

Abstract
SET Domain Bifurcated Histone Lysine Methyltransferase 1 (SETDB1, ESET, KMT1E) is a H3K9 methyltransferase involved in gene silencing. In recent years, SETDB1 has been implicated as an oncogene in various cancers, highlighting a critical need to better understand the mechanisms underlying SETDB1 amplification, overexpression, and activation. In the following review, we first examine the history of SETDB1, starting from its discovery in 1999 and ending with recent findings. We follow with an outline of the structure and subcellular location of SETDB1, as well as potential mechanisms for regulation of its nuclear transport. Subsequently, we introduce SETDB1’s various functions, including its roles in promyelocytic leukemia nuclear body (PML-NB) formation, the methylation and activation of Akt, the silencing of the androgen receptor (AR) gene, retroelement silencing, the inhibition of tumor suppressor p53, and its role in promoting intestinal differentiation and survival. The Cancer Cell Line Encyclopedia (CCLE) screened SETDB1 dependency in 796 cancer cell lines, identifying SETDB1 as a common essential gene in 531 of them, demonstrating that SETDB1 expression is critical for the survival of the majority of cancers. Therefore, we provide a detailed review of the oncogenic effects of SETDB1 overexpression in breast cancer, non-small cell lung cancer, prostate cancer, colorectal cancer, acute myeloid leukemia, glioma, melanoma, pancreatic ductal adenocarcinoma, liver cancer, nasopharyngeal carcinoma, gastric carcinoma, and endometrial cancer. Accordingly, we review several methods that have been used to target SETDB1, such as using Mithramycin A, Mithralog EC-8042, 3’-deazaneplanocin A (DZNep), and paclitaxel. Finally, we conclude by highlighting remaining gaps in knowledge and challenges surrounding SETDB1. Ultimately, our review captures the wide scope of findings on SETDB1’s history, function, its implications in cancer, and provides suggestions for future research in the field.
Keywords: SETDB1, histone methyltransferase, ATF7IP, AKT
History of SETDB1
As shown in Figure 1, SETDB1 was discovered in 1999 and was mapped to human chromosome 1q21.3, with a length of 38.6 Kb [1]. Chromosome 1q gains are common features of ovarian cancers, endometrial cancers, lung adenocarcinoma, breast cancers, and liver hepatocellular carcinoma [2,3]. The specific amplification of chromosome 1q21.3 has been linked to breast cancer recurrence [4] and 1q21.3 has been identified as a susceptibility locus in melanoma [5]. Three years after its discovery, SETDB1 was identified as a highly specific H3K9 methyltransferase that methylates euchromatic regions, resulting in gene silencing. Specifically, SETDB1 was found to bind to the plant homeodomain (PHD) of the KAP-1 corepressor for KRAB zinc-finger proteins. SETDB1-mediated histone H3 methylation promoted heterochromatin protein 1 (HP1) binding, resulting in compact heterochromatin formation and thus, gene silencing [6,7]. Furthermore, an in vitro pull-down assay revealed that SETDB1 interacts with transcription factor ERG and endogenous SETDB1 was shown to accumulate with ERG. Therefore, Yang et al. hypothesized that ERG regulates transcription through SETDB1-mediated histone methylation [7]. The finding that promoter H3K9 trimethylation causes transcriptional repression was further supported by a study in the following year which built on this conclusion, proposing that SETDB1 tightly associates with the human homolog of mAM, a murine ATFa-associated factor. This association was found to increase SETDB1’s enzymatic activity, thereby facilitating conversion of H3-K9 dimethyl to trimethyl [8]. The same year, the mouse homolog of SETDB1, ESET, was proposed to repress transcription by forming a multi-protein complex with mSin3A/B and corepressors HDAC1/2 [9]. These findings led to further study in 2006 in that Li et al. reported that SETDB1 interacts with the de novo DNA methyltransferases, DNMT3A and DNMT3B, which are important factors in heterochromatin formation and gene silencing. SETDB1-HDAC1 was proposed to be recruited to the RASSF1A promoter by transcriptional repressors, while DNMT3A was later recruited through direct interaction with SETDB1. These interactions result in DNA methylation and gene silencing [10]. Nearly ten years later, it was revealed that the post-translational K867 ubiquitination of SETDB1 was essential for its H3K9 methyltransferase activity. Furthermore, this ubiquitination and subsequent activation of its H3K9 methyltransferase activity was found to repress the expression of SETDB1 target gene SERPINE1 [11,12]. In 2011, SETDB1 was implicated in melanoma. Later studies proposed multiple roles of SETDB1 in tumorigenesis of other cancers. These findings are summarized in a later section. Then in 2018, SETDB1 was shown to inhibit epithelial-mesenchymal transition (EMT) via H3K9 promoter methylation of SNAI1, an important EMT transcription factor. This methylation repressed the Smad3/4-mediated H3K9 acetylation and activation of the SNAI1 promoter, thereby preventing EMT [13]. One year later, two papers reported that SETDB1 methylates Akt as a key step in the oncogenic activation of Akt. Specifically, SETDB1-mediated methylation was found to be essential for the phosphorylation of Akt at Thr308, which activates Akt and promotes tumorigenesis [14,15]. Details are discussed in a later section.
Figure 1.
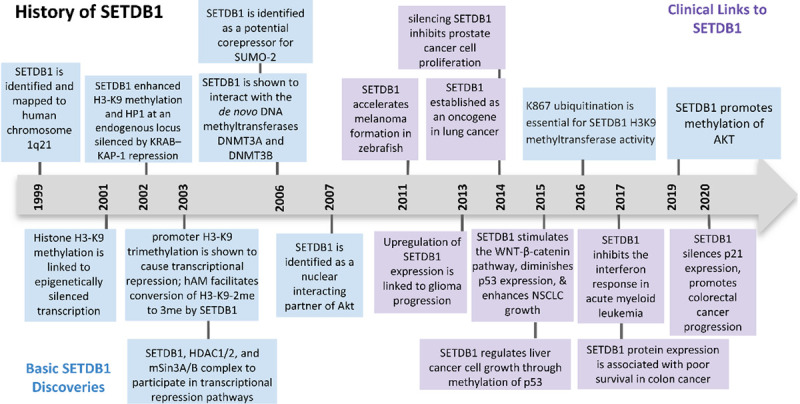
Timeline of major discoveries in SETDB1 studies, including basic science and clinical translational studies.
Structure and subcellular distribution of SETDB1
The SET domain was originally identified in the Drosophila position effect variegation suppressor SU(VAR)3-9 [16]. Several proteins that contain SET domains modulate gene activity and/or contain methyltransferase activity [17,18]. Proteins with a SET domain were later associated with lysine-specific histone methyltransferase activity [19]. SETDB1 belongs to the SET family, which has a well-known function for epigenetic regulation [20]. However, SETDB1 is an extremely distinctive SET protein; as shown in Figure 2, it has a canonical CpG DNA methyl binding domain (MBD) on its N-terminal region, which serves as a platform to bind to methylated DNA [21]. The MBD is functional; it contains two DNA-interacting arginine residues [22,23]. The MBD domain couples DNA methyl-CpG binding function with H3K9 methylation function. Therefore, the interplay between DNA methylation and histone methylation likely exists to promote epigenetic marks [24]. The C-terminal region of SETDB1 contains pre-SET, a bifurcated SET, and post-SET domains, which are essential for its methyltransferase activity [7]. While the pre-SET, SET, and post-SET domains are all crucial for histone methyltransferase activity [1,25], the 347 amino acid insertion within the SET domain was proposed to be dispensable for the catalytic activity of SETDB1. The function of this insertion has yet to be elucidated. However, within the SET insertion at lysine 867, SETDB1 is monoubiquitinated by the UBE2E family of E2 enzymes (UBE-2E1, 2E2, and 2E3) in an E3-independent manner. This ubiquitination was shown to be essential for SETDB1’s enzymatic activity in vitro and in vivo [11,12]. SETDB1 is also unique in respect to its double tudor domains; these domains complex with mSin3A/B and HDAC1/2 to participate in transcriptional repression pathways [9]. The double tudor domains also allow for (1) interaction with chromatin modification enzymes, such as Runx2 and HDAC1/2 [9,26] and (2) are essential in directing RNA processing factors and transcription factors in Cajal bodies involved in snRNP processing [27]. SETDB1 also has three small ubiquitin-related modifier (SUMO)-interacting motifs [28] to stimulate H3K9 methyltransferase activity by sumoylated interacting protein KAP1 for gene silencing.
Figure 2.
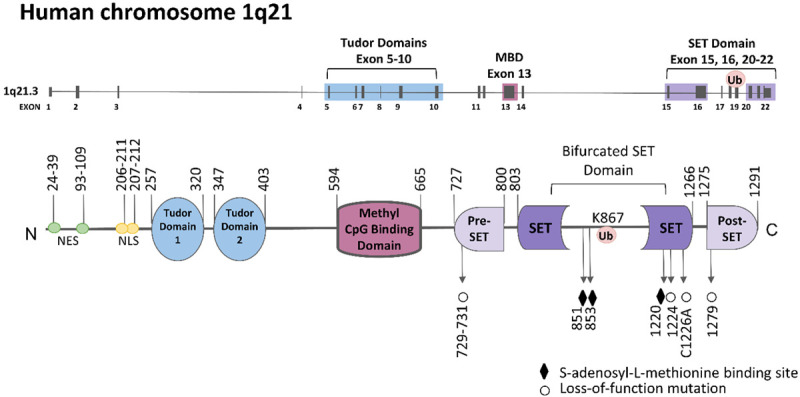
Diagram of gene and function domains of SETDB1. SETDB1 is located on chromosome 1q21.3, consisting of 22 exons. SETDB1 protein has two Tudor domains, a CpG DNA methyl binding domain (MBD), and a bifurcated SET domain.
As denoted in Figure 2, several mutations have been observed to abolish SETDB1’s methyltransferase activity: a C to Y mutation at position 1279, a C to A mutation at position 1226, an H to K mutation at position 1224, and a CDC to LDP mutation at positions 729-731 [6].
Although SETDB1 is primarily present in the cytoplasm of human cell lines, it is proposed to be also localized in the nucleus. Specifically, Tachibana et al. hypothesized that SETDB1 undergoes proteasomal degradation which results in its export to the cytoplasm, explaining the observation of SETDB1’s predominant presence in the cytosol. Their group demonstrated this by administering dual treatment of leptomycin B (a chromosome region maintenance 1 (Crm1) protein inhibitor that blocks nuclear transport) and a proteasome inhibitor, MG132. This treatment blocked the localization of SETDB1 to the cytosol and induced the nuclear accumulation of SETDB1 [29]. Nuclear entry of SETDB1 is reliant on its N-terminal region, as it contains two nuclear export signals and two nuclear localization signals. The exogenous location of SETDB1 is strictly regulated; cytoplasmic SETDB1 can successfully enter the nucleus and diffuse in the nucleoplasm or can bind to promyelocytic leukemia nuclear body (PML-NB) foci. However, SETDB1 will still be subject to export into the cytosol by a Crm1-dependent mechanism [24,30]. The culmination of these findings suggests that there is a clear regulatory mechanism in place that limits the activity of SETDB1 in the nucleus; aberrant nuclear activity would likely result in detrimental chromatin modifications.
Studies investigating potential mechanisms for the nuclear retention of SETDB1 have implicated ATF7IP, a nuclear protein that binds to SETDB1’s N-terminal region. Since the N-terminal region of SETDB1 contains the nuclear export signal motifs, it was proposed that the binding of ATF7IP inhibits the nuclear export of SETDB1. As a result, loss of ATF7IP resulted in an increase of SETDB1’s cytoplasmic signal, while the nuclear signal decreased. These findings suggest that ATF7IP may play a protective role against proteasomal degradation of SETDB1. Furthermore, Western blot analysis showed that the nuclear retention and accumulation of SETDB1 leads to an increase in its own ubiquitination; V5-SETDB1 transfection produced the expected doublet band, while co-transfection with 3xFLAG-ATF7IP produced the upper band of V5-SETDB1 (which is associated with the ubiquitinated form of SETDB1). The ubiquitinatable residue in mouse SETDB1 is lysine 885. Strikingly, when the K885R mutant was co-expressed with 3xFLAG-ATF7IP, no ubiquitination signal was observed. However, a slight shift of the upper band compared to controls was seen, suggesting that ATF7IP may induce other unknown modifications in SETDB1 [31,32].
SETDB1 functions
Although the primary role of SETDB1 is in transcriptional silencing through its H3K9 methyltransferase activity, it is a genome-wide chromatin modifier that can function in a variety of settings. SETDB1’s multiple functions are summarized in Figure 3.
Figure 3.
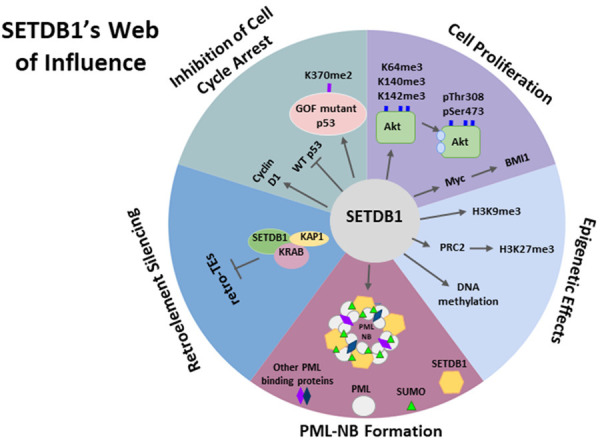
Schematic diagram of the multiple functions of SETDB1. SETDB1 has five main functions: 1) Epigenetic Effects: SETDB1 is an epigenetic modifier involved mainly in H3K9 methylation. It can also contribute to H3K27 and DNA methylation. 2) PML-NB Formation: SETDB1 is an integral component of PML-NB structures which are essential for multiple functions. 3) Retroelement silencing: SETDB1 is essential in pro-viral silencing and the silencing of ERVs. 4) Inhibition of Cell Cycle Arrest: SETDB1 can transcriptionally silence p53 and promote the stability of the oncogenic p53 mutant through its methylation, inhibiting cell cycle arrest. 5) Cell proliferation: SETDB1 can stimulate cell proliferation through the activation of Akt. This is mediated through methylation of Akt at lysine residues 64, 140, and 142 resulting in Thr-308 phosphorylation and increased activity of Akt.
