

Abstract
As an important trace element, iron plays an essential role in many biology processes like cell proliferation, metabolism, and mitochondrial function. However, the disruption of iron homeostasis tends to cells death and human diseases due to it servers as mediator to promote the production of reactive oxygen species (ROS). In this review, first we introduced the mechanism of complex iron-mediated ROS involved in apoptosis, necroptosis, ferroptosis and pyroptosis. Next, we discussed the controversial role of excess iron and iron deficiency in tumor. Finally, we discussed the anti-cancer effects of iron on both sides, and novel iron-related strategies. This review outlined the mechanisms and regulation of iron homeostasis and iron-mediated ROS in tumors, and discussed the iron-related treatments.
Keywords: Iron, metabolism, ROS, cancer therapy, homeostasis
Iron and cancer
Elemental iron is one of the most plentiful and widely used metal on Earth [1]. It is essential for proper functioning of biological systems in most eukaryotes, is distributed throughout the human body and is concentrated in the liver, spleen, and lungs [2]. Iron is also an important component of some complex proteins, such as in hemoglobin, myoglobin, heme enzymes, and nonheme compounds, which facilitates the transport and exchange of oxygen in the blood, and involves in electron transfer and other redox reactions [3]. It is crucial for cellular metabolic activity, including proliferation, metabolism, and growth [4]. Because iron and its related compounds can be oxidized or reduced, they would participate in reactions with free radicals. Generation of free radicals is concerning as they can interact cellular proteins and lipids, impair their function and cause DNA strand damage, ultimately resulting in cellular dysfunction or mutations [5-7]. In particular, lipid dysfunction can promote the iron-dependent death, ferroptosis, which is distinct from apoptosis, necrosis, and autophagy [8,9]. Because of permanent impacts on redox reactions and its role in cellular proliferation, iron has been consistently considered as a carcinogen [10,11]. Simply, as an essential and potentially toxic element in all mammals, the regulation of iron import, storage, and export in body fluids influence body health.
The dysregulation of iron homeostasis is common in malignant cancer, specifically, for cellular metabolism, and it seems that iron accumulation impels a risk of tumor [12]. Speculatively, cancer cells may have a sophisticated mechanism for acquiring, transporting, and storaging iron. Iron-related proteins such as transferrin receptor 1 (TFR1), can bind iron-bound transferrin, has a high abundance in cancer patients suggesting cancerous cells have a greater iron demand when compared with normal cells [13-16]. Due to the ferroportin (FPN) is the only protein directly functions in export iron, a number of studies have suggested that abundances of FPN are significantly lesser in tumorous cells, especially for breast cancer [17-20]. Furthermore, the stability of FPN is maintained via hepcidin, which directly interacts with FPN and induces release of iron into extracellular fluid [21], and the increased abundance of hepcidin in serum have been observed in various tumors [22]. Cancerous cells produce hepcidin inside the cell, and this can lead to the degradation of FPN, and leading to elevated concentrations of iron in cells [23-25]. Therefore, abundance of FPN and hepcidin may be used as potential predictors of some cancers. Divalent metal transporter-1 (DMT1) as the transporter intercellular endosomal membrane has reported be responsible for iron accumulation in colorectal cancer [26]. Consequently, the abundance of iron-related proteins influence cellular iron and the processes of cellular division, growth, and survival [27].
Carcinogenesis of iron was first studied in 1959, they built a sarcoma model in rats intramuscularly injected with iron, suggesting iron contributed to tumorigenesis [28]. Similarly, excessive dietary iron or direct injection of iron may produce excess reactive oxygen species (ROS) and contribute to greater risks of developing cancer [29]. A number of studies have demonstrated that high levels of iron and ferritin increase risks of developing some solid tumors [30-32]. Mechanically, the tumorigenicity of excess iron could directly inhibited the degradation of p53 signaling, which provides an insight for iron-based therapy, instead, other researchers found that Fe2+ inhibited granulosa cells proliferation through ROS-mediated p38-MAPK/p53/p21 pathway [33,34]. Besides, iron would combine with cyclin-dependent kinases 1 (CDK1) to upregulate the expression of IL-6 receptor subunit GP130 through phosphorylation of 4E-BP1, which promotes the tumorigenicity by activating the JAK/STAT3 pathway [35]. What’s more, p53 inhibits cystine into the tumor cells and suppresses the expression of solute carrier family 7 member 11 (SLC7A11) sensitizes cells to ferroptosis [36]. The main pathway of iron in cancer cells are presented in Figure 1. In contrast, the study from Jian et al. had found that excess iron improved the survival time and reduced tumor recurrence in younger patients, whereas the opposite results were observed in older patients [37]. Interestingly, Cross et al. observed that 21 male volunteers with diets high in red meat, indicated higher iron consumption, had a higher chance of developing tumors in a number of organs, but no promotion for carcinogenesis was observed in inorganic iron group [38,39]. Although evidence exists to suggest iron is a precancerous substance, few studies have been published recently. Considering the complex role of iron in tumor formation, metastasis and cell survival, a better understanding of the mechanisms and related regulations of iron in the body are warranted.
Figure 1.
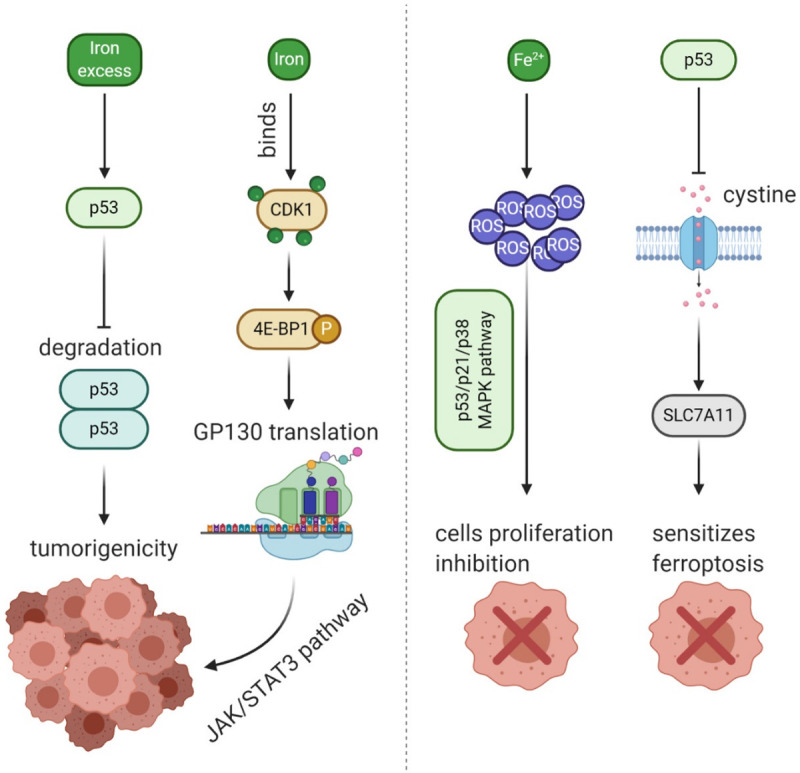
The schematic diagram of some iron-related pathway in cancer cells. The excess iron would impact the degradation of p53, then promote the tumorigenicity; iron binds the protein CDK1 activate 4E-BP1, then lead to translation of GP130 via JAK/STAT3 pathway; Fe2+ produce ROS in cancer cells which inhibits the proliferation of cells trough p53/p21/p38 MAPK pathway; p53 could also inhibit the uptake of cystine to repress expression of SLC7A11, and then promote the ferroptosis in cancer cells.
The catalysis of ROS induced by iron
Iron is involved in a number of cellular metabolic processes, the imbalance of iron homeostasis can directly or indirectly lead to generation of ROS, and induce cell damage or death [40,41]. Iron is an important component of ROS-generated enzymes such as those involved in the electron transport chain, nicotinamide adenine dinucleotide phosphate hydride (NADPH) oxidase, P450 enzymes, lipoxygenases (LOX), and xanthine oxidase. ROS-related enzymes are mainly Fe-S protein containing such as heme, can generate soluble ROS, namely the superoxide radicals (O2 ·-), hydrogen peroxides (H2O2), and hydroxyl radicals (HO·) or lipid ROS such as lipid peroxyl radicals (LO·), and lipid peroxides (LOOH) [42,43]. Overall, formation of ROS is closely related to iron throughout the body [42,44].
