

Abstract
HPV-induced cervical cancer is one of the prevalent gynecological cancers world-wide. In the present study, we determined the efficacy of Minnelide, a prodrug which is converted to its active form (Triptolide) in vivo against cervical cancer cells. Our studies show that Triptolide inhibited HPV-16 and HPV-18 positive cells at nanomolar concentrations. Tumor cells treated with Triptolide failed to grow in 3-D cultures in a concentration-dependent manner. Triptolide markedly reduced E6 and E7 transcript levels. Further studies revealed that exposure to Triptolide increased the levels of p53 and pRb. As a consequence, Caspase-3/7 activation and apoptosis was induced in cervical cancer cells by Triptolide. Subsequently, we evaluated the efficacy of Minnelide in xenotransplantation models of cervical cancer. Minnelide at very low doses effectively inhibited the growth of established cervical cancers in all the three animal models tested. Furthermore, Minnelide treatment was more effective when combined with platinum-based chemotherapy. These studies show that Minnelide can be used to inhibit the growth of cervical cancer.
Keywords: Cervical cancer, HPV, Minnelide, apoptosis
Introduction
About 527600 women are diagnosed with cervical cancer world-wide annually [1]. In the United States, 13240 new cases are reported in 2018 (NCI Cancer Statistics; <seer.cancer.gov>) of which 4,170 women died of cervical cancer. Etiology of cervical cancer is strongly associated with human papillomavirus (HPV) infection. More than 120 HPV types have been identified to date. These are broadly classified as low risk HPV and high risk HPV. There are about fifteen different high risk virus types of which HPV16 and HPV18 account for more than 70% cervical cancer cases [2,3]. Virus infection of cervical squamous epithelium leads to cellular transformation. During the early stages of infection, the virus remains episomal and later integrates into the host genome. During integration, some of the early genes (E2, E5) and late gene, such as L2 are deleted. Loss of E2 relieves repression and thereby activates the expression of HPV encoded oncoproteins, E6 and E7. E6 and E7 interfere with p53 and pRb-regulated molecular pathways respectively [4]. E6 binds to E6AP, an E3 Ubiquitin ligase and activates ubiquitination of p53 tumor suppressor protein. Polyubiquitination of p53 is then targeted for proteasomal degradation. Reduced levels of p53 affects DNA-repair, reduced apoptosis and promotes cell survival and proliferation. The second oncoprotein, E7, binds to another tumor suppressor protein, retinoblastoma protein (pRb) and structurally related proteins p107 and p130 having a ‘pocket domain’. pRb has multiple roles in regulating cell cycle progression. One of the primary functions of pRb is sequestration of E2F, a heterodimeric transcription factor. Binding of pRb with E2F prevents it from transactivating target genes. pRb is dynamically phosphorylated at several sites by cyclin dependent kinases (CDK) during cell cycle progression. Hyperphosphorylated pRb fails to bind E2F. E2F then transactivates target genes including Cyclin A and Cyclin E which are necessary for cell cycle progression [5]. Recent studies have shown that HPV infection can also interfere with oxidative stress pathways and modulate proteostasis [6].
HPV infection is prevented by vaccination (Gardasil and Cervarix). However, both premalignant lesions and established cancers are not affected by HPV vaccines. Furthermore, cervical cancers associated with rare types of HPV strains escape easily from current vaccine treatments [7]. In the present studies, we investigated the efficacy of a pro-drug, Minnelide, against cervical cancer growth. Minnelide is a semi-synthetic drug derived from the natural compound, Triptolide, a diterpene triepoxide, isolated from the Chinese herb, Tripterygium wilfordii (Thunder God Vine). Triptolide is not water soluble. Addition of a phosphate group to Triptolide (Minnelide) made it water soluble and improved its bioavailability. Minnelide is non-toxic in vitro, but is activated in vivo by phosphatases to release bioactive Triptolide. Our studies show that Triptolide treatment inhibits cervical cancer cell proliferation, migration, colony formation in vitro at nanomolar concentration. Both HPV16 and HPV18 positive cancer cells are inhibited by Triptolide. In xenograft models of cervical cancer, Minnelide treatment was very effective in inhibiting tumor growth either alone or in combination with Carboplatin. Mechanistic studies showed that Triptolide treatment affected the levels of p53 and pRb oncoproteins. These studies suggest that Minnelide can be useful in treating cervical cancers.
Materials and methods
Cell lines and reagents
Cervical Cancer cells (CaSki, SiHa and ME-180) were purchased from American Type Culture Collection. Triptolide and Carboplatin were obtained from TCI America (Thermo Fisher) and Sigma Chemicals respectively. Triptolide was dissolved in DMSO and then diluted in tissue culture medium as required. Carboplatin was dissolved in PBS. ME-180 cells (HPV-68 homology) established from metastatic site (omentum) were cultured in McCoy’s 5A Medium containing 10% Fetal Bovine Serum (FBS), 1% penicillin-streptomycin, and 1% L-glutamate. CaSki cells (HPV-16 and HPV-18) originated from a metastatic site (small intestine) and were cultured in RPMI 1640 (Gibco) containing 10% FBS (Gibco), 1% penicillin-streptomycin (Sigma), and 1% L-glutamate (sigma). SiHa cells (HPV-16) were from primary tumor tissue and were grown in DMEM (Gibco) medium containing 10% FBS and antibiotics.