SETDB1 is essential for PML-NB formation
SETDB1 has been established as an integral component of the PML-NB structure, which is essential for multiple functions, including maintenance of pluripotency in embryonic stem cells and recruitment of SUMO-1 and Sp100 proteins [30]. Furthermore, PML has been extensively linked to gene transcription, tumor suppression, apoptosis, the antiviral response, and DNA repair [33,34]. Cho et al. demonstrated that knockdown of SETDB1 resulted in PML-NB dismantling, while PML degradation via arsenic treatment caused SETDB1 foci to disappear [35]. A later study revealed that the SUMO-interaction motif (SIM) on SETDB1 was essential for interaction between SETDB1 and PML proteins, while SETDB1 proteins with a SIM mutation resulted in damage to PML-NB structures [30].
Methylation of Akt
Dysregulated Akt (protein kinase B) signaling has been linked to multiple human diseases, such as cancer, where hyperactivity of Akt drives chemotherapy resistance and is a landmark of tumor severity [36-39]. The first link between SETDB1 and Akt was established in 2007 via yeast two-hybrid screening. This nuclear interaction was demonstrated to be direct via a GST pull-down assay wherein SETDB1 specifically interacted with GST-Akt1, but not GST alone [40]. More recently, two studies from Wei and Lin groups proposed that SETDB1 not only interacts with Akt, but is essential for the activation of Akt. Using site-specific antibodies, both groups detected Akt trimethylation at three sites: K64, K140, and K142. Both groups used mass spectrometry (MS), however, only Guo et al. detected trimethylation at K140/142. While K64 rests within Akt’s pleckstrin homology (PH) domain, K140 and K142 were located in the Akt linker region [14,15]. Guo et al. proposed that K140 and K142 were major lysine trimethylated residues on Akt; mutation of both K140 and K142 resulted in a dramatic reduction of Ak1 lysine trimethylation in cells. To assess the function of trimethylation within the Akt1 linker region, Guo et al. created a methylation-deficient variant of Akt (K140R and/or K142R) in DLD1- AKT1/2-/- cells. The Akt1-K140/142R double mutant experienced the greatest reduction in activation, although Akt1-K140R and Akt1-K142R mutants also experienced reduced activation in comparison to wild type Akt1. Consequently, lack of methylation on K140 and K142 resulted in a reduction of cell colony formation, anchorage-independent growth, glucose uptake, and lactate production in vitro. In mice where the double mutation was knocked in (Akt1K1/K1), body size, weight, and Akt kinase activity decreased, when compared to control mice. The Akt1K1/K1 mice also had lower occurrence of skin tumors, and lower papilloma burden. Therefore, Akt1 methylation in its linker region was suggested to promote oncogenic Akt1 kinase activity.
Guo et al. presented a mechanism of Akt activation wherein K140/142me3 primes Akt for activation and may also prime Akt for methylation at K64. Specifically, SETDB1 was proposed to bind Akt via phosphatidylinositol (3,4,5)-triphosphate, which binds to Akt’s PH motif at the plasma membrane [15]. However, Wang et al. proposed a different mechanism that centered around K64 methylation. Specifically, Wang et al. proposed that SETDB1-mediated K64 methylation of Akt takes place in the cytosol and serves as a key platform for the recruitment of JMJD2A (also known as KDM4A), a histone demethylase that has been previously reported to drive tumorigenesis [14,41,42]. After this interaction, E3 ligases (TRAF6 and Skp2-SCF) are recruited to the Akt complex leading to promoting K63-linked ubiquitination and cell membrane recruitment of Akt. Once Akt has been recruited to the cell membrane, Akt can be activated by PDK1-mediated phosphorylation on Thr308 for activating Akt activity [14,43]. Therefore, SETDB1-mediated Akt K64 methylation was found to be crucial for the Akt-JMJD2A interaction and therefore, important for Akt’s K63-linked ubiquitination and ultimate activation. Accordingly, when JMJD2A/KDM4A was depleted, Akt ubiquitination and Thr308 phosphorylation levels decreased [14].
Since SETDB1, unlike other methyltransferases such as EZH2, was able to enhance Akt me3 in cells, Guo et al. proposed that SETDB1 could be an upstream regulator of Akt methylation. In addition to SETDB1 promoting methylation of K64, S’-adenosylmethionine (SAM)-mediated in vitro methylation assays revealed that SETDB1 could also methylate Akt1 at its K140 and K142 residues. Deletion of SETDB1 in cells reduced Akt activation and Akt-pThr308 in different cell lines. Contrastingly, KDM4B demethylated and attenuated Akt1 kinase activity. Specifically, KDM4B-WT erased Akt1-K140me3, and interactions between Akt and KDM4B predominantly occurred in the late phase of insulin stimulation. This finding suggested that KDM4B may be a bona fide Akt1 demethylase [15]. However, other studies have implicated KDM4B in gastric carcinogenesis as a coactivator of oncogene c-Jun [44].
In xenograft tumor models, SETDB1-mediated Akt K64 methylation was found to promote tumorigenesis via induction of Akt K63-linked ubiquitination and activation. The same study used IHC staining of 156 NSCLC specimens to reveal upregulated expression of SETDB1 and Akt K64 methylation, ultimately predicting poor survival of NSCLC patients. This conclusion aligned with Kaplan-Meier survival analysis. [14]. Guo et al. supported these findings, adding that Akt methylation synergizes with PI3K signaling to control Akt activation. While methylation of Akt enhanced Akt’s kinase activity, the absence of methylation attenuated Akt kinase activity, leading to repression of tumorigenesis and cell growth [15]. Therefore, in cancers that are driven by hyperactive Akt, SETDB1 may be a promising therapeutic target.
SETDB1 silences the androgen receptor gene
The androgen receptor (AR) is a ligand-activated transcription factor that is essential for sexual development in males. Alterations in AR function have been linked to several diseases, including prostate cancer [45]. Argonaute (AGO) proteins are known to be essential in the establishment of the RNA-induced silencing complex (RISC), which results in transcript degradation or inhibition of translation [46]. Specifically, Argonaute-2 (AGO2) has been described as the catalytic engine that drives mRNA cleavage. It has been previously reported that AGO2 is recruited to target sites, specifically when in the presence of promoter-targeted antigene RNAs (agRNAs) that are complementary to AR and progesterone receptor (PR) promoter sequences [47]. A later study demonstrated that SETDB1 cooperates with AGO2 to facilitate promoter-targeted agRNA-induced transcriptional gene silencing. agRNA-driven AGO2 is targeted to the promoter, then is followed by SETDB1 and the SIN3-HDAC complex. Localization of these components at the promoter resulted in H3K9 trimethylation. Without AGO2 or SETDB1, agRNA-mediated transcriptional gene silencing failed to be induced at AR and PR promoters. A dual-lock mechanism was proposed wherein the agRNA-targeted AR promoter is modified by two repressive marks: H3K9me3 via SETDB1 and H3K27me3 via EZH2 [48].
SETDB1 mediates retroelement silencing
Retrotransposition can result in genome instability. Thus, retroelements that have integrated are often transcriptionally silenced via DNA methylation and/or H3K9 trimethylation. Matsui et al. reported that SETDB1 and KAP1 are not only responsible for H3K9 trimethylation, but also are essential for the silencing of endogenous and introduced retroviruses, specifically in mouse embryonic stem (ES) cells. While H4K20 methyltransferases were found to be inessential for silencing, SETDB1 was proved to be critical for heterochromatin protein 1 (HP1) binding and efficient proviral silencing. Considering that triple knockout of DNA methyltransferases Dnmt1-/- Dnmt3a-/- Dnmt3b-/- did not affect SETDB1-mediated H3K9 trimethylation, it was proposed that proviral silencing relies on a pathway involving SETDB1 and KAP1, independent of DNA methylation [49]. Indeed, later studies showed that de novo recruitment of SETDB1 to retroelements was dependent on the KRAB-zinc finger protein/KAP1 pathway; KAP1 complexes with SETDB1 and KRAB zinc finger proteins. KAP1 contains a RING-B box-coiled coil domain, an HP1 binding domain, and a PHD-bromodomain for the recruitment of SETDB1 to gene promoters. However, while the mechanism of de novo recruitment of SETDB1 is relatively well understood, the spreading mechanisms of SETDB1-mediated H3K9me3 remain to be further elucidated [50].
SETDB1 regulates p53 stability and inhibits p53-mediated apoptosis
Previous studies have demonstrated that p53 activity is regulated by lysine methylation. Specifically, methylation of p53-K372 via Set9 was shown to stabilize a chromatin-bound fraction of p53 and regulate p53 target genes [51]. Similarly, SETDB1 was shown to dimethylate p53-K370 and subsequently be one important mode of regulating the stability of p53 [52]. Other seminal works include a study in CRC where two SETDB1 enrichment sites were found in the TP53 promoter region, -200 and -600 bp upstream from the start codon. Specifically, SETDB1 was observed to suppress apoptosis in CRC cells by inhibiting TP53 [53]. This finding was also supported in the pancreas; ChIP assays revealed that SETDB1 bound directly to the p53 promoter region, thereby directly regulating the expression of p53 in the pancreas [54]. More specific findings related to SETDB1 and its role in these specific cancers will be outlined in the following sections.
In contrast to wild-type p53, SETDB1 stabilizes the gain of function mutant p53R249S, which has a half-life greater than three times that of the wild-type. Surprisingly, the presence of the mutated form coincided with increased SETDB1 gene amplification and expression. This mutant form of p53, which was shown to promote cell proliferation and migration in lung cancer cells, was found to be considerably more methylated at its K370 than the wild-type form in liver cancer cells. Additionally, cells bearing the p53R249S mutation were significantly more susceptible and sensitive to SETDB1 loss than those with wild-type or deleted p53 [52].
SETDB1 promotes intestinal epithelium differentiation and prevents inflammation
SETDB1 was identified to be highly expressed in the intestinal epithelium [55]. Related to the findings of Fukuda & Shinkai, another group found that SETDB1 deletion resulted in the activation of endogenous retroviruses [55]. Additionally, in mice with SETDB1 deletions, DNA damage and inflammation were induced, ultimately leading to death from significant epithelial cell loss. Specifically, alkaline phosphatase levels served as a marker of differentiation and were found to be depressed in SETDB1-deleted mice. Although GLUT2 levels remained unchanged between control and SETDB1-deleted mice, the expression of SGLT1, an apical glucose transporter, decreased progressively until eventual complete loss. Ultimately, it was shown that SETDB1 is essential for the survival and differentiation of the intestinal epithelium [55].
SETDB1 & immune cell function
SETDB1 has been shown to play an important role in immune cells function and development. Rumi et al. found that SETDB1 negatively regulated the expression of TLR4-mediated pro-inflammatory cytokines, such as IL6 in macrophages. Specifically, SETDB1-mediated H3K9 methylation at the promoter of cytokines resulted in impaired recruitment of NF-κB, a transcription factor responsible for cytokine production in macrophages [56]. SETDB1 is essential in T-cell development as well [57]. Takikita et al. demonstrated that SETDB1 depletion can significantly hinder T-cell development, especially for CD8 T-cells. In the absence of SETDB1, TCR-induced ERK activation, which is necessary for T-cell development, was suppressed via increased expression of FcγRIIB. SETDB1 H3K9 methylation was shown to be responsible for regulating the transcription level of FcγRIIB [57].
SETDB1 H3K9 tri-methylation activity can be regulated by ATF7IP
The Human Silencing Hub (HUSH) complex, consisting of TASOR, MPP8, and Periphilin, can recruit SETDB1 to catalyze H3K9me3 as a regulator of heterochromatin. Additionally, ATF7IP was shown to be essential for the functioning of SETDB1 at the HUSH complex, promoting transcriptional repression. Specifically, ATF7IP can regulate SETDB1 by enhancing SETDB1 stability and its protection from proteasomal degradation inside the nucleus [31]. Timms et al. further showed that ATF7IP level can also be affected in the absence of SETDB1 as SETDB1 knocked-out cells displayed a significantly reduced level of ATF7IP protein level inside the nucleus. Comparing RNA-seq data from SETDB1-knockout HeLa cells to that of ATF7IP-knockout cells illustrated significant overlap within differentially expressed genes relative to wild type with only a few genes expressed discordantly. These findings suggest that SETDB1 and ATF7IP functions are greatly intertwined.
SETDB1 is overexpressed in multiple cancers and is correlated with worse survival
Apart from being implicated in obesity, Huntington’s disease, schizophrenia, and autism, SETDB1 has also been involved in a wide variety of cancers: breast cancer, non-small cell lung cancer, prostate cancer, colorectal cancer, acute myeloid leukemia, glioma, melanoma, pancreatic ductal adenocarcinoma, liver cancer, and endometrial cancer. These findings are summarized in Table 1. Furthermore, the Cancer Cell Line Encyclopedia (CCLE) screened SETDB1 dependency in 777 cancer cell lines, identifying SETDB1 as a common essential gene in 524 cancer cell lines. This demonstrates that SETDB1 expression is critical for the survival of the majority of cancers (DepMap Portal). Additionally, using the cBioPortal and Oncomine databases, SETDB1 overexpression was correlated with worse survival in colorectal cancer (CRC), endometrial cancer (UCEC), thyroid cancer (THCA), liver cancer (LIHC), renal cancer, and melanoma [14].
Table 1.