The process of ROS formation can be divided into two general mechanisms. Oxygen is firstly converts into superoxide radicals by ROS-generated enzymes, and then reduces into hydrogen peroxide by superoxide dismutase (SOD). Then the hydrogen peroxide can be converted into water by glutathione peroxidase (GPX), or react with Fe-S proteins or heme to generate ferrous ion. The ferrous ions and total cellular liable iron (<20 μM) can also impel to produce ROS via Fenton reactions [45,46]. Highly reactive hydroxyl peroxyl radicals were produced by a Fenton reaction are more destructive to cells and promote the DNA strand breaks, damage proteins and lipids via peroxidation, they also in turn to induce mutagenesis or inhibit cancer suppressors [29,43]. Subsequently, hydroxyl peroxyl radicals can react with lipids and O2 to form into the lipid ROS or lipid peroxyl radicals. As a result, the free iron ion and highly reactive free radicals in cells can be carried out via the Fenton reaction, and then exert cytotoxicity by destroying a variety of biomolecules.
Fe3+ + O2 ·- → Fe2+ + O2
Fe2+ + H2O2 → Fe3+ + HO- + HO·
In addition, oxygen can react with lipids to release LOOH under the catalysis of LOX, followed by conversion to LOH via GPX4, or LOOH can undergo a Fenton reaction to produce lipid peroxyl radicals. Ferrous ion and labile iron can be converted into ferric ions via lipid peroxides and hydrogen peroxides [43] (Figure 2). The intracellular labile iron pool and iron-related proteins react with the H2O2, superoxide radicals or LOOH can directly produce higher reactive radicals via Fenton reactions, whereas indirect production of ROS via catalysis of iron-containing proteins requires a series of biological reactions. Interestingly, cancerous cells have increased abundances of iron and ROS but are able to maintain intracellular ROS homeostasis and evade death [47]. Regardless, ROS is important to the proper functioning of cells as it is involved in the signaling processes of cell proliferation, immunoregulation, autophagy, inflammation and stress-related responses, while the excessive ROS leads to oxidative stress and loss of metabolic function, suppressing normal cellular growth and can directly lead to cell death by directly damaging essential proteins, DNA or lipids, or lead to development of a range of diseases including inflammation and cancer [48]. Recent studies have demonstrated that ROS plays a complicated role in cells, as it is a mediator of some proteins which induce cell death [42,49,50].
Figure 2.
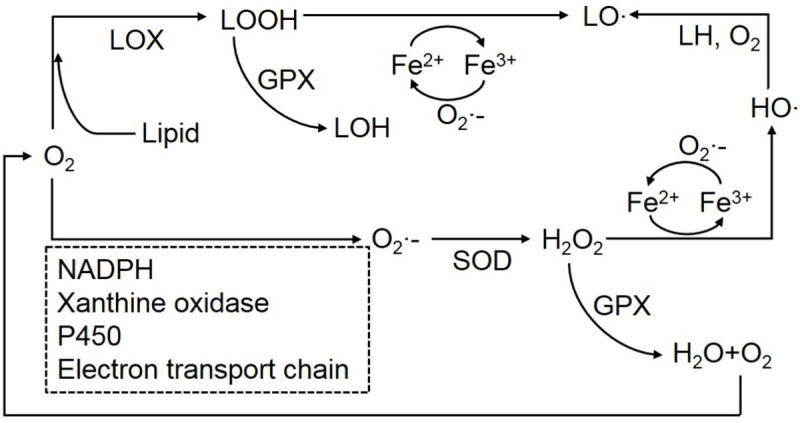
The process of ROS formation. Oxygen converts into superoxide radicals under the ROS-generated enzymes, and then consume hydrogen peroxide by superoxide dismutase (SOD). Hydrogen peroxide could be converted into water by glutathione peroxidase (GPX) instead of self-damage, or it can react with Fe-S proteins or heme to generate ferrous ion. Highly reactive hydroxyl peroxyl radicals produced by a Fenton reaction are destructive to cells. Besides, oxygen could produce LOOH company with the lipid in LOX, followed by converted into the LOH via GPX4, or LOOH can undergo a Fenton reaction to produce lipid peroxyl radicals. Ferrous ion and labile iron can be converted into ferric ions via lipid peroxides and hydrogen peroxides.
Because ROS is involved in a number of cell signaling pathways, such as suppressing signaling proteins related to cell senescence and death, activating apoptotic signaling pathways, and initiation of necroptosis [51-53]. In addtion, iron plays an important role in ROS production, iron-mediated ROS might contribute to various types of cell death including apoptosis and necrosis, including the ferroptosis and pyroptosis, the former is a type of programmed death with the properties of iron-dependent and lipid-oxidative, and the latter can be induced by iron-mediated ROS and dependent on formation of plasma membrane pores of the gasdermin (GSDM) protein family, often due to caspase activation [53,54].
Regulation of apoptosis by iron-mediated ROS
Apoptosis is characterized by a series of cellular changes including cell shrinkage, loss of adhesion, bubble formation, fragmentation of organelles and DNA degradation [55]. Apoptosis initiation can divided into an intrinsic and extrinsic pathway [53]. Generally, apoptosis is initiated by mitochondrial outer membrane permeabilization (MOMP) [56], where increased extracellular iron leads to increased concentrations of intracellular labile iron pools which promote MOMP and production of intracellular ROS. Iron-mediated ROS production can lead to intrinsic apoptosis via two pathways. Cardiolipins present on the mitochondrial outer membrane, can be oxidized by ROS, and cytochrome c released via Bak/Bax during MOMP can activate caspase-9/3 resulting in apoptosis [57,58]. In addition, ROS can depolymerize thioredoxin-apoptosis signal-regulating kinase 1 protein compound (Trx-ASK1) in the cytoplasm and dissociate ASK1 from an apoptotic signaling protein, ASK1 then mediates c-Jun N-terminal kinase (JNK)/p38 pathway to induce apoptosis [59-61]. In contrast, the extrinsic apoptotic pathway is initiated by Fas ligand (FasL) binding to Fas on the cell membrane, which is an apoptosis receptor that mediates production of apoptosis. Under high iron levels, serine and arginine-rich splicing factor 7 (SRSF7) is inhibited, thus impeding splicing and translation of Fas pre-mRNA into soluble Fas which is an antiapoptotic form. Therefore suppression of soluble Fas promotes cellular apoptosis via activation of caspase-8 [53,62,63].
Regulation of necroptosis by iron-mediated ROS
Necroptosis is a form of programmed necrosis, nonapoptotic cell death, which is triggered by death signaling stimuli such as ROS, tumor necrosis factor α (TNF-α), FasL and other stimuli [64,65]. Necroptosis is considered a cellular defensive mechanism against external pathogens or damage, which results in a cell undergoing self-destruction [65]. Because iron regulates ROS and Fas, necroptosis is closely related to iron. Similarly, although necroptosis is nonapoptotic cell death, it can also undergo extrinsic apoptosis [66]. In addition to the steps of extrinsic apoptosis described above, iron is involved in other necroptotic pathways. Ferritin heavy chain (FTH) is an important modulator of cellular necroptosis which is induced by TNF-α [67]. FTH can transform toxic labile iron in cells into non-toxic iron (Fe3+) [68], while TNF-α can increase the concentration of the iron pool and subsequent accumulation of ROS, thus it has been suggested that FTH might prevent necroptosis induced by TNF-α. In addition, studies have shown that TNF-α induced necroptosis is blocked by FTH through the JNK pathway, however contrasting results have been observed as one JNK phenotype, JNK1, promotes ROS accumulation, which suggests that the underlying mechanism of JNK on TNF-α induced necroptosis remains unclear [69,70]. Besides, other studies have suggested that autocrine TNF-α binds to TNF-α receptor (TNFR), which couples with riboflavin kinase (RFK) to mediate activation of NADPH oxidase 1 (NOX1), which incorporates iron. Activated NOX1 can induce necroptosis by activating the JNK pathway [71,72]. It is worth mentioning that heme can also participate in the process of necroptosis in macrophages, whereby free extracellular heme can enter the cell via Toll-like receptor 4 (TLR4) and result in TNF-α production [73], ultimately resulting in necroptosis. Intracellular heme is degraded into toxic labile iron under catalysis by HMOX1, which in turn generates ROS and activates JNK, inducing necroptosis [74,75].
