

Abstract
Mitochondrial homeostasis is essential for providing cellular energy, particularly in resource‐demanding neurons, defects in which cause neurodegeneration, but the function of interferons (IFNs) in regulating neuronal mitochondrial homeostasis is unknown. We found that neuronal IFN‐β is indispensable for mitochondrial homeostasis and metabolism, sustaining ATP levels and preventing excessive ROS by controlling mitochondrial fission. IFN‐β induces events that are required for mitochondrial fission, phosphorylating STAT5 and upregulating PGAM5, which phosphorylates serine 622 of Drp1. IFN‐β signaling then recruits Drp1 to mitochondria, oligomerizes it, and engages INF2 to stabilize mitochondria–endoplasmic reticulum (ER) platforms. This process tethers damaged mitochondria to the ER to separate them via fission. Lack of neuronal IFN‐β in the Ifnb –/– model of Parkinson disease (PD) disrupts STAT5‐PGAM5‐Drp1 signaling, impairing fission and causing large multibranched, damaged mitochondria with insufficient ATP production and excessive oxidative stress to accumulate. In other PD models, IFN‐β rescues dopaminergic neuronal cell death and pathology, associated with preserved mitochondrial homeostasis. Thus, IFN‐β activates mitochondrial fission in neurons through the pSTAT5/PGAM5/S622Drp1 pathway to stabilize mitochondria/ER platforms, constituting an essential neuroprotective mechanism.
Keywords: ATP, hydroxydopamine, mitochondrial metabolism, Parkinson disease, ROS
Subject Categories: Immunology, Metabolism, Neuroscience
Mitochondrial dynamics and energy supply of brain neurons are controlled by IFN‐β in vivo.
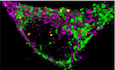
Introduction
Using up to 20% of the energy that is consumed by the entire body, the brain has the highest metabolic rate of all organs in mammals. Neurons have specific energy requirements, due to their highly compartmentalized morphology and activities, such as restoration of the gradient across the membrane after neuronal depolarization (Crotty et al, 2006), neurotransmitter recycling (Attwell & Laughlin, 2001), and axonal transport (Maday et al, 2014). Mitochondria provide most neuronal energy through oxidative metabolism. They are highly dynamic organelles that can fuse (i.e., mitochondrial fusion), divide (fission), or migrate in response to metabolic and environmental changes.
Defects in mitochondrial fusion or fission can result in gradual neurodegeneration (Burte et al, 2015). Disruptions in these processes have been implicated in familial and sporadic forms of Parkinson disease (PD) (Camilleri & Vassallo, 2014; Haelterman et al, 2014; Cieri et al, 2017), the most common progressive neurodegenerative movement disorder. Mitochondrial fusion mitigates oxidative stress by mixing the contents of partially damaged mitochondria as a form of complementation (Jahani‐Asl et al, 2007). Fission is needed to create new or smaller mitochondria for transport to axonal extremities (Fukumitsu et al, 2016). Also, fission contributes to essential quality control processes by enabling the removal of damaged mitochondria by mitophagy (Buhlman et al, 2014) and regulating apoptosis during high levels of cellular stress (Germain et al, 2005). Several factors that are necessary for mitochondrial fusion and fission have been identified, including members of the guanosine triphosphatase (GTPase) family, such as mitofusins (MFN1 and MFN2) on the outer mitochondrial membrane (OMM), and optic atrophy 1 (OPA1), which lies on the inner mitochondrial membrane (IMM) and is involved in cristae remodeling [reviewed in Bertholet et al (2016)].
Dynamin‐related protein 1 (Drp1) is another GTPase that is important in fission. On phosphorylation, Drp1 relocates to the mitochondrial surface and oligomerizes (Macdonald et al, 2014). The subsequent hydrolysis of GTP induces a conformational change in the Drp1 oligomers, generating traction forces that spawn contractile rings around mitochondria (Mears et al, 2011). Mitochondrial fission occurs at contact points with the endoplasmic reticulum (ER), which harbors the molecular machinery that is necessary for Drp1 maturation and complex formation, and coordinates actin polymerization to drive the assembly of Drp1 oligomers around mitochondria (Friedman et al, 2011; Hatch et al, 2014; Ji et al, 2015).
Interferon‐β (IFN‐β) is a cytokine that is primarily associated with immune responses during viral infections. IFN‐β signaling activates the Janus kinase (JAK)‐signal transducer and activator of transcription (STAT)1/2 pathway, leading to the transcription of IFN‐stimulated genes (ISGs). ISGs encode many antiviral effectors, controlling all of the steps in viral replication while limiting tissue damage and preventing autoimmunity (Schneider et al, 2014). Also, several members of the STAT family have nongenomic functions, such as STAT5 and its involvement in curvature of the ER membrane (Lee et al, 2013).
IFN‐β reduces the inflammatory response that is mediated by immune cell infiltration into the brain and is used to treat patients with multiple sclerosis (MS), a neuroinflammatory disease of the central nervous system (CNS) that is caused by immune cell‐induced neuronal demyelination (Teige et al, 2003; Liu et al, 2014, 2017). Mitochondrial functions are linked to microbial infections and pathogen recognition responses, including recognition of microbial double‐stranded DNA. Such events induce parallel antimicrobial immune responses through the activation of type I IFN pathway and response genes (West et al, 2015). These complementary pathways are well documented in immune cells, allowing them to fight infections. However, no direct function of the type I IFN, in particular IFN‐β in regulating mitochondrial homeostasis in general or in neurons has been described.
We have reported that Ifnb –/– mice experience age‐related motor and cognitive deficits, aggregation of α‐synuclein in intraneuronal Lewy bodies (LBs), and spontaneous neurodegeneration—collectively resembling the clinical features of PD with dementia (PDD) (Ejlerskov et al, 2015). Because dysfunctional mitochondria are a significant pathogenic factor in PD, we examined whether and how the absence of Ifnb affects neuronal mitochondrial homeostasis. Our results demonstrate that mitochondrial fission is altered in Ifnb –/– brains—a model of spontaneous PDD—and that in other PD models, induced by 6OH‐DA or by overexpression of human SCNA in the midbrain, IFN‐β regulates mitochondrial fission and mitophagy, processes that are critical in PD, thereby mitigating mitochondrial damage and preventing dopaminergic neuronal cell death.
Results
Accumulated mitochondria in Ifnb –/– neurons have enlarged and multibranched morphology
By 2D electron microscopy of thalamic sections, we observed an increase in mitochondrial accumulation in Ifnb –/– neurons, accompanied by large aggregates, none of which was detected in wild‐type neurons (Ifnb +/+) in the brain (Fig EV1A and B) (Ejlerskov et al, 2015). These findings were confirmed by the rise in MitoTracker Green (MTG) staining in primary cultures of Ifnb –/– versus Ifnb +/+ cortical neurons (CNs) (Fig EV1C and D). Treatment with recombinant IFN‐β (rIFN‐β) significantly reduced the mitochondrial accumulation in Ifnb –/– neurons (Fig EV1D).
Figure EV1. Mitochondrial dysfunction in cultured primary cortical neurons.

- TEM of thalami in 12‐month‐old Ifnb +/+ and Ifnb –/– mice. White arrows indicate inclusions. M indicates mitochondria. N denotes nuclei. Scale bars equal 2 μm.
- Mitochondrial mass, quantified as total mitochondrial area per cell body.
- MitoTracker staining in DIV6 Ifnb +/+ and Ifnb –/– CNs. Scale bars equal 50 μm.
- Total mitochondrial mass in Ifnb +/+ and Ifnb –/– primary cortical neuronal cultures, untreated or complemented with 30 U/ml rIFN‐β. Quantification was performed using MTG integrated density. Error bars mean + SEM from 3 independent cultures.
- Total cellular volume of CNs, quantified by β‐3 tubulin staining, matching the Tom‐20 staining in Fig 1.
- Mitochondrial volume, quantified from 3D imaging of Tom20 immunolabeling in Ifnb +/+ and Ifnb –/– MEFs. Scale bars equal 10 μm.
Data information: For all graphs, error bars mean SEM; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by t‐test (B; E; F) or two‐way ANOVA (D) with t‐test as post‐hoc.
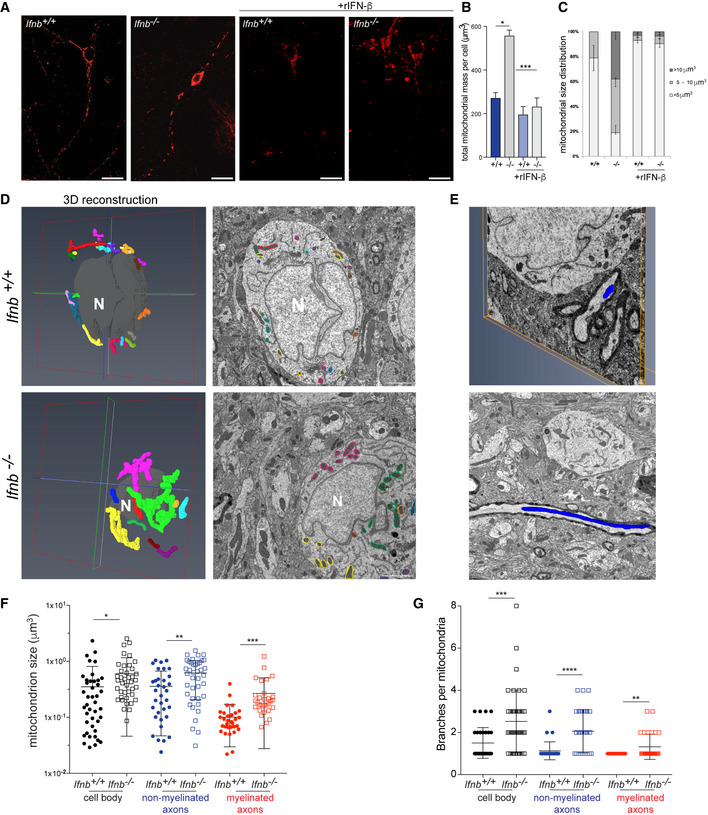
We then determined the morphological changes that occur when Ifnb is genetically deleted. By 3D immunofluorescence of the OMM protein Tom20, we verified the rise in total mitochondrial mass in Ifnb –/– primary CNs (Fig 1A and B) and found that this phenomenon was due to an increase in the size of individual mitochondria, wherein 40% of neurons had a mean mitochondrial volume above 10 μm3 (Fig 1C and D), despite the overall size of the cells remaining constant (Fig EV1E); these large mitochondria were found in cell bodies and axons. Further, MEFs that were derived from Ifnb –/– mice had nearly twice the mean volume of individual mitochondria compared with wild‐type Ifnb +/+ MEFs (Fig EV1F).
Figure 1. Mitochondria are enlarged and multibranched in Ifnb –/– CNs and thalamus tissue.
-
A–CImmunostaining of Tom20 (mitochondria) in cells positive for β3‐tubulin in DIV6 Ifnb +/+ and Ifnb –/– CNs with or without rIFN‐β for 24 h. (A) Projection of 3D image of Tom20. Scale bars equal 20 μm. (B) Quantification of total mitochondrial mass per neuron. (C) Mean individual mitochondria volume in Ifnb +/+ and Ifnb –/– CNs.
-
D–G3D electron microscopy of thalamus in 12‐month‐old Ifnb +/+ and Ifnb –/– mice. (D) Volume rendering of mitochondria (left panel) and corresponding mitochondria in 2D electron microscopy image (right panel) in cell bodies. Each individual mitochondrion is colored differently. Scale bars equal 2 μm. Voxel size equals 0.00878908x0.00878908x0.03 μm for the Ifnb +/+ neuron and 0.00439453x0.00438453x0.04 μm for Ifnb –/–. Nuclei are in gray and marked with “N’. (E) Reconstituted axonal mitochondria. Images extracted from Movies EV1 and EV2. (F) Size of individual mitochondria in cell bodies and nonmyelinated and myelinated axons. N > 30; error bars are SD. (G) Number of branches per mitochondrion. N > 30; error bars are SD.
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To confirm these findings, we reconstructed whole neurons, including axons, from the thalami of 1‐year‐old Ifnb –/– and Ifnb +/+ mice by 3D electron microscopy (EM) using serial block‐face technology (Fig 1D and E and Movies EV1 and EV2), allowing us to quantify mitochondrial size and structure at a resolution of approximately 5 nm/pixel. We observed a significant increase in mitochondrial size in all compartments of neurons in the Ifnb –/– brain, including myelinated axons (Fig 1F and G and Movies EV1 and EV2), in addition to multibranched mitochondria—i.e., mitochondria with multiple extensions and several axes. In contrast, wild‐type brains harbored few such mitochondria in nonmyelinated axons and none in myelinated axons (Fig 1G).