Summary of current SETDB1 literature in multiple cancer types
| Cancer Type | Patient Sample or Cell Line | Methods | Conclusion and References | |
|---|---|---|---|---|
| 1 | BRCA | H1299, BT549, MCF7, MD-MB231, MD-MB453, &MD-MB468 | Clonogenic assay, Western blotting (WB), immunoprecipitation (IP), RT-PCR, SQ-PCR, and yeast two-hybrid screening. | SETDB1 mediates the stabilization of p63 via interaction with ΔNp63α. 30 genes with a positive correlation to improved survival in BRC patients were identified to be repressed by ΔNp63α in a SETDB1-mediated manner [62]. |
| 2 | BRCA | n = 45 | qRT-PCR, flow cytometry, and luciferase reporter assay. | SETDB1 knockdown may suppress breast cancer growth via miR-381-3p-related regulation [58]. |
| 3 | BRCA | MCF7, T47D, BT549 and MDA-MB-231 | qRT-PCR, CHIP, mice experiments, and wound healing, migration, and invasion assays. | SETDB1 binds directly to the transcription factor, Snail, promoter, thereby acting as an EMT inducer [107]. |
| 4 | BRCA | n = 55 | qRT-PCR, IHC, soft agar assay, xenograft mice model, microarray analysis, and breast tumor tissue array. | Overexpression of SETDB1 promotes BRC tumorigenesis by enhancing translation of CCND1 and MYC mRNA. MYC was found to be essential in the ERα-mediated regulation of SETDB1 [59]. |
| 5 | BRCA | MDA-MB-231 | qRT-PCR, gene network analysis, and wound healing, migration, and invasion assays. | SMAD7 expression is regulated by SETDB1 levels; up-regulation of SMAD7 by SETDB1 knockdown inhibits BRC [60]. |
| 6 | BRCA | n = 159 | IHC, differential gene expression analysis. | SETDB1 is an oncogene and can serve as a prognostic biomarker in BRC [61]. |
| 7 | NSCLC, SCLC | A549, NCI-H1299, DMS-114, DMS-273, etc. (Total 15 cell lines) | qRT-PCR, quantitative genomic PCR, fluorescence in situ hybridization, mice experiments. | SETDB1 depletion reduces lung cancer growth in vitro and in vivo; SETDB1 overexpression increases tumor invasiveness. SETDB1 is a bona fide oncogene in lung cancer [63]. |
| 8 | NSCLC | n = 282 | Fluorescence in situ hybridization. | SETDB1 amplification was associated with an advanced cancer stage [66]. |
| 9 | NSCLC | n = 64 | qRT-PCR, univariate and multivariate Cox analyses. | SETDB1 and BRCA1 overexpression were correlated with worse disease-free survival in stage I NSCLC patients [108]. |
| 10 | NSCLC | A549 | Promoter cloning, luciferase assay, ChIP, human phosphor-MAPK kinase screening, qRT-PCR, Western blot, and scratch, cell migration, and colony formation assays. | Treatment with doxorubicin led to a decrease in SETDB1 expression and an increase in FosB expression. Luciferase reporter activity studies suggest FosB is negatively regulated by SETDB1. MEK inhibition activated ERK2 during doxorubicin treatment, inhibiting SETDB1 from regulating the FosB promoter. These findings suggest potential for an anti-therapeutic mechanism within the ERK2-FosB-SETDB1 signalling pathway [106]. |
| 11 | NSCLC | n = 30 | Luciferase reporter vector construction, dual luciferase reporter assay, qRT-PCR, and Western blotting. | SETDB1 was shown to be a target of miR-29s; ectopic expression of miR-29 decreased SETDB1 expression. TP53 was shown to upregulate expression of miR-29s, leading to a reduction in SETDB1 expression. TP53 negatively regulates SETDB1 expression by increasing miR-29 expression [109]. |
| 12 | NSCLC | Data from 1140 NSCLC cases | Gene Expression Omnibus (GEO) analysis. | Global overexpression of SETDB1 mRNA is likely a hallmark of NSCLC. SETDB1 was elevated in NSCLC patients who were current or former smokers [65]. |
| 13 | NSCLC | n = 196 | IHC, qRT-PCR, cell proliferation assay, flow cytometry, cell invasion and migration assay, luciferase reporter assay, and WB. | Oncogenic activity of SOD1 was inhibited by miR-409-3p binding to its 3’ UTR. SETDB1 was negatively correlated with miR-409-3p and positively correlated with SOD1. Thus, SETDB1 may participate in a regulatory feedforward loop involving miR-409-3p and SOD1 [110]. |
| 14 | Prostate Cancer (PCa) | n = 108 | IHC, qRT-PCR, in vitro scratch and transwell invasion assay. | SETDB1 is overexpressed in human PCa; silencing of SETDB1 inhibited PCa cell proliferation, migration and invasion [69]. |
| 15 | CRC | n = 8 (fresh CRC tissues), n = 132 (CRC paraffin sections) | IHC, CCK8 assay, ChIP assay, cell scratch assay, subcutaneous tumor implantation in BALB/C nude mice, survival analysis. | SETDB1 overexpression promoted CRC proliferation and migration in vitro and in vivo, and inhibited 5-FU induced apoptosis in CRC cells. Knocking down SETDB1 suppressed CRC growth. SETDB1 can bind the promoter region of TP53 [53]. |
| 16 | CRC | n = 90 | IHC, SurvExpress data mining, and statistical analyses. | Expression of SETDB1 on the protein level was identified to be 82% higher in colon adenocarcinoma tissue compared to paired control tissues, and was correlated with histological grade, TNM stage, T-class, N-class, and worse survival [111]. |
| 17 | CRC | n = 207 | IHC and image analysis. | Compared to normal tissues, SETDB1 expression was lower in CRC tumor tissues. In CRC tissues, no correlation was found between nuclear expression of SETDB1 and H3K9me3 [70]. |
| 18 | CRC | n = 296 | IHC, qRT-PCR, flow cytometry, IF staining, retrovirus transduction, ChIP, organoid culture, and transcription entropy analysis. | SETDB1 hinders the function of several transcription factors that commit stem-like colorectal cancer cells into a differentiated normal-like state; SETDB1 depletion converts stem-like CRC cells into postmitotic cells, restoring normal morphology [42]. |
| 19 | CRC | n = 60 | qRT-PCR, ChIP-PCR, dual luciferase reporter assay, CCK8 assay, tumor xenograft in nude mice, migration and invasion assay, colony formation assay. | SETDB1 regulates EMT in CRC cells. SETDB1 can regulate the activity of p21 via promoter binding. SETDB1 silencing inhibits CRC proliferation, migration, and invasion in vitro, and inhibits tumorigenesis in vivo [72]. |
| 20 | CRC | n = 115 | IHC, real-time PCR, CCK8 assay, flow cytometry, immunofluorescence, migration, invasion, and colony formation assays. | SETDB1 binds the STAT1 promoter region, increasing STAT1 expression. The STAT1-CCND1/CDK6 axis is a downstream effector of SETDB1-mediated CRC cell proliferation. SETDB1 overexpression promotes CRC tumorigenesis in vivo [71]. |
| 21 | CRC | n = 60 | IHC, RNA interference, transwell migration assay, subcutaneous tumor implantation in BALB/C nude mice. | SETDB1 level correlates with worse clinical outcome, SETDB1 attenuation inhibits CRC tumorigenesis and increases sensitivity to cetuximab [112]. |
| 22 | AML | THP-1 cells | CRISPR library generation, CHIP-seq, CHIP-qPCR, CRISPR/Cas9 sgRNA screen and analysis. | SETDB1 is a negative regulator of innate immunity in AML. SETDB1 disruption induces viral response gene expression. SETDB1-disruption-induced cell death in AML relies on the viral sensing machinery. Loss of SETDB1 induces retrotransposable element expression and induction of dsRNA [73]. |
| 23 | AML | AML cell lines were generated from C57Bl/6 transgenic mouse bone marrow | Proliferation, luciferase, and colony formation assays, IP, affinity purification MS, CHIP-qPCR. | SETDB1 was identified to directly bind to the PAF1c subunit CDC73, whose interaction is stabilized by mutant CDC73, resulting in repression of PAF1c downstream genes, Meis1 and HOXA9 [77]. |
| 24 | AML | MLL-AF9 and E2A-HLF | Cell line generation, mouse modeling, qPCR, sequencing libraries preparation, data mining for patient sample data. | SETDB1 expression is correlated to disease status and overall survival in acute myeloid leukemia patients. SETDB1 induces repressive changes to the promoter epigenome and downregulation of AML-linked genes [78]. |
| 25 | Glioma | n = 5 | IHC, RT-PCR, WB, siRNA gene knockdown, and cell proliferation, migration, clonogenic, and apoptosis assays. | SETDB1 and SUV39H1 are both overexpressed in glioma tissues and cells. Suppressing H3K9 HKMTs reduced glioma proliferation, migration, and colony formation [80]. |
| 1321N1, GOS-3, T98G, and U87MG | ||||
| 26 | Glioma | n = 40 | IHC, real-time PCR, and WB, and syngeneic mouse models. CCK-8 assay, colony formation assay, and wound healing and transwell assays. | SETDB1 was upregulated and related to worse progression in glioma. SETDB1 overexpression promoted cell proliferation, invasion, and migration in vitro, while SETDB1 knockdown had opposite effects. SETDB1 promoted AKT/mTOR-dependent CSF-1 secretion, resulting in recruitment of macrophages and tumor growth [79]. |
| 27 | Melanoma | n = 20 | IHC, miniCoopR assay, senescence assay, RNA extraction. | SETDB1 accelerated melanoma formation and correlated with more aggressive tumor formation in zebrafish. SETDB1 was overexpressed in human melanocytes. SETDB1 overexpression resulted in transcriptional dysregulation of Hox genes [85]. |
| 28 | Melanoma | n = 100 | SETDB1 amplification analysis, ChIP, IHC, transcriptomic analysis, migration, invasion, and proliferation assays, and mouse xenograft studies. | SETDB1 was associated with poor prognosis and more aggressive phenotypes in in vitro and in vivo. SETDB1 induces THBS1 activation via H3K4me1. Catalytic mutation of SETDB1 leads to decrease in tumorigenicity in melanoma [86]. |
| 29 | Melanoma | SK-HI-SETDB1 (subclone generated from parental SKMEL28 cells) | Plasmid/lentiviral cell transduction, PCR, tissue microarray analysis, IF, ELISA proteome profiler, transcriptional profiling assay, and cell viability, migration, and invasion assays. | Mithramycin A and Mithralog EC-8042 (1) reduced SETDB1 expression in melanoma cells, (2) induced transcriptomic, morphological, and functional changes, and (3) improved efficacy of MAPK inhibitor treatment [113]. |
| 30 | Melanoma | n = 299* | Zebrafish transgenic model, TCGA melanoma data mining, IHC, BRAFV600E MCR melanoma model. | Two genes on chromosome 1q, SETDB1 and HDGF, were identified to be commonly co-altered, resulting in progression of melanoma and overall worse patient prognoses [87]. |
| *zebrafish transgenic model | ||||
| 31 | Pancreatic ductal adenocarcinoma (PDAC) | n = 48 | IHC, ChIP, cell proliferation assay, methyltransferase activity quantification assay, qRT-PCR, microarray analysis, and mice models. | SETDB1 is essential in the development of pancreatic ductal adenocarcinoma and is activated by KRAS. SETDB1 was found to directly bind to the p53 promoter region to regulate expression of p53. SETDB1 suppression led to p53 upregulation. Further, deletion of SETDB1 was found to prevent the formation of PDAC via induction of p53-mediated apoptosis [54]. |
| 32 | Hepatocellular carcinoma (HCC) | n = 59 | IHC, TMA, cell proliferation assay, RNAi, immunoprecipitation, WB, RT-PCR, FFPE, in vitro methylation assay, TP53 mutagenesis. | GOF TP53 mutations such as R294S were shown to correlate with SETDB1 overexpression. In cell lines carrying the R294S mutation, inactivation of SETDB1 inhibited HCC growth. SETDB1 was shown to mechanistically regulate HCC growth by complexing with p53 and dimethylating p53K370 [52]. |
| 33 | Hepatocellular carcinoma (HCC) | n = 16 | RNA-Seq, qRT-PCR, IHC, WB, establishment of stable SETDB1 knockdown cells, in vivo orthotopic tumor implantation model, CHIP assay, luciferase reporter assay. | SETDB1 was identified as the greatest upregulated epigenetic regulator in HCC; its overexpression was linked to greater disease progression, aggressiveness, and worse prognosis. SETDB1 knockdown inhibited cell migration, lung metastasis, and orthotopic tumorigenicity. SP1 hyperactivation was found to transcriptionally increase SETDB1 expression, while miR-29 had the opposite effect [88]. |
| 34 | Hepatocellular carcinoma (HCC) | n = 24 | Yeast two-hybrid test, GST pull-down, CLIP assay, cell proliferation, migration, and invasion assays, xenograft studies, and establishment of stable cell lines. | SETDB1 overexpression in HCC was related to increased cell proliferation and migration. These effects were reversed by Tiam1 knockdown, leading to the conclusion that SETDB1 relies on complexing with Tiam1 for its oncogenic functions [93]. |
| 35 | Hepatocellular carcinoma (HCC) | n = 90 | IHC, ISH, TCGA data mining, qRT-PCR, WB, luciferase reporter assays, IR, colony formation and apoptosis assays, neutral comet assay, mice models. | In HCC tissues, miR-621 was found to inhibit SETDB1 expression by targeting SETDB1’s 3’ UTR. This inhibition led to an increase in HCC cell radiosensitivity, in part due to the activation of the p53 pathway [97]. |
| 36 | Endometrial Cancer (EC) | n = 95 | Copy number and somatic variant calling, methylation analysis, HotSpot3D, RNA quantification and analysis, MS data interpretation, and other proteogenomic analyses. | CDKN1A/p21 RNA levels were anticorrelated with those of SETDB1. SETDB1 was also negatively correlated with TNFRSF10B, an apoptotic protein. In general, genes such as SETDB1 that mapped to chromosome 1q showed negative correlations with p53 pathway activity in EC; however, SETDB1 exhibited the strongest anticorrelation [94]. |
| 37 | Nasopharyngeal Carcinoma (NPC) | n = 152 | IHC, qRT-PCR, WB, cell viability assay, cell cycle analysis, transwell assays, and statistical analyses. | SETDB1 was more highly expressed in NPC tissues compared to control tissues and correlated with poor prognosis in NPC patients. in vitro overexpression of SETDB1 resulted in increased cell proliferation, migration, and invasion; downregulation of SETDB1 had opposite effects [95]. |
| HONE1, SUNE1, CNE1, CNE2, 5-8F and 6-10B | ||||
| 38 | Gastric Cancer (GC) | n = 66 | qRT-PCR, luciferase assay, H. pylori-infected mouse model, ChIP, proliferation and metastasis assays, CCK-8 assay, IHC, and wound healing assays. | SETDB1 promoted tumorigenesis in GC, in part via interaction with ERG, enhancing transcription of oncogenes CCND1 and MMP9. TCF4 promoted transcriptional expression of SETDB1 [96]. |
Breast cancer
SETDB1 has been implicated in BRC from various studies. For example, Wu et al. assessed 45 BRC tissue samples via RT-PCR, finding that SETDB1 mRNA expression was markedly elevated in BRC tissues in comparison with normal tissues. Accordingly, Kaplan-Meier survival assay and log-rank test revealed that low SETDB1 mRNA expressing tissues had significantly longer survival times in comparison with the group with high SETDB1 mRNA expression. However, when SETDB1 was knocked down in MCF-7 and MDA-MB-231 cells, proliferation, cell cycle progression, and migration were inhibited. In vivo, SETDB1 knockdown resulted in significant decreases in tumor volume and tumor weight [58]. Xiao et al. supported these findings and added that SETDB1 is upregulated via c-MYC binding to the SETDB1 promoter. On the other hand, silencing of c-MYC or BMI1 hindered tumorigenic effects of SETDB1 [59]. Ryu et al. expanded on these findings, adding that SETDB1 knockdown in human breast cancer cell line MDA-MB-231 resulted in 1) regulation of actin cytoskeleton and cell adhesion molecules, 2) upregulation of epithelial-mesenchymal transition (EMT) markers (CDH1 and Claudin 1), and 3) downregulation of mesenchymal-epithelial transition (MET) markers (CHD2 and Vimentin). These findings suggested SETDB1 was associated with metastasis and cell migration in BRC. In the same study, SMAD7, an antagonist of the TGF-beta signaling pathway, was shown to be upregulated when SETDB1 was knocked down. Strikingly, the EMT markers that were upregulated by knockdown of SETDB1 were downregulated when SETDB1 and SMAD7 were both knocked down. This led to the conclusion that SETDB1 overexpression promotes metastasis via the downregulation of SMAD7 [60]. A 2020 paper studied 159 BRC tissue samples and supported these findings, demonstrating that SETDB1 protein level positively correlated with tumor size, histopathological grading, and lymph node metastasis. Ultimately, SETDB1 was proposed as an oncogene in BRC; high expression of SETDB1 correlated with shorter relapse-free survival, and overall worse survival [61].
