

Abstract
Cancer cells prone to utilize aerobic glycolysis other than oxidative phosphorylation to sustain its continuous cell activity in the stress microenvironment. Meanwhile, cancer cells generally suffer from genome instability, and both radiotherapy and chemotherapy may arouse DNA strand break, a common phenotype of genome instability. Glycolytic enzyme PFKFB3 (6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3), plays essential roles in variety physiology and pathology processes, and generally maintain high level in cancer cells. Although this protein has been reported to involve in genome instability, its role remains unclear and controversial. Here, we showed that PFK-15, a PFKFB3 inhibitor, obviously induced apoptosis, cell viability loss, and inhibited cell proliferation/migration. Besides, PFK-15 was also found to induce necroptosis, as it not only up-regulated the phosphorylated RIP1, RIP3 and MLKL, but also enhanced the interaction between RIP3 and RIP1/MLKL, all of which are characterization of necroptosis induction. Both genetically and pharmacologically deprivation of necroptosis attenuated the cytotoxic effect of PFK-15. Besides, PFK-15 increased the γ-H2AX level and micronuclei formation, markers for genome instability, and inhibition of necroptosis attenuated these phenotypes. Collectively, the presented data demonstrated that PFK-15 induced genome instability and necroptosis, and deprivation of necroptosis attenuated cytotoxicity and genotoxicity of PFK-15 in colorectal cancer cells, thereby revealing a more intimate relationship among PFKFB3, necroptosis and genome instability.
Keywords: PFKFB3, necroptosis, genome instability, colorectal cancer, cell viability
Introduction
In the past two decades, knowledge of cell death types and mechanisms have greatly elucidated, and according to distinct cell morphology and internal regulation mechanism, three major types of programmed cell death (PCD) pathways have been widely recognized: apoptosis, autophagy-dependent cell death, and necroptosis (programmed necrosis) [1,2]. Receptor interacting protein kinase 3 (RIP3) and mixed lineage kinase domain like pseudokinase (MLKL) are sequential activated and critical for necroptosis initiation [3]. Upon necroptosis initiation by TNFR1, RIP1 (receptor interacting protein kinase 1) activates RIP3 through a mechanism involving thephysical interaction between their respective RHIM (RIP homotypic interaction motif) domains [4,5]. Subsequently, phosphorylated RIP3 further renders MLKL phosphorylation and trimerization, which then translocate to the plasma membrane and cause necrotic permeabilization [6,7]. The RIP1-RIP3-MLKL complex, also known as the “necrosome”, which links the upstream cell death receptors and downstream executive molecules/events, is essential component in executing necroptosis process [8]. In addition to participating in necroptosis, RIP1 has been shown involved in complex leading to cell survival [9], and other complexes resulting in apoptosis [10]. Accordingly, RIP1 inhibitor necrostatin-1 (Nec-1) robustly inhibits TNFR1-driven necroptosis, both in vitro and in vivo [11,12]. Besides Nec-1, RIP3 kinase inhibitor GSK-872, and MLKL inhibitor necrosulfonamide (NSA) were also found to inhibit necroptosis process [13].
Due to living in the stress microenvironment, cancer cells generally alter its metabolism pattern to sustains continuous cell growth [14]. The Warburg effect points that cancer cells prefer to utilize glycolysis to catabolize glucose and generate ATP even not under hypoxia condition. While hexokinase (HK), 6-phosphofructo-1-kinase (PFK-1) and pyruvate kinase (PK) are the three rate limiting enzymes in glycolysis, the rate of glycolysis is determined by PFK-1 as it characters the lowest catalytic efficiency [14]. Bifunctional 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase or PFKFB) isoform 3 (PFKFB3) character the highest ratio of kinase to phosphatase, and control the level of fructose-2,6-bisphosphate (F2,6BP), which effectively allosteric activate PFK-1 [14,15]. Meanwhile, cancer cells generally present high level of PFKFB3, and its activity is modulated by variety factors, such as hypoxia and inflammatory stimuli [14]. Beyond regulates glycolysis in the cytoplasm, PFKFB3 has also been reported to targeted in the nuclear in several cell lines, which contribute to cell proliferation [16], and play a promotion role in modulating autophagy [15]. While hexokinase II inhibitor 3-Bromopyruvate (3BP) induces autophagy, apoptosis as well as necroptosis in colorectal cancer cells [17], our former study showed PFKFB3 inhibitor PFK-15 induced apoptosis, but inhibited autophagic flux [18,19]. However, the role of PFKFB3 in necroptosis process has not been investigated, thus we focus on the relationship between PFKFB3 and necroptosis in the current study.
PFKFB3 also plays essential roles in variety physiology and pathology processes, such as metabolism, angiogenesis and inflammation [14]. Meanwhile, PFKFB3 has been reported to involve in genome instability as well, yet its role remains unclear and controversial. The diagnostic marker of genome instability is the formation of micronuclei, which are small DNA containing membrane-enveloped structures isolated from the cell nuclei [20]. As a common type of genome instability, DNA damage is easily aroused by either reactive oxygen compounds (such as reactive oxygen species, ROS) or ionizing radiation (IR). Although double-strand breaks (DSBs) are not common occur as the other kind of lesions, they are difficult to repair and that make DSBs extremely toxic to cells. In addition to be considered as a marker for DSBs [21], γ-H2AX (phospho-H2AX, Ser139) functions as an adaptor as well, which recruits chromatin remodeling modify factors [22]. If DSBs are not appropriately repaired, cells will occur either cell cycle arrest or cell death.
In the present study, we demonstrated that PFKFB3 inhibitors induce cell viability loss, apoptosis, and necroptosis in colorectal cancer cells. Meanwhile, the level of γ-H2AX, and the formation of micronuclei were stimulated by PFKFB3 inhibitors. Interestingly, both genetically and pharmacologically inhibition of necroptosis attenuated the γ-H2AX level, and micronuclei percentage increased by PFK-15.
