

Abstract
Arhgef11 is a Rho-guanine nucleotide exchange factor that was previously implicated in kidney injury in the Dahl salt-sensitive (SS) rat, a model of hypertension-related chronic kidney disease. Reduced Arhgef11 expression in an SS-Arhgef11SHR-minimal congenic strain (spontaneously hypertensive rat allele substituted for S allele) significantly decreased proteinuria, fibrosis, and improved renal hemodynamics, without impacting blood pressure compared with the control SS (SS-wild type). Here, SS-Arhgef11−/− and SS-wild type rats were placed on either low or elevated salt (0.3% or 2% NaCl) from 4 to 12 weeks of age. On low salt, starting at week 6 and through week 12, SS-Arhgef11−/− animals demonstrated a 3-fold decrease in proteinuria compared with SS-wild type. On high salt, beginning at week 6, SS-Arhgef11−/− animals demonstrated >2-fold lower proteinuria from weeks 8 to 12 and 30 mm Hg lower BP compared with SS-wild type. To better understand the molecular mechanisms of the renal protection from loss of Arhgef11, both RNA sequencing and discovery proteomics were performed on kidneys from week 4 (before onset of renal injury/proteinuria between groups) and at week 12 (low salt). The omics data sets revealed loss of Arhgef11 (SS-Arhgef11−/−) initiates early transcriptome/protein changes in the cytoskeleton starting as early as week 4 that impact a number of cellular functions, including actin cytoskeletal regulation, mitochondrial metabolism, and solute carrier transporters. In summary, in vivo phenotyping coupled with a multi-omics approach provides strong evidence that increased Arhgef11 expression in the Dahl SS rat leads to actin cytoskeleton-mediated changes in cell morphology and cell function that promote kidney injury, hypertension, and decline in kidney function.
Keywords: alleles, blood pressure, cytoskeleton, fibrosis, proteinuria
Chronic kidney disease (CKD) is common among adults in the United States, with an estimated 30 million or 15% of adults exhibiting some degree of kidney injury or reduced renal function.1 Of these, 468 000 individuals require dialysis and 193 000 live with a functioning kidney transplant.2 Hypertension is a major driver of CKD, second only to diabetes mellitus, and both diseases combined contribute to more than two-thirds of the cases that progress to kidney failure.2,3 Other factors that contribute to kidney failure include heart disease, obesity, and family history of CKD.4 Numerous single gene causes have been identified for a variety of kidney diseases, including polycystic kidney disease, nephrotic syndrome/focal segmental glomerulosclerosis, and nephronophthisis.5 However, the vast majority of CKD is complex in nature, with multiple genetic factors as well as environmental influences contributing to disease progression.6 Over the past 10 years, many large-scale genome-wide association studies have mapped genetic loci/genes involved in CKD phenotypes.7 In total, >130 single nucleotide polymorphisms across 18 studies have been identified that associate with various traits related to CKD,8 with some of these studies involving hundreds of thousands of patient samples.9 Unfortunately, many of these studies have not been replicated, and confirming the functional role of identified variants in humans continues to be a challenge. Therefore, there is a critical need to be able to investigate genes and their associated signaling pathways as this information could provide a deeper understanding of mechanisms of onset and progression of kidney injury.
The Dahl salt-sensitive (SS) rat provides a robust model to study progressive kidney injury related to age with moderate hypertension (low salt) as well as the ability to accelerate decline in kidney function by induction of significant hypertension (high-salt diet).10,11 Previously, genetic studies using the SS rat and the spontaneously hypertensive rat (SHR), which is highly resistant to kidney injury despite being hypertensive, identified at least 9 loci throughout the genome linked to kidney injury (proteinuria).12 Positional cloning using congenic strains and recombinant progeny testing narrowed the genome region to a handful of genes, with Arhgef11 being identified as top candidate for underlying the genomic locus.13 Increased Arhgef11 expression (before injury), as well as several variants, predicted to have an impact on protein function was observed in the SS compared with SHR. Subsequently, the evaluation of an SS-Arhgef11SHR-minimal congenic strain (SHR allele substituted for SS allele) demonstrated decreased proteinuria and tubulointerstitial injury/fibrosis and improved renal hemodynamics (increased GFR, increased RBF, and decreased RVR) compared with wild-type SS (SS-WT).13 Knockdown of Arhgef11 in cell lines resulted in reduced RhoA activity, decreased activation of the Rho-ROCK pathway, and less stress fiber formation (≈2-fold reduction) versus control.14 In total, the animal studies and in vitro cell studies suggest that chronic activation of Arhgef11 (in the context of other susceptibility factors) contributes to significant tubulointerstitial injury observed in the SS rat.
