

Abstract
Mitochondria have attracted attention in cancer research as organelles associated with tumor development and response to therapy. We recently reported acquisition of resistance to cisplatin (DDP) associated with a metabolic rewiring in ovarian cancer patient-derived xenografts (PDXs) models. DDP-resistant PDXs models were obtained mimicking the clinical setting, treating mice bearing sensitive-DDP tumors with multiple cycles of DDP until the development of resistance. To further characterize the metabolic rewiring, the present study focused on tumor mitochondria. We analysed by transmission electron microscopy the mitochondria structure in two models of DDP-resistant and the corresponding DDP-sensitive PDXs and evaluated tumor mDNA content, the expression of genes and proteins involved in mitochondria functionality, and mitochondria fitness-related processes, such as autophagy. We observed a decrease in the number of mitochondria paralleled by an increased volume in DDP-resistant versus DDP-sensitive PDXs. DDP-resistant PDXs presented a higher percentage of damaged mitochondria, in particular of type 2 (concave-shape), and type 3 (cristolysis) damage. We found no difference in the mDNA content, and the expression of genes involved in mitochondrial biogenesis was similar between the sensitive and resistant PDXs. An upregulation of some genes involved in mitochondrial fitness in DDP-R versus DDP-S PDXs was observed. At protein level, no difference in the expression of proteins involved in mitochondrial function and biogenesis, and in autophagy/mitophagy was found. We here reported that the acquisition of DDP resistance is associated with morphological alterations in mitochondria, even if we couldn’t find any dysregulation in the studied genes/proteins that could explain the observed differences.
Keywords: Mitochondria, platinum resistance and ovarian carcinoma
Introduction
Resistance to therapy is still an unresolved issue [1,2], and an active field of research [1-3] in ovarian cancer. High-grade ovarian carcinomas (about 75% of all ovarian cancers) have the poorest outcome among ovarian cancers with a 5-year overall survival (OS) of 35% [1,4,5]. Standard treatment involves cytoreductive surgery followed by adjuvant chemotherapy (platinum/taxol), and even if 85% of high-grade serous patients respond to the first-line therapy, about 70% of patients will relapse within 3 years, and the majority of them will develop resistance to a further cisplatinum (DDP) challenge [3,4]. DDP targets mainly DNA, producing inter and intra-strand crosslinks, inducing cytotoxicity and oxidative stress [6-8]. Different mechanisms underlying DDP resistance have been reported, including those interfering with drug transport, with the repair of DDP-induced DNA damage, with signalling to the apoptotic machinery, with deregulated miRNAs, and others (for a recent review see [9]). Recently, the role of metabolic reprogramming in the resistance to anticancer therapy, including a DDP-based treatment, has been advocated [10-12].
In last years, mitochondria, the “powerhouse” of cells, have attracted attention as playing roles in cancer development and response to therapy [13-15]. They use multiple carbon sources to produce ATP and metabolites (i.e. pyruvate, aminoacids, and fatty acids), in order to fuel anaplerotic circuits, such as the TCA cycle and generate NADH and FADH, which deliver their electrons to the electron transport chain (ETC). Thus, the mitochondria operate as a central hub of both catabolic and anabolic metabolism [16]. Mitochondrial dysfunctions have been reported to be associated to different diseases, including cancer [17-21]. Recent evidence shows the involvement of mitochondrial metabolism in oncogenesis [22-25], tumor dissemination [26,27], as well as in tumor metabolic plasticity [28], and response to therapy [29,30]. In particular, mutations in the mitochondrial DNA (mtDNA) and mitochondrial enzymes defects (such as mutations in SDH, FH or IDH1/2) have been reported in different cancers [31,32]. Mitochondria play a central role also in the regulation of the programmed cell death, as well as in the development of primary or acquired resistance, including cisplatin resistance [33]. Different studies reported metabolic adaptation in cancer cells after a specific treatment, such as the shift from glycolysis to OXPHOS [34].
We have recently reported an increase in the oxidative metabolism associated with the acquisition of cisplatin resistance in models of ovarian cancer patient-derived xenografts (PDXs) [35]. We further investigated the involvement of mitochondria in this phenomenon, and here we reported the morphological and structural impairments at mitochondrial level in DDP-resistant ovarian cancer PDXs.