Materials and methods
Antibodies and chemicals
PFK-15 (1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one, ab145859), Necrostatin-1 (Nec-1, ab141053), the antibodies of phospho-MLKL (Ser358; ab187091) and PFKFB3 (ab181861) were purchased from abcam (Cambridge, MA, USA). 3-PO ((2E)-3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, SML1343) and PMS (P9625) were obtained from Sigma-Aldrich (St Louis, MO, USA). GSK872 (HY-101872), and Necrosulfonamide (NSA, HY-100573) were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Z-VAD-FMK (FMK001) and Recombinant Human TNF-alpha (210-TA-005) were purchased from R&D Systems (Minneapolis, MN, USA). The antibodies of phospho-RIP1 (Ser166; 65746), phospho-RIP3 (Ser227; 93654), total MLKL (14993), phospho-H2AX (Ser139, γ-H2AX, 80312), H2AX (7631), Rad51 (8875), Bcl-xL (2764), c-Myc (5605) and PARP-1 (9542) were got from Cell Signaling Technology (Beverley, MA, USA). Total RIP1 (610458) antibody was acquired from BD Biosciences (San Diego, CA, USA). Total RIP3 (sc-374639) antibody was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). The antibodies of PCNA (10205-2-AP), E-cadherin (20874-1-AP), β-Tubulin (10068-1-AP), N-cadherin (66219-1-lg), and β-Actin (66009-1-Ig) were acquired from Proteintech (Wuhan, Hubei, China). We bought MTS (G1111) from Promega Corporation (Madison, WI, USA).
Cell culture, siRNA interference and PFKFB3 knockout cell line generation
Human colorectal cancer cell lines SW480 and HT29 were obtained from ATCC (Manassas, Grand Island, VA, USA), and cultured with DMEM high glucose medium, which supplemented with 1% antibiotics, and 10% fetal bovine serum (GIBCO, Grand Island, NY, USA). For siRNA interference, 30% confluence of cells was prepared, and then transfected using DharmaFECT (Dharmacon, T2001, Denver, CO, USA) according to the manufacturer’s instructions with indicated siRNAs. Following 48 h culture, cells were trypsinized and plated in suitable plates and cultured overnight. Cells then exposed to indicate treatment for appropriated period. The siRNA specific for human RIP1 (sc-36426), and RIP3 (sc-61482) and control siRNA (sc-37007) were from Santa Cruz Biotechnology.
The designed sgRNA (single guide RNA, 5’-TCAACGTCGGGGAGTATCGC-3’) with targeted the exon 3 of PFKFB3 was ligated into the pLV-hCas9: T2A: Neo-U6 vector, and verified by direct sequencing. To generate cell lines with PFKFB3 knockout, 60% confluence of cells were transfected with PFKFB3 targeted sgRNAs using Attractene (QIAGEN, Hilden, Germany). Following transfection for 24 h, 100,000 cells were split into 6 cm-plate and cultured with 100 ug/mL G418 (HY-17561, MedChemExpress). Monoclonal cells were chosen and detected by immunoblotting analysis. Subsequently, genetic ablation of PFKFB3 was confirmed by first generation sequencing.
Immunoblotting analysis and immunoprecipitation
Cells of 70% to 80% confluence were subjected to appropriate treatments for indicate time points. Then Triton X-100/glycerol buffer was used to extract the total protein homogenates [23]. According to the protein molecular weight, different concentration of SDS-PAGE gels were used to separate the protein samples after denaturation. Following transferring to PVDF membrane and blocking by non-fat milk (5%), appropriate primary and secondary antibodies were performed in order. Enhanced chemiluminescence (Pierce Chemical, 34080, Rockford, IL, USA) was used to detect the blots, which then analyzed by Image J software (National Institutes of Health, Bethesda, MD, USA).
For immunoprecipitation, Triton X-100/glycerol buffer was used to obtain the whole cell lysates. RIP3 antibody (1:100) was used to immunoprecipitate at 4°C for 6 h. Then Protein A/G Magnetic Beads (HY-K0202, MedChemExpress) were added and incubated for 1 h. After wash three times, immunoprecipitates were collected by Magnetic Stand (HY-K0200, MedChemExpress) and then subjected to immunoblotting analysis.
Colony growth assay and EDU staining
Following seeding at a concentration of 300 cells per mL and exposure to indicated drugs for two weeks to allow colony growth (change medium and exposure to treatment same as the original every four days), 4% paraformaldehyde was added and incubated for 10 min. After staining with either crystal violet or giemsa stain for 2 h, pictures were obtained and Image J software was used to calculate the colony numbers.
Following incubation with indicated drugs for 4 h, EDU (final concentration was 10 μM) was added to each well for another 2 h. Following the manufacturer’s protocol, cells were stained using the EDU-488 cell proliferation assay kit (C0071, Beyotime, Shanhai, China) according to. Fluorescence microscopy (Nicon, Melville, NY, USA) was utilized to acquire the images.
Cell migration detection
Wound healing assay: Following adjust to 5 × 105 cells/mL with complete culture medium, 110 μL cell suspensions were applied into each well of Culture-Insert (80469, ibidi, Berlin, Germany). Following culture overnight, the Culture-Insert was removed gently by using sterile tweezers, and then the culture medium was discarded and cells were washed twice with PBS. After addition of indicated treatment diluted in fresh medium (1% FBS), the microscopy was used to capture the wound closure (8 random locations were selected for each group) at indicated time points. Utilizing the Photoshop software (adobe, San Jose, CA, USA), the migration was determined as an average closed area of the wound relative to the initial wound area. Transwell assay: Briefly, 5 × 104 cells in serum-free medium (400 μL) were added into the upper chamber of the Transwell (Millipore, MCEP24H48, Burlington, MA, USA), which was pre-coated with Matrigel (BD Biosciences, 356234, San Diego, CA, USA). Then, 800 μL DMEM (10% FBS) was added to the lower chamber. After culture for indicated period, a swab was used to remove the cells on the top side of the membrane. Following fixing and staining with 0.1% crystal violet, the basal side cells were captured via microscopy.
Cell viability assay (MTS)
Equal amount of cells were plated in 96-well plates (7500 cells per well) and cultured overnight with complete culture medium. 20 μL MTS/PMS (20:1) was added following treatment with indicated chemicals diluted in phenolic red-free complete medium for indicated period. After culture for another 2 h, Microplate reader (Cytation5, BioTek, Winooski, VT, USA) was used to monitor the cell viability via analyzing the absorbance at 490 nm.
Immunofluorescence staining
Glass coverslips were placed to 12 well-plate and allowed cells to adhere. After appropriate treatments, cells were fixed by freshly prepared 4% paraformaldehyde for 12 min. Then cells were permeabilized with Ca2+-/Mg2+-free PBS (CMF-PBS) supplement with 0.1% Triton X-100 and 0.5% bovine serum albumin (BSA) for 5 min. Following incubation with primary antibodies (1:30) for 4 h and appropriated secondary antibodies (1:100) for 1 h, cells were immersed in VECTASHIELD with DAPI (Vector laboratories, H1200, Burlingame, CA, USA). Fluorescence microscopy (Nicon, Melville, NY, USA) was utilized to capture the images.