Regulation of ferroptosis by iron-mediated ROS
Ferroptosis is a type of cell death that was recently discovered to be iron-dependent in certain cell types and differs from apoptosis, necrosis and autophagy as it results in lipid ROS formation [8,76]. Similar to the other types of programmed cell death, ROS and labile iron are important in ferroptosis. It has been demonstrated that ferroptosis can be rescued following treatment with the lipid antioxidants Trolox, ferrostatin-1 or vitamin E [8]. Interestingly, many factors could induce the ferroptosis. The ferroptosis inducers, artemisinin and iron, can be used to effectively promote ferroptosis, whereas their combination usually does not cause ferroptosis, but apoptosis [77,78]. One study has seggested that the sufficient amount of iron is the initiator of ferroptosis, and iron chelator deferoxamine can be useful to hamper the ferroptosis [8]. In brief, iron does a matter for promoting the ferroptosis through direct and indirect effects mainly via lipid ROS.
The main process of ferroptosis can be as follows, nuclear receptor coactivator 4 (NCOA4) specifically binds ferritin to form ferritinophagy, followed by release of free iron, thereby increasing the content of cellular labile iron and leading to direct production of lipid ROS [79]. In addition, ferroptosis can be initiated by peroxidation via reaction of arachidonoyl (AR) and then conversion to AR-CoA via acyl-CoA synthetase long-chain family 4 (ACSL4) catalysis. Next, AR-CoA is converted to AR-PE by lysophosphatidylcholine acyltransferase 3 (LPCAT3) and finally conversion to AR-LOOH-PE via LOXs [80-83]. After generation of lipid ROS, it can react with cellular membranes, change the permeability of the membrane, reduce thickness and sensitivity to oxides, eventually lead to cellular membrane to rupture and finally induce the ferroptosis [84].
In general, ferroptosis is tightly regulated by intracellular signaling pathways, including iron homeostasis regulatory pathways, RAS pathways, and cystine transport pathways. Ferroptosis is caused by the inactivated of glutathione peroxidase (GPX4), an enzyme for membrane lipid repairing, results in the accumulation of ROS on membrane lipids by the reaction of Fenton, which requires the participation of iron [85], thus the iron-mediated ROS plays an important role in ferroptosis. Ferroptosis can also be caused by a variety of substances and other pathways. Small molecule erastin inhibits cystine-glutamate exchange on plasma membrane, which reduces the acquisition of cystine by cells, thus impeding the synthesis of glutathione, the substrate of GPX4, and eventually triggering the accumulation of membrane lipid ROS and ferroptosis, in addition, another small molecule, RSL3, which acts as an inhibitor of GPX4, also causes ferroptosis [86]. Thus, the lipid ROS and iron play an important role in ferroptosis, however the exact mechanisms and relations among lipid ROS, iron and ferroptosis remain unknown [87,88].
Regulation of pyroptosis by iron-mediated ROS
Pyroptosis, a type of programmed cell death, is dependent on formation of plasma membrane pores by the GSDM family, including GSDMA, GSDMB GSDMC, GSDMD, and GSDME. The ability of GSDMD and GSDME to induce pyroptosis has been well studied and consists of breaking up inflammatory caspases and apoptotic caspases respectively [54,89]. Inducement of pyroptosis had been observed in melanoma cells by iron-mediated ROS [90]. In this study, carbonyl cyanide m-chlorophenyl hydrazine (CCCP) was used to initiate formation of iron-mediated ROS via interaction with exogenous iron in melanoma cancer cells which initiated mitochondrial outer membrane protein Tom20 oxidation, recruitment of BAX and inducement of cytochrome c release, thereby activating cas3/9 cleavage of the GSDM and subsequent formation of pores in the cell membrane leading to pyroptosis [90,91].
Controversial effect of iron in tumors
Due to the participation of iron in redox processes, iron directly participates in Fenton reactions to produce ROS, and can be incorporated into proteins involved in cellular metabolism [92]. Iron involves in a number of important biological processes and any imbalance in its storage, transfer or efflux can negatively affect the health of cells [93]. It is well known that excess iron plays an important role in tumorigenesis, diabetes and coronary heart disease, in contrast iron deficiency, in healthy organisms, can lead to anemia and other more serious diseases such as cancer [41,93-96].
Excess iron and carcinogenesis
Generally, total body iron loads greater than 5 g are classified as an iron overload [97]. As previously demonstrated, excess iron can lead to production of ROS, thereby increasing risks of developing cancer [10]. Iron intake can be divided into nonheme (90%) and heme (10%) iron, where iron derived from meat sources is mostly present as heme iron, and vegetables have high levels of nonheme iron [98]. Results of previous studies have demonstrated that intake of red meat or processed meat can increase risks of developing colorectal cancer when compared to white meat, whereas ingestion of white meat with equal amounts of excess iron (as red meat) does not increase risks of developing a number of forms of cancer but reduces it. It has been also suggested that the high content of heme iron in red meat contributes to the increased risk of developing colorectal cancer [39,99]. Interestingly, the role of inorganic iron in non-heme tumorigenesis remains controversial. Sesink et al. observed that equimolar amounts of inorganic iron in the diet increased colonic epithelial proliferation and fecal toxicity, whereas heme or protoporphyrin did not. In addition, some studies have observed that excess inorganic iron in the diet promotes tumorigenesis in mice, but not sufficiently to achieve tumor formation [39,100,101]. Furthermore, Cross et al., confirmed that the main cause of heme-derived carcinogenesis is due to an increase in the body’s nitrosation following heme intake which can induce tumorigenesis in a number of organs [38]. As more studies have observed that heme iron promotes production of nitrosation in the body and increased accumulation of lipid peroxides, it has been recognized that this leads to promotion of tumor development [102-106].
Iron deficiency and carcinogenesis
Iron deficiency is a public health issue and has come to the forefront as the most common nutritional deficiency globally. Iron deficiency is a cause of anemia, it has been estimated that over 35% of the global population exhibits some symptoms of iron deficiency and over 50% of pregnant women [107]. Recent studies have found that iron deficiency can lead to other more serious negative impacts. Results of a nationwide population-based study suggested that patients presenting with iron-deficiency based anemia have a higher risk of developing gastrointestinal cancer and it might be related to compromised immune activity [108]. Similarly, Dallman observed that patients presenting with iron deficiency had abnormal cell-mediated immunity responses [109]. It has confirmed that iron deficiency impacts the body’s antioxidant capacity, which in turn impacts the body’s ability to control oxidative stress, damages mitochondrial functioning, and ultimately affects cell metabolism [110]. Individuals with low iron intake or low body iron reserves have an increased risk of developing gastrointestinal tumors, and other in vivo data derived from rodent cancer models suggested early progression of gastrointestinal tumors during iron deficiency [111]. Other studies have investigated a link between iron deficiency and gastric cancer [112,113]. For example, some studies have shown that the occurrence of some precancerous lesions are most likely a result of iron deficiency [112,114]. Plummer-Vinson syndrome (PVS) is a condition characterized by iron deficiency based anemia, upper airway stenosis, and ceramic dysphasia, and seems to be related to gastric and esophageal carcinoma [115]. It has been speculated that the role of PVS in carcinogenesis was caused by iron deficiency [116]. The research team of Richie found that mild iron deficiency and low glutathione levels contributed to elevated levels of oxidative stress and an increased risk of oral cancer [117]. Besides, some researchers have demonstrated, used a mouse model, that an iron-deficient diet can lead to stability of hypoxia-inducible factor-α (HIF-α), which plays an important role in breast cancer malignancy and invasiveness [118]. Overall, the relation between iron deficiency and carcinogenesis has been extensively studied and it is recognized that iron deficiency can lead to increased oxidative stress, impact immune functioning and alter cellular oxidative metabolic conditions.
Role of double-sided iron in tumor treatment
Iron plays a vital role in mammalian cells including cellular growth and proliferation, and catalyzes production of ROS by cells, which is essential for normal physiological functioning of cells [119]. Cancerous cells require greater amounts of iron for proliferation when compared to normal cells [31,120,121]. In this regard, tumor cells produce greater amounts of ROS when compared to normal cells and thus exert greater energy in maintaining ROS homeostasis, therefore the sensitivity of cancer cells to ROS might present an opportunity for suppressing tumor growth [96,122]. In fact, tumor cells accumulate mutations throughout tumorigenesis as they evolve from a precancerous to cancerous state, ROS plays an important and varying role throughout this process depending on the stage of development [123]. For example, ROS can promote cell growth and proliferation, while slightly higher concentration of ROS can induce apoptosis or differentiation, and even higher concentration might cause initiation of necrosis [124]. Recently, oxidative therapy has been applied in the treatment of tumors with the aim of increasing production of ROS in mitochondria of tumor cells or inhibit their antioxidant capacity [122,125]. It is evident that imbalance of cellular ROS and iron can lead to development of cancerous cells or initiation of cell death processes. However, it remains unclear if changes in the abundance of intracellular iron or ROS can be used to treat certain types of cancer.