Cell viability, proliferation, migration, ROS measurement and apoptosis
Cell viability was determined using Dojindo Cell Counting Kit-8 (Dojindo Molecular Technologies) following the protocol provided by the manufacturer. Treatment-induced changes in cell proliferation and migration, were determined using the RTCA (Real-Time Cell Analyzer) Instrument (Roche Applied Science & ACEA Biosciences) as previously described [8]. Triptolide-induced apoptosis was determined in real-time using IncuCyte following manufacturers recommendations. NucLight Red Live-Cell Labeling reagent was used to directly identify viable tumor cells. Apoptotic cells were identified by active Caspase3/7. Cells were simultaneously loaded with IncuCyte® Caspase3/7 apoptosis reagent which contains a DNA-binding dye (NucView-488) which is released from the Caspase3/7 recognition motif, DEVD upon cleavage. This enables simultaneous detection of live cells (Red) and cells undergoing apoptosis (green fluorescence). Triptolide-induced ROS levels were determined by DCFDA (Invitrogen) as per Manufacturer’s protocol. In brief, 5000 cells were seeded in 96-well plates and treated with 50 nM Triptolide for 6 hours. After washings, DCFDA was added at a concentration of 10 uM and incubated for 30 min. Fluorescence was measured in SpectraMax i3X (Molecular Devices) microplate reader using 485/525 nm filter sets.
Spheroid formation
ME-180 cells were cultured in growth medium until 75% confluency, trypsinized, and counted using a Cellometer Mini Bright Field Cell Counter (Nexcelom Biosciences). Cells were then plated using the GravityPlus Hanging Drop System (InSphero Biomimetic Microtissue Technology). Five hundred cells in 40 uL of medium were plated in each well of the Hanging Drop System. After 24 hours of incubation at 37°C, cells were treated with Triptolide. After treatment, cells were incubated at 37°C for 3 days to allow spheroid formation. Spheroids were then dropped from the GravityPLUS Plate to the GravityTRAP plate. Once in the GravityTRAP plate, size and number of spheroids were determined using a Zoe Fluorescent Imager (Bio-Rad).
Spheroid formation of CaSki cells was analyzed using an alternate method. One thousand cells were plated in 2 mL of growth medium into a Corning Costar Ultra-Low Attachment 6-well plate (Sigma-Aldrich). The cells were then incubated for 24 hours at 37°C and subsequently treated with Triptolide at the concentrations indicated. Spheroids were allowed to grow for 1 week at 37°C. Growth of spheroids was determined by imaging the plate using a ZOE Fluorescent Imager (Bio-Rad).
Western blot
ME-180 and CaSki cells were grown in six 10 cm2 plates. Cultures were treated with Triptolide for 48 hours. After washing with PBS, 150 uL of Radio Immunoprecipitation Assay Buffer (Sigma-Aldrich), containing protease and phosphatase inhibitors, was added directly on top of the cells. Cells were scraped and pipetted into a 1.5 mL microfuge tube and incubated on ice for 2 hours, and cleared by centrifugation for 15 minutes at 16,000 rpm. Protein concentration was determined using a Peirce BCA Protein Assay Kit (Thermo Fisher). Following antibodies were used for western blots. Polyclonal rabbit Caspase-3 antibody #9662 (Cell Signaling Technology, Inc, Danvers MA), anti-mouse β-actin antibody, #3700, rabbit anti-human p53 antibody #9282 (Cell Signaling Technology, Inc, Danvers MA), mouse monoclonal anti-pRb #9309, rabbit polyclonal phospho-pRb #9308, and mouse monoclonal p21 Waf1/Cip1.
Reverse Phase Protein Array (RPPA) analysis
CaSki and ME-180 cells were treated with 50 nM Triptolide for 48 hours and then harvested. Cell lysates were prepared and five serial dilutions (1:2) were prepared and analyzed by MD Anderson Cancer Center, core facility. A total of 304 unique, validated antibodies were used to perform RPPA analysis. Heat maps were developed by the MD Anderson Cancer Center Department of Bioinformatics and Computational Biology, In Silico Solutions, Santeon and SRA International. All values were normalized and transformed to linear values for comparison. From the 304 spots, images of p53, pRb, Phospho pRb, and Cleaved Caspase-7 are presented.
Real-time PCR
CaSki and SiHa cells were grown in 100 mm petri dishes and sub-confluent cultures were treated with 50 nM of Triptolide. After 24-hour incubation at 37°C, cells were washed with ice cold PBS twice. Trizol method was used to isolate total RNA. cDNA was synthesized using Roche Applied Biosciences kit. E6, and E7 primers were obtained from Sigma-Aldrich and real-time quantitative PCR (RT-PCR) was performed using Light-cycler SYBR-Green 480.
Tumor growth studies
Three xenograft tumor models were used to evaluate the efficacy of Minnelide. All animal studies were carried out under approved protocols and direct supervision from veterinary staff in accordance with the regulations of the University of Miami. In one of the models, SiHa cancer cells were injected s.c. and tumors were established for 12 days before treatment with Minnelide (0.4 mg/kg, daily for 21 days). The other two tumor models used ME-180 and CaSki cells. Five million cells in 100 uL of Geltrex suspension were injected subcutaneously into the right flank of female, athymic, nude mice. Tumors (ME-180 and CaSki) were allowed to establish for 7 days and then randomized. Groups of ten mice were then treated i.p. with, Minnelide, Carboplatin, or Minnelide plus Carboplatin combination. Control mice were treated daily with injections of 200 uL of sterile, normal saline. Minnelide was injected daily at a dose of 0.2 mg/kg while Carboplatin was injected twice weekly at a dose of 20 mg/kg. The combination group was treated with 0.2 mg/kg daily injections of Minnelide along with twice weekly 20 mg/kg injections of Carboplatin. Mice were treated for five weeks. Tumor growth was monitored by caliper measurements. Mice were euthanized at the end of the experiment and tumor size along with weight were recorded. Half of the tumors were then fixed in neutral formalin for histopathology while the other half was snap frozen in liquid nitrogen to prepare frozen sections.