Flow cytometry assay
Following appropriate treatment, cells were harvested with all floating cells kept. According to the instructions of the Apoptosis Detection Kit (K101-100, Biovision Inc., Milpitas, CA, USA), the samples were incubated with fluorescein isothiocyanate-labeled annexin V (FITC) and propidium iodide (PI). Then the samples were monitored by flow cytometry (Becton Dickinson, FACSAria, Franklin Lakes, NJ, USA).
Real-time PCR analysis
TRIzol reagent (15596-018, Invitrogen, Carlsbad, CA, USA) was utilized to extract the total RNA, followed by reversely transcribing with the help of PrimeScriptTM RT reagent Kit (DRR037A, TaKaRa, Dalian, Liaoning, China). The primer pairs used are as follows (Table 1):
Table 1.
The primer pairs
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| RIP1 | GGCATTGAAGAAAAATTTAGGC | TCACAACTGCATTTTCGTTTG |
| RIP3 | GACTCCCGGCTTAGAAGGACT | CTGCTCCTGAGCTGAGACAGG |
| MLKL | AGAGCTCCAGTGGCCATAAA | TACGCAGGATGTTGGGAGAT |
| β-Actin | GCCTGACGGCCAGGTCATCAC | CGGATGTCCACGTCACACTTC |
Utilizing LightCycler 480 (Roche, Basel, Switzerland), real-time PCR was initiated with denaturation (95°C) for 10 min, and repeat the cycle profile of 95°C (15 s), 60°C (45 s) and 72°C (1 min) for 40 times. Each sample contained three repetitions, and the results were analyzed by means of 2-ΔΔCt.
Statistical analysis
Mean + S.D. (standard deviation) were shown for the normally distributed data. Statistical comparisons were done using two-tailed unpaired Student’s t-test between two groups. And one-way analyses of variance with the Student-Newman-Keuls post hoc test were used for multigroup comparisons. Values of P < 0.05 were considered as statistically significant different. All data represented at least three independent experiments.
Results
PFKFB3 inhibitors reduce cell proliferation/invasion in colorectal cancer cells
In our former research, we found that the potent small molecule antagonist of PFKFB3, PFK-15 enhanced the cytotoxic effect of oxaliplatin [19]. Here, we verified the cytotoxic effect of PFK-15 by variety methods in colorectal cancer cells. In the MTS assay, PFK-15 obviously inhibited the viability of SW480 and HT29 cells in a dose-dependent manner (Figure 1A). Besides, colony growth assay shown it decreased the colon number (Figure 1B). The inhibition on cell proliferation was also confirmed by the EDU staining results, in which PFK-15 obviously blocked the DNA synthesis (Figure 1C). Wound healing assay and transwell assay are widely used methods to detect the cell migration ability, while the latter method is also utilized to evaluate the cell invasion activity under Matrigel pre-incubation condition. In both SW480 and HT29 cells, PFK-15 significantly decreased the cell migration and invasion activity (Figure 1D and 1E). Immunoblotting results also indicated that PFK-15 inhibited the cell proliferation- and migration-related proteins (Figure 1F). 3-PO, another widely used inhibitor of PFKFB3, was also tested and similarly phenotype was observed in the 3-PO-treated SW480 cells (Figure S1A-F). The inhibitory effect of PFK-15 and 3-PO on PFKFB3 was confirmed by monitoring the PFK-1 activity, as PFKFB3 catalytic product F2,6BP effectively allosteric activate PFK-1 (Figure S1G). Lactate, the final product of glycolysis, was also decreased by PFK-15 and 3-PO treatment (Figure S1H).
Figure 1.

PFK-15 reduces cell proliferation and migration. (A) Cells were treated with indicated dose of PFK-15 for 24 h, and then cell viabilities were analyzed by MTS assay. (B) Different dose of PFK-15 were performed in colony growth assay. Scale bars = 1 cm. (C) EDU staining assay was carried in SW480 and HT29 cells with treatment of PFK-15 for 6 h. Scale bars = 0.4 mm. (D and E) Cell migration following 24 h PFK-15 treatment was monitored by wound healing assay and transwell assay. (Scale bars: D, 0.5 mm; E, 200 μm). (F) Cell lysates were prepared and subjected to immunoblotting with presented antibodies after treated with PFK-15 for 24 h. **P < 0.01 vs. control.
PFK-15 induces apoptotic cell death in colorectal cancer cells
It is interesting to notice that E-Cadherin and N-Cadherin (play opposite roles during Epithelial-Mesenchymal Transition) decreased simultaneously upon PFK-15/3-PO treatment, which may due to that these inhibitors cause acute cell stresses, and cell viability loss. Meanwhile, these observations derived us to monitor whether PFK-15 could arouse cell death in SW480 and HT29 cells. Former reports indicate that inhibition of PFKFB3 inhibits cell viability and renders cells to apoptotic cell death in variety kind of cells [18,24]. The cleavage of PARP-1, which is serving as marker of cells undergoing apoptosis [25], was increased upon PFK-15 treatment by immunoblotting analysis (Figure 2A). Meanwhile, PFK-15 also decreased the protein level of Bcl-xL (Figure 2A), which functions as inhibitor of apoptosis [26]. Similarly, 3-PO was also found to increase PARP-1 cleavage, and downregulate Bcl-xL level (Figure S2A). Utilizing flow cytometry, PFK-15 was confirmed to induce apoptotic cell death in SW480 and HT29 cells (Figure 2B). Furthermore, Z-VAD-FMK (pancaspase inhibitor) was added to examine the cell death aroused by PFKFB3 inhibitors. Interestingly, the cell viability loss aroused by PFK-15 was only partly rescued by addition of Z-VAD-FMK as shown in the MTS assay (Figure 2C). However, immunoblotting results indicated that Z-VAD-FMK effectively blocked the PFK-15-aroused apoptosis (Figure 2D). Similarly results were also obtained in SW480 cells upon 3-PO exposure (Figure S2B and S2C). These findings indicated that PFK-15 and 3-PO could induce several kind of cell death, not merely apoptosis.
Figure 2.

PFK-15 arouses apoptotic cell death. A. After 24 h exposure to PFK-15, cell lysates were collected and subjected to immunoblotting with indicated antibodies. B. Flow cytometry was performed after 24 h treatment with PFK-15 to analyze apoptosis (Annexin V-positive). C. Cell viabilities were analyzed by MTS assay in SW480 and HT29 cells after 24 h upon to PFK-15 treatment with or without Z-VAD-FMK (20 μM). D. Cell lysates were subjected to immunoblotting after treatment with 6 μM PFK-15 (unless otherwise indicated) with or without Z-VAD-FMK. **P < 0.01 versus control, and NS indicated of none significant.