Excess iron therapy
Tumor cells require additional iron and produce greater amounts of ROS, therefore strategies to negate development of tumors might focus on concentrations of intracellular iron or ROS. For example, one study observed that the iron complex ferric-sorbitol-citrate (FSC) can inhibit proliferation of a variety of tumor cells, including B16, KB, HeLa, and GHC cells, in vivo and in vitro, however no effect was observed in HBS or Vero cells. Interestingly, the same team demonstrated that ferrocene analogs can be used to inhibit a range of malignancies including in the HepG2 and B16-F10 tumor cell lines, but did not inhibit fibroblasts in human HEF or mice L929 cells. Similarly, it was found that FSC can reduce expression of Bcl-2 and mp53 proto-oncogenes in CaCo2 cells and inhibit proliferation of malignant tumor cells by inducing apoptosis [126,127]. These iron-related substances increase the abundance of intracellular labile iron pool in tumor cells leading to accumulation of ROS, and inhibition of cell growth and in some cases apoptosis. As stated above, ferroptosis is a recently identified type of programmed cell death that is characterized by iron-dependent or iron-mediated ROS production. Shenglin Fang et al. demonstrated that excess cellular iron caused by ferric ammonium citrate (FAC) and Ferric 8-hydroxyquinoline complexes can induce HT1080 fibrosarcoma cell ferroptosis and induce AML12 cell parthanatos partially activating polymerase-1 [128]. Qiao Wu et al. identified iron-mediated ROS as a causative factor driving GSDME-dependent pyroptosis, interestingly, they demonstrated that supplementing iron-deficient patients with iron can result in clinically significant ROS-induced antitumor effects via pyroptosis and inhibit xenograft tumor growth or melanoma cell metastasis [90]. Kiessling et al. reported that ferritin heavy chain is down-regulated via disruption of the NF-κB pathway in cutaneous T-cell lymphoma cells, resulting in increased concentrations of intracellular iron and production of greater amounts of ROS in cells resulting in cell death [129]. However, no effect was observed with the same treatment in isolated primary T cells [129]. And when compared to traditional iron replacement therapies, iron oxide nanoparticles might have a number of advantages in prevention of tumors. For example, inherent magnetic properties of iron oxide nanoparticles might aid in gene delivery and cell sheet formation to promote angiogenesis, and nanoparticles can be tracked using magnetic resonance imaging [130,131]. However, magnetic nanoparticles have been observed as non-toxic in vitro but to selectively kill certain tumor types in vivo [132]. Intracellular iron-dependent cell death is influenced by the rate of iron accumulation and formation of iron complexes which can selectively kill tumor cells.
Iron removal therapy
Tumor cells require higher abundances of iron to facilitate proliferation and DNA synthesis [133,134], therefore selective removal of iron might be an effective treatment method [135-137]. Iron chelators eliminate iron from cells by binding iron with high affinity, and has been demonstrated to inhibit aggressive proliferation of tumors such as neuroblastoma and breast cancer cells in rodent models and patients [138-140]. It has been confirmed in some preclinical studies that some iron chelators are structurally and pharmacologically different and can be defined as antitumor drugs which effectively resist tumor growth by chelating iron [20,141]. The drugs deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX), have been developed to fix iron into a soluble form, are safe for use, and have been used in the clinical settings for their anti-tumor efficacy [142-144]. Due to the clinical safety of DFO, it has received the majority of attention in clinical research. Studies have shown that MCF-7 and MDA-MB-231 breast cancer cells treated with excess DFO exhibited disrupted iron statuses after treatment, reduced cell viability and growth potential, and increased breast cancer apoptosis [145]. DFP has the potential to effectively inhibit proliferation of prostate cancer in clinical applications, furthermore a meta-analysis has found that a combination therapy of DFP and DFO results in better improvements in cardiac ejection when compared to monotherapy, but had no other significant effects [146,147]. DFX is considered a potential NF-κB inhibitor and can specifically induce apoptosis in myeloid leukemia cell lines. DFX has been shown to be safe in several case reports, and some common side effects are not observed, including no progressive change in the serum, intestinal and skin [148]. In other studies, iron chelators such as DFO were used in combination with some chemotherapeutic drugs to increase efficacy against advanced blastoma and neuroectodermal tumors [149]. In addition, another study has reported that chelators inhibit growth of tumors and influence the polarization of macrophages, lighted another option in the development of novel therapies [150].
Novel iron-based treatment strategies for cancer
Recently it has been observed that overloading of macrophages with iron results in unrestrained M1 phenotype in chronic venous leg ulcers [151]. Similarly, it has reported that macrophages with excess iron undergo polarization resulting in a detrimental proinflammatory-M1 type response in injured spinal cord patients [152]. In addition, the occurrence and progression of tumors are largely dependent on signals from the external environment. And studies have demonstrated iron-containing complexes, such as ironomycin, have a therapeutic effect on development of resistance to the breast cancer stem cells, ultimately resulting in generation of iron-dependent ROS and tumor cell death [153]. Results of these studies suggest that iron-mediated cell death could be used to target the tumor microenvironment (TM) [133,154]. In fact, there are many non-malignant cells in the TM, such as immune cells and blood vessels, which are closely related to tumor tissues [155]. It has been demonstrated that the abundance of iron-related proteins in the microenvironment of macrophages, lymphocytes, and other immune cells, are correlated with clinical prognosis markers of cancers in patients. Therefore, targeting the homeostasis of tumor microenvironment might be an effective and novel treatment strategy for certain cancers [156].
Macrophages play a sophisticated role in maintaining iron homeostasis by recovering iron from damaged hemoglobin or senescent erythrocytes [157]. In TM, pro-inflammatory phenotypes of M1 macrophages contribute to iron sequestration, while anti-inflammatory phenotypes of M2 macrophages release iron into the microenvironment and thus promote growth of tumor cells [158,159]. Furthermore, macrophages are involved in the pathogenesis of some diseases, and potential to be the therapeutic targets in development of treatments [160]. In this regard, the dichotomy between M1 and M2 provides an abundance of pathological targets for treatment such as macrophage elimination or reprogramming [161]. For example, tumor-associated macrophages could be reprogrammed to an anti-cancer phenotype by overloading the intracellular iron which promotes an immune responses, and directly causes tumor death [162]. Breakthroughs have been recently made in the application of nanotechnologies as immunotherapies for cancers by regulating polarization of macrophages [163,164]. Previously, Zanganeh et al. found the FDA-approved iron supplement, ferumoxytol, to inhibit tumor growth by reprogramming macrophages to increase the abundance of M1 macrophages in TM [165]. In another recent study, a magnetic iron oxide nanomaterial was reported to promote M1 macrophage polarization by elevating intracellular iron levels via the IRF5 pathway, and leading to cytotoxic T lymphocyte (CTL) activation in TM in combatting the development of tumors [166]. To improve the biocompatibility of nano-iron materials, researchers used the macrophage membranes, combined with iron materials (Fe3O4) to further improve targeted function as the photothermal properties of Fe3O4 can be used to improve the efficiency of the method for tumor treatment [167]. Thus, as an important component in tumor microenvironment, macrophages may be an important target for effective and novel anti-tumor therapies in combination with iron materials. And iron materials affect the phenotype of macrophages in TM, further changing the characteristics of TM and affecting tumor growth.
Redox homeostasis of cells plays an important role in cell survival, growth and proliferation, and new strategies to combat development of cancers have focused on the role played by ROS and iron. Generation of ROS and abundance of free iron tightly control to maintain redox homeostasis, and disruption can lead to negative impacts [124]. Therefore, among available tumor treatment strategies, controlling abundances of intracellular ROS or iron is an important consideration. Except for the therapy of iron chelators, researches have focused on elucidating the anti-tumor mechanisms observed following disorder in redox homeostasis [168-170]. For example, ascorbate therapy has been demonstrated to influence tumor cells via an iron-mediated manner, which is dependent on the production of excess H2O2 by metal catalysis in cancer cell, mainly by iron, ascorbate does the agent of antioxidant function to induce oxidative stress [171,172]. Although ascorbate therapy has been ambiguous in tumor treatment, it has been proposed in recent years as a potential antitumor therapy [173,174]. Alternatively, ferroptosis, is mainly characterized by the production of iron-dependent lipid peroxides, it can also lead to the disorder of redox [170], and many studies further combined with other drugs changes the redox homeostasis in the cells, causing ferroptosis. Recent studies have found that high concentrations of iron and artemisinin in tumors, a traditional Chinese medicine for the treatment of malaria, promote the production of free radicals and induce ferroptosis [175,176]. However, the mechanism of how artemisinate induces ferroptosis remains unclear [177,178].