Methods
Animals
The IRFMN adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorisation for persons conducting animal experiments (Quality Management System Certificate-UNI EN ISO 9001:2015-Reg. N° 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE).
Tumor fragments were subcutaneously inoculated in female CD1 nude mice obtained from Charles River Laboratories (Italy) when six- to eight-week-old. Mice were maintained under specific pathogen-free conditions, housed in isolated vented cages, and handled using aseptic procedures. Mice were constantly monitored and tumor growth was followed by measurements with a Vernier caliper and tumor volume was calculated as already reported [36]. When tumor volume reached 20% of mouse body weight, mice were sacrified, tumors were excised and fragments were fixed as described above for TEM analysis, and snap frozen for molecular (DNA, gene and protein expression) studies.
Transmission electron microscopy (TEM) analysis
Fragments of DDP-sensitive and -resistant PDXs were fixed with 4% paraformaldehyde (PFA) and 2% glutaraldehyde in phosphate buffer 0.12 M pH 7.4 for 4 hours at room temperature, followed by post-fixation with 1% OsO4 in 0.12 M cacodylate buffer at room temperature for 1 h. After dehydration in graded series of ethanol, tissue samples were cleared in propylene oxide, embedded in Epoxy medium (Epon 812 Fluka) and polymerized at 60°C for 72 h. From each sample, 1 μm-section was cut with a Leica EM UC6 ultramicrotome (Leica Microsystems,), stained with Toluidine Blue and mounted on glass slides for light microscopic identification of areas of interest. Ultra-thin 60 nm-sections were obtained and counterstained with uranyl acetate and lead citrate, and images were obtained with an energy filter transmission electron microscope (Libra120, Carl Zeiss) coupled with a yttrium aluminium garnet (YAG) scintillator slow-scan CCD camera (Sharp eye, TRS). Numerical density of mitochondria (NV, n/µm3) was estimated using morphometrical analysis using an orthogonal grid digitally superimposed to 90 digitized electron microscope pictures of cellular cytoplasm at 5000× magnification and analyzing an average of 2246 mitochondria for each experimental group. Briefly, the mitochondrial profile area density (NA) was estimated by the ratio between the number of mitochondria and the cytoplasm area traced manually and calculated with Image J (version 1.52a). Mitochondrial volume density (VV) was determined by the ratio of grid points falling over mitochondria divided by the total number of points of the grid in cellular cytoplasm. NV was then estimated for each cell using the formula: NV= (1/b) (NA 3/2/VV 1/2). Where b is the shape coefficient for ellipsoidal mitochondria, calculated from the ratio of the harmonic mean of major and minor axis of mitochondria sections measured on digital images. The mean mitochondrial volume was then calculated for each cell as the ratio of mitochondrial volume density VV and numerical density NV. Damage of each mitochondrion was then scored on the basis of morphological characteristics: Type 1= normal morphology, Type 2= intact cristae but concave shapes with flattened center and bent extremities to form C-, U- or ring-shaped form, Type 3= marked disruption of cristae.
mDNA content
DNA was extracted from snap-frozen tissues by using Maxwell (Promega), according manufacturer protocols. mDNA was quantified by real time PCR techniques amplifying ND1 gene (left primer: 5’-GCTCCTTTAACCTCTCCACCC-3’; right primer: 5’-CGGTTGGTCTCTGCTAGTGT-3’). mDNA content was normalized to nDNA content, by amplification of β-actin (left primer: 5’-AGACTCCCCATCCCAAGACC-3’; right primer: 5’-ACCATGTCACACTGGGGAAG-3’) gene.
Gene expression analysis
Total mRNA was extracted from snap-frozen tissues by using Maxwell 16 LEV SimplyRNA (Promega), according to manufacturer protocols and was retrotranscribed by High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Hene expression was evaluated by RT-PCR with ad hoc-designed primers (Primer3, http://primer3.ut.ee/) for gene of interest and housekeeping gene (β-actin). The results were analyzed using the ΔΔCt method [37].