PFK-15 stimulates necroptosis in colorectal cancer cells
In addition to apoptosis, autophagic cell death and necroptosis are two widely studied kind of programmed cell death modes. Due to that PFK-15 was reported to inhibit basal and stimulates-induced autophagy in our former studies [15,19], here we further explored the effect of PFK-15 on necroptosis. By the real-time PCR assay, PFK-15 was found to up-regulate RIP1/RIP3/MLKL at the mRNA level (Figure 3A), which are components of necrosome and play essential role in executing necroptosis process [8]. The combination of TNFα (Tumor Necrosis Factor alpha) and Z-VAD-FMK stimulated cell death was originally defined as necroptosis [11], and here we used them as the positive control for inducing necroptosis. As shown in Figure 3A, TNFα+Z-VAD-FMK promoted RIP1/RIP3/MLKL expression at mRNA levels as well. The activation of RIP3 is marked by autophosphorylation at Ser227, which then recruits and phosphorylates MLKL at Thr357 and Ser358 sites, facilitating its trimerization and translocation to the cell membrane [6,7,27]. Similarly to the positive control, PFK-15 not only increased the protein levels of RIP1/RIP3/MLKL, they also promoted the phosphorylation levels of RIP1/RIP3/MLKL (Figure 3B). Furthermore, the punctate staining of RIP1 was obviously increased upon PFK-15 treatment (Figure 3C). Similarly results were observed in 3-PO-treated SW480 cells as well (Figure S2D and S2E). We then verified whether PFK-15 treatment could increase the formation of necrosome by immunoprecipitation using the RIP3 antibody. Identical to positive control group, PFK-15 treatment enhanced the interaction among RIP1/MLKL and RIP3 when compared to the untreated Ctrl group in SW480 and HT29 cells (Figure 3D). As shown in Figure S2F, 3-PO promoted the formation of necrosome as well. In order to exclude the nonspecific binding, the loading control proteins Actin/Tubulin were immunoblotted here (Figures 3D, S2F). Nec-1, the widely used necroptosis blocker [11,28], was found to attenuate the phosphorylated-RIP1/RIP3/MLKL levels up-regulated by either PFK-15 or 3-PO (Figures 3E, S2G). Taken together, our findings here stated that PFKFB3 inhibitors could consider as inducers of necroptosis in colorectal cancer cells.
Figure 3.
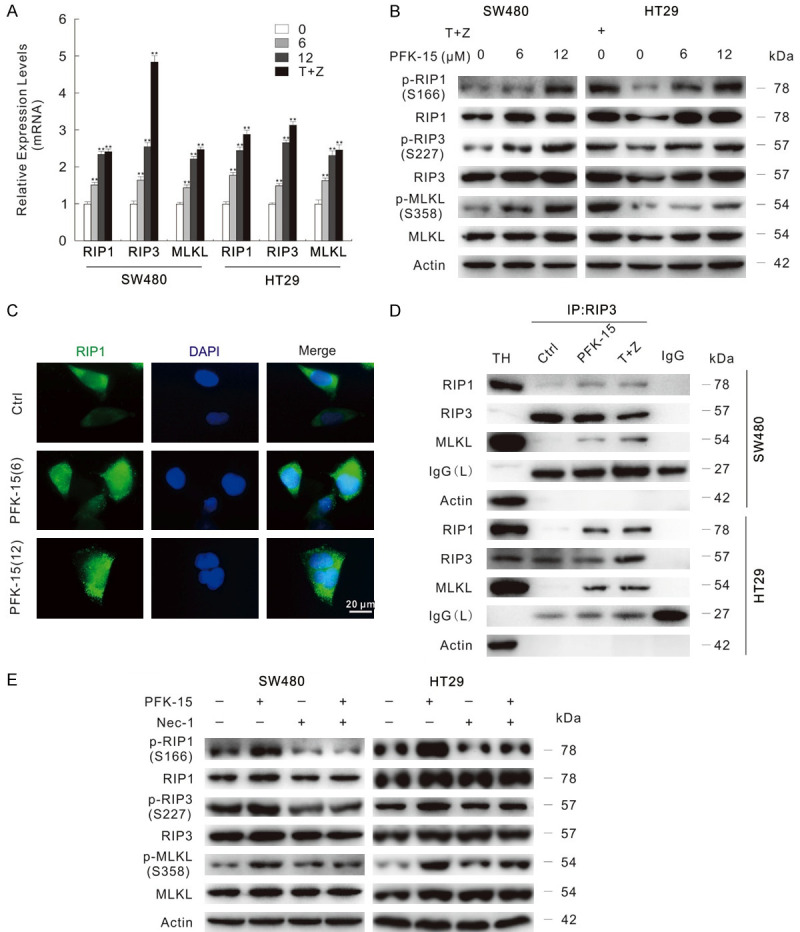
PFK-15 stimulates necroptosis in SW480 and HT29 cells. A. The mRNA expression of indicated genes were monitored by the real-time PCR following treatment with different dose of PFK-15 or T+Z for 6 h, **P < 0.01 vs. control. T+Z: hTNFα (10 ng/mL)+Z-VAD-FMK. B. Following PFK-15 treatment for 24 h, cells were lysed and then subjected to immunoblotting. C. RIP1 antibody was used in immunofluorescence staining in SW480 cells following exposure to indicated treatment for 6 h. Scale bars = 20 μm. D. SW480 cells were exposed to drugs for 24 h, cells lysates were precipitated using the RIP3 antibody. TH: The total homogenate; IgG: the negative control antibody. E. Immunoblotting was carried out following indicated treatment (Nec-1: 30 μM, unless otherwise indicated) for 24 h.