Results
Human cervical cancer cell lines were initially investigated for their sensitivity to Triptolide in vitro. Since Minnelide needs to be converted to its active form, Triptolide, in vivo by phosphatases, Triptolide was used for in vitro studies. First, we determined the effects of Triptolide on cervical cancer cells in real-time proliferation assays. Electrical impedance (Cell index) measurements were recorded every 15 min. Triptolide-treated CaSki cells showed an increase in Cell index during the first 12 hours and then decreased to near zero levels by 48 hours. The transient increase in Cell index is due to increased cell attachment and spreading. CaSki cells were highly sensitive and even 50 nM concentration, Triptolide completely inhibited cell proliferation (Figure 1A). Data in Figure 1B show concentration-dependent inhibition in Cell index of ME-180 cells. Both treated and control cultures showed progressive increase in Cell index up to 6 hours. Triptolide treatment started to show reduction in Cell index (proliferation) from this point onwards. At the end of the experiment, control cultures showed a Cell index of 1.5 and at 50 nM Cell index dropped to 0.6. Higher concentrations such as 100 and 200 nM Triptolide reduced the Cell index to 0.15. Similarly, Triptolide was also effective against a third cell line, SiHa (Supplementary Figure 1). These studies showed cervical cancer cells are sensitive to Triptolide. Then we determined the effect of Triptolide treatment on 3D-growth (anchorage independent) of cervical cancer cells. CaSki cells were grown in the presence of Triptolide in ultra-low attachment plates. Even 25 nM Triptolide treatment completely inhibited spheroid formation and clonogenic growth of CaSki cells in this assay system (Figure 1C). Parallel studies showed that Triptolide treatment completely inhibited the spheroid formation of ME-180 cells in hanging drop cultures (Figure 1D). ME-180 cells showed marked decrease in the size of spheroids at 50 and 100 nM Triptolide. Triptolide treatment also inhibited cell migration in real-time (Figure 1E and 1F).
Figure 1.

Triptolide inhibits cervical cancer cell proliferation and clonogeneic growth in vitro. (A and B) show real time cell proliferation in the presence of varying concentrations of Triptolide. (A) CaSki, and (B), ME-180. Cell index refers to mean cell density (arbitrary unit) from quadruplicate wells in xCELLigence RTCA using E-plates. (C) shows representative data from clonogeneic growth of CaSki cells seeded in low adherence tissue culture petri dishes. Images were obtained on day 7. (D) shows Me-180 cells seeded in hanging drops. Photomicrographs show images acquired on day 3 after seeding. (E) (CaSki) and (F) (SiHa) show real time cell migration in xCELLigence RTCA using CIM plates. Trp refers to Triptolide.
Triptolide inhibits E6 and E7 transcript levels of HPV and induces apoptosis of cervical cancer cells
Real-time PCR was used to determine the transcript levels of the two major oncoproteins of HPV. CaSki cells which are positive for both HPV16 and HPV18 showed more than 43-fold and 60-fold reduction in transcript levels of E6 and E7 respectively after Triptolide treatment (Figure 2A). Similarly, when SiHa cells (HPV-16) were treated with Triptolide, there was a 79-fold reduction in E6 transcript levels and 75-fold inhibition in E7 transcripts (Figure 2B). Next we determined the consequence of reduced E6 and E7 expression on the apoptotic rate of Cervical cancer cells. Two independent series of experiments were carried out. In one series, TUNEL assay was used to visually record changes in apoptotic cells. Data in Figure 2C show that both CaSki and ME-180 cells treated with Triptolide lead to apoptosis of 62 and 82% (Figure 2D) of cells respectively. In a second set of experiments, kinetics of apoptosis was recorded in real-time using IncuCyte system (Figure 2E). Control SiHa cultures did not show significant cell death during the 48 hours of observation. At 25 nM concentration, Triptolide induced 50% cell death (apoptosis) by 44 hour of continuous exposure to the drug. Triptolide accelerated the rate of apoptosis in a concentration-dependent manner. It took 32 and 30 hours to induce 50% cell death by 50 and 100 nM Triptolide respectively. Representative images of apoptotic cells (green) and viable cells (red) at the beginning of the experiment and at 36 hours after exposure to Triptolide is shown in Figure 2F. Only a few viable cells are seen in the cultures treated with 50 and 100 nM Triptolide. Similarly, ME-180 cells treated with Triptolide also showed a concentration-dependent induction of Caspase3/7 activation (apoptosis) in IncuCyte system (Supplementary Figure 3, Supplementary Videos 1, 2, 3 and 4). Apoptosis also coincided with induction of ROS. Cells treated with Triptolide (10-80 nM) for a short period of time (6 hr) showed progressive increase in ROS levels in SiHA and CaSki cells whereas, Me-180 cells did show concentration dependent changes in ROS (Supplementary Figure 2).
Figure 2.

Triptolide treatment inhibits HPV E6, E7 expression and induces apoptotic cell death. (A) (CaSki) and (B) (SiHa) show the relative levels of E6 and E7 mRNA following treatment with 50 nM Triptolide for 24 hours. mRNA expression was normalized to 18S rRNA. (C) shows representative images of TUNEL assay. Apoptotic cells are labeled red. (D) shows the ratio of apoptotic cells to total number of cells/field. Images were taken 48 hours after treatment with 50 nM of Triptolide. Ten random fields were chosen for image analyses. (E) shows the kinetics (real-time) of concentration dependent Triptolide-induced apoptosis in SiHa cells as determined by InCucyte. Number of apoptotic cells per well was measured using ZOOM software. (F) shows the representative images of control and treated cells before and 36 hours after treatment with Triptolide (Trp). Live cells are labeled with NucRed reagent from InCucyte, and apoptotic cells with active Caspase3/7 are identified by green fluorescence emitted by NucView-488, DNA binding dye which was released from DEVD peptide substrate for Caspase3/7 enzyme.