These results demonstrate that neuronal IFN‐β is essential for regulating mitochondrial structure and dynamics.
Defective mitochondria accumulate in neurons in Ifnb –/– brains
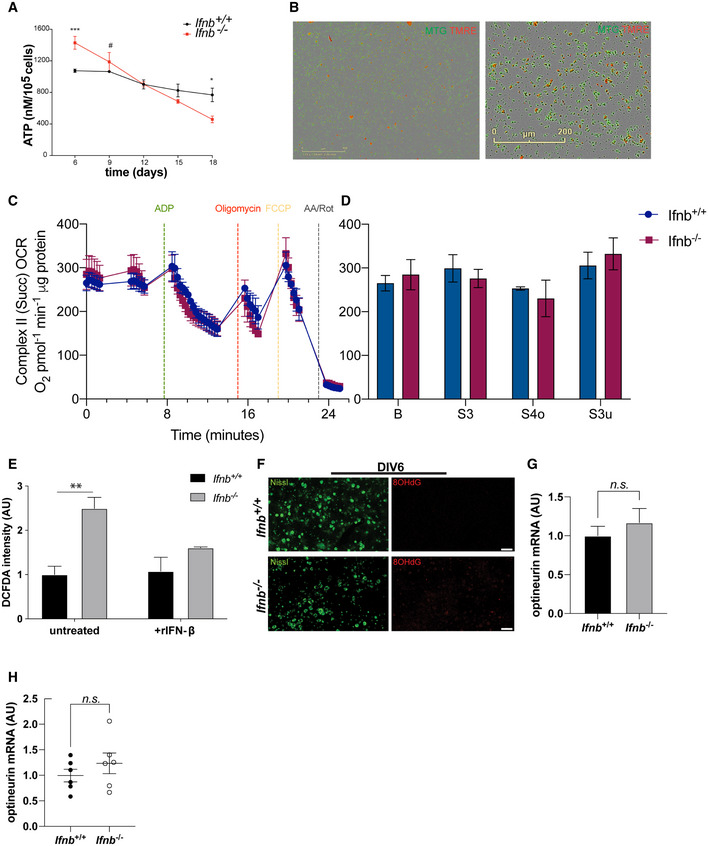
To determine whether these large mitochondria (Fig 1) were functional, we measured ATP and reactive oxygen species (ROS) levels in CNs and brain tissue. In the midbrains of Ifnb +/+ and Ifnb –/– mice, we quantified the ATP that was generated by these cells by luminescence‐based assay. We observed a slight increase in ATP content in 3‐day‐old Ifnb –/– brains, which declined significantly at age 3 months and decreased further at age 1 year (Fig 2A). We then measured the ATP content in CNs to determine whether this alteration in ATP production specifically affected neurons. Notably, young Ifnb –/– neurons—i.e., 6 days old [days in vitro (DIV)6]—contained approximately 1.5‐fold more ATP than wild‐type cells (Fig EV2A). But the ATP content decreased gradually in Ifnb –/– neurons on aging at DIV18 by roughly 70% compared with their initial content at DIV6 and was approximately 60% lower than in aged wild‐type neurons (Figs 2B and EV2A). Next, we determined whether the increase in mitochondrial mass caused the rise in ATP production. To this end, we quantified the ATP that was produced per mitochondrial unit. At DIV6, mitochondria from Ifnb –/– CNs generated approximately 40% less ATP than those from Ifnb +/+ CNs (Fig 2C). These findings demonstrate that mitochondrial deficiencies already exist in very young Ifnb −/− neurons (DIV6) and accumulate with age.
Figure 2. Deficient mitochondrial metabolism and function in Ifnb –/– CNs.

-
AATP quantification on aging from 3 days to 1 year in Ifnb +/+ and Ifnb –/– midbrains. Error bars are mean ± SEM from 6 mice.
-
BATP amounts in DIV18 CNs, untreated or complemented with 30 U/ml rIFN‐β. Error bars equal SEM from 5 (untreated cells) or 3 (treated cells) independent experiments.
-
CATP quantification on total mitochondrial mass in DIV6 CNs. Error bars show mean + SEM from 6 independent experiments.
-
D, ERespiration of isolated mitochondria from Ifnb +/+ and Ifnb –/– whole brains. OCR measurements under Complex I substrates (pyruvate/malate) were obtained at baseline (B) and on addition of ADP, oligomycin, FCCP, and rotenone/antimycin A to capture S3, S4o, S3 u, and nonmitochondrial respiration. N = 3–4 mice per group; bar graphs show mean + SE.
-
FQuantification of the respiratory control ratio (RCR), calculated as the ratio between S3 and S4o.
-
GROS levels, based on mean fluorescence intensity of DCFDA in DIV18 Ifnb +/+ and Ifnb –/– CNs, cultured with or without 30 U/mL rIFN‐β. Error bars equal SEM from 3 independent experiments.
-
HDCFDA quantifications in δIfnb N2A cells on treatment with the mitochondria‐specific antioxidant S3qet and MitoTEMPO.
-
IMitoSox quantification in DIV6 Ifnb +/+ and Ifnb –/– CNs. Rotenone was used as a positive control for the assay.
-
JExpression of oxidative stress response genes by qPCR in DIV6 Ifnb +/+ and Ifnb –/– CNs.
-
KImmunofluorescence of 8OHdG in CN cultures. Neurons were labeled with Nissl. Scale bars equal 50 μm.
-
LQuantification of 8OHdG by ELISA in brain tissues in young (aged 1.5–3 months) and old (aged 6–12 months) Ifnb +/+ and Ifnb –/– mice.
-
MImmunoblot for optineurin in 6‐week‐old midbrains of Ifnb +/+ and Ifnb –/– mice.
-
NImmunoblot for optineurin in DIV6 Ifnb +/+ and Ifnb –/– CNs with or without 30 U/ml rIFN‐β.
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Figure EV2. Mitochondria are dysfunctional in Ifnb−/− neuron and brain, but this is not due to alterations of complex II function.
-
AQuantification of ATP on aging from 6 to 18 days in Ifnb +/+ and Ifnb –/– CNs. Error bars are mean + SEM from 3 independent experiments.
-
BMonolayer of mitochondria after isolation, labeled with MitoTracker Green and TMRE, showing intact and polarized mitochondria.
-
C, DRespiration of isolated mitochondria from Ifnb +/+ and Ifnb –/– whole brains. OCR measurements under Complex II substrate (succinate supplemented with rotenone to inhibit Complex 1 activity) were obtained at baseline (B) and on addition of ADP, oligomycin, FCCP, and rotenone/antimycin A to capture S3, S4o, S3u, and nonmitochondrial respiration. N = 3–4 mice per group. Bar graphs show mean + SEM.
-
EROS levels based on mean fluorescence intensity of DCFDA in DIV6 Ifnb +/+ and Ifnb –/– CNs.
-
FImmunofluorescence of 8OHdG in DIV6 CN cultures. Neurons were labeled with Nissl. Scale bars equal 50 μm.
-
GExpression of optineurin mRNA in DIV6 Ifnb +/+ and Ifnb –/– CNs, quantified by qPCR.
-
HExpression of optineurin mRNA in Ifnb +/+ and Ifnb –/– brainstems, quantified by qPCR.
Data information: For all graphs, error bars mean SEM; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by t‐test (A; G; H) or two‐way ANOVA (E) with t‐test as post‐hoc.
We tested whether rIFN‐β restored the defects in Ifnb –/– neurons and whether it could preserve mitochondrial function on aging. We quantified the ATP in aged CNs, at DIV18, and noted that rIFN‐β rescued the mitochondrial dysfunction in Ifnb –/– CNs by enhancing ATP production by approximately 1.5‐fold (Fig 2B). Treatment of wild‐type neurons with rIFN‐β increased ATP production to the levels in DIV6 (Fig 2B).
To determine the cause of the decrease in ATP levels in the neurons and brain of Ifnb −/− mice, we performed Seahorse assay using isolated mitochondria from the brain (Figs 2D–F and EV2C and D). Seahorse coupling assay can be used to assess critical mitochondrial parameters, including coupled and uncoupled oxygen consumption rates (OCRs) and the respiratory control ratio (RCR), the ratio of oxidation rates in the presence of excess substrate and adenosine diphosphate (State 3) versus after ADP has been phosphorylated to a steady‐state concentration (State 4) (Brand & Nicholls, 2011).
We observed that basal respiration from Complex I (Fig 2D and E), activated by the downstream glycolysis products pyruvate and malate, decreased slightly in Ifnb −/− while rising modestly in CII (Fig EV2C and D). ADP was then added to the mitochondria to induce coupled respiration (State S3) by prompting ATP synthesis. The ability of Ifnb −/− mitochondria to stimulate this ATP synthesis declined by two‐thirds. Uncoupled respiration that was induced by the mitochondrial uncoupler FCCP (State S3 u), which circumvents Complex V, was unaffected in mitochondria from Ifnb −/− brains. Finally, the RCR (Fig 2F), representing the effectiveness of oxidative phosphorylation, decreased by over 25% in isolated mitochondria from Ifnb −/− brains, demonstrating a defect in ATP production.
The impairments in ATP production correlated inversely with the rise in ROS levels in Ifnb –/– neurons, based on staining with the ROS‐sensitive dye 2′,7′‐dichlorodihydrofluorescein diacetate (DCFDA) in DIV6 (Fig EV2E) and DIV18 CNs (Fig 2G). Long‐term rIFN‐β treatment (30 U/ml added every third day of culture) or treatment with the mitochondria‐specific antioxidants S3qel and MitoTempo significantly lowered ROS in Ifnb –/– neurons (Figs 2G and H and EV2E). Further, by MitoSox staining in CN cultures at DIV6, we observed a rise in mitochondrial superoxide (SOX) and the ability of rIFN‐β to counterbalance the mild increase in SOX that was induced by rotenone (Fig 2I).
To confirm the increase in ROS levels, we examined the oxidative stress response genes catalase (encoding catalase), gsr (glutathione reductase), park7 (the protein deglycase DJ‐1), ucp2 (mitochondrial uncoupler 2; UCP‐2), and sod1 and sod2 (superoxide dismutase 1 and 2; SOD1 and SOD2) by qPCR, of which gsr, sod2, and ucp2 were significantly upregulated in Ifnb –/– CNs (Fig 2J). To determine whether the rise in oxidative stress was causing damage to neurons, we stained cortical neurons for 8‐hydroxy‐2’deoxyguanosine (8OHdG), one of the major base lesions that are induced by oxidative stress and which has been linked to PD (Abe et al, 2003). In DIV18 primary CN cultures, we observed a robust increase in Ifnb –/– compared with wild‐type neurons (Fig 2K). A moderate increase was already present at DIV6 (Fig EV2F).
To determine whether excessive ROS were also produced in vivo, we measured 8OHdG in brain lysates of young (aged 6 weeks to 3 months) and old mice (aged 6 months to 1 year) by ELISA. As a result, we found a 50% increase in 8OHdG in young Ifnb –/– mice, rising further on aging (Fig 2L).
Finally, we observed the accumulation of optineurin, a mitophagy adaptor that recognizes damaged mitochondria (Wong & Holzbaur, 2014), in CNs and brain tissue (Fig 2M and N). This change was not due to altered optineurin mRNA expression (Fig EV2H).
These results suggest that IFN‐β is essential for mitochondrial homeostasis, energy production, and the prevention of excessive oxidative stress and accumulation of damaged mitochondria.
IFN‐β promotes mitochondrial fission by inducing Drp1 phosphorylation on serine 622 and its localization to mitochondria
Our results demonstrate that neuronal IFN‐β is essential for regulating mitochondrial morphology and activity. The large size of mitochondria and their multibranched structure—as opposed to an increase in the number of smaller mitochondria—indicates a defect in fission in neurons that lack Ifnb. To determine whether and how IFN‐β regulates mitochondrial fission, we first labeled the key fission protein Drp1 by immunofluorescence in CRISPR/Cas9‐engineered Ifnb‐depleted (δIfnb) versus wild‐type nontargeting control (NTC) N2A neuroblastoma cells (Fig 3A). Drp1 was found primarily in dense puncta that localized to the mitochondria, whereas the deletion of Ifnb excluded Drp1 from mitochondria. To quantify this difference, we fractionated mitochondria from the cytoplasmic contents of wild‐type NTC and Ifnb N2A neurons. In wild‐type and NTC N2A cells, Drp1 abound in the mitochondrial fractions, whereas the levels of mitochondria‐associated Drp1 were substantially reduced on depletion of Ifnb in 2 independently generated clones (Fig 3B and C). These data suggest that IFN‐β activates Drp1 for mitochondrial translocation and that the lack of IFN‐β in neurons causes its exclusion from mitochondria.