Inhibition of necroptosis rescues PFK-15-induced cell viability loss
As PFK-15-stimulated cell deaths contain necroptosis, the inhibition of necroptosis probably rescues the cell proliferation and invasion reduced by PFK-15. To verify this hypothesis, we carried out the following experiment. Though Nec-1 alone failed to obviously influence the cell viability, it showed significant protection role in the PFK-15 induced cell viability loss (Figure 4A). Meanwhile, GSK872 (an inhibitor of RIP3) and NSA (an inhibitor of MLKL), which were shown to restrain the formation of necrosome (Figure S3A), also partly reversed the cell viability loss aroused by PFK-15 (Figure 4B). The effect of necroptosis inhibitors on cell proliferation and migration inhibited by PFK-15 were then monitored. As expected, the combination of Nec-1/GSK872/NSA and PFK-15 group showed higher migration rate and more colony numbers than the PFK-15 alone treated group (Figures 4C and 4D, S3B and S3C). Accordingly, Nec-1 reversed the cell proliferation related proteins levels that were reduced by PFK-15 treatment (Figure 4E). Interestingly, Nec-1 enhanced the ability of PFK-15 to arouse apoptosis as observing the accumulation of cleavaed-PARP-1 and reduction of Bcl-XL (Figure 4E). Meanwhile, GSK872 and NSA were also found to strengthen the cell apoptosis susceptible of PFK-15 (Figure S3D). Similarly, Nec-1 rescued 3-PO-inhibited cell viability, cell proliferation and cell migration (Figure S4A-C); it also enhanced the apoptosis stimulated by 3-PO (Figure S4D).
Figure 4.
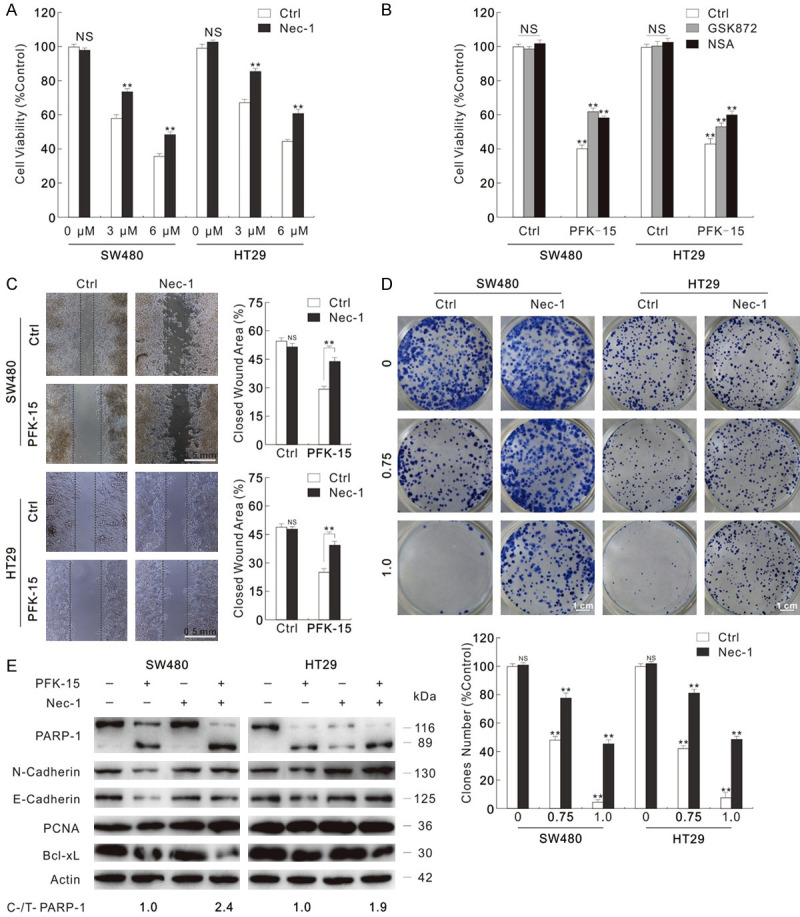
Necroptosis inhibitors attenuate the cell viability loss aroused by PFK-15. (A and B) MTS assays were performed in SW480 and HT29 cells exposed to PFK-15 treatment with or without necroptosis inhibitors (3 μM GSK872, 3 μM NSA). (C and D) Wound healing assay and colony growth assay were performed with PFK-15 (C: 6 μM; D: 1.5 μM) with or without Nec-1. (Scale bars: D, 0.5 mm; E, 1 cm). (E) Following indicated treatment for 24 h, cell lysates of SW480 and HT29 cells were subjected to immunoblotting with apporpriated antibodies. **P < 0.01 versus control, and NS indicated of none significant.
To clarity the effect of necroptosis inhibitors, RIP1 and RIP3 were genetically deprived using targeted siRNAs, separately (Figure 5A). Silencing of both RIP1 and RIP3 partly rescued the cell viability loss stimulated by PFK-15 in SW480 and HT29 cells (Figure 5B and 5C). Although neither RIP1 nor RIP3 deprivation alone significantly affected the proliferation/migration, the silenced cells resisted the cytotoxic effect of PFK-15 (Figure 5D and 5E). Meanwhile, both RIP1 and RIP3 silencing enhanced PFK-15-stimulated PARP-1 cleavage, and up-regulated the cell proliferation related proteins levels (Figure 5F). Similar phenotype was also observed in 3-PO-treated cells (Figure S4E-H). These findings indicated that inhibition of necroptosis enhanced the apoptotic cell death, but rescued the cell viability loss upon exposure to PFK-15.
Figure 5.

Siliencing of either RIP1 or RIP3 lessens the cytotoxicity of PFK-15. Cells were transfected with the RIP1, RIP3 target siRNAs or control siRNA for 48 h. (A) Immunoblotting with indicated antibodies was performed to verify the knockdown efficiency. (B and C) MTS assay was performed to monitor the cell viability after 24 h treatment (B: SW480; C: HT29). (D and E) Colony growth assay and wound healing assay were performed with PFK-15 (D: 1.5 μM; E: 6 μM). (Scale bars: D, 1 cm; E, 0.5 mm). (F) Following PFK-15 treatment for 24 h, cell lysates were subjected to immunoblotting. **P < 0.01 versus control, and NS indicated of none significant.
Knockout of PFKFB3 promotes the necroptosis and cell viability loss aroused by TNFα+Z-VAD-FMK
To verify the results acquired from PFK-15 treatment, PFKFB3 knockout HT29 cell line was obtained using target sgRNA as described. Although knockout of PFKFB3 alone failed to noteworthy increase the phosphorylation levels of RIP1/RIP3/MLKL, TNFα+Z-VAD-FMK showed higher capacity in inducing necroptosis in PFKFB3-knockout cells (Figure S5A). The addition of Nec-1 blocked the phosphorylated-RIP1/RIP3/MLKL levels increased upon TNFα+Z-VAD-FMK treatment in both wild type (WT) control and PFKFB3-knockout cells (Figure S5A), indicating that knockout of PFKFB3 fascinate cells to occur necroptosis. Moreover, TNFα+Z-VAD-FMK exposure showed lower colony numbers, migration rate and cell viability in PFKFB3-deprived cells than the WT cells, and Nec-1 reversed the cytotoxic effect of TNFα+Z-VAD-FMK to approximate level in these two group cells (Figure S5B-D).