Since E6 and E7 target tumor suppressor proteins p53 and pRb respectively, we then investigated the effect of Triptolide on the levels of tumor suppressor proteins. Western blot analyses of CaSki cells (Figure 3A) treated with Triptolide showed total pRb and phosphorylated pRb levels significantly increased in treated cells (3 and 10-fold respectively). Quantitation of images from three independent experiments are shown in Figure 3B. Increase in pRb and phosphorylated pRb were statistically significant (P=0.029 and P=0.046 respectively). Concurrently, cleaved caspase-3 levels increased by 5-fold in the Triptolide treated cells. Interestingly, Triptolide treatment decreased (P=0.051) p53 levels in CaSki cells (Figure 3A). In contrast to CaSki cells, ME-180 cells treated with Triptolide showed increased levels of p53. About 70-fold increase in p53 levels were seen in Triptolide treated cells (Figure 3C and 3D). However, there was no change in p21, one of the downstream targets of p53. In parallel, the pRb levels increased by 66% in Triptolide treated cells. Phosphorylated pRb levels showed marginal increase which was not statistically significant (P=0.2). Cleaved Caspase-3 did not show significant increase in ME-180 cells. To further confirm these results, Reverse Phase Protein Array (RPPA) analyses were carried out. A total of 304 cellular proteins including phosphorylated proteins were investigated. Data in Figure 3E show heat map of selected markers related to p53 and pRb. These studies showed Triptolide treatment increased the levels of p53 in both the cell lines. pRb increased in CaSki cells after Triptolide treatment whereas, ME-180 cells showed no significant change. Function of pRb is regulated by phosphorylation. Therefore, phosphorylated Serine 807 and Serine 811 at the C-terminal domain pRb was studied. Results showed pRb was highly phosphorylated even in HPV-infected control cells. Triptolide treatment only marginally increased phosphorylated pRb. Interestingly, cleaved, executor Caspase-7 was differentially increased by Triptolide treatment only in the ME-180 cells. These studies show that Triptolide induces apoptosis in cervical cancer cells by differentially modulating p53 and phosphorylated pRb levels to either activate Caspase-3 or Caspase-7.
Figure 3.

Triptolide-induced changes in p53 and pRb proteins. (A and C) show protein levels of p53, p21, pRb, phosphorylated pRb, and Cleaved Caspase3 in control and Triptolide treated cells. Cell lysates were prepared following 48 hours of 50 nM of Triptolide treatment. (A) CaSki; (C) ME-180. (B and D) show the quantification of the changes from three independent Western blots following Triptolide treatment. (B) CaSki, (D) ME-180. Statistically significant differences are denoted by asterisks. (E) Reverse Phase Protein Array analysis (RPPA) of control and Triptolide treated cultures is shown as heatmap. Lanes 1 and 2 show cell lysates from CaSki cells and lanes 3 and 4 show cell lysates from ME-180 cells. Lanes 1 and 3 are control and lanes 2 and 4 are Triptolide treated samples. (F and G) show histogram of fold-changes in protein levels as determined by RPPA analyses. (F) CaSki and (G), ME-180 cells.
Inhibition of cervical cancer growth by Minnelide as a monotherapy
For in vivo studies, Minnelide was used. Female, athymic nude mice were subcutaneously injected with SiHa cells. Tumors were allowed to establish for 12 days and then treated with Minnelide at a dose of 0.4 mg/kg daily for 21 days. Control group of mice received intraperitoneal injections of sterile saline under similar conditions. Tumor growth was followed for 48 days (Figure 4A). As early as one week into treatment with Minnelide, there was about a two-fold reduction in tumor volume (78 mm3 Minnelide group Vs 136 mm3 control group). Suppression of tumor growth continued throughout the experiment. Tumor volume did not change until Day 35 (62 mm3) in the treated animals while, the control group showed a mean tumor volume of 209 mm3 on day 35. Tumors started to grow albeit at a slower pace after the termination of Minnelide treatment. There was a 55 % reduction in tumor volume at the end of the experiment. These results were further validated by determining the wet weight of the resected tumors (Figure 4B). Control animals showed a mean tumor weight of 0.473 g and Minnelide treatment resulted in 56% reduction in tumor weight (mean weight 0.197 g).
Figure 4.
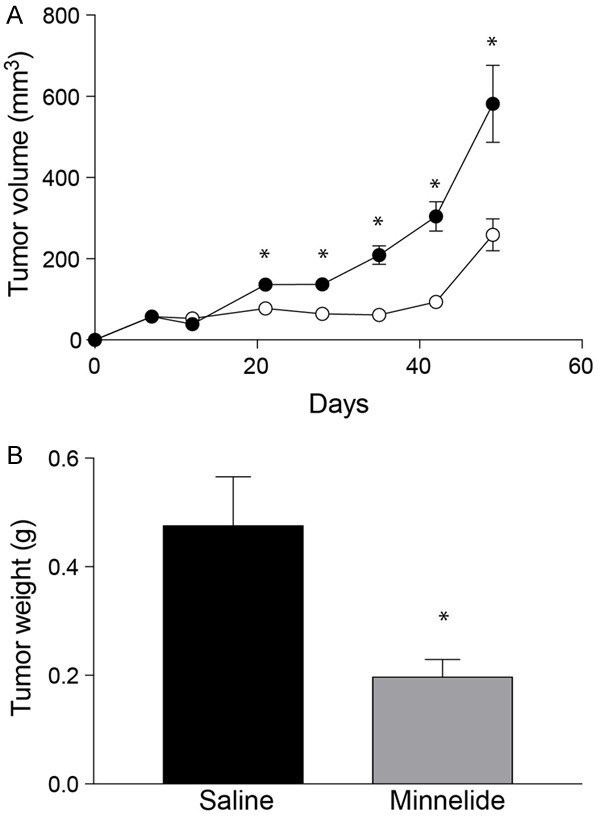
Inhibition of Cervical cancer growth by Minnelide monotherapy. (A) shows the effect of Minnelide as a monotherapy in treating subcutaneous tumors established in female, athymic mice. SiHa cells were injected into the right flank and 0.4 mg/kg of Minnelide was injected daily starting from day 12 after tumor cell implantation. Treatment was continued for 21 days. Tumor volume was measured by calipers every week. Closed circles, control and open circles, minnelide-treated group. Open circles, Control and closed circles minnelide treated group. (B) effects of Minnelide on SiHa tumor weight at the end of experiment. Statistically significant differences are denoted by asterisks.