Figure 3. IFN‐β promotes the mitochondrial localization and S622 phosphorylation of Drp1.
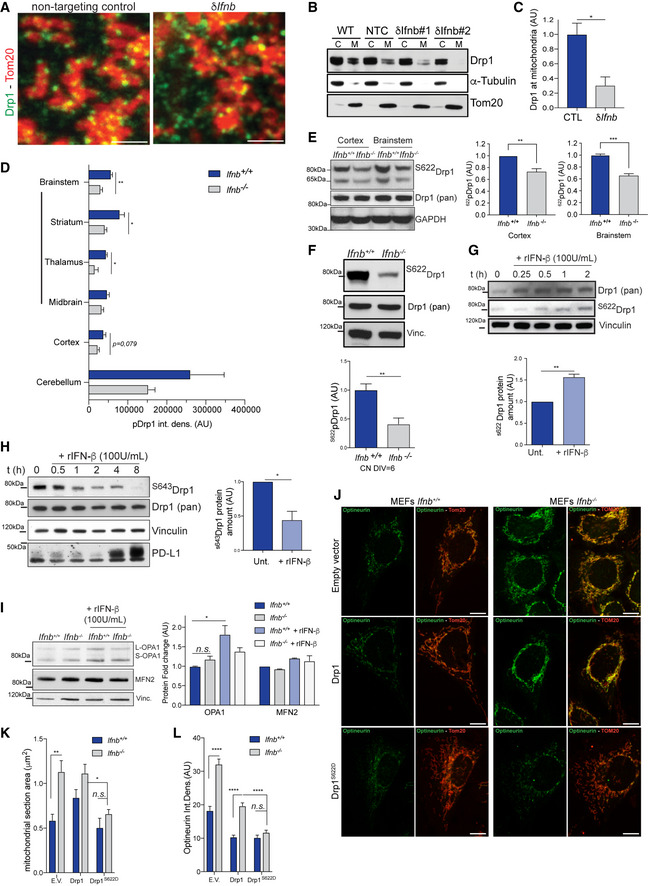
- Immunostaining of Tom20 and Drp1 in N2As depleted of Ifnb (δIfnb) by CRISPR/Cas9 and matching nontargeting control (NTC).
- Immunoblot of Drp1 from cell fractions of wild‐type (WT), δIfnb, and NTC N2As. Purity of cytoplasmic (C) and mitochondrial fractions (M) were evaluated by immunoblotting of tubulin and Tom20, respectively.
- Quantification of (B).
- Quantification of anti‐pDrp1 immunohistostains in different regions of the brain. Images are shown in Fig EV3.
- Immunoblot of phospho‐S622 Drp1 in cortices and brainstem of 3‐month‐old Ifnb +/+ and Ifnb –/– mice and quantification.
- Phospho‐S622‐Drp1 immunoblot and quantification in Ifnb +/+ and Ifnb –/– CN cultures. Vinculin was used as a loading control. Error bars are SEM of 3 independent experiments.
- Kinetics of Drp1 phosphorylation on S622 on rIFN‐β treatment in wild‐type CN cultures. Error bars are SEM from 3 independent experiments.
- Kinetics of Drp1 dephosphorylation at S643 on rIFN‐β treatment in wild‐type CN cultures. PD‐L1 was used as a positive control of rIFN‐β activity.
- MFN2 and OPA1 immunoblots and quantification in Ifnb +/+ and Ifnb –/– CN cultures and quantification. Vinculin was used as a loading control. Error bars are SEM from 3 independent experiments.
- Optineurin and Tom20 immunofluorescence in Ifnb +/+ and Ifnb –/– MEFs transfected with Drp1 or phosphomimetic Drp1.
- Mean mitochondrial section area in Ifnb +/+ and Ifnb –/– MEFs transfected with Drp1 or phosphomimetic Drp1. Error bars are SEM from 10 transfectants.
- Quantification of optineurin staining in (J).
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The localization of Drp1 to mitochondria is central for mitochondrial fission(Smirnova et al, 2001). The lack of this localization in Ifnb –/– neurons could thus be a major failure event that impairs mitochondrial fission, explaining our observation of large multibranched mitochondria (Figs 1 and EV1) and indicating that mitochondria are incapable of completing fission. To determine the molecular basis of this impairment in fission, we examined whether IFN‐β affects Drp1 activity directly by regulating its phosphorylation. Intracellular localization of Drp1 has been shown to be controlled in part by its phosphorylation on serine (S)622—the equivalent of serine 616 in the human homolog—which has been implicated as its active form during fission (Taguchi et al, 2007).
Overall, phosphorylated S622Drp1 levels decreased in the brains of 6‐week‐old to 3‐month‐old Ifnb –/– mice, particularly in the brainstem region, which encompasses the striatum, thalamus, and midbrain (Figs 3D and E, and EV3A and B). In primary CNs, the phosphorylation pattern of Drp1 was altered, reflected by a 50% decline in the phosphorylation of Drp1 on S622 in Ifnb –/– neurons compared with their wild‐type counterpart (Fig 3F). To determine whether IFN‐β promoted the phosphorylation of Drp1, we treated cultured CNs with rIFN‐β. rIFN‐β significantly induced neuronal Drp1 phosphorylation at S622 (Figs 3G and EV3B), as it did in N2A cells (Fig EV3A), and substantially decreased its S643‐phosphorylated form within 1 h of treatment (Fig 3H). The rIFN‐β‐induced phosphorylation of Drp1 on S622 in CNs peaked between 1 and 2 h post‐treatment and then declined rapidly, reaching baseline levels at 4–6 h (Fig EV3C). The lack of Ifnb did not have a significant impact on Drp1 mRNA (Fig EV3D) or total protein levels (Fig 3E and F).
Figure EV3. Drp1 phosphorylation on S622 is impaired in Ifnb –/– cells.
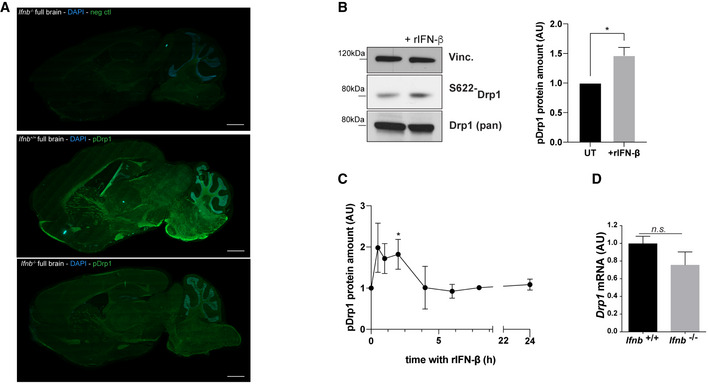
- Phospho‐S622‐Drp1 immunostaining in Ifnb +/+ and Ifnb –/–whole brains. Scale bars equal 1 mm. Control indicates incubation with secondary antibodies only.
- Phospho‐S622‐Drp1 immunoblots and quantification in Ifnb +/+ and Ifnb –/– CN cultures. Vinculin was used as a loading control. Error bars are SEM from 3 independent experiments.
- Kinetics of S622 phosphorylation on treatment with rIFN‐β.
- Drp1 mRNA levels in Ifnb +/+ and Ifnb –/– CN cultures by qPCR. Error bars equal SEM from 5 independent experiments.
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by t‐test.
We also examined whether IFN‐β functions in mitochondrial fusion. Knockout of Ifnb and treatment with rIFN‐β had no effects on the level of the profusion (Lee et al, 2012) and calcium uptake regulator (de Brito & Scorrano, 2008) MFN2 (Fig 3I). In addition, there was no shift between OPA1 isoforms, but total OPA1 levels rose in Ifnb knockout cells and on rIFN‐β treatment (Fig 3I).
To determine whether the absence of S622 phosphorylation in Drp1 mediates the increase in mitochondrial size, we overexpressed Drp1 or its phosphomimetic mutant form, S622D, in MEFs and measured mitochondrial size and optineurin levels. The overexpression of phosphomimetic Drp1 decreased the size of mitochondria and cleared the optineurin that had accumulated in Ifnb –/– cells, whereas overexpression of Drp1 had no effect (Fig 3J–L).
These data demonstrate that IFN‐β stimulates Drp1 by regulating its phosphorylation, which in turn promotes mitochondrial fission and its subsequent clearance.
IFN‐β promotes Drp1 oligomerization to stabilize ER–mitochondria interactions that are critical for fission
Drp1 is maintained in a targeted equilibrium on the OMM. For successful fission, a signal at sites of ER–mitochondria contact induces actin polymerization, which enhances Drp1 oligomerization. In parallel, the key mitochondrial fission ER protein inverted formin 2 (INF2) (Korobova et al, 2013) and the mitochondrial protein Spire 1C bind to actin and myosin for mitochondrial constriction (Ji et al, 2015). To determine whether IFN‐β is involved in this step of fission, we treated CNs with rIFN‐β with or without a biochemical cross‐linker, followed by electrophoresis under reducing conditions with DTT. Without rIFN‐β, Drp1 was found primarily in dimer form, whereas 2 h after rIFN‐β treatment, high‐molecular‐weight Drp1 oligomer levels rose significantly (Fig 4A and B), demonstrating that rIFN‐β induces Drp1 oligomerization and activity. Further, rIFN‐β increased the amounts of Drp1 and INF2 in the mitochondrial fraction of CNs (Fig 4C).
Figure 4. Lack of Ifnb alters ER/mitochondria platforms and mitochondrial fission.
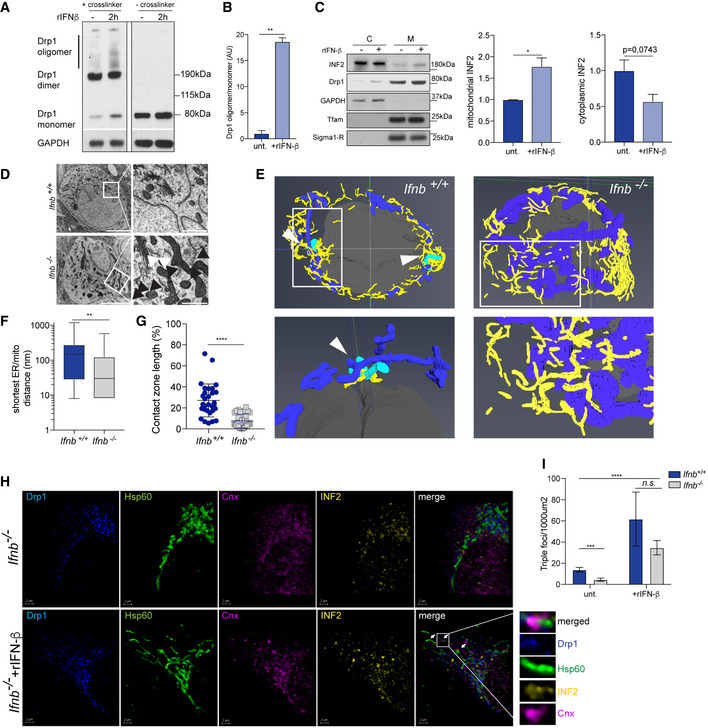
- Drp1 oligomerization by chemical cross‐linking in wild‐type CNs with or without rIFN‐β for 2 h.
- Quantification of (A).
- Immunoblot of INF2 and quantification from cell fractionation of Ifnb +/+ and Ifnb –/– CNs with or without rIFN‐β for 2 h.
- EM of Ifnb +/+ and Ifnb –/– thalami from 12‐month‐old mice. Scale bars equal 5 μm. Black arrows indicate mitochondria at a distance < 200 nm, and white arrows indicate distance > 200 nm.
- Volume rendering from 3D TEM images of Ifnb +/+ and Ifnb –/– thalami. Mitochondria are blue, the ER is yellow, and nuclei are gray. Mitochondria undergoing fission are cyan and indicated with a white arrow. 2D pictures are shown in Fig 2.
- ER/mitochondria closeness, evaluated as the distance from a mitochondrion to the nearest ER structure, extracted from EM images.
- Contact zone length, quantified as the percentage of the perimeter of a mitochondrion in close contact (< 20 nm) with ER, extracted from EM images.
- Projections of 3D images from immunostaining of Drp1 (blue), Hsp60 (mitochondria, green), INF2 (yellow), and calnexin (ER, magenta) in Ifnb –/– MEFs with or without rIFN‐β treatment for 6 h. Scale bar equal 2 μm. Arrows indicate fission foci.
- Quantifications of triple‐positive MT‐DR+Drp1+Cnx+ in MEFs. Images are shown in Fig EV3.