The objective of the current study was to investigate the molecular and physiological mechanisms by which loss of Arhgef11 impacts kidney injury exhibited by the Dahl SS rat. A temporal study of renal and cardiovascular measurements was performed between the SS-WT and the SS-Arhgef11−/− on both low and elevated salt intake. In addition, both genomic and proteomic techniques were employed to investigate early molecular changes occurring in the kidney between SS-Arhgef11−/−and SS-WT, before phenotypic differences were detected (week 4), as well as molecular changes occurring once significant differences in renal injury and function are observed (week 12). It is anticipated that greater insight into the molecular factors that contribute to hypertensive CKD in the Dahl SS model may provide a means to target drug development as well as provide important predictive biomarkers predictive of renal injury progression.
Material and Methods
The detailed methods are described in an expanded Material and Methods in the online-only Data Supplement. The authors declare that all supporting data are available within the article and in the online-only Data Supplement.
Animals
All experimental procedures were approved by the University of Mississippi Medical Center Institutional Animal Care and Use Committee. The Dahl SS (SS/Jr) and SS-Arhgef11−/− are maintained at our institutional animal facility. The SS-Arhgef11−/− (SS/JrHsdMcw) model was developed using CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat–associated 9) system (via Medical College of Wisconsin), which resulted in a 17 bp deletion in exon 2 of the Arthgef11 gene, resulting in premature stop codon and protein truncation (Figure S1 in the online-only Data Supplement). The SS-Arhgef11−/− (SS/JrHsdMcw) was backcrossed to the SS/Jr at University of Mississippi Medical Center 5× before being fixed in the homozygous state. All rats were housed in a 12-hour light-dark cycle, maintained on low-salt rodent chow (0.3% NaCl Teklad 7034) and provided water ad libitum. All animals studied were male.
Phenotyping
Study 1: Temporal Study of Renal Injury and Impact of Salt Loading
At 4 weeks of age, age-matched male SS-WT and SS-Arhgef11−/− were weaned onto a low-salt diet (0.3% NaCl; TD7034; Harlan Teklad). Twenty-four-hour urine collections were done at 4 and 6 weeks of age on low-salt diet for the determination of proteinuria.13 After urine collections were performed at week 6, each group was randomly divided and either continued on low-salt diet (0.3% NaCl) or placed on elevated salt diet (2% NaCl, TD 94217, Harlan Teklad). The following groups were studied: low salt, SS-WT (n=16) and SS-Arhgef11−/− (n=17); and high salt, SS-WT (n=18) and SS-Arhgef11−/− (n=18). Urine collections were performed at weeks 8, 10, and 12 for the determination of proteinuria. After week 12 urine collections, a terminal blood pressure measurement was performed as done previously under isoflurane anesthesia.13 Serum samples were obtained from cardiac puncture to measure blood parameters as previously described.15 On euthanasia, kidney and heart weights were taken, followed by snap freezing half kidney in liquid nitrogen for downstream -omics analyses and formalin fixing the other half kidney for histological examination.