Conclusions
Iron homeostasis is important to the functioning of normal and tumor cells. When compared to normal cells, tumor cells require greater amounts of iron to ensure survival, maintenance of TM and proliferation. Aberrant expression of iron-related proteins is a common feature of malignant tumors. In addition, tumors exhibit altered iron-related pathways and physiological processes. Due to the role of iron in tumor formation and proliferation, iron-related gene or protein expression have been highlighted for their prognostic value and as a reference for clinical decision-making. However, changes in iron-related proteins are cancerous type specific, thus limiting their prognostic value. Generally, it is believed that iron metabolism imbalances are related to metastases [133].
The dependence of tumor cells on iron provides an opportunity for development of anti-tumor treatments. For example, treatments can focus on limiting iron utilization by tumor cells, thus inhibiting tumor growth. Thus, removal of iron is a promising prospect for treatment of tumors. Anti-cancer drugs can be designed to target free iron near tumor cells or to target iron-related proteins important to metabolism, and greatly influence tumor growth. In addition, elimination of intracellular iron by chelating agents can elicit production of cytotoxic ROS in tumor cells. However, cancer stem cells (CSCs) have robust reactive oxygen defense systems [179]. Therefore, treatment methods must increase ROS levels and target the oxidative response system of CSCs to inhibit their growth. Moreover, ferroptosis caused by excess iron has been recently discovered and offers a unique opportunity for development of targeted therapies. Selective targeting of tumor cells via ferroptosis might become an attractive anti-cancer strategy, however a greater understanding of its mechanism is required.
Despite greater amounts of studies investigated the role of iron in tumor development, more studies are required. For example, metabolic mechanisms of tumor cells, mechanisms of iron-mediated ROS production, mechanisms of increased iron-mediated ROS induced cell death, mechanisms of iron-mediated ROS effects on the tumor microenvironment require further research. In addition, the safety and efficiency of iron-related anti-tumor preclinical drugs need to be established. Iron-related threpies have some basic and clinical application cases in the treatment of cancer, they are gradually becoming one of the new methods for the treatment of cancer, and has a great prospect. We expect these therapies to be effective against the tumor. However, as mentioned above, there are still some limitations and unknowability in the study of iron related therapeutic mechanism, which requires further research and exploration, eventually enriches the means of anti-tumor therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81672430 to XZM).
Disclosure of conflict of interest
None.
References
- 1.Frey PA, Reed GH. The ubiquity of iron. ACS Chem Biol. 2012;7:1477–1481. doi: 10.1021/cb300323q. [DOI] [PubMed] [Google Scholar]
- 2.Gupta A. Iron Metabolism in Human Body. Nutritional Anemia in Preschool Children. Singapore: Springer Singapore; 2017. pp. 29–46. [Google Scholar]
- 3.Hurrell RF. Bioavailability of iron. Eur J Clin Nutr. 1997;51(Suppl 1):S4–8. [PubMed] [Google Scholar]
- 4.Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov. 2017;16:400–423. doi: 10.1038/nrd.2016.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue S, Kawanishi S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987;47:6522–6527. [PubMed] [Google Scholar]
- 6.Pfeifhofer-Obermair C, Tymoszuk P, Petzer V, Weiss G, Nairz M. Iron in the tumor microenvironment-connecting the dots. Front Oncol. 2018;8:549. doi: 10.3389/fonc.2018.00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storr SJ, Woolston CM, Zhang Y, Martin SG. Redox environment, free radical, and oxidative DNA damage. Antioxid Redox Signal. 2013;18:2399–2408. doi: 10.1089/ars.2012.4920. [DOI] [PubMed] [Google Scholar]
- 8.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2017;8:992. doi: 10.3389/fphar.2017.00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Yu L, Ding J, Chen Y. Iron metabolism in cancer. Int J Mol Sci. 2018;20:95. doi: 10.3390/ijms20010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou L, Zhao B, Zhang L, Wang S, Dong D, Lv H, Shang P. Alterations in cellular iron metabolism provide more therapeutic opportunities for cancer. Int J Mol Sci. 2018;19:1545. doi: 10.3390/ijms19051545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung M, Mertens C, Tomat E, Brüne B. Iron as a central player and promising target in cancer progression. Int J Mol Sci. 2019;20:273. doi: 10.3390/ijms20020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung M, Mertens C, Brune B. Macrophage iron homeostasis and polarization in the context of cancer. Immunobiology. 2015;220:295–304. doi: 10.1016/j.imbio.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Gao W, Ye G, Duan X, Yang X, Yang VC. Transferrin receptor-targeted pH-sensitive micellar system for diminution of drug resistance and targetable delivery in multidrug-resistant breast cancer. Int J Nanomedicine. 2017;12:1047–1064. doi: 10.2147/IJN.S115215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, Karl RT, Rosager AM, Nixon AM, Tesar PJ, Hamerlik P, Kristensen BW, Horbinski C, Connor JR, Fox PL, Lathia JD, Rich JN. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, Xian W, McKeon F, Lynch M, Crum CP, Hegde P, Brewer M, Wang X, Miller LD, Dyment N, Torti FM, Torti SV. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36:4089–4099. doi: 10.1038/onc.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang S, Wang X, Guo W, Wang L, Zhang D, Yuan L, Zhang Z, Xu Y, Liu S. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell Signal. 2015;27:168–176. doi: 10.1016/j.cellsig.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1:155–157. doi: 10.1016/j.cmet.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Xue D, Zhou CX, Shi YB, Lu H, He XZ. Decreased expression of ferroportin in prostate cancer. Oncol Lett. 2015;10:913–916. doi: 10.3892/ol.2015.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng Z, Manz DH, Torti SV, Torti FM. Effects of ferroportin-mediated iron depletion in cells representative of different histological subtypes of prostate cancer. Antioxid Redox Signal. 2019;30:1043–1061. doi: 10.1089/ars.2017.7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- 22.Dong D, Zhang G, Yang J, Zhao B, Wang S, Wang L, Zhang G, Shang P. The role of iron metabolism in cancer therapy focusing on tumor-associated macrophages. J Cell Physiol. 2019;234:8028–8039. doi: 10.1002/jcp.27569. [DOI] [PubMed] [Google Scholar]
- 23.Ward DG, Roberts K, Brookes MJ, Joy H, Martin A, Ismail T, Spychal R, Iqbal T, Tselepis C. Increased hepcidin expression in colorectal carcinogenesis. World J Gastroenterol. 2008;14:1339–1345. doi: 10.3748/wjg.14.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu XN, Su D, Wang L, Yu FL. Roles of the hepcidin-ferroportin axis and iron in cancer. Eur J Cancer Prev. 2014;23:122–133. doi: 10.1097/CEJ.0b013e3283627f14. [DOI] [PubMed] [Google Scholar]
- 26.Brown RAM, Richardson KL, Kabir TD, Trinder D, Ganss R, Leedman PJ. Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front Oncol. 2020;10:476. doi: 10.3389/fonc.2020.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwok JC, Richardson DR. The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002;42:65–78. doi: 10.1016/s1040-8428(01)00213-x. [DOI] [PubMed] [Google Scholar]
- 28.Richmond HG. Induction of sarcoma in the rat by iron-dextran complex. Br Med J. 1959;1:947–949. doi: 10.1136/bmj.1.5127.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi JY, Neuhouser ML, Barnett MJ, Hong CC, Kristal AR, Thornquist MD, King IB, Goodman GE, Ambrosone CB. Iron intake, oxidative stress-related genes (MnSOD and MPO) and prostate cancer risk in CARET cohort. Carcinogenesis. 2008;29:964–970. doi: 10.1093/carcin/bgn056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintana Pacheco DA, Sookthai D, Graf ME, Schubel R, Johnson T, Katzke VA, Kaaks R, Kuhn T. Iron status in relation to cancer risk and mortality: findings from a population-based prospective study. Int J Cancer. 2018;143:561–569. doi: 10.1002/ijc.31384. [DOI] [PubMed] [Google Scholar]