After establishing the efficacy of Minnelide as a monotherapy, we determined potential synergy between an established chemotherapeutic agent, Carboplatin and Minnelide as a combination therapy. These studies were carried out in two other Cervical cancer cell lines, CaSki and ME-180. Data in Figure 5A show real-time changes in CaSki cell proliferation (Cell Index) when treated with 50 nM Triptolide either alone or in combination with Carboplatin (50 mM). Both control and treated cultures showed a steady increase in Cell Index until 15 hours after seeding. At the low concentration tested, Carboplatin did not inhibit CaSki cells. IC50 of CaSki cells is around 200 uM of Carboplatin (data not shown). Triptolide was very effective in inhibiting CaSki cell proliferation. Combination of Carboplatin and Triptolide showed better inhibition of cell proliferation. Data on Figure 5B show effect of combination treatment on ME-180 cells. Control ME-180 cultures grew steadily with an increase in Cell index over a period of 36 hours (Figure 5B). Carboplatin treated cultures showed plateauing Cell index values after 20 hours of treatment and started to decrease progressively after that. Triptolide treated cultures exhibited attenuated increase in Cell index as early as 5 hours into treatment. Triptolide was more effective in reducing ME-180 cell proliferation when compared to Carboplatin. A combination of both Triptolide and Carboplatin showed greater inhibition of ME-180 cells. Cell index values started to decrease only after 24 hours of treatment with either Carboplatin or Triptolide. In contrast, combination therapy decreased Cell index as early as 7-hours after treatment. At the end of the experiment, combination therapy resulted in more than additive inhibition in cell growth.
Figure 5.
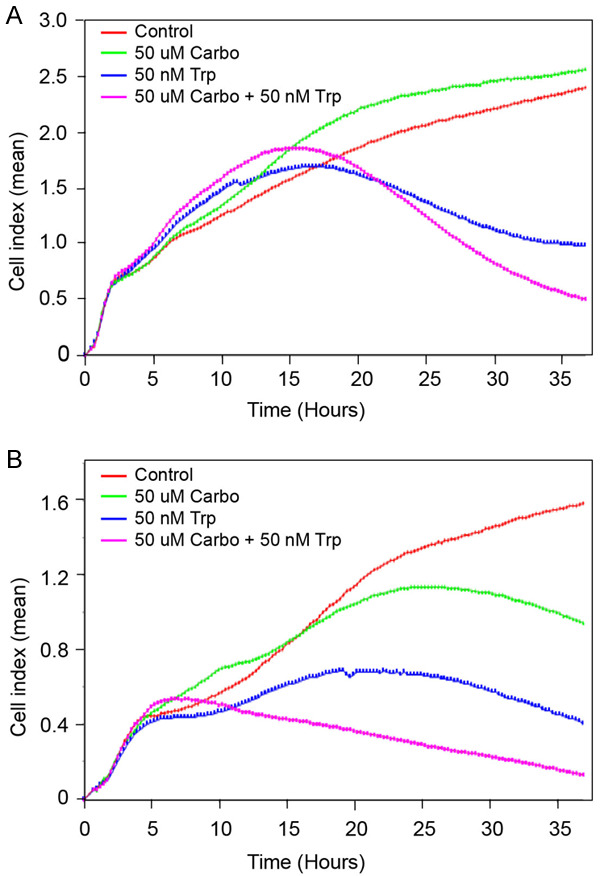
Enhanced inhibition of Cervical cancer cells in vitro by combination treatment with Triptolide and Carboplatin. Changes in Cell Index (real-time cell proliferation) in the presence of Triptolide, Carboplatin, and a combination of Triptolide and Carboplatin are shown. (A) CaSki, (B) ME-180. Cell index refers to mean values (arbitrary unit) from quadruplicate wells.
The encouraging results from in vitro studies led to evaluation of combination therapy in vivo. Data in Figure 6A show treatment-related changes in the growth of CaSki tumors. Control mice showed progressive tumor growth reaching a mean tumor volume of 2000 mm3 by day 42. Carboplatin treatment showed statistically significant inhibition of tumor growth. Differences in growth rate was visible by two weeks into treatment. At the end of the experiment, Carboplatin-treated mice showed a 75% reduction in tumor volume when compared to control animals. Minnelide at 0.2 mg/kg dose was more effective than Carboplatin in inhibiting CaSki tumors. Changes in tumor growth was visible even after one week into treatment. Tumors remained small throughout the experiment. Minnelide treated animals showed a mean tumor volume of 50 mm3 (a forty-fold inhibition when compared to control group). Combination of Minnelide and Carboplatin showed further improvement in tumor growth inhibition. These observations were further reflected in the wet weight of tumors resected from the animals at the end of the experiment (Figure 6B). Control mice showed a mean tumor weight of 1.75 g. Carboplatin treatment reduced the tumor burden to 0.8 gm. Minnelide treated animals showed a mean tumor weight of 0.1 gm. Combination treatment further reduced the tumor weight to 0.03 gm. These data suggest that Minnelide at the given dose was more effective than Carboplatin in inhibiting CaSki tumors and combination therapy showed further improvement in inhibiting tumor growth. In a separate experiment, survival of CaSki tumor bearing mice were determined. Data in Figure 6C show Kaplan-Meir plot of treated mice. All the control group of mice died of tumor burden by day 52. Carboplatin treatment even though reduced the tumor size, there was no survival advantage at the doses given. Mean survival was very similar between control and Carboplatin treated animals. Minnelide treatment increased the survival of animals and all Minnelide treated animals died by day 75. Combination treatment with Minnelide and Carboplatin showed survival of 100% of the animals even at day 110.