Study 2: Temporal Study of Renal Injury and Telemetric Blood Pressure Measurements
At 4 weeks of age, age-matched male SS-WT (n=9) and SS-Arhgef11−/− (n=10) were weaned onto a low-salt diet (0.3% NaCl). Twenty-four-hour urine collections were done at 6 weeks of age on low salt and then each group was placed on high salt (2.0% NaCl) for the remainder of the study. Albuminuria was measured using a rat albumin ELISA kit (Abcam, ab108789). At week 7, a subset of animals from each group (n=8) were implanted with telemetry transmitters (model HD-S10, Data Sciences International) as previously described.13,16 After 1 week of recovery, blood pressure was measured for 24 hours one day each week from weeks 8 to 15, followed by 24-hour urine collections were made for the determination of proteinuria and albuminuria. Euthanasia was performed after week 15 blood pressure, and urine collections were complete. Organ weights, serum, and kidney samples for histological examination were collected.
Study 3: Measurement of Renal Function, Survival, and Assessment of Renal Injury at an Advanced Age
An additional group of age-matched male SS-WT (n=16) and SS-Arhgef11−/− (n=17) were weaned onto a low-salt diet (0.3% NaCl) at 4 weeks of age. Similar to study 2, 24-hour urine collections were done at 6 weeks of age on low salt and then each group was placed on high salt (2.0% NaCl) for the remainder of the study. Tail vein blood was collected immediately after the urine collection for the measurement of serum creatinine and calculation of creatinine clearance at weeks 6, 9, 12, and 15. Subsequently, these animals were aged to perform a survival study as done previously.17 To provide some indication of renal injury and function between groups in older animals, measurements were collected at weeks 25 and 35.
RNA Sequencing and Real-Time PCR
RNA was extracted using the Pure Link RNA Mini Kit (Invitrogen) from archived study 1 kidney samples according to manufacturer’s instructions and assessed for quality control parameters of minimum concentration and fidelity (ie, 18S and 28S bands, RQI >8).18 Libraries were prepared, and RNA sequencing was performed as previously described. RNA sequencing was performed using n=4 per group (WT and KO) for week 4 samples and n=6 per group (WT and KO) for week 12 samples. Gene expression differences are denoted as Log2 (ratio) and Q>0.05. Gene set enrichment analysis was performed using Enrichr (http://amp.pharm.mssm.edu/Enrichr/),19,20 which provides multiple tools for gene list enrichment analysis. Raw reads can be accessed under BioProject PRJNA600744 and short read archive accession numbers SRR10874291–SRR10874310. Validation of RNAseq data was performed using SYBR-green dye chemistry on Bio-Rad real-time polymerase chain reaction (PCR) platform as done previously.15 Statistical analysis of real-time PCR data was performed by Bio-Rad Maestro Software. The data are presented as mean+SEM.
Mass Spectrometry Proteomics
Total protein was isolated from kidney using protein extraction reagent type 4. Proteins were reduced, alkylated, and trypsin-digested into peptides. Peptides were cleaned using Sep-Pak Vac C18 cartridge (Waters, Milford, MA) and analyzed label-free by liquid chromatography-tandem mass spectrometry using a Q Exactive (ThermoFisher, Waltham, MA) coupled with a 15 cm×75 μm C18 column (5 μm particles with 100 Å pore size) as done previously.18 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD011758.
Flow Cytometry
Single-cell suspensions from the kidney cortex were prepared in 5 mL RPMI (Roswell Park Memorial Institute) media containing 200 U/mL DNase and 10 mg/mL collagenase IV using the Gentle MACS dissociator (Milltenyi Biotec, Bergisch Gladbach, Germany) using a user-defined protocol for rodent kidney as done previously.21 Samples were analyzed on a Gallios flow cytometer (Beckman Coulter, Indianapolis, IN), and a total of 25 000 events were acquired for each sample. Data were analyzed using FCS express software (DeNovo Software, Glendale, CA).
Histological Analysis
Kidneys were fixed in 10% buffered formalin, embedded in paraffin, cut into 4-μm sections, and either stained with Periodic acid Schiff or Masson’s trichrome. For glomeruli, morphometric analysis (diameter [μm] and area [μm2]) was performed on 20 randomly selected images (Periodic acid Schiff stained at 40×) per section. Tubulointerstitial injury was determined by evaluation of slides stained with Masson’s trichrome to quantify the percent fibrosis (blue staining) compared with background in 20 randomly selected images from renal cortex as previously done.15,17 Tubulointerstitial injury was evaluated separately on a semi-quantitative scale from 0 (normal) to 4 (severe) using a minimum of 20 randomly selected images (Masson Trichrome at 20×).16 All histology analyses were performed with the analyst blinded to the animal/ID/category. Images were captured using SeBaP4-PH1 Brightfield/Phase contrast microscope (Laxco, Mill Creek, WA) and analyzed using Nikon Elements image analysis software.