Western blot analysis
Cell pellets were lysed in the lysis buffer [38] added with protease and phosphatase inhibitor cocktail for 30 min on ice. Insoluble material was pelleted at 10,000×g for 15 min at 4°C, and the protein concentration was determined using a BioRad assay kit (BioRad, Hercules, CA, USA). Thirty μg of total cellular proteins were separated on SDS-PAGE and electro-transferred to nitrocellulose membrane (Merck Millipore, Burlington, MA, USA). Immuno-blotting was carried out with anti-OXPHOS (Abcam, 1:200), anti-cytc (Santa Cruz Biotechnology, 1:3000), anti-PGC1α (Abcam, 1:2000), anti-p62 (Cell Signaling, 1:1000), anti-LC3 (MBL, 1:1000), and anti-PINK1 (Santa Cruz Biotechnology, 1:200) primary antibodies, and peroxidase-labelled secondary antibodies (BioRad). Horseradish-peroxidase substrate (ECL Western Blotting Detection, Amersham-Life Science, Little Chalfont, UK) was added and the signal was revealed through an Odyssey Fc instrument (LI-COR, Lincoln, NE, USA).
Statistical analysis
All statistical analyses were done using Prism (V8, GraphPad, San Diego, CA, USA), and non-parametric Wilcoxon Mann-Whitney test.
Results
Electron microscopy studies of the mitochondria morphology in DDP-sensitive and -resistant ovarian cancer PDXs
Starting from DDP-sensitive PDXs, we obtained two DDP-resistant models through multiple cycles of in vivo DDP treatment and we showed that the DDP-resistance was associated with an increased oxidative metabolism [35] (Supplementary Figure 1). Given the importance of mitochondria in cellular metabolism, we further investigate the mitochondria at a structural level. Studies performed by transmission electron microscopy (TEM) were undertaken in the two different DDP resistant and their sensitive counterparts (MNHOC124 and MNHOC239, from now referred as #124R, #239R and #124S and #239S, respectively). We observed that both #124-R and #239-R PDXs showed a statistically significant lower number of mitochondria per µm3 than PDX-S tumors (0.7 vs 1.1 n/µ3 P=0.01, and 0.8 vs 0.9 n/µ3 P=0.01, in PDX #124 and PDX #239 respectively, Figure 1A), and a twice bigger mitochondrial volume (0.14 vs 0.06 µ3 P=0.02, and 0.12 vs 0.08 µ3 P=0.01 in #124 and #239 PDXs respectively, Figure 1B).
Figure 1.
Electron microscopy studies in #124 DDP-S (□) and -R (■), and #239 DDP-S (Mark 1) and -R (Mark 2). For each PDX histograms represent the number of mitochondria per µm3 (A), the mitochondrial volume (µm3, B), the percentage of normal mitochondria (Type I), % of Type II and Type III mitochondria (damaged), (C). Bars represents the mean ± SE of three different biological samples, done in triplicates. Statistical analysis was performed by t-test.
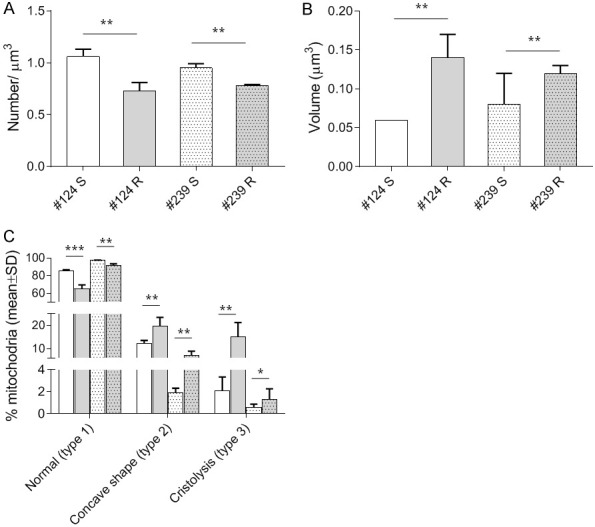
We further investigated the structure of mitochondria, in terms of normal (type 1) or damaged (type 2 or type 3 damage) mitochondria, as an indication of their function. We found that both PDXs-R showed a lower percentage of normal mitochondria (type 1, displayed in Figure 2A), respect to their S counterpart (65.3+4.3% vs 85.7+1.2%, P=0.001, and 91.6+2.0+20.3% vs 97.6+0.3% P=0.02 in #124 and #239 respectively, Figure 1C).
