT, Hickey MJ et al (2018) Glucose homeostasis is important for immune cell viability during candida challenge and host survival of systemic fungal infection. Cell Metab 27: 988–1006.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17‐cell‐mediated autoimmunity to environmental toxins. Nature 453: 106–109 [DOI] [PubMed] [Google Scholar]
- Wang T, Pan D, Zhou Z, You Y, Jiang C, Zhao X, Lin X (2016) Dectin‐3 deficiency promotes colitis development due to impaired antifungal innate immune responses in the gut. PLoS Pathog 12: e1005662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Fan C, Yao A, Xu X, Zheng G, You Y, Jiang C, Zhao X, Hou Y, Hung MC et al (2018) The adaptor protein CARD9 protects against colon cancer by restricting mycobiota‐mediated expansion of myeloid‐derived suppressor cells. Immunity 49: 504–514.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L (2010) A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139: 1844–1854.e1 [DOI] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF (2007) Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546 [DOI] [PubMed] [Google Scholar]
- Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N et al (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170: 548–563.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, Jiang YY, Jia XM, Lin X (2013) C‐type lectin receptors Dectin‐3 and Dectin‐2 form a heterodimeric pattern‐recognition receptor for host defense against fungal infection. Immunity 39: 324–334 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The microbiota sequencing data from this publication have been deposited to the NCBI SRA‡ ‡ database and assigned the identifier accession number (PRJNA661172, PRJNA661186, and PRJNA661617).