A 2016 study identified interaction between ΔNp63α (the amino-deleted form of p63 and a potent oncogene in head and neck squamous cell carcinomas) and SETDB1 in H1299 and MCF-7 cells. A reciprocal regulatory relationship was identified in which silencing of SETDB1 led to a marked reduction in p63, while p63 silencing reduced SETDB1 expression, albeit to a lesser degree. Regarding the effect of this interaction, 30 genes were identified to be repressed by ΔNp63α in a SETDB1-dependent manner, which may in part explain oncogenic effects of ΔNp63α in BRC [62].
Lung cancer
In a 2014 study, Rodriguez-Paredes et al. observed over four-fold changes in SETDB1 copy number in two non-small cell lung cancer (NSCLC) cell lines, NCI-H1437 and NCI-H1395. Similar overexpression was also seen in a small cell lung cancer line, DMS-273. Further investigation showed that depletion of SETDB1 expression hindered cancer growth in vitro and in vivo. Specifically, SETDB1 shRNA-transfected DMS-273 and NCI-H1437 cells had lower tumorigenicity in nude mice compared with scramble shRNA-transfected cells, which rapidly formed tumors. Using a matrigel invasion assay, SETDB1 overexpression was shown to increase tumor invasiveness [63].
Of all cases of lung cancer, NSCLC accounts for approximately 80-85% of them, presenting a critical need to understand NSCLC specifically [64]. In 2018, Chen et al. observed that miR-29s targeted SETDB1; when miR-29 was ectopically expressed, a reduction in SETDB1 mRNA and protein followed. TP53 upregulated miR-29 expression, leading to the hypothesis that p53 acts through mir-29 to negatively regulate SETDB1 level. The following year, Cruz-Tapias et al. analyzed 1140 NSCLC cases, finding that SETDB1 was overexpressed in NSCLC tissues compared to normal lung tissues. Subgroup analyses showed that SETDB1 mRNA was increased in both adenocarcinoma (ADC) and squamous cell carcinoma (SCC) NSCLC subtypes, suggesting that global overexpression of SETDB1 mRNA is likely a hallmark of NSCLC. Interestingly, SETDB1 was elevated in NSCLC patients who were current or former smokers, compared to NSCLC patients who were non-smokers. Analysis of NSCLC microarray datasets that contained pathological characteristics of NSCLC patients provided evidence that the overexpression of SETDB1 is not reliant on patient age nor gender. Furthermore, there was no statistical evidence of an association between clinical stages of the carcinogenic process and expression levels of SETDB1 [65]. However, findings from a 2015 paper reported that SETDB1 amplification in ADC was associated with an advanced cancer stage [66]. The culmination of these findings provides a basis for future studies to evaluate SETDB1 as both a diagnostic marker and a therapeutic target in human lung tumorigenesis.
In a 2020 study conducted by Kang & Min, a genome and transcriptomic analysis was performed in ADC and SCC subtypes of lung cancer samples acquired from the TCGA data set. ADC samples were determined to have higher SETDB1 gene copy numbers relative to SCCs. SETDB1 expression was also correlated with the gene copy number in both subtypes. Comparing samples with high levels of SETDB1 to samples with low levels of SETDB1 in ADC and SCC revealed distinct gene signatures and markers specific for each individual subtype. Specifically, pathways related to immune responses and EMT processes were largely diminished in high level SETDB1 populations among both subtypes. Conversely, RNA interference genes, chromatin-modifying genes, DNA methyltransferase genes, histone lysine methylation-related genes, and genes involved in the G2M and DNA replication checkpoints were present at higher levels in SETDB1 high groups. CpG methylation levels were significantly decreased in SETDB1 high samples in ADCs, while the reverse was observed in SCCs. Within differentially expressed CpG methylation sites between high and low SETDB1 groups, lower methylation levels were observed in high SETDB1 ADCs compared to SCCs [67]. Further genome-wide methylation analysis is needed to explain this inverse correlation between DNMT expression and DNA methylation level in ADCs.
Prostate cancer
Hormonal therapy can be extremely effective in prostate cancer (PCa); however, many PCa become hormone-refractory and metastatic [68], demonstrating the need to elucidate causes of PCa. In a 2014 study, SETDB1 overexpression was seen in 19 of the 25 PCa tissues studied. siRNA was used to downregulate SETDB1 in PCa cells; SETDB1 depletion resulted in marked reductions in cell proliferation compared to control cells. In an in vitro scratch assay, SETDB1-depleted cells had lower migratory ability in comparison with the control cells. Matrigel invasion assays showed that siRNA-SETDB1 transfected cells were 1) less able to digest the Matrigel and penetrate through the transwell polycarbonate filter and 2) decreased in invasiveness. Data also suggested that silencing of SETDB1 induces cell cycle arrest in 22RV1 cells; flow cytometric analysis showed that siRNA-control transfection reduced the number of cells in S phase and increased the percentage of cells in G0/G1 [69]. Therefore, SETDB1 is a candidate oncogene in PCa and possesses great potential as a therapeutic target.
Colorectal cancer
Ho et al. found that SETDB1 protein expression was 82% higher in colon adenocarcinoma tissues compared to paired control tissues, and was correlated with histological grade, TNM stage, T-class, N-class, and worse survival. Using IHC in 30 Colorectal cancer (CRC) tissues, Chen et al. found that 1) SETDB1 was highly expressed in CRC tissues, 2) upregulation of SETDB1 promoted proliferation and migration in vitro and tumor growth in vivo, and 3) over-expression of SETDB1 in CRC cells inhibited 5-fluorouracil-induced apoptosis. Furthermore, TP53 expression was significantly decreased in DLD1-SETDB1 cells. This led to the hypothesis that SETDB1 is recruited to the promoter of tumor suppressor TP53, which results in the inhibition of apoptosis. Conversely, knockout of SETDB1 resulted in the inhibition of CRC proliferation, migration, and tumor growth [53]. However, a study in 2018 challenged these findings, asserting that SETDB1 expression was lower in CRC tissues (n = 207) compared to normal tissues [70].
Ultimately, following studies supported the original findings of Ho et al. and Chen et al. For example, Yu et al. supported that SETDB1 is more highly expressed in CRC, is associated with worse outcomes, and operates through the STAT1-CCND1/CDK6 axis to increase CRC proliferation. In a ChIP-PCR assay, SETDB1 was observed to bind directly to the STAT1 promoter, leading to upregulation of STAT1. Real-time PCR showed that CCND1 and CDK6 were markedly upregulated upon SETDB1 overexpression, while opposite results were observed upon SETDB1 knockout. Western blot using SW480 and SW620 cells showed that STAT1, CCND1, CDK6, and cell colony formation ability were positively correlated with SETDB1 expression. A dual luciferase assay showed that STAT1 specifically increased CCND1 promoter activity, leading to the conclusion that STAT1 directly binds to the promoter of CCND1 and promotes its transcription. Dual luciferase assays confirmed that SETDB1 binds directly to the promoter region of STAT1, -240 bp upstream from the start codon, thereby regulating the STAT1-CCND1/CDK6 axis [71].
A different study investigating the molecular mechanism in CRC found that SETDB1 hinders the function of five transcription factors (CDX2, ELF3, HNF4G, PPARG, and VDR) that convert stem-like CRC cells into a differentiated normal-like state. Knockdown of SETDB1 led to the downregulation of oncogene Myc, and the upregulation of four markers of differentiated colon epithelial cells: KRT20, FABP1, FABP2, and CEACAM5. Using single-cell RNA sequencing, higher expression of SETDB1 was seen in stem cell-like cancer cells compared to SETDB1 levels in normal stem cells. Using CRC patient tissue microarrays, high SETDB1 expression was observed in poorly differentiated CRC cells that did not have typical gland-forming histology. In patient derived organoid lines, organoids with depletion of SETDB1 showed higher levels of KRT20+ cells, a marker of differentiated colon cells. Ultimately, these findings suggested that depletion of SETDB1 allows for the conversion of stem-like cells into postmitotic cells, overcoming the CRC oncogenic process, and restoring normal morphology [42].
SETDB1 knockdown was also shown to cause cell cycle arrest at G1 and enhance apoptosis. SETDB1 was found to transcriptionally silence p21 expression by H3K9 methylation at the p21 promoter. When SETDB1 was knocked down in BALB/c nude mice, decreases in tumor growth and CRC proliferation was observed while p21 expression was upregulated [72].
Acute myeloid leukemia
Cuellar et al. developed a loss-of-function (LOF) cell viability screen in the MLL-AF9 translocation-driven THP-1 AML cell line, which was designed to stably express Cas9. These cells were infected with a sgRNA pool that targeted 350 known human epigenetic and transcriptional modifiers to determine those most crucial for the regulation of AML growth and malignancy. Of all the individual sgRNAs targeting specific genes in the screen, SETDB1 sgRNAs decreased cell viability the most, and SETDB1 sgRNAs had the highest overall fold change. Further study revealed that SETDB1 knockdown led to reduced protein levels, loss of viability, and induction of apoptosis markers in THP-1 cells. Similar findings of reduced cell growth were also seen in MOLM-13, ML-2, HL-60, OCI-AML-3, and MV-4-11 cells after treatment with SETDB1-specific sgRNAs. However, UKE-1 and EOL-1 cell lines were insensitive [73].
Cuellar et al. made SETDB1 mutant THP-1 lines with two SETDB1-specific sgRNAs for comparison against a non-targeted control (NTC) sgRNA-treated line. Using RNA-seq, robust activation of interferon (IFN)-stimulated genes (ISGs) was observed in SETDB1 mutant cells. Furthermore, type I IFN signaling genes (IFN-β, IFIT1-3, RIG-I/DDX58, OAS3, and MDA5) and antiviral response genes were the most upregulated pathways, while viral transcription and gene expression genes were the greatest downregulated genes. Therefore, whether directly and/or indirectly, SETDB1 likely represses type I IFN induction.
Upregulated expression of endogenous retroviruses (ERVs) has been observed following deletion of SETDB1 in embryonic stem cells (ESCs) and B cells [49,73-76]. Using RNA-seq, Cuellar et al. observed upregulation of ERV3-1 in the SETDB1 mutant THP-1 cells and three other AML cell lines that were treated with SETDB1-specific sgRNAs or shRNAs. Furthermore, the RNA-seq data was used to align repeat masked reads and revealed long interspersed nuclear elements (LINEs) and satellite repeats that were frequently upregulated in SETDB1 sgRNA-treated cells, but not in the NTC cells. These findings suggested that, in AML cells, SETDB1 plays a pivotal role in the repression of retrotransposable elements.