PFKFB3 inhibitors increase the frequency of micronuclei and γ-H2AX level
During our previous research process, abnormal nuclear structure was prone to observed in PFK-15 treated cells than control cells. γ-H2AX, the commonly used marker for DSBs [21], is generally elevated upon DNA damage elements. In addition, γ-H2AX accumulates at the DSBs sites, and functions as an adaptor for recruiting chromatin remodeling modifying factors. Utilizing fluorescence microscopy, PFK-15 treatment obviously caused an accumulation of γ-H2AX punctate staining in both SW480 and HT29 cells (Figure 6A and 6B). Analogously, both the intensity and punctate staining were increased in 3-PO-treated SW480 cells (Figure S6A). We then counted and statistics analyzed the micronuclei percentage following indicated treatment. Obviously, both PFK-15 and 3-PO significantly raised the percentage of micronuclei (Figures 6C and 6D, S6B and S6C). Consistently, the protein level of γ-H2AX was notably up-regulated upon either PFK-15 or 3-PO treatment as monitored by immunoblotting (Figures 6E, S6D). Interestingly, PFKFB3 inhibitors tested also decreased the protein level of Rad51, which plays pivotal role in DNA repair (Figures 6E, S6D).
Figure 6.
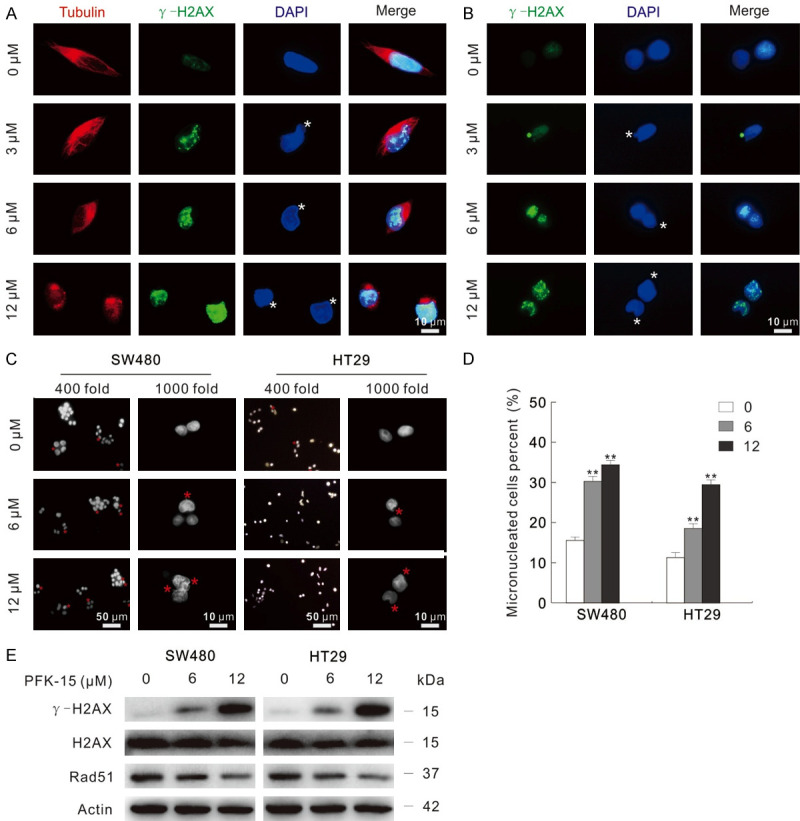
PFK-15 induces genome instability. (A and B) Immunofluorescence using the antibodies of Tubulin and γ-H2AX were performedafter PFK-15 treatment for 6 h in SW480 (A) and HT29 (B) cells, respectively. (C and D) Cells were treated with indicated dose of PFK-15 for 6 h, and then the images were obtained by fluorescence microscopy after DAPI staining with both 400 and 1000 magnification. Micronuclei percentages were analyzed and shown as mean + S.D. in graphs (D), **P < 0.01. Each group included at least 50 cells, and sterisk indicated micronuclei. (E) Following treatment of the cells with PFK-15 for appropriated periods, cell lysates were subjected to immunoblotting. The scale bars were shown in the figure.
Blockage of necroptosis reduced micronuclei percent and γ-H2AX level enhanced by PFK-15
As PFK-15 functions as an inducer of necroptosis, and inhibition of necroptosis attenuates the cytotoxicity of PFK-15, thus the effect of necroptosis inhibition on the PFK-15-induced nuclear instability was further explored. Nec-1 obviously attenuated the γ-H2AX punctate accumulated by PFK-15 treatment under fluorescence microscopy in SW480 cells (Figure 7A), and the protein level of γ-H2AX in Nec-1+PFK-15 treated group was lower than the PFK-15 treated alone group (Figure 7B). Similarly results were also obtained with both GSK872 and NSA treatment (Figure S7A and S7B). Meanwhile, the addition of necroptosis inhibitors significantly attenuated the micronuclei percent that up-regulated by PFK-15 treatment (Figure 7C and 7D). When it came to SW480 cells under 3-PO treatment condition, Nec-1 was also found to not only decrease the γ-H2AX punctate and protein level, but also reduce the percentage of micronuclei (Figure S8A-C). Beyond inhibitors, we then tested the nuclear instability in either RIP1 or RIP3 silenced cells. Coincident with former observation, PFK-15 and 3-PO increased less γ-H2AX punctate, its protein level and micronuclei percent in the RIP1/RIP3 silenced cells than the Mock control group (Figures 7E-G, S8D and S8E). Taken together, deprivation of necroptosis attenuates PFKFB3 inhibitors-induced genome instability and cell viability loss.
Figure 7.