- 31.Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and cancer. Annu Rev Nutr. 2018;38:97–125. doi: 10.1146/annurev-nutr-082117-051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sukiennicki GM, Marciniak W, Muszynska M, Baszuk P, Gupta S, Bialkowska K, Jaworska-Bieniek K, Durda K, Lener M, Pietrzak S, Gromowski T, Prajzendanc K, Lukomska A, Waloszczyk P, Wojcik JZ, Scott R, Lubinski J, Jakubowska A. Iron levels, genes involved in iron metabolism and antioxidative processes and lung cancer incidence. PLoS One. 2019;14:e0208610. doi: 10.1371/journal.pone.0208610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, Hu H, Huang Y, Hui L, Xie D, Wang F, Hu R. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014;7:180–193. doi: 10.1016/j.celrep.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen MJ, Chou CH, Shun CT, Mao TL, Wen WF, Chen CD, Chen SU, Yang YS, Ho HN. Iron suppresses ovarian granulosa cell proliferation and arrests cell cycle through regulating p38 mitogen-activated protein kinase/p53/p21 pathway. Biol Reprod. 2017;97:438–448. doi: 10.1093/biolre/iox099. [DOI] [PubMed] [Google Scholar]
- 35.Kuang Y, Guo W, Ling J, Xu D, Liao Y, Zhao H, Du X, Wang H, Xu M, Song H, Wang T, Jing B, Li K, Hu M, Wu W, Deng J, Wang Q. Iron-dependent CDK1 activity promotes lung carcinogenesis via activation of the GP130/STAT3 signaling pathway. Cell Death Dis. 2019;10:297. doi: 10.1038/s41419-019-1528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jian J, Yang Q, Shao Y, Axelrod D, Smith J, Singh B, Krauter S, Chiriboga L, Yang Z, Li J, Huang X. A link between premenopausal iron deficiency and breast cancer malignancy. BMC Cancer. 2013;13:307. doi: 10.1186/1471-2407-13-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cross AJ, Pollock JR, Bingham SA. Haem, not protein or inorganic iron, is responsible for endogenous intestinal N-nitrosation arising from red meat. Cancer Res. 2003;63:2358–2360. [PubMed] [Google Scholar]
- 39.Corpet DE. Red meat and colon cancer: should we become vegetarians, or can we make meat safer? Meat Sci. 2011;89:310–316. doi: 10.1016/j.meatsci.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Huang X. Does iron have a role in breast cancer? Lancet Oncol. 2008;9:803–807. doi: 10.1016/S1470-2045(08)70200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 43.Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci. 2017;38:592–607. doi: 10.1016/j.tips.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 44.Worwood M. The laboratory assessment of iron status--an update. Clin Chim Acta. 1997;259:3–23. doi: 10.1016/s0009-8981(96)06488-1. [DOI] [PubMed] [Google Scholar]
- 45.Petrat F, de Groot H, Sustmann R, Rauen U. The chelatable iron pool in living cells: a methodically defined quantity. Biol Chem. 2002;383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 46.Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- 47.Kim J, Kim J, Bae JS. ROS homeostasis and metabolism: a critical liaison for cancer therapy. Exp Mol Med. 2016;48:e269. doi: 10.1038/emm.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, Ashrafi K, Glickman MH, Rutter J. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40:465–480. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ludtmann MHR, Arber C, Bartolome F, de Vicente M, Preza E, Carro E, Houlden H, Gandhi S, Wray S, Abramov AY. Mutations in valosin-containing protein (VCP) decrease ADP/ATP translocation across the mitochondrial membrane and impair energy metabolism in human neurons. J Biol Chem. 2017;292:8907–8917. doi: 10.1074/jbc.M116.762898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–767. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 53.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 56.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 57.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 58.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 61.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 63.Cascino I, Fiucci G, Papoff G, Ruberti G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol. 1995;154:2706–2713. [PubMed] [Google Scholar]
- 64.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 66.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie C, Zhang N, Zhou H, Li J, Li Q, Zarubin T, Lin SC, Han J. Distinct roles of basal steady-state and induced H-ferritin in tumor necrosis factor-induced death in L929 cells. Mol Cell Biol. 2005;25:6673–6681. doi: 10.1128/MCB.25.15.6673-6681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruzzenenti P, Asperti M, Mitola S, Crescini E, Maccarinelli F, Gryzik M, Regoni M, Finazzi D, Arosio P, Poli M. The Ferritin-Heavy-Polypeptide-Like-17 (FTHL17) gene encodes a ferritin with low stability and no ferroxidase activity and with a partial nuclear localization. Biochim Biophys Acta. 2015;1850:1267–1273. doi: 10.1016/j.bbagen.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 69.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 70.Antosiewicz J, Ziolkowski W, Kaczor JJ, Herman-Antosiewicz A. Tumor necrosis factor-alpha-induced reactive oxygen species formation is mediated by JNK1-dependent ferritin degradation and elevation of labile iron pool. Free Radic Biol Med. 2007;43:265–270. doi: 10.1016/j.freeradbiomed.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 71.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 72.Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermohlen O, Bruning JC, Schutze S, Kronke M. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature. 2009;460:1159–1163. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 73.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 74.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. 2012;119:2368–2375. doi: 10.1182/blood-2011-08-375303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 76.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Efferth T, Benakis A, Romero MR, Tomicic M, Rauh R, Steinbach D, Hafer R, Stamminger T, Oesch F, Kaina B, Marschall M. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med. 2004;37:998–1009. doi: 10.1016/j.freeradbiomed.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 78.Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, Wang L, Liang G. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381:165–175. doi: 10.1016/j.canlet.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 79.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, Torii S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017;108:2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8:5155. doi: 10.1038/s41598-018-23408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Rådmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Förster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16:e2006203. doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol. 2019;10:139. doi: 10.3389/fphys.2019.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zheng Z, Li G. Mechanisms and therapeutic regulation of pyroptosis in inflammatory diseases and cancer. Int J Mol Sci. 2020;21:1456. doi: 10.3390/ijms21041456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, Sun RY, Zhou D, Han J, Wu Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–1185. doi: 10.1038/s41422-018-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 92.Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 93.Cherayil BJ. Iron and immunity: immunological consequences of iron deficiency and overload. Arch Immunol Ther Exp (Warsz) 2010;58:407–415. doi: 10.1007/s00005-010-0095-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bird CL, Witte JS, Swendseid ME, Shikany JM, Hunt IF, Frankl HD, Lee ER, Longnecker MP, Haile RW. Plasma ferritin, iron intake, and the risk of colorectal polyps. Am J Epidemiol. 1996;144:34–41. doi: 10.1093/oxfordjournals.aje.a008852. [DOI] [PubMed] [Google Scholar]
- 95.Jian J, Pelle E, Huang X. Iron and menopause: does increased iron affect the health of postmenopausal women? Antioxid Redox Signal. 2009;11:2939–2943. doi: 10.1089/ars.2009.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang X. Iron overload and its association with cancer risk in humans: evidence for iron as a carcinogenic metal. Mutat Res. 2003;533:153–171. doi: 10.1016/j.mrfmmm.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 98.Munoz M, Villar I, Garcia-Erce JA. An update on iron physiology. World J Gastroenterol. 2009;15:4617–4626. doi: 10.3748/wjg.15.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Daniel CR, Cross AJ, Graubard BI, Hollenbeck AR, Park Y, Sinha R. Prospective investigation of poultry and fish intake in relation to cancer risk. Cancer Prev Res (Phila) 2011;4:1903–1911. doi: 10.1158/1940-6207.CAPR-11-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sesink AL, Termont DS, Kleibeuker JH, Van der Meer R. Red meat and colon cancer: the cytotoxic and hyperproliferative effects of dietary heme. Cancer Res. 1999;59:5704–5709. [PubMed] [Google Scholar]