CRISPR was used to knock out cytosolic RNA sensors of innate immunity (IFIH1/MDA5, DDX58/RIG-I, and MAVS), then SETDB1, MDA5, MAVS, and RIG-I were disrupted. These cytosolic sensors were shown to contribute to IFIT2 induction. Therefore, cell death induced by the disruption of SETDB1 was shown to be dependent on viral sensing machinery. Considering that the desilencing of TEs is concurrent with IFN-induced apoptosis, SETDB1 was proposed to suppress innate immunity by limiting levels of TE expression in AML cells. In the absence of SETDB1, a cytosolic nucleic acid-sensing cascade and IFN-mediated cell death is induced [73].
The following year, Ropa et al. found that SETDB1 plays a role in regulating the growth of MLL-fusion driven AML cells. Specifically, SETDB1 was identified to directly bind to the Polymerase Associated Factor 1 Complex (PAF1c) subunit CDC73, whose interaction is stabilized by mutant CDC73. This interaction promoted H3K9 methylation at PAF1c target genes (Hoxa9 and Meis1) promoter sites responsible for AML cell growth and leukemogenesis. SETDB1-mediated H3K9 methylation resulted in transcriptional silencing of Hoxa9 and Meis1 and reduced cell growth. In human AML samples, SETDB1 expression was correlated greatly with Meis1 expression. Albeit to a lesser degree, the same trend was observed for Hoxa9 [77].
In 2020, Ropa et al. contrasted the 2017 findings of Cuellar et al., proposing that SETDB1 may suppress AML by making repressive changes to the promoter epigenome of pro-leukemic target genes (Dock1, Hoxa9, Six1, and others). SETDB1 overexpression in MLL-AF9 and E2A-HLF fusion driven AML cells resulted in reduced colony formation and proliferation, while differentiation and apoptosis increased. Decreased leukemic progression was also seen in primary mouse MLL-AF9 AML transplants in mice with SETDB1 overexpression [78]. Therefore, SETDB1 can both repress and promote AML proliferation. These contradicting findings may suggest that a narrow expression level of SETDB1 is required for the survival of AML cells.
Glioma
Han et al. used IHC and real-time PCR to assess 40 GBM tissues and 40 adjacent healthy tissues; SETDB1 was elevated in tumor samples in comparison with the surrounding control tissue. Furthermore, in U87 and U251 cells, increased SETDB1 expression caused 1) increased proliferation and clone formation and 2) a reduction in the levels of the cleaved (active) form of caspase-8 and caspase-3. Silencing of SETDB1 resulted in the opposite results [79]. These findings were consistent with a similar study in 2014, where IHC of grades II, III, and IV gliomas showed increased expression of SETDB1 in comparison with normal brain tissue. Specifically, SETDB1 expression was positively correlated with glioma histological grade [80].
Ectopic expression of SETDB1 via injection of U251-EV and U251-SETDB1 in nude mice resulted in tumor growth. Accordingly, Ki-67 assays revealed increased cell proliferation in U251-SETDB1 xenografts. SETDB1 knockdown resulted in decreased tumor size and showed decreased cell proliferation according to Ki-67 staining. In U87 and U251 cells that were subjected to SETDB1 upregulation, Western blotting revealed upregulation of mesenchymal markers (Slug and Vimentin) while the epithelial marker, E-cadherin, was downregulated. Opposite results were seen in the same cell lines where SETDB1 was silenced. Therefore, SETDB1 was shown to regulate migration, invasion, and EMT in GBM cells [79].
Colony-stimulating factor 1 (CSF-1) is a cytokine that has been associated with tumor-associated macrophage (TAM) infiltration and the progression of endometrial cancer [81]. When Han et al. overexpressed SETDB1 in U87 and U251 cells, higher levels of CSF-1 mRNA and proteins were observed. Furthermore, ELISA assays revealed that SETDB1 promoted CSF-1 secretion, while knockdown of SETDB1 suppressed the secretion of the cytokine. The AKT/mTOR signaling pathway was implicated as the underlying pathway behind CSF-1 secretion. Specifically, when SETDB1 was overexpressed, AKT and mTOR phosphorylation increased. AKT knockdown via siRNA attenuated the SETDB1-mediated increase of CSF-1. Western blotting using xenograft tumor tissue revealed that SETDB1 knockdown markedly reduced AKT activation and CSF-1 protein expression. Contrastingly, SETDB1 overexpression promoted AKT activation and elevated protein levels of CSF-1.
An in vitro migration assay revealed that that conditioned media (CM) from glioblastoma cells overexpressing SETDB1 1) promoted the recruitment of macrophages and 2) caused elevated expression of CD163, an M2 macrophage marker, in comparison with control CM. When a CSF-1 antagonist was present, recruitment of macrophages was inhibited, leading to the conclusion that macrophage migration is mediated by SETDB1 via the induction of CSF-1. Furthermore, the cytokine mRNAs and proteins that are representative of TAMs (IL-10, CCL17, and CCL22) were elevated in TAMs that were incubated with CM from GBM cells overexpressing SETDB1. M1-related genes (IL-12, IL-23, and CXCL10) were not observed to be affected. Ultimately, these findings proposed that SETDB1 and CSF-1 may contribute to macrophage entry and differentiation [79].
Clodronate-liposome treatment has been shown to deplete mature macrophages in a wide range of malignancies, including multiple myeloma and ovarian cancer [82,83]. To confirm that SETDB1 promotes oncogenic activity in a TAM-dependent manner, liposomal clodronate treatment was administered in a syngeneic mouse model, resulting in the suppression of macrophage infiltration. Tumor burden induced by SETDB1 overexpression in the mice was attenuated by clodronate liposomes, suggesting that macrophage infiltration of the tumor microenvironment mediate SETDB1’s tumor-promoting effects [79]. Ultimately, in patients with SETDB1-overexpressing tumors, CSF-1 and/or its receptor may be a promising target.
Melanoma
Although melanoma only accounts for approximately 1% of all skin cancer cases, melanoma results in the majority of skin cancer-related deaths. Furthermore, the overall rates of melanoma have been rapidly rising over recent decades [84], illustrating a need for efficient and effective interventions. SETDB1 was first established as an oncogene in melanoma in 2011. Ceol et al. demonstrated that melanomas overexpressing SETDB1 were 1) more aggressive than control tumors, 2) more locally invasive, and 3) had more extensive nuclear pleomorphism and larger nuclei. Microarray analysis of zebrafish melanomas revealed that SETDB1 overexpression led to a broad pattern of transcriptional changes, including conserved downregulation of a group of genes enriched for Hox genes and transcriptional regulators [85]. A later study investigating the underlying mechanism(s) behind these findings proposed that SETDB1 implements its oncogenic functions via regulation of thrombospondin 1 (THBS1). Specifically, in transcriptome analyses, a loss of H3K4me1 peaks in the THBS1 promoter and gene body regions of SETDB1-overexpressing cells was seen. On the other hand, silencing of SETDB1 led to the gain of H3K4me1 peaks in THBS1 regulatory regions. ChIP-qPCR validated these findings, leading to the hypothesis that a SETDB1-THBS1 regulatory axis exists. Indeed, IHC using the tumor secretions of NOD SCID mice injected with melanoma cells overexpressing SETDB1 revealed that SETDB1 and THBS1 were positively correlated in metastases and tumors; THBS1 and global H3K9me3 abundance were directly correlated. These findings highlight the importance of the chromatin state for THBS1 expression and pose the hypothesis that SETDB1 may be exerting its effects via regulation of THBS1 [86]. A 2020 study using a transgenic zebrafish model found that SETDB1 and Heparin Binding Growth Factor (HDGF), a gene also on chromosome 1q, were frequently co-altered. Their co-alteration resulted in disease progression and overall worse patient prognoses [87].
Pancreatic ductal adenocarcinoma
In 2020, Ogawa et al. [54] observed overexpression of SETDB1 in acinar-to-ductal metaplasia (ADM), pancreatic intraepithelial neoplasia (PanIN), and PDAC. Ptf1aCre; KrasG12D is a well-established mouse model of PDAC. PtF1aCre; SETDB1f/f mice were generated to assess if SETDB1 was required for the maintenance and development of the pancreas. Since no pancreatic abnormalities were observed in the PtF1aCre; SETDB1f/f mice, it was concluded that SETDB1 was dispensable for the development of the pancreas. However, in response to cerulein-induced acute pancreatitis, PtF1aCre; SETDB1f/f mice experienced major pancreatic atrophy and a significant reduction in pancreas to body weight ratio, compared to controls. Furthermore, seven days post-cerulein treatment, control mice had nearly fully recovered whereas mice with the pancreatic SETDB1 deletion experienced prolonged pancreatitis. Therefore, SETDB1 was deemed indispensable for exocrine pancreatic regeneration. Immunostaining for cleaved caspase 3 revealed elevated levels of apoptotic cells in PtF1aCre; SETDB1f/f mice compared to controls. However, immunostaining for Ki67 showed no differences in cell proliferation between the two groups.
Ogawa et al. next investigated the impact of a SETDB1 deletion in the context of oncogenic Kras activation. Ptf1aCre; KrasG12D; Setdb1f/f (KCS) mice were generated and compared to Ptf1aCre; KrasG12D (KC) control mice. The SETDB1 deletion was found to (1) accelerate the formation of ADM/PanIN and 2) promote pancreatic atrophy with increased apoptosis. Furthermore, SETDB1 deletion was also observed to increase apoptosis and elevate p53 expression; microarray analysis of mRNA from KC and KCS pancreata revealed upregulation of genes related to apoptotic pathways (such as Tns4, p19, Serpina3g, Noxa, Trfrs11b, and S100a8) in KCS mice. qRT-PCR confirmed these results, and decreased expression of Bcl2 (an anti-apoptotic gene) was also observed in KCS pancreata. ChIP assays using isolated acinar cells were used to confirm that SETDB1 bound directly to p53 promoter regions.
In the context of heterozygous p53 deletion, SETDB1 deletion protected against PDAC formation. Specifically, mice with the heterozygous p53 deletion and the SETDB1 deletion (KPheteroCS) showed more pancreatic atrophy compared to control mice that only had the heterozygous p53 deletion (KPheteroC). Ki67 immunostaining of 20 weeks old control mice revealed prominent cell proliferation. Contrastingly, cleaved caspase 3 and p53 immunostaining revealed upregulation of p53 in KPheteroCS mice, and an increased number of apoptotic cells compared to control mice. Ultimately, all control mice developed PDAC as early as 20 weeks while none of the KPheteroCS mice developed PDAC as old as 24 weeks. However, in experiments with homozygous p53 deletion, the results of increased apoptosis and protection against PDAC formation were not observed. Therefore, the homozygous p53 deletion offset the effects of the SETDB1 deletion and suggested that the SETDB1 deletion suppresses PDAC formation by inducing p53-mediated apoptosis.
When SETDB1 was silenced in human PDAC cells that have a wild type TP53 (PK59 and KP4), qRT-PCR revealed elevated TP53 expression and decreased SETDB1 expression, consistent with mice models. In SETDB1 silenced PK59 orthotopic PDAC models, 1) tumor size was reduced, 2) p53 was upregulated, 3) SETDB1 was downregulated, and 4) H3K9me3 expression was decreased, in comparison control models. Using ChIP, lower levels of SETDB1 and H3K9me3 binding to TP53 promoter regions were observed in SETDB1 silenced PK59 cells [54]. Therefore, in human PDAC cells, SETDB1 may epigenetically regulate TP53 in part via H3K9me3. Together, these findings demonstrate SETDB1’s value as a therapeutic target in PDAC.
Liver cancer
In a 2015 study, SETDB1 was identified as the greatest upregulated epigenetic regulator. SETDB1 overexpression was linked to hepatocellular carcinoma (HCC) tumorigenesis: greater disease progression, aggressiveness, and worse overall prognosis. SETDB1 knockdown inhibited cell migration, lung metastasis, and orthotopic tumorigenicity. Specificity protein 1 (Sp1) hyperactivation was found to transcriptionally increase SETDB1 expression, while miR-29 hyperactivation downregulated expression of SETDB1 [88].
The same year, Fei et al. assayed SETDB1 expression in six liver cancer samples with adjacent normal tissue. RT-qPCR revealed that SETDB1 expression was elevated in four of the six pairs of tumor tissue, compared with control tissue. Exome sequencing of 84 Asian HCC tissue samples returned 12 liver cancer cases where TP53 mutations were associated with SETDB1 copy number gain or overexpression. Four of these cases carried the R249S mutation, a well known GOF TP53 hotspot mutation [52,89] that has been proposed to increase cell migration and proliferation in lung and liver cancer [52,61,90,91]. In HCC cell line HCCLM3, an endogenous R249S mutation was identified along with a SETDB1 copy number gain and high SETDB1 expression. SETDB1 inhibition via both siRNAs or shRNAs suppressed HCCLM3 cell proliferation.
Fei et al. found that knockdown of SETDB1 in both p53 null Hep3B cells and p53-restored Hep3B cells did not result in growth inhibition. This data suggested that wild-type p53 does not dictate SETDB1 dependence in liver cancer. However, expression of p53R249S in p53 null Hep3B cells resulted in sensitivity to SETDB1 knockdown. Wild-type p53 control cells and the p53R249S-restored Hep3B cells were treated with doxorubicin to induce DNA damage; only the p53R249S-expressing cells were inhibited, suggesting that the R249S mutation confers cell growth sensitivity to SETDB1 knockdown.