Figure 6.
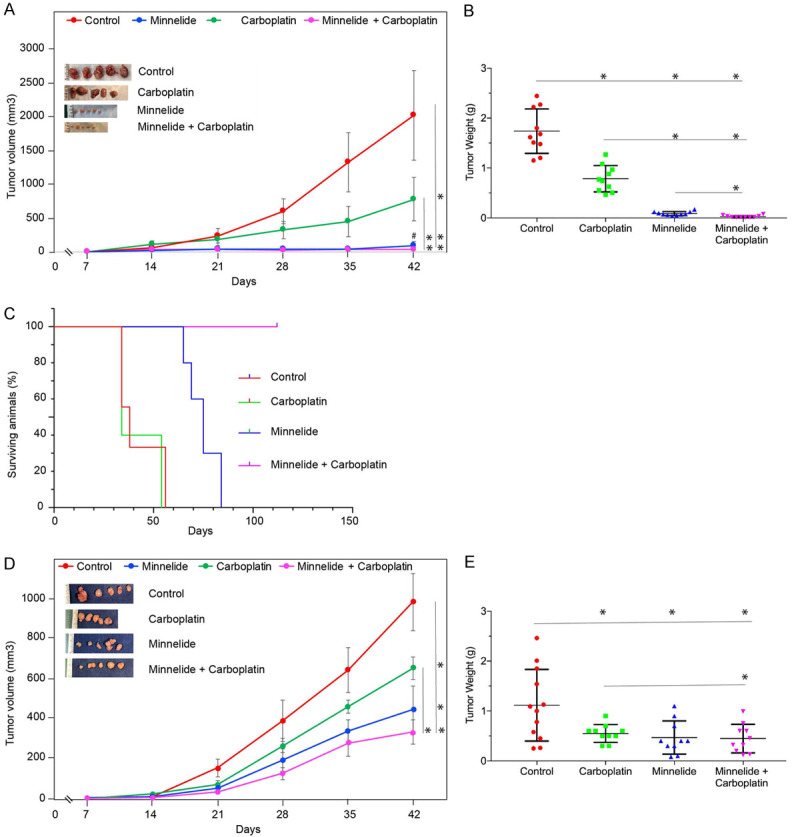
Improved inhibition of Cervical cancer growth by combination treatment. (A and D) show the effect of 0.2 mg/kg Minnelide, 20 mg/kg Carboplatin, and a combination of both treatments on subcutaneous tumor volume over the course of experiment. Treatment started 7 days after tumor cell implantation and continued until day 42. (A), CaSki, (D), ME-180. (B and E) show the effect of 0.2 mg/kg Minnelide, 20 mg/kg Carboplatin, and a combination of both treatments on tumor weight at the end of the experiment. Each dot in the scatter plot represents one mouse. (B), CaSki, (E), ME-180. (C) shows the effect of 0.2 mg/kg Minnelide, 20 mg/kg Carboplatin, and a combination of both treatments on the survival of athymic mice bearing subcutaneous tumors (CaSki) from an independent experiment. Statistically significant differences are denoted by asterisks. Group comparisons are indicated by lines. ‘#’ in (A) shows comparison between Minnelide and combination group which was statistically significant.
Then we evaluated the efficacy of Minnelide on ME-180 tumors. Treatment started one week after tumor cell implantation and terminated on week 6. ME-180 tumors grew to a mean volume of 900 mm3 by day 42. Carboplatin treatment resulted in 30% reduction in tumor volume. Minnelide treatment again was more effective than Carboplatin in inhibiting ME-180 tumors. Minnelide treated mice showed a mean tumor volume of 300 mm3. Combination treatment with Minnelide and Carboplatin showed further inhibition than monotherapies. Mean tumor volume of combination therapy group showed a value of 230 mm3 (Figure 6D). Tumor wet weight at the time of sacrifice showed that control group of mice had a mean tumor burden of 1.1 gm. Minnelide as a monotherapy reduced the tumor burden to 0.47 gm. Carboplatin monotherapy reduced the mean tumor weight to 0.55 gm (Figure 6E). Combination treatment, in this model did not show any additive effect. Mean tumor burden of mice treated with a combination of Minnelide and Carboplatin was 0.448 gm. Monotherapy with Minnelide was as effective as combination treatment with Carboplatin in this model. Minnelide treatment (0.2 mg/kg) did not affect the body weight of the animals when compared to control animals during the course of the study indicating safety of the Minnelide dose used (Supplementary Figure 4).
Discussion
In the present studies, we have investigated the efficacy of Minnelide against cervical cancers in three different model systems. Minnelide is inactive in vitro and needs to be converted to its active form, Triptolide, by phosphatases in vivo. Therefore, Triptolide was used for in vitro studies to determine cytotoxicity against human cervical cancer cell lines. Our studies show that Triptolide is very effective not only against HPV-16 (SiHa), HPV-16/18 (CaSki) positive cells but also inhibited ME-180 cells (HPV-68). HPV-68 is associated with only a minority of cervical cancer patients. However, it is an important cervical cancer model as HPV-68 induced cervical cancers will escape currently available vaccines against HPV. ME-180 cells were completely inhibited even at 100 nM concentration. Cervical cancer cells were as sensitive as other tumor cells to Triptolide [9-12]. Triptolide also inhibited three dimensional growth of cancer cells, a hall mark of aggressive cancer cells in vitro [13].