Statistical Analysis
All physiological data are presented as mean+SE. Time course experimental data (eg, proteinuria and telemetry blood pressure) was evaluated by 1-way or 2-way ANOVA followed by either Dunnett or Tukey post-test (GraphPad Prism 6, La Jolla, CA).22 Data for single time point measures (eg, organ weight, terminal blood pressure, etc) were analyzed using Student t test or ANOVA when >2 groups. Survival was evaluated by the Kaplan-Meier method (GraphPad Prism 5, La Jolla, CA). A P<0.05 was considered to be statistically significant.
Results
Temporal Measurement of Cardiovascular and Renal Traits
At week 4, proteinuria between SS-WT and the SS-Arhgef11−/− (KO) animals on a low salt (0.3% NaCl diet) was similar (Figure 1A). Starting as early as week 6, KO animals demonstrated a significant decrease in proteinuria compared with WT, and by week 12, KO animals exhibited a 3-fold reduction in proteinuria compared with SS-WT. On an elevated salt diet (2% NaCl), both groups demonstrated increased proteinuria compared with their low-salt counterparts, with KO animals demonstrating a larger fold change (1.7 versus 1.3) compared with WT. By week 12, proteinuria in SS-WT animals (201±19 mg/24 hours) was 2.5-fold higher than KO animals (83±9 mg/24 hours) after high salt intake (Figure 1A). A 2-way ANOVA identified a significant difference between strains (P<0.0001), salt-loading (P<0.0001), and strain by time interaction (P<0.01) over the time course. At the end of study 1, KO animals had mean arterial pressure on low-salt diet that was slightly lower and marginally significant (P=0.05) compared with SS-WT. On the 2% NaCl diet, WT animals exhibited a large increase (+27±4.7 mm Hg, P<0.0001) in mean arterial pressure compared with low-salt SS-WT group