Data information: For all graphs, error bars mean SEM; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Next, we examined whether the lack of IFN‐β signaling caused a defect in mitochondria–ER fission by EM. We observed more interconnections between mitochondria and the ER in Ifnb –/– thalamic neurons as a result of the accumulation of mitochondria and ER compared with wild‐type cells (Fig 4D). In a reconstruction of the ER and mitochondrial networks by 3D transmission electron microscopy (TEM), we noted mitochondria that were surrounded by ER that were undergoing fission in wild‐type neurons, but such structures were absent in the reconstructions with Ifnb –/– thalamic neurons (Fig 4E). Although the mitochondria established closer contact with the ER in the Ifnb –/– thalamus (Fig 4E and F), the contact zone length at < 20 nm, which is the range of contact that is necessary for mitochondrial fission, decreased by approximately threefold (Fig 4G).
Then, we determined whether IFN‐β induces fission and ER remodeling. Treatment of Ifnb –/– MEFs with rIFN‐β effected the translocation of Drp1 to the mitochondria and increased the number of INF2/mitochondria, detected as positive co‐association of calnexin+ ER/Drp1/INF2/mitochondria foci (Fig 4H and I, and EV4).
Figure EV4. rIFN‐β induces mitochondrial fission in Ifnb +/+ and Ifnb –/– MEFs.
- Negative control for quadruple INF2‐Drp1‐Cnx and MitoTracker Deep Red (MT‐DR). Upper panel without MT‐DR staining and lower panel with MT‐DR staining.
- 3D projections of quadruple positivity for INF2‐Drp1‐Cnx and MT‐DR. Scale bars equal 5 μm. White arrows indicate fission points.
These results establish that neuronal IFN‐β is required to activate S622Drp1, which, on oligomerization, is necessary to create tight INF2–ER–mitochondrial platforms that are vital for Drp1‐dependent mitochondrial fission.
STAT5 is essential for IFN‐β–mediated PGAM5 and Drp1 phosphorylation in neurons
Based on the induction of Drp1 phosphorylation by IFN‐β, we examined the immediate molecular signaling events downstream of IFN‐β receptor during this activity. On binding to its receptor, IFN‐β activates STAT—in particular, STAT1/2 but also STAT3 and STAT5 (Uddin et al, 2003; Tanabe et al, 2005). Thus, we initially determined whether activation via IFN‐β signaling and consequent S622Drp1 phosphorylation are STAT1/2/3‐dependent, especially because it has recently been shown that certain STAT2‐deficient patients have lower levels of phosphorylated Drp1 and subsequent impairments in mitochondrial fission (Shahni et al, 2015). Moreover, these patients experience severe neurological deterioration following viral infection. By siRNA, we silenced the STAT2‐encoding gene in N2A cells (Fig EV5A) and CNs (Fig EV5B) and measured Drp1 phosphorylation on S622. We did not observe any change in Drp1 phosphorylation in these cells compared with wild‐type and nontargeting siRNA (NTC)‐transfected cells. Further, in primary CNs, STAT1 had no impact on Drp1 phosphorylation, and STAT3 had a moderate effect (Fig EV5B).
Figure EV5. Impact of modulation in STATs and PGAM5 on IFN‐β‐dependent Drp1 phosphorylation and mitochondrial health.
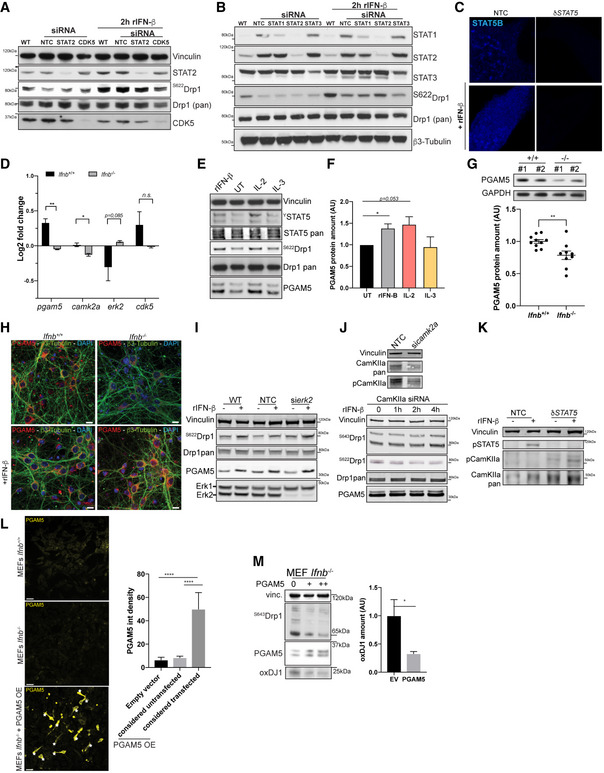
- Immunoblot of total and phospho‐S622‐Drp1 in wild‐type N2A cells or N2A cells silenced for STAT‐2 or CDK5. Vinculin was used as a loading control. CDK5 was used as a positive control for the loss of Drp1 phosphorylation.
- Immunoblot of total or phospho‐S622‐Drp1 in wild‐type CNs or CNs silenced for STAT1, STAT2, or STAT3. β3‐tubulin was used as a loading control.
- STAT5B immunostaining in N2A cells depleted for STAT5 A and B by CRISPR/Cas9 (δStat5) or with nontargeting control (NTC).
- PGAM5, CamKIIa, ERK2, and Cdk5 expression in Ifnb +/+ and Ifnb –/– CGNs extracted from microarrays.
- Immunoblot for STAT5, pan‐ and phospho‐Drp1, and PGAM5 on rIFN‐β, IL‐2, and IL‐3 treatment in wild‐type CNs at DIV6.
- Quantification of (E).
- Immunoblot for PGAM5 and quantification in Ifnb +/+ and Ifnb –/– CNs at DIV6.
- immunostaining for PGAM5 and β3‐tubulin in Ifnb +/+ and Ifnb –/– CNs with or without rIFN‐β. Nuclei stained with DAPI.
- Immunoblot in N2A control or N2A cells transfected with si‐NTC or si‐ERK2 with or without rIFN‐β for 2 h.
- Immunoblot for pan‐ and phosphor‐Drp1 and PGAM5 in N2A cells transfected with si‐CamKIIa with rIFN‐β for 0 to 4 h. Matching si‐NTC control is presented in Fig 6, and side‐by‐side blots with anti‐CamKIIa pan and phosphorylated from the same untreated samples are provided in the upper panel.
- Immunoblot for STAT5 and pan‐ and phospho‐CamKIIa in NTC or δSTAT5 N2A cells.
- Large‐field overviews of PGAM5 overexpression in Ifnb +/+ and Ifnb –/– MEFs. Stars indicate transfected cells. Scale bars equal 50 μm. Quantification in right panel of PGAM5 intensity in cells transfected with empty vector or cells overexpressing PGAM5 or not. Graph shows mean + SD; N = 10. Statistics analyzed by ANOVA and post hoc t‐test.
- Immunoblots and quantification of overexpression of PGAM5 in Ifnb −/− MEFs. Vinculin was used as a loading control.
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by t‐test (D; G; L; M) or one‐way ANOVA (F) with t‐test as post‐hoc.
Next, we examined STAT5, which only 1 report has suggested to relocate to mitochondria on stimulation with interleukin 2/3 in a leukemic T‐cell line in vitro [(Chueh et al, 2010) and reviewed in Meier and Larner (2014)]. The function of STAT5 in the phosphorylation of Drp1 under IFN‐β signaling and its consequent translocation to mitochondria in noncancer cells have not been studied. Notably, IFN‐β signaling induced the phosphorylation of STAT5 and S622Drp1 in primary CNs (Fig 5A). Further, on treatment of neurons with pimozide, a STAT5‐specific tyrosine phosphorylation inhibitor, for 16 h, phospho‐S622Drp1 was dose‐dependently suppressed—rIFN‐β effected the phosphorylation of STAT5 and S622Drp1 at a low dose of pimozide but not that of the latter at 50 μM (Fig 5A).
Figure 5. STAT5 controls Drp1 phosphorylation in neurons by promoting PGAM5 expression.

- Immunoblot of phospho‐S622 and pan‐Drp1 in wild‐type CNs treated with or without 10 or 50 μM pimozide and with or without 100 U/ml rIFNβ. *denotes a potential Drp1 cleavage product of approximatively 50 kDa.
- Immunoblot of phospho‐S622 and pan‐Drp1 in N2A cells depleted of STAT5 A and B by CRISPR/Cas9 (DKO) or with nontargeting control (NTC). Vinculin was used as a loading control.
- Quantification of (B).
- Projections of 3D images from immunostaining for phospho‐S622 Drp1 (yellow), Hsp60 (mitochondria, green), and calnexin (ER, magenta) in N2A cells depleted of STAT5 A and B (δStat5) or IFN‐β (δIfnb) or with nontargeting control (NTC). Cells with or without rIFN‐β treatment for 6 h. Scale bar equals 5 μm. Arrows indicate triple‐positive foci.
- Quantification of (D).
- Pgam5 promotor contains STAT5 binding motifs. Schematic of the upstream region of pgam5, encompassing the pgam5 promotor and pxmp2 with its terminator. The TTCT/CnA/GGAA motif is the optimal STAT5A and B binding motif and is specific to STAT5. OR indicates the origin of replication, as predicted by GPminer. Distances are in bp.
- Kinetics of the expression of pgam5 by qPCR in CNs on rIFN‐β treatment.
- PGAM5 levels by rtPCR in NTC, δIfnb, and δStat5 N2A cells with and without rIFN‐β for 6 h. Error bars are SD from 1 representative graph of 3 independent experiments.
- Immunoblotting against PGAM5 in Ifnb +/+ and Ifnb –/– CNs with or without rIFN‐β treatment for 2 h and quantification.
- Quantification of the impact of rIFN‐β on Drp1 phosphorylation after silencing of genes encoding the kinases CDK5, Camk2a, and ERK2 and the phosphorylase PGAM5.
- Kinetics of the impact of rIFN‐β on S622 and S643 phosphorylation. Quantified in (J).
- Immunostaining of PGAM5 and Hsp60 in NTC and δSTAT5 N2As transfected with PGAM5 or empty vector control and quantification of mitochondrial section area. Scale bars equal 5 μm. Error bars mean SEM from 10 individual cells.
- Optineurin immunostaining in Ifnb +/+ and Ifnb –/– MEFs overexpressing PGAM5 and quantification. Empty vector was used as control. Additional controls are shown in Fig EV5L. Error bars are SD from N = 30 cells.
- Oxidative stress in δIfnb N2A cells decreases on overexpression of PGAM5 and phosphomimetic Drp1 but not wild‐type Drp1. Immunoblots of PGAM5. Vinculin was used as a loading control.
- Loss of mitochondrial membrane potential in δIfnb N2A cells. Quantification of TMRE by automated Incucyte‐based imaging and analysis. Bars are mean + SEM.
Data information: For all graphs, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
These findings prompted us to determine the function of STAT5 in the phosphorylation of S622 and mitochondrial fission. We depleted both isoforms of STAT5 (δSTAT5)—STAT5A and STAT5B (double‐knockout; DKO)—in N2A neurons by CRISPR/Cas9, eliminating rIFN‐β‐mediated induction of STAT5 (Fig EV5C). rIFN‐β was unable to induce Drp1 phosphorylation on S622 when STAT5A/B were absent (Fig 5B and C). STAT5 has been suggested to influence ER remodeling in human pulmonary arterial endothelial cells (Lee et al, 2013), but it has not been linked to mitochondrial fission, particularly in neurons. In N2As that were depleted of Stat5A/B, we observed a significant decrease in phosphorylated S622Drp1 and less calnexin+ ER that was associated with Hsp60+ mitochondria, mimicking the cellular events that occur as a result of Ifnb deletion (Fig 5D). However, complementing the defect in δIfnb neurons with rIFN‐β resulted in more triple foci of phosphorylated S622Drp1 that relocated to the mitochondria and ER and consequently decreased the size of mitochondria, whereas rIFN‐β treatment of δStat5 N2A neurons failed to mend these defects (Fig 5D and E).