Figure 2.
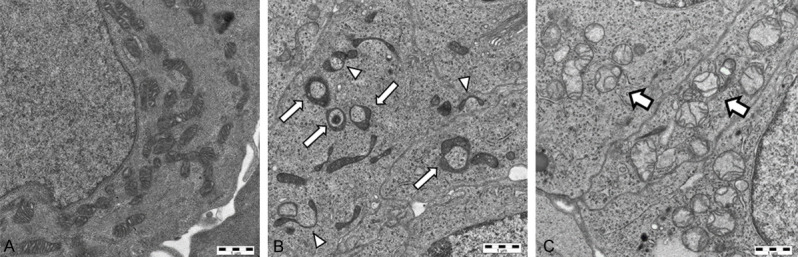
Representative figures captured by electron microscopy of mitochondrial Type 1 (normal, A), Type 2 (C-shaped (white arrowheads) and ring-shaped (white arrows), B), and Type 3 (cristolysis (white arrows), C) found in DDP-R PDX #124.
Altered mitochondria in PDX-R were higher in number and were of two types: mitochondria with abnormal concave shape (i.e. C-, U- or ring-shaped) (type 2) or with evident cristolysis (type 3). Type 2 mitochondria (displayed in Figure 2B) were found 12.2±1.4% and 2.1±1.23% as compared to 19.7±3.8% and 7.1±1.8%, respectively in sensitive and resistant #124 (P=0.09) and #239 models (P=0.02) (Figure 1C). Type 3 mitochondria, (displayed in Figure 2C) were higher in both PDX-R than in PDX-S (respectively in #124: 15.0±6.3% vs 2.1±1.2%, P=0.07; and in #239: 1.3±0.96% vs 0.6±0.27%, P=0.5) (Figure 1C).
Molecular investigations of mitochondrial markers
The increased number of damaged mitochondria observed in R-PDXs by TEM prompted us to study mitochondrial fitness at gene and protein level, to understand if the differences found in mitochondrial morphology might have a functional role.
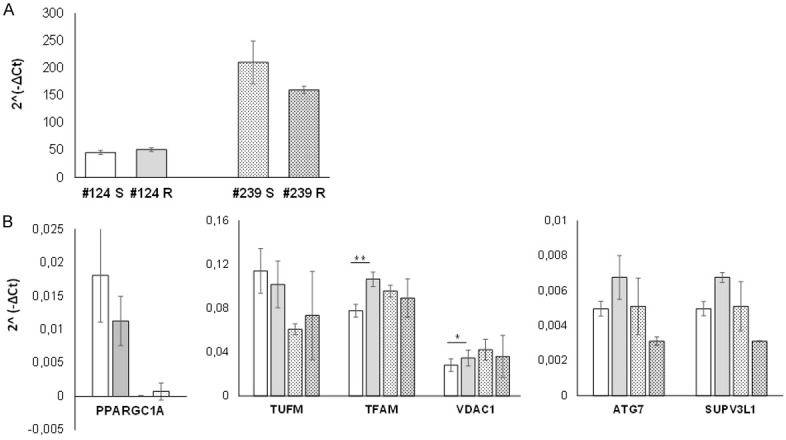
The differences found in the number and volume of mitochondria could be due to a different regulation of mitochondrial biogenesis and clearance. At first, we investigate the content of mitochondrial (mDNA) respect to nuclear DNA (nDNA), and as shown in Figure 3A, we found no significant differences in mDNA content between R and S models in both PDX models. Then, we investigated the expression of PPARGC1A gene, coding for the master regulator of mitochondrial biogenesis and mass, PGC1α, of genes coding for mitochondrial transcription factors (i.e. TUFM, TFAM), as well as for mitochondrial autophagy and fitness (i.e. ATG7, a regulator of mitophagy, and SUPV3L1, an helicase involved in mitochondrial RNA metabolism). Among genes involved in mitochondrial biogenesis, we found a statistically significant increase in the expression of TFAM (P=0.0012), and VDAC1 (P=0.047) genes only in #124-R vs #124 S PDXs. We found no statistical significant difference in genes involved in mitophagy and mitochondrial RNA metabolism (Figure 3B).