as well as KO animals on high salt (Figure 1B). KO animals on 2% NaCl diet demonstrated no significant increase in mean arterial pressure over the low-salt group. Consistent with mean arterial pressure, no significant differences were observed in heart weight (mg/g body weight) between low-salt groups, while KO animals demonstrated significantly reduced heart weights compared with SS-WT on elevated salt diet (Table S1). Overall, there was a strong correlation (r=0.68, P<0.0001) between heart weight and blood pressure (eg, greater the blood pressure, larger the heart).
Figure 1.
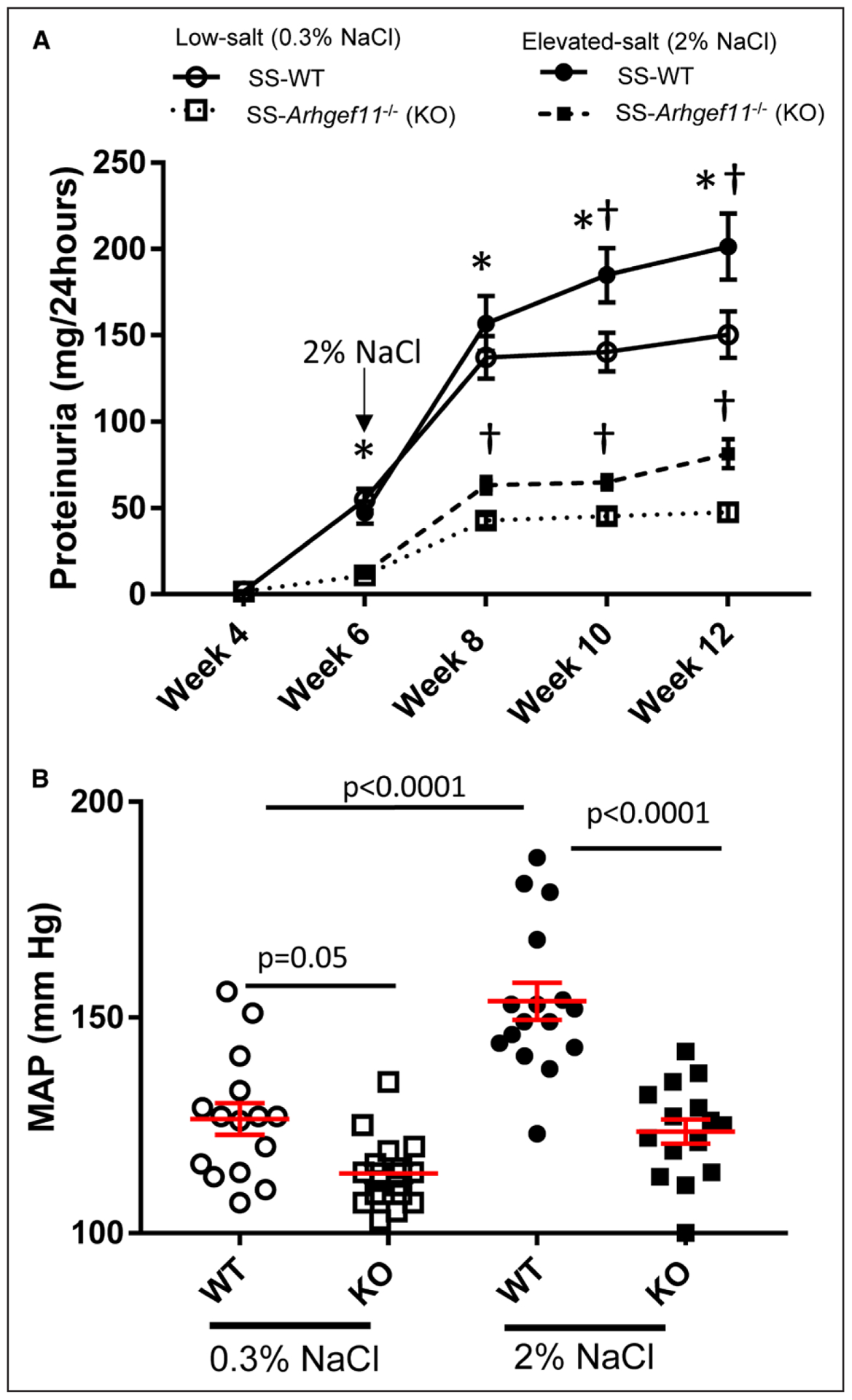
Attenuated proteinuria and blood pressure in Dahl salt-sensitive-(SS)-Arhgef11−/− (KO) compared with wild-type SS (SS-WT) rats on low salt (LS, 0.3% NaCl) and elevated-salt diet (ES, 2% NaCl). A, Proteinuria was assessed in the WT and KO rats from 4 to 12 wk on LS and ES diets. B, Mean arterial pressure (MAP) was measured at week 12. Low salt, SS-WT (n=16), KO (n=17) and elevated salt, SS-WT (n=18), KO (n=18). *P<0.05 KO vs WT, either LS or ES, † LS vs ES, either WT or KO. Two-way ANOVA with Tukey multiple comparison (strain, time, and strain time all P<0.0001) mean values±SE are presented.
At week 12, there was no significant difference in creatinine clearance (CrCl) between SS-WT and KO on low-salt groups; however, blood urea nitrogen was significantly lower in KO animals (Table S1). On the 2% NaCl diet, KO animals demonstrated a significant reduction in CrCl and blood urea nitrogen compared with SS-WT animals. No significant changes in other clinical chemistries were observed (electrolytes, liver enzymes, serum protein, and albumin), except KO demonstrated reduced total cholesterol and HDL (high-density lipoprotein) compared with WT.