We aimed to identify downstream IFN‐β/STAT5 signaling molecules that regulate the phosphorylation of Drp1 and promote mitochondrial fission. Analyzing our previous Affymetrix data on Ifnb –/– CGNs (Ejlerskov et al, 2015), we detected a loss in phosphoglycerate mutase family member 5 (PGAM5), a mitochondrial serine/threonine phosphatase that mediates the dephosphorylation of S643 in Drp1, a precondition for phosphorylation of S622 (Wang et al, 2012), and in calcium/calmodulin‐dependent protein kinase type II subunit alpha (CaMKIIa), a kinase that can phosphorylate Drp1 directly on S622 (Bo et al, 2018) (Fig EV5D). Our analysis of the pgam5 promotor revealed 4 optimal binding sites for STAT5A/B (Fig 5F). We observed that treatment of CNs with rIFN‐β increased pgam5 mRNA (Fig 5G) and protein (Figs EV5E and H, and 5I). Similar to the deletion of Ifnb (δIfnb), depletion of STAT5 (δSTAT5) was essential for the induction of pgam5 expression (Fig 5H). However, in contrast to its ability to rescue defective pgam5 expression in δIfnb neurons, rIFN‐β failed to upregulate pgam5 when STAT5 was deleted in neurons (Fig 5H). Also, the activation of STAT5, using other cytokines that have been previously reported in this context (Chueh et al, 2010; Meier & Larner, 2014) as controls, revealed that only interleukin‐2 (IL‐2) could promote Drp1 phosphorylation on S622 and PGAM5 expression, whereas IL‐3 had no impact (Fig EV5E and F). The loss in PGAM5 was confirmed at the protein level in CNs and tissue (Figs 5I and EV5G and H).
Finally, we silenced pgam5 by siRNA and found that rIFN‐β‐induced dephosphorylation of Drp1 on S643 and phosphorylation on S622 were abrogated (Fig 5J and K). Notably, while testing kinases of S622 that could synergize with PGAM5 on IFN‐β regulation, we found that, in contrast to Erk2 and Cdk5, silencing CamKIIa inhibited this mechanism (Figs 5J and EV5A, I–K). However, CamKIIa phosphorylation of Drp1 is most likely STAT5‐independent, because CamKIIa was upregulated in response to depletion of STAT5 (Fig EV5K).
Next, to determine whether the loss in PGAM5 was associated with the defects in mitochondria due to the absence of Ifnb and IFNβ/STAT5 signaling, we overexpressed PGAM5 in STAT5 DKO N2A cells (Fig 5L), Ifnb –/– MEFs (Figs 5M and N, and EV5L and M), and δIfnb N2A cells. Similarly, overexpression of PGAM5 or phosphomimetic Drp1 in δIfnb N2A cells and Ifnb –/– MEFs cleared optineurin (Fig 5M), reduced the amounts of oxidized DJ1 (Figs 5N and EV5M), and restored mitochondrial function, as indicated by the increase in membrane potential (Fig 5O).
These results establish that IFN‐β is essential for mitochondrial homeostasis, controlling PGAM5 expression through a STAT5‐dependent pathway and the subsequent clearance of optineurin, likely through the phosphorylation of Drp1.
IFN‐β–PGAM5 in PD models is associated with preventing neurodegeneration
Based on the requirement of IFN‐β for mitochondrial homeostasis in neurons, we examined whether rIFN‐β maintains or restores mitochondrial integrity in other neurodegenerative processes. We induced neurodegeneration in mice using 6‐hydroxydopamine (6OH‐DA) by intracranial injection into the striatum, a model that is mediated by interference of mitochondrial homeostasis (Mazzio et al, 2004). We observed a loss of tyrosine hydroxylase‐positive (TH+) neurons in the brains of mice that were injected with 6OH‐DA (45% reduction compared with control, P < 0.0001, N = 4/group), but treatment of these mice with rIFN‐β resulted in significant partial rescue of the loss of TH+ neurons in the striatum (Fig 6A and B) and substantia nigra (Fig 6C and D).
Figure 6. Increased expression of pgam5 by rIFN‐β treatment is beneficial in 6OH‐DA‐ and hSCNA‐induced models of neurodegeneration.

-
A, BImmunohistochemistry of TH in coronal sections of the striatal region in mice treated with 6OH‐DA and rIFN‐β and quantification.
-
C, DImmunohistochemistry of TH in coronal sections of the substantia nigra in mice treated with 6OH‐DA, with, or without rIFN‐β and quantification.
-
E–KImmunofluorescence in the striatal region in mice treated with 6OH‐DA and rIFN‐β for optineurin (E), quantified in (F) and Tom20 (E), with quantification of individual mitochondrial size by mitochondrial section area (G). 8OHdG‐ (H, I), OxDJ1‐ (J‐K), and DAPI‐labeled nuclei. Scale bars equal 10 μm.
-
L, MPGAM5 and TH immunostaining (L) and quantification (M) in striatum from mice treated with 6OH‐DA and rIFN‐β.
-
NPercentage of right versus left forepaw use before and 21 days after injection with hSNCA (left hemisphere) and control lentivirus (CTL) or hSNCA and lenti‐Ifnb (right hemisphere). Bars are mean + SEM. ΨΨΨP < 0.001 by ANOVA and **P < 0.01 by post hoc Tukey’s multiple comparison correction. N = 6/hSCNA‐CTL and 8/hSCNA‐Ifnb.
-
OImmunostaining of TH and human α‐synuclein in SN. Scale bars equal 20 μm.
-
PTH immunostaining in brains of rats injected with hSNCA (left hemisphere) and control lentivirus (CTL) or hSNCA and lenti‐Ifnb (right hemisphere). Scale bar equals 1 mm.
-
QImmunostaining of PGAM5 in brains of rats injected with hSCNA and control or hSCNA and Ifnb. Scale bars equal 100 μm.
-
RQuantification of (P).
-
SQuantification of (Q).
-
TCorrelation of levels of PGAM5 and TH in rat brains injected with hSNCA (left hemisphere) and control lentivirus (CTL) or hSNCA and lenti‐Ifnb (right hemisphere) by Pearson correlation.
Data information: For all graphs, error bars show mean SEM; N = 4, unless otherwise stated. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Notably, optineurin was upregulated in the brains of 6OH‐DA‐injected mice, but its levels fell significantly on administration of rIFN‐β (Fig 6E and F). Further, mitochondria decreased in size in the brains of rIFN‐β‐treated mice (Fig 6E and G), consistent with the ability of rIFN‐β to impact mitochondrial dynamics. 6OH‐DA effects massive oxidative stress in neurons (Tieu, 2011; Galindo et al, 2012). We observed that 6OH‐DA induced a significant increase in 8OHdG (Fig 6H and I) and oxidized DJ1 (Fig 6J and K), another marker of ROS that is associated with PD (Saito, 2014). Treatment with rIFN‐β significantly downregulated both markers in the brains of 6OH‐DA‐injected mice (Fig 6H–K).
Thus, rIFN‐β partially reverts the loss in dopaminergic neurons by maintaining mitochondrial quality and alleviating mitochondrial oxidative stress in a chemically induced model of PD.
Finally, we addressed whether the upregulation in PGAM5 was relevant in neurodegenerative models of PD. In the 6OH‐DA mouse model, we observed low levels of PGAM5, and the lack of PGAM5 in the substantia nigra was compensated by rIFN‐β treatment (Fig 6L and M).
We have reported that Ifnb gene therapy prevents dopaminergic neuron loss in a genetic PD model, in which human α‐synuclein (hSNCA) is overexpressed in the rat brain (Ejlerskov et al, 2015). Here, we found that the administration of Ifnb gene therapy to these mice precluded the loss in motor function, as evidenced by the maintenance of forepaw use (Fig 6N), and protected TH+ neurons (Fig 6O–R). In support of this protective role for IFN‐β, PGAM5 levels were higher in the substantia nigra on overexpression of Ifnb (Fig 5Q–S), correlating with the survival of TH+ dopaminergic neurons (Fig 6T).
In conclusion, we have established that a lack of neuronal Ifnb, which causes spontaneous PD‐like pathology with motor and cognitive defects in mice (Ejlerskov et al, 2015), also impairs neuronal mitochondrial fission, a process that is upstream of mitophagy. This defect is associated with disrupted mitochondrial ATP production and regulation of oxidative stress. Additionally, we have demonstrated that IFN‐β promotes the oligomerization of Drp1 via the activation of STAT5 and PGAM5. The initiation of this cascade of events stabilizes the ER/mitochondrial platform, which in turn promotes the tethering of mitochondria by the ER for successful mitochondrial fission and the removal of damaged mitochondria. This model is supported by our findings that IFN‐β reverses 6OH‐DA‐ and hSCNA‐induced Parkinson‐like pathology, dopaminergic neuronal death, and excessive oxidative stress and promotes mitochondrial fission, thereby rescuing mitochondrial defects.
Discussion
IFN‐β‐dependent mitochondrial fission is essential for maintaining neuronal homeostasis
Neurons are postmitotic cells—i.e., they are unable to regenerate—with high metabolic activity and must produce and regulate energy rapidly to conduct presynaptic signals. These properties render mitochondrial homeostasis a critical process for neuronal signaling and survival, because mitochondria provide most neuronal energy through oxidative metabolism (Devine & Kittler, 2018). Disruptions in mitochondrial homeostasis, including the regulation of oxidative metabolism, dynamic movements, fusion, and fission, could have detrimental outcomes, such as the aggregation of senescent mitochondria and gradual neurodegeneration (Burte et al, 2015). Such mitochondrial defects are common in PD (Camilleri & Vassallo, 2014; Haelterman et al, 2014; Cieri et al, 2017).
We have reported that genomic deletion of Ifnb or its receptor, Ifnar, in the mouse brain precipitates neurodegeneration (Ejlerskov et al, 2015). The relevance of the lack of Ifnb signaling to PD might be attributed to the pleiotropic effects of IFNβ through its receptor. IFNβ signaling induces classical neuronal pPI3K/pAKT pathways to maintain neuronal innate immunity and sustain the neuronal capacity to regulate the brain microenvironment and neuroinflammation (Liu et al, 2013, 2014, 2017). IFNβ signaling activates classical JAK‐STAT signaling to regulate autophagy (Ambjorn et al, 2013), and we have demonstrated here that it is also involved in mitochondrial homeostasis by initiating mitochondrial fission.
Because Ifnb –/– mice present with unexplained mitochondrial alterations, we examined how IFN‐β regulates mitochondrial dynamics and function in neurons in the current study. Our data establish that IFN‐β governs mitochondrial fission in neurons by regulating the phosphorylation and activation of the STAT5/PGAM5/Drp1 pathway, which in turn stabilizes mitochondria/ER platforms to complete the fission process.
The defect in mitochondrial fission due to the deletion of Ifnb was also associated with impaired oxidative metabolism. Genomic lack or CRISPR/Cas9‐mediated deletion of Ifnb in brain tissue, cultured primary cortical neurons, and N2A neurons led to aging‐related accumulation of oxidative stress and deficient Complex I activity, impairing ATP production. These events correlated with an increase in mitochondrial mass, attributed to larger, multibranched mitochondria, whereas phosphorylation of S622 in the fission protein Drp1 fell significantly. Although the translocation of Drp1 toward mitochondria generally follows S622 phosphorylation, it is not absolute, particularly in postmitotic neurons (Cho et al, 2014). Our results, however, show that the inability of Drp1 to translocate to mitochondria and the accumulation of large mitochondria correlate with the loss of Drp1 phosphorylation at S622 and the inactivation of fission.
IFN‐β‐STAT5 signaling regulates the posttranslational modifications of Drp1
Mice that have a neuron‐specific deficiency in Dnml1, the gene that encodes Drp1, die postnatally due to impaired brain development (Ishihara et al, 2009). Primary cortical neurons from these mice harbored aggregated mitochondria that failed to distribute properly throughout the cell processes, decreasing the number of neurites and impairing synapse formation, characteristics that were also observed in the current study in Ifnb –/– brains and primary cortical neurons. We expect the large mitochondria in Ifnb –/– brains, particularly in myelinated axons, to have a major impact on their transport along axons, because they occupy most of the space in the axon.
Notably, we did not observe any direct function for STAT2/3 in mitochondrial homeostasis, especially given that STAT2 is an important signaling molecule in the IFN‐β pathway (Steen & Gamero, 2013), which was recently reported to be associated with virally induced neurodegeneration in a family with STAT2 mutations (Shahni et al, 2015). Instead, we found STAT5 activation and signaling to be essential for IFN‐β‐mediated mitochondrial fission—specifically by regulating the posttranslational modifications of Drp1. This finding was not due to alterations in Dnml1 expression, despite STAT5 being a transcription factor. Similarly, total Drp1 protein levels in Ifnb‐deficient neurons and on IFN‐β treatment remained unchanged; although IFN‐β substantially decreased S643‐phosphorylated Drp1 levels, it induced the phosphorylation of S622. Moreover, we observed that STAT5 regulates PGAM5 expression in neurons, which in turn governs Drp1 phosphorylation; PGAM5 has been reported to mediate the dephosphorylation Drp1 at S643, a prerequisite for phosphorylation of S622 (Wang et al, 2012). Both events were transient, returning to their initial levels 4–6 h postinduction, consistent with the tight regulation of the JAK/STAT pathway (Murray, 2007).