Figure 3.
A. Mitochondrial DNA content was calculated as target ND1 gene and the reference β-actin nuclear gene. B. The expression of the genes reported was evaluated by real time PCR. The bars represent the average of the ratio (calculated as 2^(-ΔCt)) ± SD of values obtained. All the data are the mean of three different biological replicates, done in triplicate [#124 DDP-S (□), -R (■), and #239 DDP-S (Mark 3) and -R (Mark 4)]. Statistical analysis was performed by Mann-Whitney test.
At protein level, the expression of all the OXPHOS complexes, cytochrome C (a protein essential in the mitochondrial electronic transport chain, and involved in apoptosis), and PGC1α proteins was similar in DDP-R and DDP-S in both PDXs, as reported in Figure 4A, arguing against a basal difference in mitochondrial function [35].
Figure 4.

Western blot analysis of oxidative phosphorylation system (OXPHOS), cytochrome c, PGC1α proteins (A), and autophagy and mitophagy related proteins p62, LC3-I and LC3-II, and PINK1 (B) in the different PDXs. The bars represent the mean ± SD of the ratio calculated for three biological replicates between densitometric signal of interest and control proteins.
One of the major mechanisms that controls mitochondrial fitness is the autophagic removal of damaged mitochondria [39]. As we observed an accumulation of damaged mitochondria in DDP-R PDXs, we investigated the basal level of autophagy, studying the expression of proteins involved in autophagy (i.e. p62, LC3 I-II) and in the control and regulation of mitophagy (i.e. PINK). No difference in the basal levels of p62 were found between S and R PDXs; higher levels of LC3-I were found in 124-R vs 124-S but, on the contrary, lower levels of LC3-I were observed in the resistant #239-R as compared to #239-S. LC3-II protein (the autophagosome-associated form) was barely detected in all the PDXs samples (Figure 4B). PINK1 levels were found upregulated in #124-R vs #124-S PDXs, while it was downregulated in #239-R as compared to #239-S (Figure 4B). In aggregate, no a clear-cut upregulation of autophagy could be demonstrated in the resistant PDXs.
Discussion
The use of DDP resistant models obtained after multiple in vivo treatments starting from DDP sensitive ones, represents a valid preclinical tool to study the development of resistance [40,41]. We have recently isolated DDP-resistant PDXs after in vivo DDP multiple treatments of mice bearing DDP-sensitive PDXs [35], reporting that resistance was associated to a rewiring of tumor metabolism, with a shift towards an increased oxidative metabolism. These data suggested a role of mitochondria in the development of DDP resistance, and prompted us to better investigate these organelles.
The present work shows that DDP-resistance is associated with an altered mitochondria morphology. In particular, by TEM we observed that DDP-R PDXs present a lower number mitochondria per µm3, and an increased volume.
In addition, an higher percentage of mitochondria with abnormal concave shapes (type 2) were observed in the DDP-R PDXs. While these structural changes, triggered by loss of mitochondrial membrane potential, are considered transient and easily reversible with no clear significance on mitochondrial function [42], mitochondria with abundant disrupted cristae (cristolysis, type 3) are unable to produce an adequate amount of ATP by oxidative phosphorylation [43]. Since cristolysis was significantly increased in the DDP-R PDXs and a similar two-three fold increase of all altered mitochondria was observed respectively in the two different resistant PDXs, we decided to search for markers of mitochondrial damage and dysfunction.