Triptolide has been shown to act at multiple cellular pathways [9,14-18], including inhibition of transcription by inducing proteasomal degradation of RNA polymerase II [14]. Our studies show that Triptolide treatment resulted in marked decrease in the transcript levels of HPV encoded oncoproteins E6 and E7. As a consequence, molecular targets of E6 and E7 are affected by Triptolide treatment. E6 and E7 target p53 and pRb respectively. These interactions are known to down regulate the levels of p53 and pRb tumor suppressor proteins. Triptolide treatment differentially affected the levels of p53 and pRb in a cell dependent manner. CaSki cells showed increased levels of pRb and phosphorylated pRb. In contrast, ME-180 cells showed marked increase in p53 levels after Triptolide treatment whereas the pRb levels were not affected. In both cases Triptolide treatment lead to apoptosis through an increase in Caspase3/7 activity in InCucyte assays. However, in western blots, cleaved Caspase3 was increased only in the CaSki cells but not in ME-180 cells indicating that Caspase7 activation may be responsible for executing apoptosis. RPPA analyses confirmed activation of Caspase7 by Triptolide treatment in ME-180 cells. Further studies are needed to understand the differential activation of executor caspases by Triptolide treatment. While in cervical cancer cells HPV-mediated destabilization of p53 and pRb is important for transformed phenotype and tumorigenesis, p53 levels and function are modulated in other cancer cells by affecting MDM2 or mutations. Triptolide treatment has been shown to induce apoptosis in acute lymphoblastic leukemia cells by directly affecting MDM2 levels in a p53-independent mechanism [15]. Previous studies from our laboratory have shown that Triptolide induces autophagy (programmed cell death pathway II) and apoptosis in pancreatic cancer cells [10]. Changes in p53 and pRb following Triptolide treatment significantly increased apoptotic cell death of cervical cancer cells. Increased levels of pRb is known to complex with E2F1 and turn off S-phase genes leading to cell cycle arrest. In addition, we and others have shown that triptolide can induce mitochondrial ROS generation [19] and DNA-damage [20]. Under these conditions, increased levels of p53 and pRb will trigger programmed cell death [21]. Furthermore, our previous studies have shown that while Triptolide does not inhibit interaction between pRb and E2F1 but decrease the transactivation of target genes (functional activity after DNA binding) such as CDC2, Cyclin A and Orc-1 [22]. Thus Triptolide induces cell cycle arrest and apoptosis by reducing E6/E7 levels and by inhibiting E2F1 activity and p53-mediated cell death.
For in vivo studies, we used Minnelide. Intraperitoneal injections of Minnelide inhibited the growth of established SiHa tumors very effectively. In these studies, 0.4 mg/kg dose was used. Previous studies from our laboratory have demonstrated efficacy of this dose against established pancreatic cancers in xenograft models and in patient-derived tumor models [12,16,17]. Minnelide was very effective as monotherapy and also improved the efficacy of an established chemotherapeutic drug, Carboplatin. Earlier studies have shown that TRAIL-induced activation of death receptors in pancreatic cancer cells was improved by the addition of Triptolide [11]. Triptolide in combination with cis-platin was more effective in inhibiting nasopharyngeal cancer cells [18]. In a recent study, Minnelide was found to overcome oxaliplatin resistance in prostate cancer models [23]. In the previous studies, higher doses of Minnelide were used. Current studies showed efficacy of Minnelide even at 0.2 mg/kg both in monotherapy and in combination with Carboplatin. Co-administration of Carboplatin and Minnelide showed statistically significant improvement in tumor growth inhibition of CaSki tumors. The improvement seen in the efficacy is likely due to multiple contributing factors such as increased DNA damage, decrease in DNA repair enzymes and higher intracellular accumulation of the chemotherapeutic drugs. Furthermore, Minnelide-induced changes in the tumor microenvironment can also play a role in the improved anti-tumor activity [24,25]. In summary, our studies demonstrate that Minnelide is an effective drug against cervical cancers and can be used in combination with platinum drugs to improve therapeutic efficacy.
Acknowledgements
We thank Manju Saluja, and Shamsudheen Moidunny for technical assistance. This work was supported in part by the following grants R01CA170946, R01CA124723, R01DA047089 from the NIH and funding from the Sylvester Comprehensive Cancer Center, Miller School of Medicine, University of Miami. C.D was supported by the Training Grant T32CA211034-03 from the NIH.
Disclosure of conflict of interest
The University of Minnesota has a patent for Minnelide, which has been licensed to Minneamrita Therapeutics, LLC. Ashok Saluja is the co-founder and the Chief Scientific Officer of this company and this relationship is managed by the University of Miami as per the University regulations.