IFN‐β/STAT5 signaling recruited another mitochondrial fission ER protein, INF2 (Korobova et al, 2013), to the mitochondria/ER platform. Although STAT5 localizes to the mitochondria on stimulation with interleukins 2 and 3 [reviewed in Meier and Larner (2014)], this mechanism has not been demonstrated in neurons, and the importance of STAT5 in mitochondrial fission has not been established, nor has its involvement in the regulation of PGAM5 or Drp1. STAT5 promotes remodeling of the ER in human pulmonary arterial endothelial cells (Lee et al, 2013) through interaction with the GTPase atlastin 3 (ATL3, yeast Sey1p). Remodeling of the ER must occur for the fission activity of Drp1. The ER coordinates and accompanies the polymerization of actin filaments, which is necessary for Drp1 to oligomerize, which then forms constriction rings around mitochondria (Korobova et al, 2013; Ugarte‐Uribe et al, 2018). The function of IFN‐β‐STAT5 in Drp1 phosphorylation facilitates its stabilization at the OMM, maintaining it in a platform with INF2‐ER to allow it to surround the mitochondria and ensure successful fission. This model is supported by our finding that the ER is closely associated with mitochondria in Ifnb –/– primary neurons and brain tissue but is unable to envelop mitochondria, in contrast to IFN‐β‐sufficient neurons. Loss of these platforms impairs mitochondrial fission and mitochondrial homeostasis, potentiating neuronal cell death (Grimm, 2012).
Ifnb –/– neurons initially compensate for deficient ATP synthesis but are exhausted by early aging
Although mitochondria from Ifnb –/– mice produced less ATP, their increased overall mass initially compensated for this loss. In myelinated axons, the greater size of the mitochondria could also have resulted concomitantly from the preservation of large mitochondria to offset the decline in ATP supply, as has been reported during demyelination (Campbell & Mahad, 2011), which is pertinent to demyelinating diseases, including MS. We did not observe any direct impact of Ifnb deficiency or rIFN‐β treatment on mitochondrial fusion in CNs, which was unexpected, because fission and fusion are believed to be tightly co‐regulated. Further, we noted a mild increase in OPA1 in the absence of Ifnb and on rIFN‐β treatment. OPA1 has been implicated in fusion and fission as an important modulator of the morphology of mitochondrial cristae (Del Dotto et al, 2017; Quintana‐Cabrera et al, 2017). OPA1 determines respiratory efficiency by controlling the assembly and stability of respiratory chain super‐complexes, which synthesize ATP (Cogliati et al, 2013). Consequently, mild overexpression of OPA1 increases respiratory efficiency and can contribute to harmful phenotypes in genetic mouse models of mitochondrial diseases (Varanita et al, 2015). The upregulation in OPA1 protein that we observed might sustain the early synthesis of ATP in Ifnb knockout animals; however, it is likely to be attributed to a compensatory mechanism—not linked directly to IFN‐β.
IFN‐β protects against oxidative stress and neurodegeneration
In addition to a deficiency in ATP synthesis, we observed an increase in ROS levels in Ifnb –/– neurons and brains, accompanied by impaired fission. Consistent with our findings, mitochondrial degradation is reported to be a fission‐dependent phenomenon (Frank et al, 2012; Burman et al, 2017), the lack of which contributes to the accumulation of senescent mitochondria on aging, resulting in high ROS levels, which in turn could amplify the inability to degrade senescent mitochondria in neurons (Meng et al, 2011). Although we noted a defect in ATP synthesis by mitochondria and greater ROS levels in 6‐day‐old Ifnb –/– cortical neuron cultures, aging compounded these metabolic disruptions. Notably, rIFN‐β maintained ATP synthesis and lowered ROS levels by mitochondria in aged neurons, implicating it as an important factor and a potential therapeutic target in mitochondrial integrity.
In vivo, in the 6OH‐DA‐ and hSCNA‐induced neurodegenerative models of PD, IFN‐β prevented dopaminergic neuronal loss by reducing the damage that was caused by extensive oxidative products, thus maintaining mitochondrial health. Further, IFN‐β restored mitochondrial fission, based on the normalization in mitochondrial size. 6OH‐DA—like other chemicals that induce dopaminergic neuronal cell death, such as rotenone—causes mitochondrial defects by disrupting Drp1‐dependent fission (Galindo et al, 2012). Whereas extensive mitochondrial fission is harmful for neurons (Lassus et al, 2016), normal mitochondrial fission is a prerequisite for successful mitophagy. Consistent with this function, rIFN‐β or Ifnb gene therapy restored fission and mitochondrial metabolism, thus promoting the survival of dopaminergic neurons.
IFN‐β‐mediated signaling regulates mitochondrial metabolism and homeostasis
Collectively, our results establish that neuronal IFN‐β is central in the maintenance of mitochondrial homeostasis in the absence of stress or infection, initiating a series of events. First, IFN‐β is required for mitochondrial fission—by inducing phosphorylated STAT5, it upregulates PGAM5 to phosphorylate S622 of Drp1, which then recruits Drp1 to mitochondria, oligomerizes it, and engages INF2 to stabilize the mitochondrion‐ER platform. This process then leads the ER to envelop the damaged mitochondrion to facilitate its separation by fission. As a result, IFN‐β promotes the removal of damaged and senescent mitochondria to maintain the balance between mitochondrial ATP and oxidative metabolites. Finally, IFN‐β‐mediated regulation of mitochondrial metabolism and homeostasis prevents neuronal cell death—particularly in dopaminergic neurons, as shown in our study by in vivo treatment in the 6OH‐DA‐ and hSCNA‐induced neurodegenerative models of PD and as reported in a familial model of PD (Ejlerskov et al, 2015).
We have demonstrated that Ifnb –/– mice—a model for PD and LB with dementia (Abou‐Sleiman et al, 2006; Ejlerskov et al, 2015)—experience significant mitochondrial alterations at age 4–6 weeks, before they exhibit any obvious neurological deficits or LB pathologies. Such changes are significant in the pathogenesis of PD, and in this study, we have shown that they precede the other PD‐related physiopathological criteria in the Ifnb –/– brain, implicating IFN‐β‐STAT5‐mediated molecular cascades as potential therapeutic targets in neurodegenerative diseases, such as PD/LB dementia, and other mitochondrial diseases.
Materials and Methods
Mice and cell culture
Ifnb –/– mice (Erlandsson et al, 1998) were backcrossed 20 generations to B10.RIII mice. The wild‐type controls were Ifnb +/+ littermates. Mice were housed in standard facilities. Sex‐ and weight‐matched mice were used in experiments, performed in accordance with the ethical committees in Denmark, and approved by our institutional review boards (ethical permission #2013‐15‐2934‐00807, 2018‐15‐0201‐01572). Cortical neurons (CNs) were isolated from the cortex of 1‐day‐old mice and cultured for the indicated times. Neurons are fully differentiated by DIV6 and then gradually undergo neurodegenerative‐like cell death at approximately DIV15 (Kim et al, 2007).
Immortalized mouse embryonic fibroblasts (MEFs) were generated using a pBabe‐neo LargeTcDNA retrovirus (Hahn et al, 1999), transduced into MEFs from Ifnb +/+ or Ifnb –/– mice. SV40‐expressing cells were selected with G418.
Overexpression vectors for PGAM5 (Wang et al, 2012), Drp1 (addgene #44599), and Drp1 S622D (corresponding to S579D) (addgene #46343) (Loson et al, 2013) were transfected using Lipofectamine 2000 (Invitrogen) per the manufacturer’s recommendations. The transfection efficiency was 75–90% in N2As; thus, no selection was performed. Transfected MEFs were selected by immunostaining, as shown in Fig EV5L.
6‐Hydroxydopamine lesions were generated in mice (age 8–15 weeks, body weight 28.4 g ± 2.2 g) as described (Francardo et al, 2011). Briefly, mice were anesthetized with isoflurane and placed in a stereotactic frame in the flat‐skull position. 6‐Hydroxydopamine (3.2 µg/µl free base concentration in 0.02% ascorbate‐saline) was injected at the following coordinates, in mm relative to the bregma, sagittal suture, and dural surface: AP = +0.3, L = 2.3; DV = 2.9. Toxin solution (1 µl) was injected at each site. Sham‐operated mice were injected with 1 µl 0.02% ascorbate‐saline solution. Mice received rIFN‐β (100 U in DMSO or DMSO only for controls) with 6OH‐DA and then every other day for 10 days by intraperitoneal injection. Injections were assigned to the animals through block randomization, and the number of injected animals was defined, based on the robust effect of 6OH‐DA with respect to the 3R ethical guidelines. On Day 10, mice were deeply anesthetized with isoflurane (4%) and perfused intracardially with saline solution, followed by ice‐cold 4% PFA (pH 7.4, Sigma‐Aldrich). Brains were removed, postfixed with 4% PFA for 24 h, and immersed in 25% sucrose solution (Sigma‐Aldrich). Then, 30‐μm‐thick coronal sections were generated on a microtome (SM 2000R, Leica Microsystems, Wetzlar, Germany).
CRISPR/Cas9 gene editing
The Neuro2A (N2A) neuroblastoma cell line was cultured in DMEM, containing Gluta‐Max that was supplemented with 10% FBS. Ifnb and STAT5A‐ and B‐depleted N2A cells were generated by CRISPR/Cas9 editing. Two sets of 24‐nt oligo pairs were designed for Ifnb (first exon, Table 1), Stat5a (second exon, Table 1), and Stat5b (first exon, Table 1) using the CRISPR Design Tool (http://crispr.mit.edu/). SgRNA oligos were cloned into LentiCRISPR.V2, and constructs were validated by sequencing. Plasmids that harbored the cloned or nontargeting sgRNAs and the packaging plasmids (Pmd2G and PsPAX2) were transfected into HEK293 FT cells for lentiviral production. N2A cells were transduced with the viruses, and Cas9 expression and knockout efficiency were assessed by Western blot.
Table 1.
List and sequences of the primers used in this study.
| qPCR primers | |||||
|---|---|---|---|---|---|
| Gene | Position | Specie | Sequence | Sense | Reference |
| catalase | 203 | ms | GAGGCGGGAACCCAATAG | F | Shanmugam et al (2017) |
| catalase | 304 | ms | GTGTGCCATATCGTCAGTGAA | R | Shanmugam et al (2017) |
| DJ1 | 285 | ms | GTGCAGTGTAGCCGTGATGT | F | Yu et al (2016) |
| DJ1 | 379 | ms | CCTCCTGGAAGAACCACCAC | R | Yu et al (2016) |
| Drp1 | 273 | ms | GTGGGAAGAGCTCATTGCTGGAAAGC | F | Uo et al (2009) |
| Drp1 | 512 | ms | CTTGTCGAATTTCATCAAAATCTGTGTAAAG | R | Uo et al (2009) |
| gsr | 747 | ms | CACGGCTATGCAACATTCGC | F | Shanmugam et al (2017) |
| gsr | 815 | ms | GTGTGGAAGCGGTAAACTTTTTC | R | Shanmugam et al (2017) |
| PGAM5 | 294 | ms | ATCTGGAGAAGACGAGTTGACA | F | Lu et al (2014) |
| PGAM5 | 439 | ms | CCTGTTCCCGACCTAATGGT | R | Lu et al (2014) |
| SOD1 | 561 | ms | GTGATTGGGATTGCGCAGTA | F | Allister et al (2013) |
| SOD1 | 631 | ms | TGGTTTGAGGGTAGCAGATGAGT | R | Allister et al (2013) |
| SOD2 | 2852 | ms | TGGACAAACCTGAGCCCTAAG | F | Shanmugam et al (2017) |
| SOD2 | 3372 | ms | CCCAAAGTCACGCTTGATAGC | R | Shanmugam et al (2017) |
| UCP2 | 522 | ms | CAGCCAGCGCCCAGTACC | F | Joseph et al (2002) |
| UCP2 | 653 | ms | CAATGCGGACGGAGGCAAAGC | R | Joseph et al (2002) |
| Primers for CRISPR/Cas9 editing | |||
|---|---|---|---|
| Gene | Specie | Sequence | Sense |
| STAT5a oligo pair 1 | ms | CACCGGTGGCCTGACCTCGGTCCT | F |
| STAT5a oligo pair 1 | ms | AAACAGGACCGAGGTCAGGCCACC | R |
| STAT5a pair 2 with 5′ extension | ms | CACCgTAATCCCCAGGACCGAGGTC | F |
| STAT5a pair 2 with 5′ extension | ms | AAACGACCTCGGTCCTGGGGATTAC | R |
| STAT5b oligo pair 1 | ms | CACCGGGGAAATGCTGGCCGTACA | F |
| STAT5b oligo pair 1 | ms | AAACTGTACGGCCAGCATTTCCCC | R |
| STAT5b Oligo pair 2 with 5′ extension | ms | CACCgTCTGGTGAAGGGCATCGCCC | F |
| STAT5b Oligo pair 2 with 5′ extension | ms | AAACGGGCGATGCCCTTCACCAGAC | R |
| Ifnb CRISPR | ms | CACCGGATCTTGAAGTCCGCCCTGT | F |
| Ifnb CRISPR compl | ms |
AAACACAGGGCGGACTTCAAGATCC |
R |
Immunostaining and microscopy
Immunostainings were performed as described (Kim et al, 2013). Antibody references and concentrations are given in Table 2. CNs were identified using β3‐tubulin or Nissl (NeuroTrace 515/535 and 640/660; Invitrogen) at 1:200. Donkey secondary antibodies were used at 1:500 (Jackson ImmunoResearch for anti‐rabbit, anti‐goat, and anti‐chicken) or 1:1,000 (Abcam for anti‐mouse). Wherever specified, cells were incubated with MitoTracker Deep Red (Invitrogen, M22426) for 20 min at 100 nM in FluoroBrite DMEM medium (GIBCO), supplemented with 10% FBS, prior to washes and fixation in 4% PFA for 12 min. Signals were quantified in FIJI. For mitochondrial sections, binary and watershed filters were applied. Settings were tightly controlled for all fluorescent images that required quantification and adjusted to the brightest observed condition to avoid misinterpretation due to potential oversaturation, applied similarly to all conditions in an experiment.