- 101.Ilsley JN, Belinsky GS, Guda K, Zhang Q, Huang X, Blumberg JB, Milbury PE, Roberts LJ 2nd, Stevens RG, Rosenberg DW. Dietary iron promotes azoxymethane-induced colon tumors in mice. Nutr Cancer. 2004;49:162–169. doi: 10.1207/s15327914nc4902_7. [DOI] [PubMed] [Google Scholar]
- 102.Kabat GC, Cross AJ, Park Y, Schatzkin A, Hollenbeck AR, Rohan TE, Sinha R. Intakes of dietary iron and heme-iron and risk of postmenopausal breast cancer in the National institutes of health-AARP diet and health study. Am J Clin Nutr. 2010;92:1478–1483. doi: 10.3945/ajcn.2010.29753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kabat GC, Miller AB, Jain M, Rohan TE. Dietary iron and haem iron intake and risk of endometrial cancer: a prospective cohort study. Br J Cancer. 2008;98:194–198. doi: 10.1038/sj.bjc.6604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kabat GC, Miller AB, Jain M, Rohan TE. A cohort study of dietary iron and heme iron intake and risk of colorectal cancer in women. Br J Cancer. 2007;97:118–122. doi: 10.1038/sj.bjc.6603837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luqman M, Javed MM, Daud S, Raheem N, Ahmad J, Khan AU. Risk factors for lung cancer in the Pakistani population. Asian Pac J Cancer Prev. 2014;15:3035–3039. doi: 10.7314/apjcp.2014.15.7.3035. [DOI] [PubMed] [Google Scholar]
- 106.Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, Romero-Gallo J, Suarez G, Loh J, Slaughter JC, Tan S, Morgan DR, Wilson KT, Bravo LE, Correa P, Cover TL, Amieva MR, Peek RM Jr. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest. 2013;123:479–492. doi: 10.1172/JCI64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bailey RL, West KP Jr, Black RE. The epidemiology of global micronutrient deficiencies. Ann Nutr Metab. 2015;66(Suppl 2):22–33. doi: 10.1159/000371618. [DOI] [PubMed] [Google Scholar]
- 108.Hung N, Shen CC, Hu YW, Hu LY, Yeh CM, Teng CJ, Kuan AS, Chen SC, Chen TJ, Liu CJ. Risk of cancer in patients with iron deficiency anemia: a nationwide population-based study. PLoS One. 2015;10:e0119647. doi: 10.1371/journal.pone.0119647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dallman PR. Iron deficiency and the immune response. Am J Clin Nutr. 1987;46:329–334. doi: 10.1093/ajcn/46.2.329. [DOI] [PubMed] [Google Scholar]
- 110.Imam MU, Zhang S, Ma J, Wang H, Wang F. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients. 2017;9:671. doi: 10.3390/nu9070671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pra D, Rech Franke SI, Pegas Henriques JA, Fenech M. A possible link between iron deficiency and gastrointestinal carcinogenesis. Nutr Cancer. 2009;61:415–426. doi: 10.1080/01635580902803701. [DOI] [PubMed] [Google Scholar]
- 112.Broitman SA, Velez H, Vitale JJ. A possible role of iron deficiency in gastric cancer in Colombia. Adv Exp Med Biol. 1981;135:155–181. doi: 10.1007/978-1-4615-9200-6_9. [DOI] [PubMed] [Google Scholar]
- 113.Tabatabaei SA, Hashemi SM, Kelidari B. Transhiatal esophagectomy without mediastinal manipulation for lower third esophageal and cardial cancers: the first experience of a new technique. Iran J Cancer Prev. 2015;8:89–93. [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang YW, Eom SY, Kim YD, Song YJ, Yun HY, Park JS, Youn SJ, Kim BS, Kim H, Hein DW. Effects of dietary factors and the NAT2 acetylator status on gastric cancer in Koreans. Int J Cancer. 2009;125:139–145. doi: 10.1002/ijc.24328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bredenkamp JK, Castro DJ, Mickel RA. Importance of iron repletion in the management of Plummer-Vinson syndrome. Ann Otol Rhinol Laryngol. 1990;99:51–54. doi: 10.1177/000348949009900109. [DOI] [PubMed] [Google Scholar]
- 116.Kim KH, Kim MC, Jung GJ. Gastric cancer occurring in a patient with Plummer-Vinson syndrome: a case report. World J Gastroenterol. 2005;11:7048–7050. doi: 10.3748/wjg.v11.i44.7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Richie JP Jr, Kleinman W, Marina P, Abraham P, Wynder EL, Muscat JE. Blood iron, glutathione, and micronutrient levels and the risk of oral cancer. Nutr Cancer. 2008;60:474–482. doi: 10.1080/01635580801956477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu LZ, Jing Y, Jiang LL, Jiang XE, Jiang Y, Rojanasakul Y, Jiang BH. Acacetin inhibits VEGF expression, tumor angiogenesis and growth through AKT/HIF-1alpha pathway. Biochem Biophys Res Commun. 2011;413:299–305. doi: 10.1016/j.bbrc.2011.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 120.Wu T, Sempos CT, Freudenheim JL, Muti P, Smit E. Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults. Ann Epidemiol. 2004;14:195–201. doi: 10.1016/S1047-2797(03)00119-4. [DOI] [PubMed] [Google Scholar]
- 121.Bae YJ, Yeon JY, Sung CJ, Kim HS, Sung MK. Dietary intake and serum levels of iron in relation to oxidative stress in breast cancer patients. J Clin Biochem Nutr. 2009;45:355–360. doi: 10.3164/jcbn.09-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 123.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 124.Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal. 2014;20:1917–1924. doi: 10.1089/ars.2012.5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pilkington GJ, Parker K, Murray SA. Approaches to mitochondrially mediated cancer therapy. Semin Cancer Biol. 2008;18:226–235. doi: 10.1016/j.semcancer.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 126.Poljak-Blazi M, Kralj M, Hadzija MP, Zarkovic N, Zarkovic K, Waeg G. Involvement of lipid peroxidation, oncogene expression and induction of apoptosis in the antitumorous activity of ferric-sorbitol-citrate. Cancer Biother Radiopharm. 2000;15:285–293. doi: 10.1089/108497800414383. [DOI] [PubMed] [Google Scholar]
- 127.Ferle-Vidovic A, Poljak-Blazi M, Rapic V, Skare D. Ferrocenes (F168, F169) and fero-sorbitol-citrate (FSC): potential anticancer drugs. Cancer Biother Radiopharm. 2000;15:617–624. doi: 10.1089/cbr.2000.15.617. [DOI] [PubMed] [Google Scholar]
- 128.Fang S, Yu X, Ding H, Han J, Feng J. Effects of intracellular iron overload on cell death and identification of potent cell death inhibitors. Biochem Biophys Res Commun. 2018;503:297–303. doi: 10.1016/j.bbrc.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 129.Kiessling MK, Klemke CD, Kaminski MM, Galani IE, Krammer PH, Gulow K. Inhibition of constitutively activated nuclear factor-kappaB induces reactive oxygen species- and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res. 2009;69:2365–2374. doi: 10.1158/0008-5472.CAN-08-3221. [DOI] [PubMed] [Google Scholar]
- 130.Akiyama H, Ito A, Kawabe Y, Kamihira M. Genetically engineered angiogenic cell sheets using magnetic force-based gene delivery and tissue fabrication techniques. Biomaterials. 2010;31:1251–1259. doi: 10.1016/j.biomaterials.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 131.Srinivas M, Aarntzen EH, Bulte JW, Oyen WJ, Heerschap A, de Vries IJ, Figdor CG. Imaging of cellular therapies. Adv Drug Deliv Rev. 2010;62:1080–1093. doi: 10.1016/j.addr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 132.Foy SP, Labhasetwar V. Oh the irony: iron as a cancer cause or cure? Biomaterials. 2011;32:9155–9158. doi: 10.1016/j.biomaterials.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.El Hout M, Dos Santos L, Hamaï A, Mehrpour M. A promising new approach to cancer therapy: targeting iron metabolism in cancer stem cells. Semin Cancer Biol. 2018;53:125–138. doi: 10.1016/j.semcancer.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 134.Thévenod F. Iron and its role in cancer defense: a double-edged sword. Met Ions Life Sci. 2018;18 doi: 10.1515/9783110470734-021. /books/9783110470734/9783110470734-9783110470021/9783110470734-9783110470021.xml. [DOI] [PubMed] [Google Scholar]
- 135.Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–1994. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- 136.Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603:31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 137.Kovacevic Z, Kalinowski DS, Lovejoy DB, Yu Y, Suryo Rahmanto Y, Sharpe PC, Bernhardt PV, Richardson DR. The medicinal chemistry of novel iron chelators for the treatment of cancer. Curr Top Med Chem. 2011;11:483–499. doi: 10.2174/156802611794785190. [DOI] [PubMed] [Google Scholar]
- 138.Jiao Y, Wilkinson J 4th, Di X, Wang W, Hatcher H, Kock ND, D’Agostino R Jr, Knovich MA, Torti FM, Torti SV. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113:462–469. doi: 10.1182/blood-2008-05-155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shen L, Zhao HY, Du J, Wang F. Anti-tumor activities of four chelating agents against human neuroblastoma cells. In Vivo. 2005;19:233–236. [PubMed] [Google Scholar]