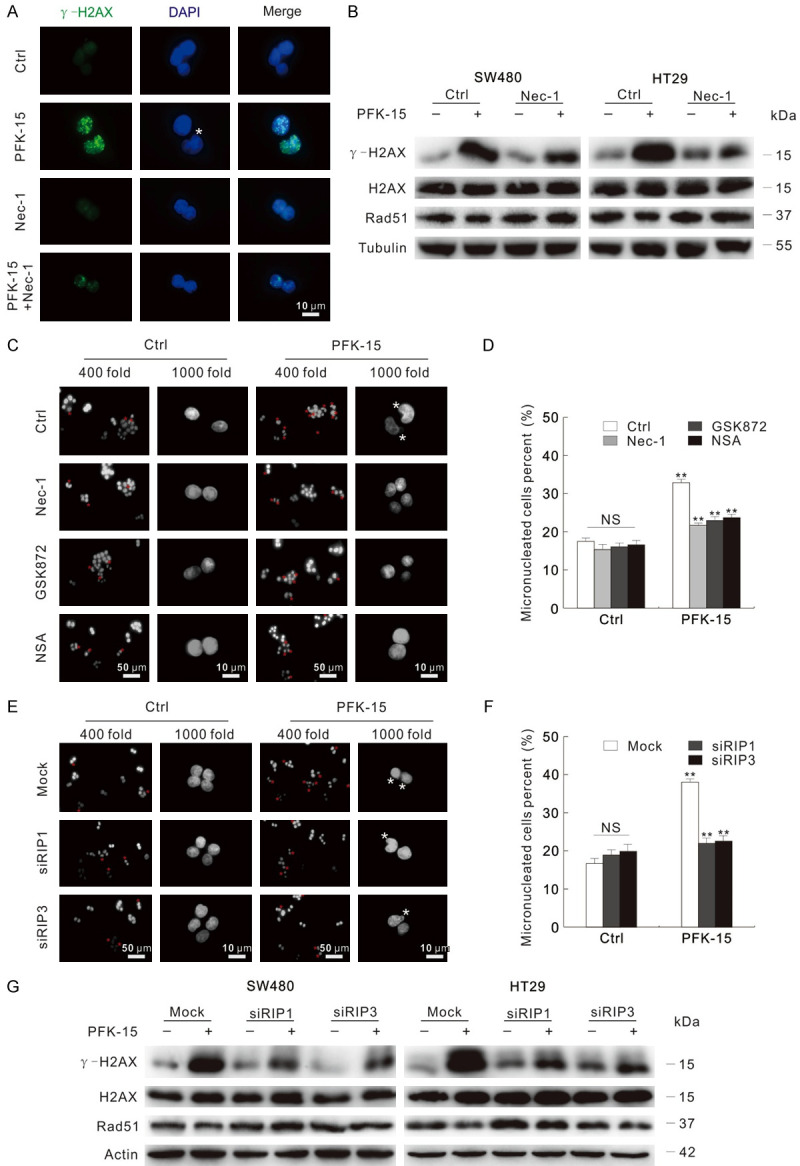
Inhibition of necroptosis depresses the genome instability induced by PFK-15. (A) Immunofluorescence using the antibodies of Tubulin and γ-H2AX were performed after exposed to indicated drugs for 6 h in SW480. (B) Following apporpriate treatment for 6 h, immunoblotting with indicated antibodies was carried out. (C and D) Following exposure to apporpriate drugs for 6 h, the images were captured by fluorescence microscopy after DAPI staining with both 400 and 1000 magnification. Micronuclei percentages were analyzed and shown as mean + S.D. in graphs (D), **P < 0.01. Each group included at least 50 cells, and sterisk indicated micronuclei. (E-G) Cells were transfected with the RIP1, RIP3 target siRNAs or control siRNA for 48 h. Following treatment with indicated chemicals for 6 h, fluorescence microscopy was used to acquire images after DAPI staining with both 400 and 1000 magnification (E); and the analyzed micronuclei percents were presented in (F); cell lysates were prepared and then monitored by immunoblotting after exposure to PFK-15 for 6 h (G). **P < 0.01 versus control, and NS indicated of none significant. The scale bars were shown in the figure. Asterisk indicated micronuclei.
Discussion
We show here that PFKFB3 inhibitors induce necrosome related protein expression in SW480 and HT29 colorectal cancer cells at both mRNA and protein levels. And PFK-15 treatment increases the interaction among RIP1, RIP3 and MLKL. Meanwhile, inhibition of necroptosis via both genetic and pharmacological methods partly attenuates the cytotoxic effect of PFK-15. PFK-15 also increases the γ-H2AX level and micronuclei formation, and necroptosis deprivation reduces its effect on influencing genome instability. Thus, PFKFB3 participates in regulating necroptosis and genome instability in colorectal cancer cells.
For a long decade, necrosis has been considered as an unregulated process which is only an incidental cell death mechanism. However, more and more studies illuminate that necrosis can appear in a regulated manner, during which involves the indispensable RIP1-RIP3-MLKL complex, also known as the “necrosome” [8]. Nevertheless, some necroptosis inducers may directly activate RIP3 or MLKL without the help of RIP1, such as poly I: C and interferons effectively induce necroptosis in RIP1 deficient cells [10]. Interestingly, there are controversial understandings of necroptosis occur in some kind of colorectal cancer cells. Moriwaki et al. and Metzig et al. reported that SW480 and HCT116 cells are insensitive to necroptosis due to lack expression of RIP3, while HT29 cells were believed in which necroptosis can occur [29,30]. However, resibufogenin was showed to suppress cell growth/metastasis through RIP3-regulated necroptosis in SW480 and HCT116 cells [31], and 3-Bromopyruvate (3BP) induced cell death partly through necroptosis in SW480 cells [17]. Here, we found that RIP3 could be detected in SW480 cells by both immunoblotting and real-time PCR assay. Meanwhile, not only in HT29 cells, TNFα+Z-VAD-FMK treatment also primed the formation of necrosome in SW480 cells, indicating that necroptosis can occur in SW480 cells as well. On the one hand, PFKFB3 inhibitors increased the protein level and phosphorylation level of RIP1/RIP3/MLKL, and enhanced the interaction between RIP1/MLKL and RIP3. On the other hand, inhibition of necroptosis significantly rescued the cell viability loss induced by PFK-15 and 3-PO. These observations demonstrated that the PFKFB3 inhibitors could be considered as the necroptosis inducers.
It is worth noticing that PFKFB3 knockout alone in HT29 cells failed to obviously increase the phosphorylation levels of RIP1/RIP3/MLKL, and its effect on cell viability loss was not comparable to PFKFB3 inhibitors, thereby PFKFB3 knockout was not sufficient enough to arouse necroptosis. This may due to that inhibitors cause acute cell stresses, prone to programmed cell death to avoid acute inflammation result from uncontrolled cell death manner. In addition, there are several PFKFB isoforms expressed in cells, and PFKFB3 knockout may stimulate the activity of other kind of PFKFB to maintain the cell homeostasis. However, under TNFα+Z-VAD-FMK treatment condition, knockout of PFKFB3 enhanced the necroptosis content when compared to the wild type cells. These observations were similarly with the report that loss function of PFKFB3 fails to arouse apoptosis itself, but renders cell apoptosis susceptible [32]. Different from 3BP induces autophagy, apoptosis and necroptosis, our former studies showed that PFK-15 inhibited autophagy process in renal cancer cells, and here we demonstrated PFK-15 could be considered as the necroptosis inducer. Thus, the relationships among autophagy, apoptosis and necroptosis are quite complicated, and they are not limited to concomitant occurrence.