Table 2.
List of antibodies used in this study.
| Protein | Supplier | Use |
|---|---|---|
| Calnexin | Abcam—ab140818 | IF 1:500 |
| CamKIIa | Santa Cruz—sc‐13141 | WB 1:100 |
| CamKIIa (pThr286) | CST—12716 | WB 1:500 |
| CDK5 | Abcam—ab40773 | WB 1:500 |
| DJ1 | Millipore—AB9718 | IF 1:500 |
| DJ1 (oxidized) | Millipore—MABN1773 | IF 1:200 |
| Drp1(pan) | Abcam—ab56788 | IF 1:200; WB 1:1,000 |
| Drp1(pS616) | CST—4355 | IF 1:200; WB 1:1,000 |
| Drp1(pS637) | CST—4867 | WB 1:1,000 |
| ERK | CST—9102 | WB 1:1,000 |
| GAPDH | Abcam—ab9484 | WB 1:5,000 |
| dGuanine (8 hydroxy‐) | Santa Cruz—sc‐139586 | IF 1:100 |
| Hsp60 | Santa Cruz—sc‐1052; ENZO ADI‐SPA‐828 | IF 1:100 both |
| INF2 | Bethyl—A303.427A‐T | IF 1:200; WB 1:1,000 |
| MFN2 | Abcam—ab56889 | WB 1:1,000 |
| OPA1 | Abcam—ab157457 | WB 1:500 |
| Optineurin | Santa Cruz—sc‐166576 | IF 1:500; WB 1:500 |
| PGAM5 | Abcam—1ab26534 | IF 1:500; WB 1:5,000 |
| STAT1 | CST—9172 | WB 1:1,000 |
| STAT2 | CST—72604 | WB 1:1,000 |
| STAT3 | LSBio—LS‐C10383/38518 | WB 1:1,000 |
| STAT5A | Life Technologies—133600 | IF 1:200, WB 1:1,000 |
| STAT5B | Life Technologies—135300 | IF 1:200, WB 1:1,000 |
| STAT5A/B (pTyr) | Abcam—ab83212 | IF 1:200, WB 1:500 |
| TH | Pel‐Freeze—P40101 | IHC 1:1,000 |
| Tom20 | Santa Cruz—sc‐11415; Abcam—ab186734 | IF 1:100; WB 1:1,000 |
| β3‐Tubulin | Santa Cruz—sc‐58888 | IF 1:1,000 |
| Vinculin | Sigma—V9131 | WB 1:10,000 |
Mitochondrial shape was analyzed by 3D imaging, performed in 0.22‐μm increments and with a pinhole < 0.8 airy unit under a Leica TCS Sp8 X confocal microscope with HyD decreasing dark noise and Huygens deconvolution (minimal resolution = 1,034 × 1,034; 2,068 × 2,068 for fission point imaging, bidirectional acquisition, frame average = 4, constant laser power and gain < 150% on HyD, and constant between replicates). Images were analyzed in VoloCity using an intensity‐based filter. For 3D reconstruction of multicolor images, a Gaussian filter was applied (Sigma 1.0) using LAX. Sixfold digital zooming with a resolution of 2,068 × 2,068 was performed to examine mitochondrial localization of Drp1, and eightfold digital zooming, with the same resolution, was used to image fission points. Costes Pearson correlation was calculated from the 3D images with a 0.22‐μm step in VoloCity.
For 3D reconstruction of multicolor images, a Gaussian filter was applied (Sigma 1.0) using LAX.
Immunohistochemistry was performed as described (Ejlerskov et al, 2015) and visualized by confocal microscopy as described above. For murine models of PD, stains were performed, blinded to the injection combination by another scientist from the one who performed the injections.
TEM was performed as described (Ejlerskov et al, 2015).
For serial block‐face scanning electron microscopy, 12‐month‐old mice were cardiac‐perfused with 2% PFA and 2% glutaraldehyde. Paratenial and central medial thalamic nuclei were dissected and processed for Epon embedding and trimming as described (Parkyn Schneider et al, 2017). Serial block‐face SEM was performed using a Teneo VS electron microscope. For each condition, 15 tiles of at least 250 slices were acquired every 30–40 nm. Amira 3D was used for segmentation, 3D reconstruction, and volume quantification. ER closeness, defined as the distance from the mitochondria to the nearest ER, was measured using FIJI. Contact length was determined as the ratio of the portion of the total mitochondrial perimeter that was in contact with ER at a distance < 20 nm. Measurements were performed in FIJI.
Settings were tightly controlled for all fluorescent images that required quantification. Settings were adjusted to the brightest observed condition to avoid misinterpretation due to potential oversaturation and applied similarly to all conditions in an experiment.
Quantitative PCR
RNA was purified with the RNeasy Micro kit (Qiagen), and cDNA was generated using the QuantiTect reverse‐transcription kit. qPCR was performed using Maxima SYBR Green/ROX qPCR Master Mix (Thermo). GAPDH and Nefh were used as housekeeping genes. All primer sequences are listed in Table 1. The primer sets for GAPDH, Nefh, and optineurin were from the RT2 qPCR Primer Assay for Mouse (Qiagen; PPM02946E‐200; PPM38954F‐200, and PPM30862B‐200, respectively).
Seahorse assay and ATP quantification
Seahorse assay
Isolation of mitochondria for respiration measurements
Mitochondria were isolated as described in Wang et al (2011), using method “B” with modifications. Briefly, brains were harvested from 3 to 4 5‐month‐old BR Ifnb +/+ and Ifnb −/− mice per group within 1 min of sacrifice and placed in 3 ml IBA on ice. All subsequent steps were performed on ice. One hemisphere of brain tissue was minced and homogenized in a 7‐ml Dounce homogenizer with 5 ml cold IBA [225 mM mannitol, 75 mM sucrose, 1 mM EGTA, 5 mM HEPES‐KOH, pH 7.2, 1 mg/ml fatty acid‐free bovine serum albumin (BSA)].
After centrifugation at 1,000 g for 5′, the supernatant was pelleted at 10,000 g for 15′ and resuspended in 12% Percoll, which was layered on top of a 19%/40% Percoll gradient. The gradient was centrifuged for 20′ at 25,000 g in an SW70Ti rotor, and the mitochondrial fraction (Fig EV2A) was collected. Mitochondria were washed with IBB and pelleted at 16,300 g for 12′, and the pellet was precipitated with BSA (7,400 g for 10′). The final mitochondrial pellet was resuspended in MAS buffer without BSA for subsequent protein quantification with BCA.
Respiration of isolated mitochondria
Respiratory measurements of isolated mitochondria were performed as described in Varkuti et al (2020), with modifications. Mitochondria were resuspended to 2 μg/20 μl in cold MAS‐BSA buffer and plated (20 μl per well) in PDL‐coated 96‐well XFeSeahorse plates. Wells without mitochondria were used for background correction. The plate was centrifuged at 2,000 g for 20 min at 4°C to attach the mitochondria. After centrifugation, the mitochondria were checked under a microscope to ensure that a homogeneous mitochondrial monolayer had formed (Fig EV2B). An additional 160 μl of MAS‐BSA buffer + substrates was added to the wells and incubated at 37°C for 10 min immediately before running the assay. The plates were further equilibrated for 8 min by 2 cycles of a 1‐min mix and a 3‐min rest before basal respiration was measured.
Two baseline measurements were obtained before the first injection, and 1 response measurement was made after each injection, followed by an additional 30‐s mix. The final concentrations of compounds after the injections were as follows: 10 mM pyruvate/malate or succinate, 1 mM ADP, 2 μM oligomycin, 4 μM FCCP, and 1.5 μM rotenone/antimycin A. Each measurement cycle consisted of a 30‐s mix and a 3‐min data acquisition step, except for the measurement after ADP injection, which lasted 6 min to observe the transition from State 3 to State 4 due to the depletion of ADP. Mitochondria that were supplemented with Complex 1 (pyruvate/malate) and Complex II (succinate) substrates were run simultaneously. Complex 2 substrate was supplemented with 2 μM rotenone to inhibit Complex 1 activity. OCR was calculated using Wave software (Agilent). OCR data on isolated brain mitochondria are displayed in “point‐to‐point” mode.
ATP quantification
CNs were plated into 96‐well semi‐opaque plates at 100,000 per well. ATP was quantified by CellTiter‐Glo Luminescent Cell Viability Assay (Promega) according to a standard curve for each quantification, and cell number was normalized to Crystal Violet staining. To quantify ATP production per mitochondrion, we quantified MTG in a matching well, as described below. To quantify ATP from tissue, microdissected midbrains were snap‐frozen directly in liquid nitrogen and stored for tissue banking from 1 week to 96 months. On being processed for ATP quantification, the tissues were reduced to powder and resuspended in ice‐cold PBS with protease and phosphatase inhibitors at 100 mg/ml, and tissue lysates were further homogenized using a mortar and pestle. The samples were then centrifuged for 1 min at high speed. The supernatants were diluted 1:10 and 1:100 and quantified using Cell Titer‐Glo using the value that corresponded to the exponential linear portion of a standard curve for ATP.
Live cell stains for oxidative stress and mitochondrial mass
MTG was added directly to plates at a final concentration of 200 nM and incubated for 20 min. DCFDA (Sigma; D6883‐50MG) was added directly to the plates at a final concentration of 5 μM for 15 min. Cells were then washed twice with PBS. TMRE was used at 0.2 nM. Cells were imaged under an Olympus IX71 fluorescent microscope or Incucyte S3 when specified. Intensity was quantified as the mean intensity of automatically thresholded images using FIJI or Incucyte.
Immunoblotting
Whole‐cell extracts were prepared as described (Tresse et al, 2010). Antibody references and concentrations are given in Table 2.
For cell fractionation, cells were scraped in 1× PBS and pelleted at 300 g for 3 min. The cell pellet was resuspended in mitochondrial isolation buffer, containing 5 mM HEPES (Sigma‐Aldrich, H3375‐100G), 210 mM mannitol (Fluka. 63560), 70 mM sucrose (Fluka, 84100), 0.2 mM EGTA (Sigma‐Aldrich, E3889‐100G), and 3 mM MgCl2 (Sigma‐Aldrich, M8266‐100G). Phosphatase inhibitor cocktail (Sigma‐Aldrich, P5726‐5ML) and protease inhibitor cocktail (Sigma, P8340) were added to the buffer at 1:500. Cells were lysed in mitochondrial isolation buffer by being passed through a 27G needle (BD Microlance, 302200) 10 times, incubated on ice for 20 min, and passed again through a 27G needle 10 times. Cells were centrifuged at 800 g for 5 min to pellet the nucleus. The supernatant was isolated and centrifuged a second time to clear it of nuclear contamination.
The supernatant was then centrifuged at 10,000 g for 10 min to pellet the mitochondrial fraction. The resulting supernatant, contacting the cytosolic fraction, was isolated and centrifuged again to remove mitochondrial contamination. The 2 mitochondrial pellets were resuspended in mitochondrial isolation buffer, pooled, centrifuged at 10,000 g for 10 min, and resuspended in RIPA buffer to form the mitochondrial fraction. The nuclear pellet was resuspended in mitochondrial isolation buffer that contained 0.1% Triton X‐100 (Sigma‐Aldrich, X100‐500ML) for 7 min and then pelleted by centrifugation at 800 g for 5 min. This pellet was washed in mitochondrial isolation buffer and pelleted again at 800 g for 5 min. The nuclear pellet was resuspended in RIPA buffer to form the nuclear fraction. All centrifugation steps were performed at 4°C.