TEM analysis performed in 16 ovarian cancer and 18 ovarian normal tissue demonstrated an increase in mitochondrial number in cancer tissues, altered mitochondrial biogenesis and increased in mDNA content [44]. We found that mitochondria showing marked cristolysis (type 3) were present in both R-PDX, supporting the presence of damaged mitochondria in cancers [44]. However, the two fold increment in the percentage of type 3 mitochondria did not associate with modification of mDNA content. This could be due to the fact that the proportion of these damaged mitocondria is not sufficiently high. We cannot, however, exclude that mutations in mDNA, not altering the absolute mDNA content, could be present, and possibly related to the observed altered mitochondrial morphology as reported [45,46]. In line with our data, Zampieri et al. that show that platinum treatment causes a shift towards a more oxidative metabolism, but, on the contrary, a decrease in number of mitochondria after platinum treatment [47].
The expression of genes and proteins involved in mitochondrial biogenesis did not shown any difference (i.e. PPARGC1A), but we found an upregulation of some genes involved in mitochondrial function and fitness (i.e. TFAM, and VDAC1) in DDP-R vs DDP-S in one PDX model (#124). When we studied the role of autophagy, the most described pathway associated with a mitochondrial dysfunction, while an upregulation of the expression of ATG7 in 124-R vs 124-S was observed, all the other genes and proteins investigated were similarly expressed, arguing against a different basal level of authophagy. Zampieri et al. found that the resistance to platinum relied on mitophagy, and inhibition of autophagy could sensitize cells to DDP treatment [47]. In cancer cells, mitophagy protected from death in a PINK1-dependent manner and increased resistance to treatment [48]. However, we could not find any statistical significance difference in PINK1 expression level. While all these data suggest a similar basal level of autophagy and mitophagy, we cannot exclude that both these processes could be activated differentially in sensitive and resistant PDXs upon cellular stress (i.e. drug treatment).
In conclusion, we have here combined quantitative mitochondrial parameters and qualitative observation of a large number of mitochondria (more than 2000 mitochondria per experimental condition) to show that acquired in vivo resistance to DDP is associated with morphological alterations of tumor mitochondria (i.e. lower number, higher mitochondrial mass and an higher percentage of damaged mitochondria). The observed alterations are unlike to be the result of a direct drug effect as were observed in tumor samples that were not under drug selection since six passages, but still resistant to the drug [35]. To our knowledge, this is the first reporting of mitochondrial ultrastructural alteration in ovarian cancer with acquired DDP resistance. This finding unexpectedly did not correlate with any of the mechanisms we investigated and this lack of correlation could be due to absolute number of severely damaged mitochondria with cristolysis (at maximum reaching 15% of the total). While it is possible that these alterations might not be associated with a global altered mitochondrial function, these were present in two different PDX models undergoing the same type of in vivo DDP-induced resistance, further supporting the involvement of mitochondrial alteration in DDP resistance.
Acknowledgements
The generous contribution of AIRC (The Italian Association for Cancer Research) is gratefully acknowledged. The research was supported by a grant from the The Italian Association for Cancer Research (Giovanna Damia IG 19797).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Christie EL, Bowtell DDL. Acquired chemotherapy resistance in ovarian cancer. Ann Oncol. 2017;28:viii13–viii15. doi: 10.1093/annonc/mdx446. [DOI] [PubMed] [Google Scholar]
- 2.van Zyl B, Tang D, Bowden NA. Biomarkers of platinum resistance in ovarian cancer: what can we use to improve treatment. Endocr Relat Cancer. 2018;25:R303–R318. doi: 10.1530/ERC-17-0336. [DOI] [PubMed] [Google Scholar]