Kidney Hypertrophy, Glomerular Morphology and Tubulointerstitial Injury on 0.3% and 2% NaCl Diets
Both KO and SS-WT groups exhibited a significant increase in kidney weight on 2% NaCl compared with low-salt diet, but the SS-WT change was significantly greater than KO (26% versus 12%, respectively; Table S1). The severity of kidney injury in SS-WT and KO was assessed at weeks 4 and 12. As expected, no significant histological injury was observed at 4 weeks of age in either strain. An unbiased morphometric analysis of glomerular area demonstrated no significant difference between strains on low salt at week 4 or week 12; however, KO animals (6651±227 um2) demonstrated less glomerular hypertrophy compared with SS-WT (7832±350 um2) on 2% NaCl diet at week 12 (Figure 2A). A slight degree of mesangial matrix expansion and glomerulosclerosis were similarly observed in both strains at week 12 using a semi-quantitative scoring system (scale 1–4, WT=0.71±0.04 and KO=0.66±0.04) on low salt (Figure S2). On the 2% NaCl diet, SS-WT animals demonstrated a significant increase in glomerular injury (1.3±0.05) compared with the low-salt group as well as the KO on high salt (1.0±0.04). Kidneys from SS-WT rats demonstrated a greater number of glomeruli with more severe injury compared with those in KO animals (Figure 2B). The severity of tubular injury was evaluated by tubular atrophy, dilation, and presence of protein casts. SS-WT rats exhibited more injury compared to KO rats at week 12 on both low salt and 2% NaCl (Figure S2). For the 2% NaCl diet, WT injury score (1.2±0.12) was ≈2-fold higher compared with KO (0.7±0.04). Consistent with tubular injury, the degree of interstitial fibrosis was similar between the SS-WT and KO on low salt, while on 2% NaCl diet, WT animals exhibited a significant degree of fibrosis (6.0%±0.44%) compared with KO (4.1%±0.39%; Figure 2C). Similar to glomerular injury, kidneys from SS-WT animals demonstrated more severe tubulointerstitial fibrosis compared with KO (Figure 2D)
Figure 2.
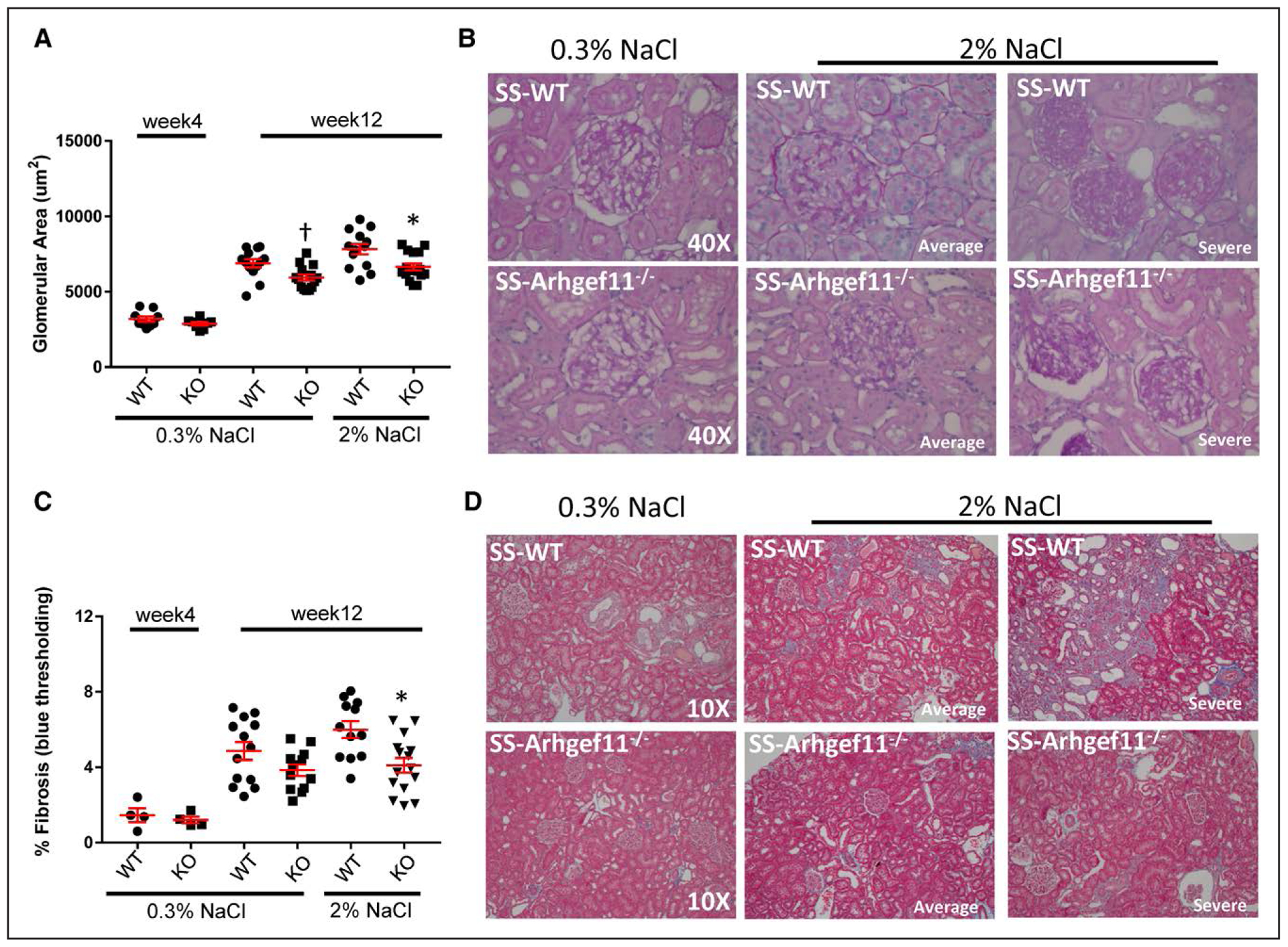