Supplementary Figures 1-4
Supplementary Video 1
Supplementary Video 2
Supplementary Video 3
Supplementary Video 4
References
- 1.Torre LA, Islami F, Siegel RL, Ward EM, Jemal A. Global cancer in women: burden and trends. Cancer Epidemiol Biomarkers Prev. 2017;26:444–457. doi: 10.1158/1055-9965.EPI-16-0858. [DOI] [PubMed] [Google Scholar]
- 2.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006;24(Suppl 3) doi: 10.1016/j.vaccine.2006.05.115. S3/1-10. [DOI] [PubMed] [Google Scholar]
- 4.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 5.Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol. 2001;14:684–691. doi: 10.1016/s0955-0674(02)00388-5. [DOI] [PubMed] [Google Scholar]
- 6.Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks-Skiba KE, Swaney DL, Johnson TL, Jang GM, Shah PS, Brand TM, Archambault J, Kreisberg JF, Grandis JR, Ideker T, Krogan NJ. Multiple routes to oncogenesis are promoted by the human papillomavirus-host protein network. Cancer Discov. 2018;8:1474–1489. doi: 10.1158/2159-8290.CD-17-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Freitas AC, Gomes Leitão Mda C, Coimbra EC. Prospects of molecularly-targeted therapies for cervical cancer treatment. Curr Drug Targets. 2015;16:77–91. doi: 10.2174/1389450116666141205150942. [DOI] [PubMed] [Google Scholar]
- 8.Kir D, Saluja M, Modi S, Venkatachalam A, Schnettler E, Roy S, Ramakrishnan S. Cell-permeable iron inhibits vascular endothelial growth factor receptor-2 signaling and tumor angiogenesis. Oncotarget. 2016;7:65348–65363. doi: 10.18632/oncotarget.11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips PA, Dudeja V, McCarroll JA, Borja-Cacho D, Dawra RK, Grizzle WE, Vickers SM, Saluja AK. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–9416. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 10.Mujumdar N, Mackenzie TN, Dudeja V, Chugh R, Antonoff MB, Borja-Cacho D, Sangwan V, Dawra R, Vickers SM, Saluja AK. Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology. 2010;139:598–608. doi: 10.1053/j.gastro.2010.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Sangwan V, Banerjee S, Chugh R, Dudeja V, Vickers SM, Saluja AK. Triptolide sensitizes pancreatic cancer cells to TRAIL-induced activation of the death receptor pathway. Cancer Lett. 2014;348:156–166. doi: 10.1016/j.canlet.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isharwal S, Modi S, Arora N, Uhlrich C 3rd, Giri B, Barlass U, Soubra A, Chugh R, Dehm SM, Dudeja V, Saluja A, Banerjee S, Konety B. Minnelide inhibits androgen dependent, castration resistant prostate cancer growth by decreasing expression of androgen receptor full length and splice variants. Prostate. 2017;77:584–596. doi: 10.1002/pros.23298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461–463. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Lu JJ, He L, Yu Q. Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II) PLoS One. 2011;6:e23993. doi: 10.1371/journal.pone.0023993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang M, Zhang H, Liu T, Tian D, Gu L, Zhou M. Triptolide inhibits MDM2 and induces apoptosis in acute lymphoblastic leukemia cells through a p53-independent pathway. Mol Cancer Ther. 2013;12:184–194. doi: 10.1158/1535-7163.MCT-12-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers SM, Saluja AK. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. 2012;4:156ra139. doi: 10.1126/scitranslmed.3004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivard C, Geller M, Schnettler E, Saluja M, Vogel RI, Saluja A, Ramakrishnan S. Inhibition of epithelial ovarian cancer by Minnelide, a water-soluble pro-drug. Gynecol Oncol. 2014;135:318–324. doi: 10.1016/j.ygyno.2014.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Zhang JJ, Sun YM, Zhang J, Wang LR, Li JC, Liu H. Triptolide induces apoptosis and synergizes with cisplatin in cisplatin-resistant HNE1/DDP nasopharyngeal cancer cells. Folia Biol (Praha) 2015;61:195–202. doi: 10.14712/fb2015061050195. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Corey C, Scott I, Shiva S, D’Cunha J. Minnelide/triptolide impairs mitochondrial function by regulating SIRT3 in P53-dependent manner in non-small cell lung cancer. PLoS One. 2016;11:e0160783. doi: 10.1371/journal.pone.0160783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chueh FS, Chen YL, Hsu SC, Yang JS, Hsueh SC, Ji BC, Lu HF, Chung JG. Triptolide induced DNA damage in A375.S2 human malignant melanoma cells is mediated via reduction of DNA repair genes. Oncol Rep. 2013;29:613–618. doi: 10.3892/or.2012.2170. [DOI] [PubMed] [Google Scholar]
- 21.Kasten MM, Giordano A. pRb and the cdks in apoptosis and the cell cycle. Cell Death Differ. 1998;5:132–140. doi: 10.1038/sj.cdd.4400323. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira A, Beyer G, Chugh R, Skube SJ, Majumder K, Banerjee S, Sangwan V, Li L, Dawra R, Subramanian S, Saluja A, Dudeja V. Triptolide abrogates growth of colon cancer and induces cell cycle arrest by inhibiting transcriptional activation of E2F. Lab Invest. 2015;95:648–659. doi: 10.1038/labinvest.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modi S, Kir D, Giri B, Majumder K, Arora N, Dudeja V, Banerjee S, Saluja AK. Minnelide overcomes oxaliplatin resistance by downregulating the DNA repair pathway in pancreatic cancer. J Gastrointest Surg. 2016;20:13–23. doi: 10.1007/s11605-015-3000-3. discussion 23-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu W, He S, Li Y, Qiu P, Shu M, Ou Y, Zhou Y, Leng T, Xie J, Zheng X, Xu D, Su X, Yan G. Anti-angiogenic activity of triptolide in anaplastic thyroid carcinoma is mediated by targeting vascular endothelial and tumor cells. Vascul Pharmacol. 2010;52:46–54. doi: 10.1016/j.vph.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee S, Modi S, McGinn O, Zhao X, Dudeja V, Ramakrishnan S, Saluja AK. Impaired synthesis of stromal components in response to minnelide improves vascular function, drug delivery, and survival in pancreatic cancer. Clin Cancer Res. 2016;22:415–425. doi: 10.1158/1078-0432.CCR-15-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.