Immunoprecipitation experiments revealed that SETDB1 can complex with both wild-type p53 and mutant p53, although SETDB1 more strongly associates with mutant p53. To assess if SETDB1 methylates p53, p53 peptide was used as a substrate and SAM was used as the methyl donor for SETDB1. Only small levels of p53 mono-methylation at K360 were detected, however, substantial K370me2 was detected when synthetic K370me1 peptide was used as the substrate. Endogenous experiments showed that knockdown of SETDB1 resulted in lower K370me2 levels in HCCLM3 cells. Furthermore, SETDB1 was found to methylate both mutant and wildtype p53, however, mutant p53 was methylated at higher levels. Importantly, SETDB1’s SET domain was found to be essential for p53K370 methylation; SET domain-truncated SETDB1 did not affect p53 methylation. These findings suggest that SETDB1 is likely a p53 methyltransferase that aids in the methylation of K370me1, resulting in K370me2 [52].
To assess SETDB1’s ability to regulate protein stability of p53 in liver cancer, HCT116 p53-null cells were introduced to wild type or mutated p53, then treated with cycloheximide (a protein synthesis inhibitor). Wild type p53 rapidly disappeared, while R249S mutant p53 displayed greater stability. When SETDB1 was knocked down, p53R249S turned over at an accelerated rate. Furthermore, inhibition of SETDB1 reduced S15 phosphorylation of p53, which has been shown to stabilize p53 by preventing it from ubiquitination [52,92]. SETDB1 knockdown increased p53 protein ubiquitination and increased the association of MDM2 with p53. Therefore, SETDB1 may confer stability of GOF p53 mutants via the dimethylation of K370, resulting in enhanced oncogenic activity of GOF mutant p53. Using a HCCLM3 xenograft model in nude mice, the authors also demonstrated that SETDB1 knockdown by shRNA inhibited in vivo tumor growth and increased cell differentiation, without significant reductions in mouse body weight [52].
Zhang et al. built on the findings of Wong et al. and Fei et al., adding that oncogenic effects of SETDB1 overexpression could be reversed by T-Cell Lymphoma Invasion and Metastasis 1 (Tiam1) knockdown. Specifically, knockdown of Tiam1 decreased SETDB1-mediated colony formation. SETDB1 and Tiam1 were observed to be positively correlated and more highly expressed in HCC tissues [93].
Endometrial cancer
SETDB1 has not been well studied in EC. However, proteogenomic characterization of EC identified anticorrelation between SETDB1 protein levels and tumor suppressor CDKN1A/p21 RNA levels. Furthermore, a strong negative correlation between SETDB1 and tumor necrosis factor receptor superfamily member 10B (TNFRSF10B), an apoptotic protein, was also identified. Dou et al. also observed that genes mapping to chromosome 1q exhibited strong negative correlations with the activity of the p53 pathway in EC. Strikingly, when ranking genes on chromosome 1q by their anticorrelation between protein level and p53 activity, SETDB1 was the strongest candidate [94]. These findings provide a strong rationale for the future study of SETDB1 in EC.
Nasopharyngeal cancer
A 2018 study proposed SETDB1 as an independent prognostic factor for NPC. Specifically, Huang et al. found that SETDB1 was amplified in 126 of the 152 NPC patient samples studied, its overexpression was linked to worse survival of NPC patients. Overexpression of SETDB1 led to enhanced proliferation, migration, and invasion ability of NPC cells, thereby promoting NPC tumorigenesis [95]. Nevertheless, mechanisms and genes involved in this process remain to be studied.
Gastric cancer
Although SETDB1 has not been extensively studied in GC, a 2020 study proposed SETDB1 as a potential oncogene in GC [96]. Specifically, Shang et al. observed that SETDB1 overexpression promoted GC tumorigenesis in vitro and in vivo and was correlated with poor prognosis. Knockdown of SETDB1 had the opposite effects, and CCND1 and MMP9 expression decreased. Further study revealed that ERG (ETS transcription factor) enhanced transcription of CCND1 and MMP9 by acting as an upstream modulator. ChIP and luciferase reporter assays suggested a potential mechanism explaining these findings; SETDB1 was found to interact with ERG and accumulate at promoter regions of CCND1 and MMP9. These findings led to the conclusion that SETDB1 enhances transcription of oncogenes CCND1 and MMP9, in an ERG-dependent manner.
One potential mechanism underlying SETDB1 overexpression in GC involves Transcription Factor 4 (TCF4), an important factor involved in Wnt signaling. TCF4 was proposed to bind a classical TCF4 binding motif on the promoter of SETDB1; this hypothesis was supported by ChIP assays. TCF4 depletion resulted in decreased expression of SETDB1 at the level of transcription and translation. Furthermore, infection with H. pylori was found to upregulate SETDB1 expression in a TCF4-dependent manner [96].
Regulating and targeting SETDB1
Mechanisms regulating SETDB1 expression have been mainly linked to transcriptional and post-transcriptional regulation. Transcription factors that target the SETDB1 promoter site, such as SP1, SP3, TCF4, and c-Myc, have been studied so far to be key factors responsible for the transcriptional regulation of SETDB1. This information has already been summarized in previous sections. In all cases, they have been shown to induce SETDB1 expression upon promoter binding. Additionally, post-transcriptional regulation, such as microRNA targeting of SETDB1 RNA transcripts, has also been widely acknowledged. Specifically, microRNAs such as miRNA-621, 381-3p, and 29 have been reported as negative regulators of SETDB1. A 2019 study in HCC revealed that miR-621 expression was lower in HCC tissues and lower expression was correlated with worse survival. Furthermore, in an independent set of 50 HCC tissues and also in a TCGA dataset of 367 HCC tissues, a negative correlation was observed between SETDB1 and miR-621. Strikingly, upregulation of miR-621 led to reduced expression of SETDB1 mRNA and protein. A dual luciferase assay confirmed that miR-621 directly targets SETDB1. Following transfection with miR-621, decreased survival fraction, increased apoptosis, and an increase of γ-H2AX (an indicator of cell response to DNA damage) was observed, ultimately suggesting that miR-621 could serve as a tumor radiosensitizer in HCC. The underlying mechanism behind the increased radiosensitivity was proposed to be via activation of the p53 pathway and subsequent repression of SETDB1. Overexpression of miR-621 activated the p53 pathway and upregulated the expression of p53 pathway effectors (p21, PUMA, and Gadd45). Contrastingly, SETDB1 inhibited the effects of miR-621 [97].
One proposed mechanism by which SETDB1 regulates tumor growth in BRC involves miR-381-3p, a miRNA that was previously confirmed to contribute to BRC development and progression [58,98]. A luciferase reporter assay revealed that the 3’-UTR on SETDB1 that is partially complementary to miR-381-3p was reduced by miR-381-3p overexpression and enhanced by miR-381-3p depletion. This finding led to the hypothesis that miR-381-3p directly targets SETDB1 [58].
Although there is no specific SETDB1 inhibitor available currently, some studies have attempted to target SETDB1 using general histone lysine methyltransferase inhibitors such as mithramycin A, 3’-deazaneplanocin A (DZNep), paclitaxel, and microRNAs miR-621 and miR-381-3p.
Mithramycin is a guanosine-cytosine-rich DNA binding antitumor antibiotic approved for clinical use. It binds to DNA’s minor groove, thereby displacing transcriptional activators that bind to GC-rich binding sites [99]. A 2006 study demonstrated that in Huntington’s disease, mithramycin interfered with specificity proteins Sp1 and Sp3, which serve as transcriptional activators of SETDB1. Combination treatment of mithramycin and cystamine resulted in downregulation of SETDB1 gene expression and the reduction of hyper-trimethylation of histone H3K9 [100]. Similar findings were observed in a 2014 study investigating SETDB1 in human lung tumorigenesis. Higher gene dosage of SETDB1 correlated with greater sensitivity to mithramycin, resulting in greater mithramycin-mediated growth inhibition. Specifically, mithramycin was able to inhibit SETDB1 in three lung cancer cell lines, DMS-273, NCI-H1437, and NCI-H1395, in a dose-dependent manner [63]. However, targeting Sp1 by blocking its ability to bind to the DNA may lead to off-target effects if important transcription factors are also unable to bind [101]. A 2020 study in melanoma found that Mithramycin A and its analog, Mithralog EC-8042, 1) reduced SETDB1 expression in melanoma cells, 2) induced transcriptomic, morphological, and functional changes, and 3) improved efficacy of MAPK inhibitor treatment.
DZNep is an epigenetic anticancer drug that is a cyclopentanyl analog of 3-deazaadenosine and serves as a SAM inhibitor that alters the genomic and epigenetic landscape. In addition to targeting degradation of EZH2 and H3K27me3 HMTase, DZNep was also identified to downregulate expression of SETDB1 in human lung cancer. Furthermore, DZNep was observed to inhibit many repressive chromatin markers and induce cell death. These findings led to the hypothesis that DZNep targets multiple HMTases during the death of lung cancer cells [102]. Karanth et al. suspect that DZNep regulates SETDB1 at the level of transcription, and treatment with DZNep likely leads to the formation of a repressive complex that binds to the SETDB1 promoter [103].
Paclitaxel (PTX) is a taxane that has been extensively used as a chemotherapy medication in a variety of cancers due to its ability to regulate death-related genes and induce tubulin stability. In lung cancer cells, Noh et al. observed that PTX could induce cell death via G2/M arrest and induce p53 expression, which led to the downregulation of SETDB1 expression [104].
A 2016 study revealed that during anticancer drug therapy, SETDB1 mediates the expression of FosB. Specifically, treatment with doxorubicin, taxol, and siSETDB1 led to downregulation of SETDB1 but enhanced expression of FosB, which was correlated with an enhanced rate of cancer proliferation. Therefore, this relationship may in part explain why cancers continue to proliferate during anticancer drug treatment [105]. A later study in NSCLC revealed that inhibition of MEK activated ERK2 during doxorubicin treatment, thereby inhibiting SETDB1 from regulating the FosB promoter [106].
Perspectives and new challenges for SETDB1
Thus far, great progress has been achieved to elucidate underlying mechanisms of SETDB1 regulation and its functional implications in different cancers. Functioning as a key H3K9 methyltransferase, SETDB1 has been established to be an important regulator of a variety of pathways responsible for cell proliferation, cell cycle progression, migration, survival, and apoptosis. SETDB1 upregulation in a wide variety of cancers was almost always (with the exception in AML) associated with enhanced tumorigenesis, cell growth, and decreased apoptosis, leading to poor patient outcomes in cancer. Particularly, SETDB1 promotes this through promoter H3K9 methylation and suppression of many important tumor suppressors such as p53 and p21. Aside from its main function as a histone H3K9 methyltransferase, SETDB1 contributes greatly to the methylation of different proteins, such as p53 and Akt, exerting its oncogenic effect through their stability and activation respectively. As SETDB1 overexpression and its oncogenic role is evident in a variety of cancers, targeting SETDB1 remains to be an important path forward. Currently, multiple groups listed in the 2021 NIH RePORTER are focusing on characterizing SETDB1 functions in the context of tissue regeneration and breast cancer. Specifically, the Chen group from the University of Texas seeks to understand the role of SETDB1 in intestinal stem cell (ISCs) self-renewal capacity, tissue regeneration, and homeostasis. Additionally, the Wei group from the Harvard Medical Center is eager to explore the connection between aberrant SETDB1 expression and hyperactivated PI3K/Akt/mTOR signaling in breast cancer, as an effort to develop new therapeutics. As outlined in the previous section, designing a specific SETDB1 inhibitor is a challenge that remains to be unmet; off-target effects and resistance can occur when using non-specific inhibitors. However, several other challenges exist for future studies involving SETDB1. Despite SETDB1 being an attractive therapeutic target, there are a lack of well-documented findings on what underlying mechanisms cause SETDB1 amplification, overexpression, and activity. As these gaps in knowledge become better understood, a clearer insight into SETDB1’s role and function in cancer can be achieved and would contribute toward better survival rates for patients in a multitude of cancers.
Acknowledgements
This project was supported by NIH R37CA238274 (SY) and the Department of Pathology Start-Up Fund (SY). The authors gratefully thank Dr. Hui-Kuan Lin for his insightful suggestions and editing contributions. We also acknowledge Jason Gao, Sean Gomendoza, and Matthew Wells for their editing contributions.
Disclosure of conflict of interest
None.