- 3.Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, Lord CJ, Lengyel E, McNeish IA, Menon U, Mills GB, Nephew KP, Oza AM, Sood AK, Stronach EA, Walczak H, Bowtell DD, Balkwill FR. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719–725. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowtell DD, Böhm S, Ahmed AA, Aspuria PJ, Bast RC, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, Brenton JD, Chiappinelli KB, Martins FC, Coukos G, Drapkin R, Edmondson R, Fotopoulou C, Gabra H, Galon J, Gourley C, Heong V, Huntsman DG, Iwanicki M, Karlan BY, Kaye A, Lengyel E, Levine DA, Lu KH, McNeish IA, Menon U, Narod SA, Nelson BH, Nephew KP, Pharoah P, Powell DJ, Ramos P, Romero IL, Scott CL, Sood AK, Stronach EA, Balkwill FR. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–679. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet (London, England) 2019;393:1240–1253. doi: 10.1016/S0140-6736(18)32552-2. [DOI] [PubMed] [Google Scholar]
- 6.Bratasz A, Weir NM, Parinandi NL, Zweier JL, Sridhar R, Ignarro LJ, Kuppusamy P. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc Natl Acad Sci U S A. 2006;103:3914–3919. doi: 10.1073/pnas.0511250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodisman J, Hagrman D, Tacka KA, Souid AK. Analysis of cytotoxicities of platinum compounds. Cancer Chemother Pharmacol. 2006;57:257–267. doi: 10.1007/s00280-005-0041-4. [DOI] [PubMed] [Google Scholar]
- 9.Damia G, Broggini M. Platinum resistance in ovarian cancer: role of DNA repair. Cancers. 2019;11:119. doi: 10.3390/cancers11010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavill R, Kamburov A, Ellis JK, Athersuch TJ, Blagrove MS, Herwig R, Ebbels TM, Keun HC. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput Biol. 2011;7:e1001113. doi: 10.1371/journal.pcbi.1001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alirol E, Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene. 2006;25:4706–4716. doi: 10.1038/sj.onc.1209600. [DOI] [PubMed] [Google Scholar]
- 14.Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer. 2017;3:857–870. doi: 10.1016/j.trecan.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maycotte P, Marín-Hernández A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortés-Hernández P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39:1010428317698391. doi: 10.1177/1010428317698391. [DOI] [PubMed] [Google Scholar]
- 16.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in gliomas and their vascular microenvironment. Biochim Biophys Acta. 2011;1807:602–8. doi: 10.1016/j.bbabio.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Daghistani HM, Rajab BS, Kitmitto A. Three-dimensional electron microscopy techniques for unravelling mitochondrial dysfunction in heart failure and identification of new pharmacological targets. Br J Pharmacol. 2019;176:4340–4359. doi: 10.1111/bph.14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamogashira T, Hayashi K, Fujimoto C, Iwasaki S, Yamasoba T. Functionally and morphologically damaged mitochondria observed in auditory cells under senescence-inducing stress. NPJ Aging Mech Dis. 2017;3:2. doi: 10.1038/s41514-017-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rybka V, Suzuki YJ, Gavrish AS, Dibrova VA, Gychka SG, Shults NV. Transmission electron microscopy study of mitochondria in aging brain synapses. Antioxidants. 2019;8:171. doi: 10.3390/antiox8060171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verschoor ML, Ungard R, Harbottle A, Jakupciak JP, Parr RL, Singh G. Mitochondria and cancer: past, present, and future. BioMed Research International. 2013;2013:612369. doi: 10.1155/2013/612369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:10. doi: 10.1186/2049-3002-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trend Cell Biol. 2016;26:655–667. doi: 10.1016/j.tcb.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Liou GY, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325–2336. doi: 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, Rajeeve V, Cardaci S, Foster S, Yun H, Cutillas P, Warren A, Gnanapragasam V, Gottlieb E, Franze K, Huntly B, Maher ER, Maxwell PH, Saez-Rodriguez J, Frezza C. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544–547. doi: 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi JI. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 27.Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B, Sonveaux P. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 28.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosc C, Selak MA, Sarry JE. Resistance is futile: targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab. 2017;26:705–707. doi: 10.1016/j.cmet.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochimica Et Biophysica Acta Bioenergetics. 