Drp1 was oligomerized as described (Zhu et al, 2004). Loading buffer that contains DTT does not interfere with BS3 cross‐linking, allowing the visualization of cross‐linked protein oligomers.
Immunoblots were quantified in FIJI and normalized against a housekeeping gene or pan‐Drp1 when assessing Drp1 phosphorylation.
SiRNA silencing
SiRNA silencing was performed as described (Tresse et al, 2010) using Dharmacon ref 058881 for STAT1, 012064 for STAT2, 040794 for STAT3, 040613 for MAPK1/ERK2, 052506 for PGAM5, 059173 for Camk2a, and 040544 for Cdk5.
Statistical analysis
Data were analyzed by unpaired and paired two‐tailed Student’s t‐tests and ANOVA using Prism. P < 0.05 was significant. Error bars are SEM.
Author contributions
ET and SI‐N conceived the experiments, interpreted the results, wrote the manuscript, and secured funding. EJ, WQGS, LR‐P, KR, and ET developed and conducted the experiments and performed the statistical analysis of the data. All authors read and contributed to the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Acknowledgements
We would like to thank Natasha Fauerby for help with the animal work, Klaus Qvortrup for assistance with 3D electron microscopy, Zhigao Wang for generously providing the PGAM5 overexpression vector, Clara Mayer for help with tissue preparation, Sara Mandatori for help with Seahorse experiments, Erika Villanueva and Henrik Hasseldam for reviewing the manuscript, and Sean Kim for reviewing and editing the manuscript. This project received funding from the Lundbeck Foundation (LF ID#R291‐2016‐536, R199‐2015‐2368, and R210‐2015‐3372), Danish Council for Independent Research‐Medicine (DFF‐6110‐00658), and the European Union’s Horizon 2020 Research and Innovation Programme under Marie Skłodowska‐Curie grant agreement no. 703217.
The EMBO Journal (2021) 40: e106868.
References
- Abe T, Isobe C, Murata T, Sato C, Tohgi H (2003) Alteration of 8‐hydroxyguanosine concentrations in the cerebrospinal fluid and serum from patients with Parkinson's disease. Neurosci Lett 336: 105–108 [DOI] [PubMed] [Google Scholar]
- Abou‐Sleiman PM, Muqit MM, Wood NW (2006) Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci 7: 207–219 [DOI] [PubMed] [Google Scholar]
- Allister EM, Robson‐Doucette CA, Prentice KJ, Hardy AB, Sultan S, Gaisano HY, Kong D, Gilon P, Herrera PL, Lowell BB et al (2013) UCP2 regulates the glucagon response to fasting and starvation. Diabetes 62: 1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambjorn M, Ejlerskov P, Liu Y, Lees M, Jaattela M, Issazadeh‐Navikas S (2013) IFNB1/interferon‐beta‐induced autophagy in MCF‐7 breast cancer cells counteracts its proapoptotic function. Autophagy 9: 287–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune‐Pelloquin L, Davezac N, Mils V, Miquel MC et al (2016) Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis 90: 3–19 [DOI] [PubMed] [Google Scholar]
- Bo T, Yamamori T, Suzuki M, Sakai Y, Yamamoto K, Inanami O (2018) Calmodulin‐dependent protein kinase II (CaMKII) mediates radiation‐induced mitochondrial fission by regulating the phosphorylation of dynamin‐related protein 1 (Drp1) at serine 616. Biochem Biophys Res Commun 495: 1601–1607 [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Buhlman L, Damiano M, Bertolin G, Ferrando‐Miguel R, Lombes A, Brice A, Corti O (2014) Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim Biophys Acta 1843: 2012–2026 [DOI] [PubMed] [Google Scholar]
- Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA et al (2017) Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 216: 3231–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu‐Wai‐Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11: 11–24 [DOI] [PubMed] [Google Scholar]
- Camilleri A, Vassallo N (2014) The centrality of mitochondria in the pathogenesis and treatment of Parkinson's disease. CNS Neurosci Ther 20: 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Mahad DJ (2011) Mitochondria as crucial players in demyelinated axons: lessons from neuropathology and experimental demyelination. Autoimmune Dis 2011: 262847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, Park JY, Kim WR, Kim H, Sun W (2014) CDK5‐dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med 46: e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chueh FY, Leong KF, Yu CL (2010) Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine‐stimulated cells. Biochem Biophys Res Commun 402: 778–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri D, Brini M, Cali T (2017) Emerging (and converging) pathways in Parkinson's disease: keeping mitochondrial wellness. Biochem Biophys Res Commun 483: 1020–1030 [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana‐Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC et al (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty P, Sangrey T, Levy WB (2006) Metabolic energy cost of action potential velocity. J Neurophysiol 96: 1237–1246 [DOI] [PubMed] [Google Scholar]
- Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, McCaffery JM, Cappelletti M, Baruffini E, Lenaers G et al (2017) OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep 19: 2557–2571 [DOI] [PubMed] [Google Scholar]
- Devine MJ, Kittler JT (2018) Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19: 63–80 [DOI] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C et al (2015) Lack of neuronal IFN‐beta‐IFNAR causes lewy body‐ and Parkinson's disease‐like dementia. Cell 163: 324–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, Leanderson T (1998) Interferon‐beta is required for interferon‐alpha production in mouse fibroblasts. Curr Biol 8: 223–226 [DOI] [PubMed] [Google Scholar]
- Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, Cenci MA (2011) Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L‐DOPA in the 6‐hydroxydopamine mouse model of Parkinson's disease. Neurobiol Dis 42: 327–340 [DOI] [PubMed] [Google Scholar]
- Frank M, Duvezin‐Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M (2016) Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol Cell Neurosci 71: 56–65 [DOI] [PubMed] [Google Scholar]
- Galindo MF, Solesio ME, Atienzar‐Aroca S, Zamora MJ, Jordan Bueso J (2012) Mitochondrial dynamics and mitophagy in the 6‐hydroxydopamine preclinical model of Parkinson's disease. Parkinsons Dis 2012: 131058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1‐regulated remodelling of mitochondrial cristae during apoptosis. EMBO J 24: 1546–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S (2012) The ER‐mitochondria interface: the social network of cell death. Biochim Biophys Acta 1823: 327–334 [DOI] [PubMed] [Google Scholar]
- Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ (2014) A mitocentric view of Parkinson's disease. Annu Rev Neurosci 37: 137–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468 [DOI] [PubMed] [Google Scholar]
- Hatch AL, Gurel PS, Higgs HN (2014) Novel roles for actin in mitochondrial fission. J Cell Sci 127: 4549–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y et al (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11: 958–966 [DOI] [PubMed] [Google Scholar]
- Jahani‐Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS (2007) Mitofusin 2 protects cerebellar granule neurons against injury‐induced cell death. J Biol Chem 282: 23788–23798 [DOI] [PubMed] [Google Scholar]
- Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4: e11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Koshkin V, Zhang CY, Wang J, Lowell BB, Chan CB, Wheeler MB (2002) Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high‐fat diet. Diabetes 51: 3211–3219 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Oh SJ, Park SH, Kang HJ, Won MH, Kang TC, Park JB, Kim JI, Kim J, Lee JY (2007) Neuronal loss in primary long‐term cortical culture involves neurodegeneration‐like cell death via calpain and p35 processing, but not developmental apoptosis or aging. Exp Mol Med 39: 14–26 [DOI] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP et al (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Ramabhadran V, Higgs HN (2013) An actin‐dependent step in mitochondrial fission mediated by the ER‐associated formin INF2. Science 339: 464–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassus B, Magnifico S, Pignon S, Belenguer P, Miquel MC, Peyrin JM (2016) Alterations of mitochondrial dynamics allow retrograde propagation of locally initiated axonal insults. Sci Rep 6: 32777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Yang YM, Yuan H, Sehgal PB (2013) Definitive evidence using enucleated cytoplasts for a nongenomic basis for the cystic change in endoplasmic reticulum structure caused by STAT5a/b siRNAs. Am J Physiol Cell Physiol 304: C312–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG (2012) Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet 21: 4827–4835 [DOI] [PubMed] [Google Scholar]
- Liu Y, Carlsson R, Ambjørn M, Hasan M, Badn W, Darabi A, Siesjö P, Issazadeh‐Navikas S (2013) PD‐L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J Neurosci 33: 14231–14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Carlsson R, Comabella M, Wang J, Kosicki M, Carrion B, Hasan M, Wu X, Montalban X, Dziegiel MH et al (2014) FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med 20: 272–282 [DOI] [PubMed] [Google Scholar]
- Liu Y, Marin A, Ejlerskov P, Rasmussen LM, Prinz M, Issazadeh‐Navikas S (2017) Neuronal IFN‐beta‐induced PI3K/Akt‐FoxA1 signalling is essential for generation of FoxA1(+)Treg cells. Nat Commun 8: 14709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M (2014) Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's‐like movement disorder. Nat Commun 5: 4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, Ramachandran R (2014) A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell 25: 1905–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL (2014) Axonal transport: cargo‐specific mechanisms of motility and regulation. Neuron 84: 292–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzio EA, Reams RR, Soliman KF (2004) The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6‐hydroxydopamine in vitro . Brain Res 1004: 29–44 [DOI] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 18: 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JA, Larner AC (2014) Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 26: 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, Ma Y, Moosmann B, Masliah E, Lipton SA et al (2011) Oxidation of the cysteine‐rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener 6: 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ (2007) The JAK‐STAT signaling pathway: input and output integration. J Immunol 178: 2623–2629 [DOI] [PubMed] [Google Scholar]
- Parkyn Schneider M, Liu B, Glock P, Suttie A, McHugh E, Andrew D, Batinovic S, Williamson N, Hanssen E, McMillan P et al (2017) Disrupting assembly of the inner membrane complex blocks Plasmodium falciparum sexual stage development. PLoS Pathog 13: e1006659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana‐Cabrera R, Mehrotra A, Rigoni G, Soriano ME (2017) Who and how in the regulation of mitochondrial cristae shape and function. Biochem Biophys Res Commun 500: 94–101 [DOI] [PubMed] [Google Scholar]
- Saito Y (2014) Oxidized DJ‐1 as a possible biomarker of Parkinson's disease. J Clin Biochem Nutr 54: 138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahni R, Cale CM, Anderson G, Osellame LD, Hambleton S, Jacques TS, Wedatilake Y, Taanman JW, Chan E, Qasim W et al (2015) Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain 138: 2834–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam G, Narasimhan M, Tamowski S, Darley‐Usmar V, Rajasekaran NS (2017) Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol 12: 937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin‐related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen HC, Gamero AM (2013) STAT2 phosphorylation and signaling. JAKSTAT 2: e25790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521–11529 [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M (2005) Cutting edge: role of STAT1, STAT3, and STAT5 in IFN‐alpha beta responses in T lymphocytes. J Immunol 174: 609–613 [DOI] [PubMed] [Google Scholar]
- Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, Holmdahl R, Issazadeh‐Navikas S (2003) IFN‐beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol 170: 4776–4784 [DOI] [PubMed] [Google Scholar]
- Tieu K (2011) A guide to neurotoxic animal models of Parkinson's disease. Cold Spring Harb Perspect Med 1: a009316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin‐containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin S, Lekmine F, Sassano A, Rui H, Fish EN, Platanias LC (2003) Role of Stat5 in type I interferon‐signaling and transcriptional regulation. Biochem Biophys Res Commun 308: 325–330 [DOI] [PubMed] [Google Scholar]
- Ugarte‐Uribe B, Prevost C, Das KK, Bassereau P, Garcia‐Saez AJ (2018) Drp1 polymerization stabilizes curved tubular membranes similar to those of constricted mitochondria. J Cell Sci 132: jcs208603 [DOI] [PubMed] [Google Scholar]
- Uo T, Dworzak J, Kinoshita C, Inman DM, Kinoshita Y, Horner PJ, Morrison RS (2009) Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Exp Neurol 218: 274–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana‐Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P et al (2015) The OPA1‐dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21: 834–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkuti BH, Kepiro M, Liu Z, Vick K, Avchalumov Y, Pacifico R, MacMullen CM, Kamenecka TM, Puthanveettil SV, Davis RL (2020) Neuron‐based high‐content assay and screen for CNS active mitotherapeutics. Sci Adv 6: eaaw8702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Leverin AL, Han W, Zhu C, Johansson BR, Jacotot E, Ten VS, Sims NR, Hagberg H (2011) Isolation of brain mitochondria from neonatal mice. J Neurochem 119: 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148: 228–243 [DOI] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, Xia Q, Kong X (2016) Deficiency of DJ‐1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. Int J Biol Sci 12: 1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C (2004) Intra‐ and intermolecular domain interactions of the C‐terminal GTPase effector domain of the multimeric dynamin‐like GTPase Drp1. J Biol Chem 279: 35967–35974 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File