References
- 1.Harte PJ, Wu W, Carrasquillo MM, Matera AG. Assignment of a novel bifurcated SET domain gene, SETDB1, to human chromosome band 1q21 by in situ hybridization and radiation hybrids. Cytogenet Cell Genet. 1999;84:83–86. doi: 10.1159/000015220. [DOI] [PubMed] [Google Scholar]
- 2.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ Cancer Genome Atlas Research Network. Cherniack AD, Beroukhim R, Meyerson M. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33:676–689. e673. doi: 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kou F, Wu L, Ren X, Yang L. Chromosome abnormalities: new insights into their clinical significance in cancer. Mol Ther Oncolytics. 2020;17:562–570. doi: 10.1016/j.omto.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goh JY, Feng M, Wang W, Oguz G, Yatim S, Lee PL, Bao Y, Lim TH, Wang P, Tam WL, Kodahl AR, Lyng MB, Sarma S, Lin SY, Lezhava A, Yap YS, Lim AST, Hoon DSB, Ditzel HJ, Lee SC, Tan EY, Yu Q. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med. 2017;23:1319–1330. doi: 10.1038/nm.4405. [DOI] [PubMed] [Google Scholar]
- 5.Macgregor S, Montgomery GW, Liu JZ, Zhao ZZ, Henders AK, Stark M, Schmid H, Holland EA, Duffy DL, Zhang M, Painter JN, Nyholt DR, Maskiell JA, Jetann J, Ferguson M, Cust AE, Jenkins MA, Whiteman DC, Olsson H, Puig S, Bianchi-Scarra G, Hansson J, Demenais F, Landi MT, Debniak T, Mackie R, Azizi E, Bressac-de Paillerets B, Goldstein AM, Kanetsky PA, Gruis NA, Elder DE, Newton-Bishop JA, Bishop DT, Iles MM, Helsing P, Amos CI, Wei Q, Wang LE, Lee JE, Qureshi AA, Kefford RF, Giles GG, Armstrong BK, Aitken JF, Han J, Hopper JL, Trent JM, Brown KM, Martin NG, Mann GJ, Hayward NK. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet. 2011;43:1114–1118. doi: 10.1038/ng.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21:148–152. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Mei Q, Zielinska-Kwiatkowska A, Matsui Y, Blackburn ML, Benedetti D, Krumm AA, Taborsky GJ Jr, Chansky HA. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem J. 2003;369:651–657. doi: 10.1042/BJ20020854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 11.Ishimoto K, Kawamata N, Uchihara Y, Okubo M, Fujimoto R, Gotoh E, Kakinouchi K, Mizohata E, Hino N, Okada Y, Mochizuki Y, Tanaka T, Hamakubo T, Sakai J, Kodama T, Inoue T, Tachibana K, Doi T. Ubiquitination of lysine 867 of the human SETDB1 protein upregulates its histone H3 lysine 9 (H3K9) methyltransferase activity. PLoS One. 2016;11:e0165766. doi: 10.1371/journal.pone.0165766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Fang J. E3-independent constitutive monoubiquitination complements histone methyltransferase activity of SETDB1. Mol Cell. 2016;62:958–966. doi: 10.1016/j.molcel.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du D, Katsuno Y, Meyer D, Budi EH, Chen SH, Koeppen H, Wang H, Akhurst RJ, Derynck R. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 2018;19:135–155. doi: 10.15252/embr.201744250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, Sun L, Yang SC, Lan J, Hou Z, Cai Z, Jin G, Hsu CC, Wang YH, Hu J, Chen TY, Li H, Lee MG, Lin HK. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat Cell Biol. 2019;21:214–225. doi: 10.1038/s41556-018-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J, Dai X, Laurent B, Zheng N, Gan W, Zhang J, Guo A, Yuan M, Liu P, Asara JM, Toker A, Shi Y, Pandolfi PP, Wei W. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat Cell Biol. 2019;21:226–237. doi: 10.1038/s41556-018-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, Reuter G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994;13:3822–3831. doi: 10.1002/j.1460-2075.1994.tb06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenuwein T, Laible G, Dorn R, Reuter G. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell Mol Life Sci. 1998;54:80–93. doi: 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Q, Simel EJ, Klein PE, Royer MT, Houtz RL. Expression, purification, and characterization of recombinant ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit nepsilon-methyltransferase. Protein Expr Purif. 1998;14:104–112. doi: 10.1006/prep.1998.0936. [DOI] [PubMed] [Google Scholar]
- 19.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 20.Bennett RL, Swaroop A, Troche C, Licht JD. The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb Perspect Med. 2017;7:a026708. doi: 10.1101/cshperspect.a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wade PA, Wolffe AP. ReCoGnizing methylated DNA. Nat Struct Biol. 2001;8:575–577. doi: 10.1038/89593. [DOI] [PubMed] [Google Scholar]
- 22.Ohki I, Shimotake N, Fujita N, Jee J, Ikegami T, Nakao M, Shirakawa M. Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell. 2001;105:487–497. doi: 10.1016/s0092-8674(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 23.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 24.Kang YK. SETDB1 in early embryos and embryonic stem cells. Curr Issues Mol Biol. 2015;17:1–10. [PubMed] [Google Scholar]
- 25.Blackburn ML, Chansky HA, Zielinska-Kwiatkowska A, Matsui Y, Yang L. Genomic structure and expression of the mouse ESET gene encoding an ERG-associated histone methyltransferase with a SET domain. Biochim Biophys Acta. 2003;1629:8–14. doi: 10.1016/s0167-4781(03)00155-6. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Lawson KA, Teteak CJ, Zou J, Hacquebord J, Patterson D, Ghatan AC, Mei Q, Zielinska-Kwiatkowska A, Bain SD, Fernandes RJ, Chansky HA. ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev Biol. 2013;380:99–110. doi: 10.1016/j.ydbio.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terns MP, Terns RM. Macromolecular complexes: SMN--the master assembler. Curr Biol. 2001;11:R862–864. doi: 10.1016/s0960-9822(01)00517-6. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ 3rd. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tachibana K, Gotoh E, Kawamata N, Ishimoto K, Uchihara Y, Iwanari H, Sugiyama A, Kawamura T, Mochizuki Y, Tanaka T, Sakai J, Hamakubo T, Kodama T, Doi T. Analysis of the subcellular localization of the human histone methyltransferase SETDB1. Biochem Biophys Res Commun. 2015;465:725–731. doi: 10.1016/j.bbrc.2015.08.065. [DOI] [PubMed] [Google Scholar]
- 30.Cho S, Park JS, Kang YK. Regulated nuclear entry of over-expressed Setdb1. Genes Cells. 2013;18:694–703. doi: 10.1111/gtc.12068. [DOI] [PubMed] [Google Scholar]
- 31.Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep. 2016;17:653–659. doi: 10.1016/j.celrep.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsusaka T, Shimura C, Shinkai Y. ATF7IP regulates SETDB1 nuclear localization and increases its ubiquitination. EMBO Rep. 2019;20:e48297. doi: 10.15252/embr.201948297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26:963–977. doi: 10.1002/bies.20089. [DOI] [PubMed] [Google Scholar]
- 34.Travis J, Varma A, duPlessis D, Turnbull I, Vilar FJ. Immune reconstitution associated with progressive multifocal leukoencephalopathy in human immunodeficiency virus: a case discussion and review of the literature. Neurologist. 2008;14:321–326. doi: 10.1097/NRL.0b013e31816e2f13. [DOI] [PubMed] [Google Scholar]
- 35.Cho S, Park JS, Kang YK. Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): maintaining PML-NB structure and regulating the expression of its associated genes. J Biol Chem. 2011;286:41115–41124. doi: 10.1074/jbc.M111.248534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakakido M, Deng Z, Suzuki T, Dohmae N, Nakamura Y, Hamamoto R. Dysregulation of AKT pathway by SMYD2-mediated lysine methylation on PTEN. Neoplasia. 2015;17:367–373. doi: 10.1016/j.neo.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark C, Shah S, Herman-Ferdinandez L, Ekshyyan O, Abreo F, Rong X, McLarty J, Lurie A, Milligan EJ, Nathan CO. Teasing out the best molecular marker in the AKT/mTOR pathway in head and neck squamous cell cancer patients. Laryngoscope. 2010;120:1159–1165. doi: 10.1002/lary.20917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SH, Juhnn YS, Song YS. Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann N Y Acad Sci. 2007;1095:82–89. doi: 10.1196/annals.1397.012. [DOI] [PubMed] [Google Scholar]
- 39.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 40.Gao H, Yu Z, Bi D, Jiang L, Cui Y, Sun J, Ma R. Akt/PKB interacts with the histone H3 methyltransferase SETDB1 and coordinates to silence gene expression. Mol Cell Biochem. 2007;305:35–44. doi: 10.1007/s11010-007-9525-3. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Zhao H, Dan J, Kim S, Hardikar S, Hollowell D, Lin K, Lu Y, Takata Y, Shen J, Chen T. Maternal setdb1 is required for meiotic progression and preimplantation development in mouse. PLoS Genet. 2016;12:e1005970. doi: 10.1371/journal.pgen.1005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S, Lee C, Hwang CY, Kim D, Han Y, Hong SN, Kim SH, Cho KH. Network inference analysis identifies SETDB1 as a key regulator for reverting colorectal cancer cells into differentiated normal-like cells. Mol Cancer Res. 2020;18:118–129. doi: 10.1158/1541-7786.MCR-19-0450. [DOI] [PubMed] [Google Scholar]
- 43.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 44.Wu J, Gao FX, Wang C, Qin M, Han F, Xu T, Hu Z, Long Y, He XM, Deng X, Ren DL, Dai TY. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J Ex Clin Cancer Res. 2019;38:321. doi: 10.1186/s13046-019-1310-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2015;36:3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 47.Janowski BA, Huffman KE, Schwartz JC, Ram R, Nordsell R, Shames DS, Minna JD, Corey DR. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat Struct Mol Biol. 2006;13:787–792. doi: 10.1038/nsmb1140. [DOI] [PubMed] [Google Scholar]
- 48.Cho S, Park JS, Kang YK. AGO2 and SETDB1 cooperate in promoter-targeted transcriptional silencing of the androgen receptor gene. Nucleic Acids Res. 2014;42:13545–13556. doi: 10.1093/nar/gku788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- 50.Fukuda K, Shinkai Y. SETDB1-mediated silencing of retroelements. Viruses. 2020;12:596. doi: 10.3390/v12060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 52.Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, Fu S, Liang Y, Li C, Chen Z, Zhao Y, Yu Z, Huang Z, Hu M, Ying H, Chen Z, Zhang Y, Xing F, Zhu J, Xu H, Zhao K, Lu C, Atadja P, Xiao ZX, Li E, Shou J. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen K, Zhang F, Ding J, Liang Y, Zhan Z, Zhan Y, Chen LH, Ding Y. Histone methyltransferase SETDB1 promotes the progression of colorectal cancer by inhibiting the expression of TP53. J Cancer. 2017;8:3318–3330. doi: 10.7150/jca.20482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogawa S, Fukuda A, Matsumoto Y, Hanyu Y, Sono M, Fukunaga Y, Masuda T, Araki O, Nagao M, Yoshikawa T, Goto N, Hiramatsu Y, Tsuda M, Maruno T, Nakanishi Y, Hussein MS, Tsuruyama T, Takaori K, Uemoto S, Seno H. SETDB1 inhibits p53-mediated apoptosis and is required for formation of pancreatic ductal adenocarcinomas in mice. Gastroenterology. 2020;159:682–696. e613. doi: 10.1053/j.gastro.2020.04.047. [DOI] [PubMed] [Google Scholar]
- 55.Juznic L, Peuker K, Strigli A, Brosch M, Herrmann A, Hasler R, Koch M, Matthiesen L, Zeissig Y, Loscher BS, Nuber A, Schotta G, Neumeister V, Chavakis T, Kurth T, Lesche M, Dahl A, von Massenhausen A, Linkermann A, Schreiber S, Aden K, Rosenstiel PC, Franke A, Hampe J, Zeissig S. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut. 2021;70:485–498. doi: 10.1136/gutjnl-2020-321339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hachiya R, Shiihashi T, Shirakawa I, Iwasaki Y, Matsumura Y, Oishi Y, Nakayama Y, Miyamoto Y, Manabe I, Ochi K, Tanaka M, Goda N, Sakai J, Suganami T, Ogawa Y. The H3K9 methyltransferase Setdb1 regulates TLR4-mediated inflammatory responses in macrophages. Sci Rep. 2016;6:28845. doi: 10.1038/srep28845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takikita S, Muro R, Takai T, Otsubo T, Kawamura YI, Dohi T, Oda H, Kitajima M, Oshima K, Hattori M, Endo TA, Toyoda T, Weis J, Shinkai Y, Suzuki H. A histone methyltransferase ESET is critical for t cell development. J Immunol. 2016;197:2269–2279. doi: 10.4049/jimmunol.1502486. [DOI] [PubMed] [Google Scholar]
- 58.Wu M, Fan B, Guo Q, Li Y, Chen R, Lv N, Diao Y, Luo Y. Knockdown of SETDB1 inhibits breast cancer progression by miR-381-3p-related regulation. Biol Res. 2018;51:39. doi: 10.1186/s40659-018-0189-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao JF, Sun QY, Ding LW, Chien W, Liu XY, Mayakonda A, Jiang YY, Loh XY, Ran XB, Doan NB, Castor B, Chia D, Said JW, Tan KT, Yang H, Fu XY, Lin DC, Koeffler HP. The c-MYC-BMI1 axis is essential for SETDB1-mediated breast tumourigenesis. J Pathol. 2018;246:89–102. doi: 10.1002/path.5126. [DOI] [PubMed] [Google Scholar]