- 140.Li P, Zheng X, Shou K, Niu Y, Jian C, Zhao Y, Yi W, Hu X, Yu A. The iron chelator Dp44mT suppresses osteosarcoma’s proliferation, invasion and migration: in vitro and in vivo. Am J Transl Res. 2016;8:5370–5385. [PMC free article] [PubMed] [Google Scholar]
- 141.Liang SX, Richardson DR. The effect of potent iron chelators on the regulation of p53: examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis. 2003;24:1601–1614. doi: 10.1093/carcin/bgg116. [DOI] [PubMed] [Google Scholar]
- 142.Gattermann N, Finelli C, Porta MD, Fenaux P, Ganser A, Guerci-Bresler A, Schmid M, Taylor K, Vassilieff D, Habr D, Domokos G, Roubert B, Rose C EPIC study investigators. Deferasirox in iron-overloaded patients with transfusion-dependent myelodysplastic syndromes: results from the large 1-year EPIC study. Leuk Res. 2010;34:1143–1150. doi: 10.1016/j.leukres.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 143.Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med. 2011;365:576–578. doi: 10.1056/NEJMc1105726. [DOI] [PubMed] [Google Scholar]
- 144.Cohen AR, Galanello R, Piga A, Dipalma A, Vullo C, Tricta F. Safety profile of the oral iron chelator deferiprone: a multicentre study. Br J Haematol. 2000;108:305–312. doi: 10.1046/j.1365-2141.2000.01866.x. [DOI] [PubMed] [Google Scholar]
- 145.Bajbouj K, Shafarin J, Hamad M. High-dose deferoxamine treatment disrupts intracellular iron homeostasis, reduces growth, and induces apoptosis in metastatic and nonmetastatic breast cancer cell lines. Technol Cancer Res Treat. 2018;17:1533033818764470. doi: 10.1177/1533033818764470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kuo KH, Mrkobrada M. A systematic review and meta-analysis of deferiprone monotherapy and in combination with deferoxamine for reduction of iron overload in chronically transfused patients with beta-thalassemia. Hemoglobin. 2014;38:409–421. doi: 10.3109/03630269.2014.965781. [DOI] [PubMed] [Google Scholar]
- 147.Simoes RV, Veeraperumal S, Serganova IS, Kruchevsky N, Varshavsky J, Blasberg RG, Ackerstaff E, Koutcher JA. Inhibition of prostate cancer proliferation by deferiprone. NMR Biomed. 2017;30:10. doi: 10.1002/nbm.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Breccia M, Alimena G. Efficacy and safety of deferasirox in myelodysplastic syndromes. Ann Hematol. 2013;92:863–870. doi: 10.1007/s00277-013-1703-7. [DOI] [PubMed] [Google Scholar]
- 149.Donfrancesco A, Deb G, Angioni A, Maurizio C, Cozza R, Jenkner A, Landolfo A, Boglino C, Helson L. D-CECaT: a breakthrough for patients with neuroblastoma. Anticancer Drugs. 1993;4:317–321. [PubMed] [Google Scholar]
- 150.Mertens C, Akam EA, Rehwald C, Brune B, Tomat E, Jung M. Intracellular iron chelation modulates the macrophage iron phenotype with consequences on tumor progression. PLoS One. 2016;11:e0166164. doi: 10.1371/journal.pone.0166164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 152.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, Weiss JM, Wlaschek M, Sunderkotter C, Scharffetter-Kochanek K. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985–997. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, Cabaud O, Leroy C, David A, Acevedo V, Ryo A, Ginestier C, Birnbaum D, Charafe-Jauffret E, Codogno P, Mehrpour M, Rodriguez R. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025–1033. doi: 10.1038/nchem.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhao B, Li X, Wang Y, Shang P. Iron-dependent cell death as executioner of cancer stem cells. J Exp Clin Cancer Res. 2018;37:79. doi: 10.1186/s13046-018-0733-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–510. doi: 10.1038/nri3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Marques O, Porto G, Rema A, Faria F, Cruz Paula A, Gomez-Lazaro M, Silva P, Martins da Silva B, Lopes C. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer. 2016;16:187. doi: 10.1186/s12885-016-2228-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kong WN, Lei YH, Chang YZ. The regulation of iron metabolism in the mononuclear phagocyte system. Expert Rev Hematol. 2013;6:411–418. doi: 10.1586/17474086.2013.814840. [DOI] [PubMed] [Google Scholar]
- 158.Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, Zammataro L, Girelli D, Cairo G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. 2010;40:824–835. doi: 10.1002/eji.200939889. [DOI] [PubMed] [Google Scholar]
- 159.Recalcati S, Locati M, Gammella E, Invernizzi P, Cairo G. Iron levels in polarized macrophages: regulation of immunity and autoimmunity. Autoimmun Rev. 2012;11:883–889. doi: 10.1016/j.autrev.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 160.Ponzoni M, Pastorino F, Di Paolo D, Perri P, Brignole C. Targeting macrophages as a potential therapeutic intervention: impact on inflammatory diseases and cancer. Int J Mol Sci. 2018;19:1953. doi: 10.3390/ijms19071953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schultze JL. Reprogramming of macrophages--new opportunities for therapeutic targeting. Curr Opin Pharmacol. 2016;26:10–15. doi: 10.1016/j.coph.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 162.Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, Meister M, Muley T, Warth A, Platten M, Hentze MW, Cerwenka A, Muckenthaler MU. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol. 2017;8:1479. doi: 10.3389/fimmu.2017.01479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Singh Y, Pawar VK, Meher JG, Raval K, Kumar A, Shrivastava R, Bhadauria S, Chourasia MK. Targeting tumor associated macrophages (TAMs) via nanocarriers. J Control Release. 2017;254:92–106. doi: 10.1016/j.jconrel.2017.03.395. [DOI] [PubMed] [Google Scholar]
- 164.Andon FT, Digifico E, Maeda A, Erreni M, Mantovani A, Alonso MJ, Allavena P. Targeting tumor associated macrophages: the new challenge for nanomedicine. Semin Immunol. 2017;34:103–113. doi: 10.1016/j.smim.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 165.Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gu Z, Liu T, Tang J, Yang Y, Song H, Tuong ZK, Fu J, Yu C. Mechanism of iron oxide-induced macrophage activation: the impact of composition and the underlying signaling pathway. J Am Chem Soc. 2019;141:6122–6126. doi: 10.1021/jacs.8b10904. [DOI] [PubMed] [Google Scholar]
- 167.Meng QF, Rao L, Zan M, Chen M, Yu GT, Wei X, Wu Z, Sun Y, Guo SS, Zhao XZ, Wang FB, Liu W. Macrophage membrane-coated iron oxide nanoparticles for enhanced photothermal tumor therapy. Nanotechnology. 2018;29:134004. doi: 10.1088/1361-6528/aaa7c7. [DOI] [PubMed] [Google Scholar]
- 168.Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A. 1978;75:4538–4542. doi: 10.1073/pnas.75.9.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6:222ra218. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 170.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cieslak JA, Cullen JJ. Treatment of pancreatic cancer with pharmacological ascorbate. Curr Pharm Biotechnol. 2015;16:759–770. doi: 10.2174/138920101609150715135921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T, Berg DJ, Zhang J, Keech J, Parekh KR, Bhatia S, Monga V, Bodeker KL, Ahmann L, Vollstedt S, Brown H, Shanahan Kauffman EP, Schall ME, Hohl RJ, Clamon GH, Greenlee JD, Howard MA, Schultz MK, Smith BJ, Riley DP, Domann FE, Cullen JJ, Buettner GR, Buatti JM, Spitz DR, Allen BG. O2(-) and H2O2-mediated disruption of fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell. 2017;31:487–500. e488. doi: 10.1016/j.ccell.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Polireddy K, Dong R, Reed G, Yu J, Chen P, Williamson S, Violet PC, Pessetto Z, Godwin AK, Fan F, Levine M, Drisko JA, Chen Q. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: mechanisms and a phase I/IIa study. Sci Rep. 2017;7:17188. doi: 10.1038/s41598-017-17568-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang F, Gosser DK Jr, Meshnick SR. Hemin-catalyzed decomposition of artemisinin (qinghaosu) Biochem Pharmacol. 1992;43:1805–1809. doi: 10.1016/0006-2952(92)90713-s. [DOI] [PubMed] [Google Scholar]
- 176.Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- 177.Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2017;56:75–93. doi: 10.1002/mc.22474. [DOI] [PubMed] [Google Scholar]
- 178.Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K, Janah R, Greten HJ, Efferth T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22:1045–1054. doi: 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 179.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]