2017;1858:686–699. doi: 10.1016/j.bbabio.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 32.Pereira L, Soares P, Máximo V, Samuels DC. Somatic mitochondrial DNA mutations in cancer escape purifying selection and high pathogenicity mutations lead to the oncocytic phenotype: pathogenicity analysis of reported somatic mtDNA mutations in tumors. BMC Cancer. 2012;12:53. doi: 10.1186/1471-2407-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, Seo JH, Kossenkov AV, Michalek RD, Schultz DC, Bosari S, Languino LR, Altieri DC. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst. 2015;107:dju502. doi: 10.1093/jnci/dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ricci F, Brunelli L, Affatato R, Chilà R, Verza M, Indraccolo S, Falcetta F, Fratelli M, Fruscio R, Pastorelli R, Damia G. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther Adv Med Oncol. 2019;11:1758835919839543. doi: 10.1177/1758835919839543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ricci F, Fratelli M, Guffanti F, Porcu L, Spriano F, Dell’Anna T, Fruscio R, Damia G. Patient-derived ovarian cancer xenografts re-growing after a cisplatinum treatment are less responsive to a second drug re-challenge: a new experimental setting to study response to therapy. Oncotarget. 2017;8:7441–7451. doi: 10.18632/oncotarget.7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Caiola E, Brunelli L, Marabese M, Broggini M, Lupi M, Pastorelli R. Different metabolic responses to PI3K inhibition in NSCLC cells harboring wild-type and G12C mutant KRAS. Oncotarget. 2016;7:51462–51472. doi: 10.18632/oncotarget.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ornatowski W, Lu Q, Yegambaram M, Garcia AE, Zemskov EA, Maltepe E, Fineman JR, Wang T, Black SM. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020;36:101679. doi: 10.1016/j.redox.2020.101679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Page C, Amuzu S, Rahimi K, Gotlieb W, Ragoussis J, Tonin PN. Lessons learned from understanding chemotherapy resistance in epithelial tubo-ovarian carcinoma from BRCA1 and BRCA2 mutation carriers. Semin Cancer Biol. 2020 doi: 10.1016/j.semcancer.2020.08.005. S1044579X20301772. [DOI] [PubMed] [Google Scholar]
- 41.Yu K, Chen B, Aran D, Charalel J, Yau C, Wolf DM, van ‘t Veer LJ, Butte AJ, Goldstein T, Sirota M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat Commun. 2019;10:3574. doi: 10.1038/s41467-019-11415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding WX, Li M, Biazik JM, Morgan DG, Guo F, Ni HM, Goheen M, Eskelinen EL, Yin XM. Electron microscopic analysis of a spherical mitochondrial structure. J Biol Chem. 2012;287:42373–42378. doi: 10.1074/jbc.M112.413674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in human cancer. Int J Biochem Cell Biol. 2009;41:2062–2068. doi: 10.1016/j.biocel.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Signorile A, De Rasmo D, Cormio A, Musicco C, Rossi R, Fortarezza F, Palese LL, Loizzi V, Resta L, Scillitani G, Cicinelli E, Simonetti F, Ferretta A, Russo S, Tufaro A, Cormio G. Human ovarian cancer tissue exhibits increase of mitochondrial biogenesis and cristae remodeling. Cancers. 2019;11:1350. doi: 10.3390/cancers11091350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGeehan RE, Cockram LA, Littlewood DTJ, Keatley K, Eccles DM, An Q. Deep sequencing reveals the mitochondrial DNA variation landscapes of breast-to-brain metastasis blood samples. Mitochondrial DNA A DNA Mapp Seq Anal. 2018;29:703–713. doi: 10.1080/24701394.2017.1350950. [DOI] [PubMed] [Google Scholar]
- 46.Peralta S, Goffart S, Williams SL, Diaz F, Garcia S, Nissanka N, Area-Gomez E, Pohjoismäki J, Moraes CT. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J Cell Sci. 2018;131:jcs217075. doi: 10.1242/jcs.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zampieri LX, Grasso D, Bouzin C, Brusa D, Rossignol R, Sonveaux P. Mitochondria participate in chemoresistance to cisplatin in human ovarian cancer cells. Mol Cancer Res. 2020;18:1379–1391. doi: 10.1158/1541-7786.MCR-19-1145. [DOI] [PubMed] [Google Scholar]
- 48.Villa E, Proïcs E, Rubio-Patiño C, Obba S, Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragón L, Riley JS, Marchetti S, Verhoeyen E, Tait SWG, Ricci JE. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 2017;20:2846–2859. doi: 10.1016/j.celrep.2017.08.087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.