- 60.Ryu TY, Kim K, Kim SK, Oh JH, Min JK, Jung CR, Son MY, Kim DS, Cho HS. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 2019;52:139–144. doi: 10.5483/BMBRep.2019.52.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Z, Wu B, Tang X, Yang W, Zou Q, Wang H. High SET domain bifurcated 1 (SETDB1) expression predicts poor prognosis in breast carcinoma. Med Sci Monit. 2020;26:e922982. doi: 10.12659/MSM.922982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Regina C, Compagnone M, Peschiaroli A, Lena A, Annicchiarico-Petruzzelli M, Piro MC, Melino G, Candi E. Setdb1, a novel interactor of DeltaNp63, is involved in breast tumorigenesis. Oncotarget. 2016;7:28836–28848. doi: 10.18632/oncotarget.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodriguez-Paredes M, Martinez de Paz A, Simo-Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vazquez-Cedeira M, Lazo PA, Carneiro F, Moura CS, Vieira J, Teixeira MR, Esteller M. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014;33:2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 65.Cruz-Tapias P, Zakharova V, Perez-Fernandez OM, Mantilla W, RamÍRez-Clavijo S, Ait-Si-Ali S. Expression of the major and pro-oncogenic H3K9 lysine methyltransferase SETDB1 in non-small cell lung cancer. Cancers (Basel) 2019;11:1134. doi: 10.3390/cancers11081134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inoue Y, Matsuura S, Kurabe N, Kahyo T, Mori H, Kawase A, Karayama M, Inui N, Funai K, Shinmura K, Suda T, Sugimura H. Clinicopathological and survival analysis of japanese patients with resected non-small-cell lung cancer harboring NKX2-1, SETDB1, MET, HER2, SOX2, FGFR1, or PIK3CA gene amplification. J Thorac Oncol. 2015;10:1590–1600. doi: 10.1097/JTO.0000000000000685. [DOI] [PubMed] [Google Scholar]
- 67.Kang YK, Min B. SETDB1 overexpression sets an intertumoral transcriptomic divergence in non-small cell lung carcinoma. Front Genet. 2020;11:573515. doi: 10.3389/fgene.2020.573515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. 2010;17(Suppl 2):S72–79. doi: 10.3747/co.v17i0.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun Y, Wei M, Ren SC, Chen R, Xu WD, Wang FB, Lu J, Shen J, Yu YW, Hou JG, Xu CL, Huang JT, Sun YH. Histone methyltransferase SETDB1 is required for prostate cancer cell proliferation, migration and invasion. Asian J Androl. 2014;16:319–324. doi: 10.4103/1008-682X.122812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carvalho S, Freitas M, Antunes L, Monteiro-Reis S, Vieira-Coimbra M, Tavares A, Paulino S, Videira JF, Jeronimo C, Henrique R. Prognostic value of histone marks H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD-1 in colorectal cancer. J Cancer Res Clin Oncol. 2018;144:2127–2137. doi: 10.1007/s00432-018-2733-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu L, Ye F, Li YY, Zhan YZ, Liu Y, Yan HM, Fang Y, Xie YW, Zhang FJ, Chen LH, Ding Y, Chen KL. Histone methyltransferase SETDB1 promotes colorectal cancer proliferation through the STAT1-CCND1/CDK6 axis. Carcinogenesis. 2020;41:678–688. doi: 10.1093/carcin/bgz131. [DOI] [PubMed] [Google Scholar]
- 72.Cao N, Yu Y, Zhu H, Chen M, Chen P, Zhuo M, Mao Y, Li L, Zhao Q, Wu M, Ye M. SETDB1 promotes the progression of colorectal cancer via epigenetically silencing p21 expression. Cell Death Dis. 2020;11:351. doi: 10.1038/s41419-020-2561-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D, Cheung TK, Chaudhuri S, Modrusan Z, Doerr JM, Classon M, Haley B. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216:3535–3549. doi: 10.1083/jcb.201612160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, Lorincz MC. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collins PL, Kyle KE, Egawa T, Shinkai Y, Oltz EM. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc Natl Acad Sci U S A. 2015;112:8367–8372. doi: 10.1073/pnas.1422187112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, Dougan G, Dawson MA, Lehner PJ. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. 2015;348:1481–1485. doi: 10.1126/science.aaa7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ropa J, Saha N, Chen Z, Serio J, Chen W, Mellacheruvu D, Zhao L, Basrur V, Nesvizhskii AI, Muntean AG. PAF1 complex interactions with SETDB1 mediate promoter H3K9 methylation and transcriptional repression of Hoxa9 and Meis1 in acute myeloid leukemia. Oncotarget. 2018;9:22123–22136. doi: 10.18632/oncotarget.25204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ropa J, Trinh T, Aljoufi A, Broxmeyer HE. SETDB1 mediated histone H3 lysine 9 methylation suppresses MLL-fusion target expression and leukemic transformation. Haematologica. 2020;105:2273–2285. doi: 10.3324/haematol.2019.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Han S, Zhen W, Guo T, Zou J, Li F. SETDB1 promotes glioblastoma growth via CSF-1-dependent macrophage recruitment by activating the AKT/mTOR signaling pathway. J Exp Clin Cancer Res. 2020;39:218. doi: 10.1186/s13046-020-01730-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Spyropoulou A, Gargalionis A, Dalagiorgou G, Adamopoulos C, Papavassiliou KA, Lea RW, Piperi C, Papavassiliou AG. Role of histone lysine methyltransferases SUV39H1 and SETDB1 in gliomagenesis: modulation of cell proliferation, migration, and colony formation. Neuromolecular Med. 2014;16:70–82. doi: 10.1007/s12017-013-8254-x. [DOI] [PubMed] [Google Scholar]
- 81.Hua F, Tian Y, Gao Y, Li C, Liu X. Colonystimulating factor 1 receptor inhibition blocks macrophage infiltration and endometrial cancer cell proliferation. Mol Med Rep. 2019;19:3139–3147. doi: 10.3892/mmr.2019.9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Opperman KS, Vandyke K, Clark KC, Coulter EA, Hewett DR, Mrozik KM, Schwarz N, Evdokiou A, Croucher PI, Psaltis PJ, Noll JE, Zannettino AC. Clodronate-liposome mediated macrophage depletion abrogates multiple myeloma tumor establishment in vivo. Neoplasia. 2019;21:777–787. doi: 10.1016/j.neo.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reusser NM, Dalton HJ, Pradeep S, Gonzalez-Villasana V, Jennings NB, Vasquez HG, Wen Y, Rupaimoole R, Nagaraja AS, Gharpure K, Miyake T, Huang J, Hu W, Lopez-Berestein G, Sood AK. Clodronate inhibits tumor angiogenesis in mouse models of ovarian cancer. Cancer Biol Ther. 2014;15:1061–1067. doi: 10.4161/cbt.29184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 85.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Orouji E, Federico A, Larribere L, Novak D, Lipka DB, Assenov Y, Sachindra S, Huser L, Granados K, Gebhardt C, Plass C, Umansky V, Utikal J. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer. 2019;145:3462–3477. doi: 10.1002/ijc.32432. [DOI] [PubMed] [Google Scholar]
- 87.Fazio M, van Rooijen E, Mito JK, Modhurima R, Weiskopf E, Yang S, Zon LI. Recurrent co-alteration of HDGF and SETDB1 on chromosome 1q drives cutaneous melanoma progression and poor prognosis. Pigment Cell Melanoma Res. 2021;34:641–647. doi: 10.1111/pcmr.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wong CM, Wei L, Law CT, Ho DW, Tsang FH, Au SL, Sze KM, Lee JM, Wong CC, Ng IO. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. 2016;63:474–487. doi: 10.1002/hep.28304. [DOI] [PubMed] [Google Scholar]
- 89.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woo HG, Wang XW, Budhu A, Kim YH, Kwon SM, Tang ZY, Sun Z, Harris CC, Thorgeirsson SS. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology. 2011;140:1063–1070. doi: 10.1053/j.gastro.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vaughan CA, Singh S, Windle B, Sankala HM, Graves PR, Andrew Yeudall W, Deb SP, Deb S. p53 mutants induce transcription of NF-kappaB2 in H1299 cells through CBP and STAT binding on the NF-kappaB2 promoter and gain of function activity. Arch Biochem Biophys. 2012;518:79–88. doi: 10.1016/j.abb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Y, Huang J, Li Q, Chen K, Liang Y, Zhan Z, Ye F, Ni W, Chen L, Ding Y. Histone methyltransferase SETDB1 promotes cells proliferation and migration by interacting withTiam1 in hepatocellular carcinoma. BMC Cancer. 2018;18:539. doi: 10.1186/s12885-018-4464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, Liu W, Wu Y, Tsai CF, Wen B, Li Z, Cao S, Moon J, Shi Z, Cornwell M, Wyczalkowski MA, Chu RK, Vasaikar S, Zhou H, Gao Q, Moore RJ, Li K, Sethuraman S, Monroe ME, Zhao R, Heiman D, Krug K, Clauser K, Kothadia R, Maruvka Y, Pico AR, Oliphant AE, Hoskins EL, Pugh SL, Beecroft SJI, Adams DW, Jarman JC, Kong A, Chang HY, Reva B, Liao Y, Rykunov D, Colaprico A, Chen XS, Czekański A, Jędryka M, Matkowski R, Wiznerowicz M, Hiltke T, Boja E, Kinsinger CR, Mesri M, Robles AI, Rodriguez H, Mutch D, Fuh K, Ellis MJ, DeLair D, Thiagarajan M, Mani DR, Getz G, Noble M, Nesvizhskii AI, Wang P, Anderson ML, Levine DA, Smith RD, Payne SH, Ruggles KV, Rodland KD, Ding L, Zhang B, Liu T, Fenyö D Clinical Proteomic Tumor Analysis Consortium. Proteogenomic characterization of endometrial carcinoma. Cell. 2020;180:729–748. e726. doi: 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang J, Huang W, Liu M, Zhu J, Jiang D, Xiong Y, Zhen Y, Yang D, Chen Z, Peng L, Yu Z. Enhanced expression of SETDB1 possesses prognostic value and promotes cell proliferation, migration and invasion in nasopharyngeal carcinoma. Oncol Rep. 2018;40:1017–1025. doi: 10.3892/or.2018.6490. [DOI] [PubMed] [Google Scholar]
- 96.Shang W, Wang Y, Liang X, Li T, Shao W, Liu F, Cui X, Wang Y, Lv L, Chai L, Qu L, Zheng L, Jia J. SETDB1 promotes gastric carcinogenesis and metastasis via upregulation of CCND1 and MMP9 expression. J Pathol. 2020;253:148–159. doi: 10.1002/path.5568. [DOI] [PubMed] [Google Scholar]
- 97.Shao Y, Song X, Jiang W, Chen Y, Ning Z, Gu W, Jiang J. MicroRNA-621 acts as a tumor radiosensitizer by directly targeting SETDB1 in hepatocellular carcinoma. Mol Ther. 2019;27:355–364. doi: 10.1016/j.ymthe.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xue Y, Xu W, Zhao W, Wang W, Zhang D, Wu P. miR-381 inhibited breast cancer cells proliferation, epithelial-to-mesenchymal transition and metastasis by targeting CXCR4. Biomed Pharmacother. 2017;86:426–433. doi: 10.1016/j.biopha.2016.12.051. [DOI] [PubMed] [Google Scholar]
- 99.Jones DE Jr, Cui DM, Miller DM. Expression of beta-galactosidase under the control of the human c-myc promoter in transgenic mice is inhibited by mithramycin. Oncogene. 1995;10:2323–2330. [PubMed] [Google Scholar]
- 100.Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc Natl Acad Sci U S A. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vizcaíno C, Rodríguez-Sánchez MA, Núñez LE, Morís F, Portugal J. Novel mithramycins abrogate the involvement of protein factors in the transcription of cell cycle control genes. Biochem Pharmacol. 2012;84:1133–1142. doi: 10.1016/j.bcp.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 102.Lee JK, Kim KC. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem Biophys Res Commun. 2013;438:647–652. doi: 10.1016/j.bbrc.2013.07.128. [DOI] [PubMed] [Google Scholar]
- 103.Karanth AV, Maniswami RR, Prashanth S, Govindaraj H, Padmavathy R, Jegatheesan SK, Mullangi R, Rajagopal S. Emerging role of SETDB1 as a therapeutic target. Expert Opin Ther Targets. 2017;21:319–331. doi: 10.1080/14728222.2017.1279604. [DOI] [PubMed] [Google Scholar]
- 104.Noh HJ, Kim KA, Kim KC. p53 down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem Biophys Res Commun. 2014;446:43–48. doi: 10.1016/j.bbrc.2014.02.053. [DOI] [PubMed] [Google Scholar]
- 105.Na HH, Noh HJ, Cheong HM, Kang Y, Kim KC. SETDB1 mediated FosB expression increases the cell proliferation rate during anticancer drug therapy. BMB Rep. 2016;49:238–243. doi: 10.5483/BMBRep.2016.49.4.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Na HH, Kim KC. SETDB1-mediated FosB regulation via ERK2 is associated with an increase in cell invasiveness during anticancer drug treatment of A549 human lung cancer cells. Biochem Biophys Res Commun. 2018;495:512–518. doi: 10.1016/j.bbrc.2017.10.176. [DOI] [PubMed] [Google Scholar]
- 107.Yang W, Su Y, Hou C, Chen L, Zhou D, Ren K, Zhou Z, Zhang R, Liu X. SETDB1 induces epithelialmesenchymal transition in breast carcinoma by directly binding with Snail promoter. Oncol Rep. 2019;41:1284–1292. doi: 10.3892/or.2018.6871. [DOI] [PubMed] [Google Scholar]
- 108.Lafuente-Sanchis A, Zuniga A, Galbis JM, Cremades A, Estors M, Martinez-Hernandez NJ, Carretero J. Prognostic value of ERCC1, RRM1, BRCA1 and SETDB1 in early stage of non-small cell lung cancer. Clin Transl Oncol. 2016;18:798–804. doi: 10.1007/s12094-015-1440-6. [DOI] [PubMed] [Google Scholar]
- 109.Chen B, Wang J, Wang J, Wang H, Gu X, Tang L, Feng X. A regulatory circuitry comprising TP53, miR-29 family, and SETDB1 in non-small cell lung cancer. Biosci Rep. 2018;38:BSR20180678. doi: 10.1042/BSR20180678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu S, Li B, Xu J, Hu S, Zhan N, Wang H, Gao C, Li J, Xu X. SOD1 promotes cell proliferation and metastasis in non-small cell lung cancer via an miR-409-3p/SOD1/SETDB1 epigenetic regulatory feedforward loop. Front Cell Dev Biol. 2020;8:213. doi: 10.3389/fcell.2020.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ho YJ, Lin YM, Huang YC, Chang J, Yeh KT, Lin LI, Gong Z, Tzeng TY, Lu JW. Significance of histone methyltransferase SETDB1 expression in colon adenocarcinoma. APMIS. 2017;125:985–995. doi: 10.1111/apm.12745. [DOI] [PubMed] [Google Scholar]
- 112.Hou Z, Sun L, Xu F, Hu F, Lan J, Song D, Feng Y, Wang J, Luo X, Hu J, Wang G. Blocking histone methyltransferase SETDB1 inhibits tumorigenesis and enhances cetuximab sensitivity in colorectal cancer. Cancer Lett. 2020;487:63–73. doi: 10.1016/j.canlet.2020.05.029. [DOI] [PubMed] [Google Scholar]
- 113.Federico A, Steinfass T, Larribere L, Novak D, Moris F, Nunez LE, Umansky V, Utikal J. Mithramycin A and mithralog EC-8042 inhibit SETDB1 expression and its oncogenic activity in malignant melanoma. Mol Ther Oncolytics. 2020;18:83–99. doi: 10.1016/j.omto.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]