A variety of genotoxic stimuli induce the formation of micronuclei, which can be used to screen genotoxic compounds [33]. Besides micronuclei, nuclear buds, and nucleoplasmic bridges are also considered as the biomarkers of genotoxic and chromosomal instabilities [20]. Our previous results showed that both sunitinib (chemotherapy drugs for renal cell carcinoma) and rasfonin simultaneously induced all three kind of phenotypes [34]. For the convenience of presentation, we statistical all three types of phenotype and labeled as micronuclei in the present study. Our results displayed that PFKFB3 inhibitors functioned as genotoxic reagents, as both the micronuclei percentage and γ-H2AX level were increased when exposure to them. In fact, the role of PFKFB3 in affecting genome instability is also complicated. Accumulating of PFKFB3 in cytoplasm protects cells from cisplatin-induced DNA damage [35], and inhibition of PFKFB3 impairs DNA repair through AKT signaling pathway [36]. However, it has been reported that p53-mediated suppression of PFKFB3 shut the glucose to PPP (pentose phosphate pathway), and the PPP product nucleotide played an essential role in promoting DNA damage repair [37]. Thus, the function of PFKFB3 during this process needs further exploration.
In conclusion, our results clearly showed that PFKFB3 inhibitors decreased cell proliferation/migration, cell viability, and stimulated apoptosis and necroptosis in SW480 and HT29 colorectal cancer cells. Both genetically and pharmacologically inhibition of necroptosis partly rescued the cytotoxic effect of PFKFB3 inhibitors. Meanwhile, PFK-15 aroused the genome instability, and necroptosis deprivation attenuated this process. These results shed light on the intrinsic relationships among PFKFB3, necroptosis and genome instability, and provide theoretical basis regarding PFKFB3 as potential therapeutic target for colorectal cancer.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31801169 to Yan S.), The Faculty Start-up Funds from Jining Medical University (to Yan S.), the Teacher Research Support Foundation in Jining medical university (JYFC2018KJ065 to Yan S.), and The Student Innovation Training Program in Jining Medical University (cx2019010, 201702010332).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bras M, Yuste VJ, Roue G, Barbier S, Sancho P, Virely C, Rubio M, Baudet S, Esquerda JE, Merle-Beral H, Sarfati M, Susin SA. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol Cell Biol. 2007;27:7073–7088. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23:748–756. doi: 10.1038/cdd.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 10.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 12.LaRocca TJ, Sosunov SA, Shakerley NL, Ten VS, Ratner AJ. Hyperglycemic conditions prime cells for RIP1-dependent necroptosis. J Biol Chem. 2016;291:13753–13761. doi: 10.1074/jbc.M116.716027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol. 2014;35:2–13. doi: 10.1016/j.semcdb.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan S, Wei X, Xu S, Sun H, Wang W, Liu L, Jiang X, Zhang Y, Che Y. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 spatially mediates autophagy through the AMPK signaling pathway. Oncotarget. 2017;8:80909–80922. doi: 10.18632/oncotarget.20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, Brock E, Siow D, Wattenberg B, Telang S, Chesney J. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem. 2009;284:24223–24232. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y, Liu Z, Zou X, Lan Y, Sun X, Wang X, Zhao S, Jiang C, Liu H. Mechanisms underlying 3-bromopyruvate-induced cell death in colon cancer. J Bioenerg Biomembr. 2015;47:319–329. doi: 10.1007/s10863-015-9612-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Q, Yan S, Sun H, Wang W, Li Y, Yang X, Jiang X, Che Y, Xi Z. Akt inhibition attenuates rasfonin-induced autophagy and apoptosis through the glycolytic pathway in renal cancer cells. Cell Death Dis. 2015;6:e2005. doi: 10.1038/cddis.2015.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan S, Zhou N, Zhang D, Zhang K, Zheng W, Bao Y, Yang W. PFKFB3 inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int J Mol Sci. 2019;20:5415. doi: 10.3390/ijms20215415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisurina-Evgenieva OP, Sutiagina OI, Onishchenko GE. Biogenesis of micronuclei. Biochemistry (Mosc) 2016;81:453–464. doi: 10.1134/S0006297916050035. [DOI] [PubMed] [Google Scholar]
- 21.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan J, Adamski R, Chen J. Focus on histone variant H2AX: to be or not to be. FEBS Lett. 2010;584:3717–3724. doi: 10.1016/j.febslet.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014;21:188–193. doi: 10.1038/cgt.2014.16. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210:2119–2134. doi: 10.1084/jem.20130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 27.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 28.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015;6:e1636. doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliver Metzig M, Fuchs D, Tagscherer KE, Grone HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene. 2016;35:3399–3409. doi: 10.1038/onc.2015.398. [DOI] [PubMed] [Google Scholar]
- 31.Han Q, Ma Y, Wang H, Dai Y, Chen C, Liu Y, Jing L, Sun X. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J Transl Med. 2018;16:201. doi: 10.1186/s12967-018-1580-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, Kamata A, Sakamoto K, Nakanishi T, Kubo A, Hishiki T, Suematsu M. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480. doi: 10.1038/ncomms4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balmus G, Karp NA, Ng BL, Jackson SP, Adams DJ, McIntyre RE. A high-throughput in vivo micronucleus assay for genome instability screening in mice. Nat Protoc. 2015;10:205–215. doi: 10.1038/nprot.2015.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan S, Liu L, Ren F, Gao Q, Xu S, Hou B, Wang Y, Jiang X, Che Y. Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis. 2017;8:e2988. doi: 10.1038/cddis.2017.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li FL, Liu JP, Bao RX, Yan G, Feng X, Xu YP, Sun YP, Yan W, Ling ZQ, Xiong Y, Guan KL, Yuan HX. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun. 2018;9:508. doi: 10.1038/s41467-018-02950-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi WK, Zhu XD, Wang CH, Zhang YY, Cai H, Li XL, Cao MQ, Zhang SZ, Li KS, Sun HC. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis. 2018;9:428. doi: 10.1038/s41419-018-0435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, Zhang Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. 2016;6:38067. doi: 10.1038/srep38067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.