Reduced renal injury in Dahl salt-sensitive-(SS)-Arhgef11−/− (KO) rats compared with wild-type SS (SS-WT) on low salt (LS, 0.3% NaCl) and elevated-salt diet (ES, 2% NaCl) at week 4 and week 12. A, Morphometric analysis of glomerular area (um2) performed on 20 randomly selected images (Periodic acid Schiff [PAS] at 40×) per group/section. B, Representative image of glomeruli for SS-WT and KO on LS and ES diet at week 12. Representative images of week 4 kidney are provided in Figure S2. For ES diet, one image represents the average injury observed based on semi-quantitative scoring (Figure S2) and one image illustrates the severity of injury observed. C, Tubulointerstitial injury at week 12 was determined by evaluation of slides stained with Masson trichrome to quantify the percent fibrosis (blue staining). D, Representative image of renal cortex/tubulointerstitial region from SS-WT and KO on low salt and ES diet. For ES diet, one image represents the average injury observed based on semi-quantitative scoring (Figure S2) and one image selected to illustrate the severity of injury observed. One-way ANOVA Tukey multiple comparisons, P<0.0001. Both SS-WT and KO groups at week 12 on either LS or ES were significantly different compared with week 4 (P<0.0001) *P<0.05 KO vs WT. One-way ANOVA Tukey multiple comparisons only including week 12 data (LS and ES), †P=0.009. Mean values±SE of diff are presented.
Telemetry Measured Blood Pressure, Renal Injury, and Survival on 2% NaCl
A second study with rats raised on 2% NaCl diet was performed to 15 weeks of age. KO animals again demonstrated a significant attenuation of proteinuria (2.5-fold) and albuminuria (2-fold) compared with SS-WT from weeks 6 to 15 (Figure 3A). By week 8, SS-WT animals demonstrated a significant increase in systolic blood pressure measured by telemetry, and by week 15, blood pressure of KO was 54±8 mm Hg below that of SS-WT animals (Figure 3B). The blood pressure measurement was corroborated by a significant correlation between increased heart weight and elevated blood pressure (r=0.9252, P<0.0001), with KO animals demonstrating a significant reduction in heart weight compared with WT (Table S2).
Figure 3.
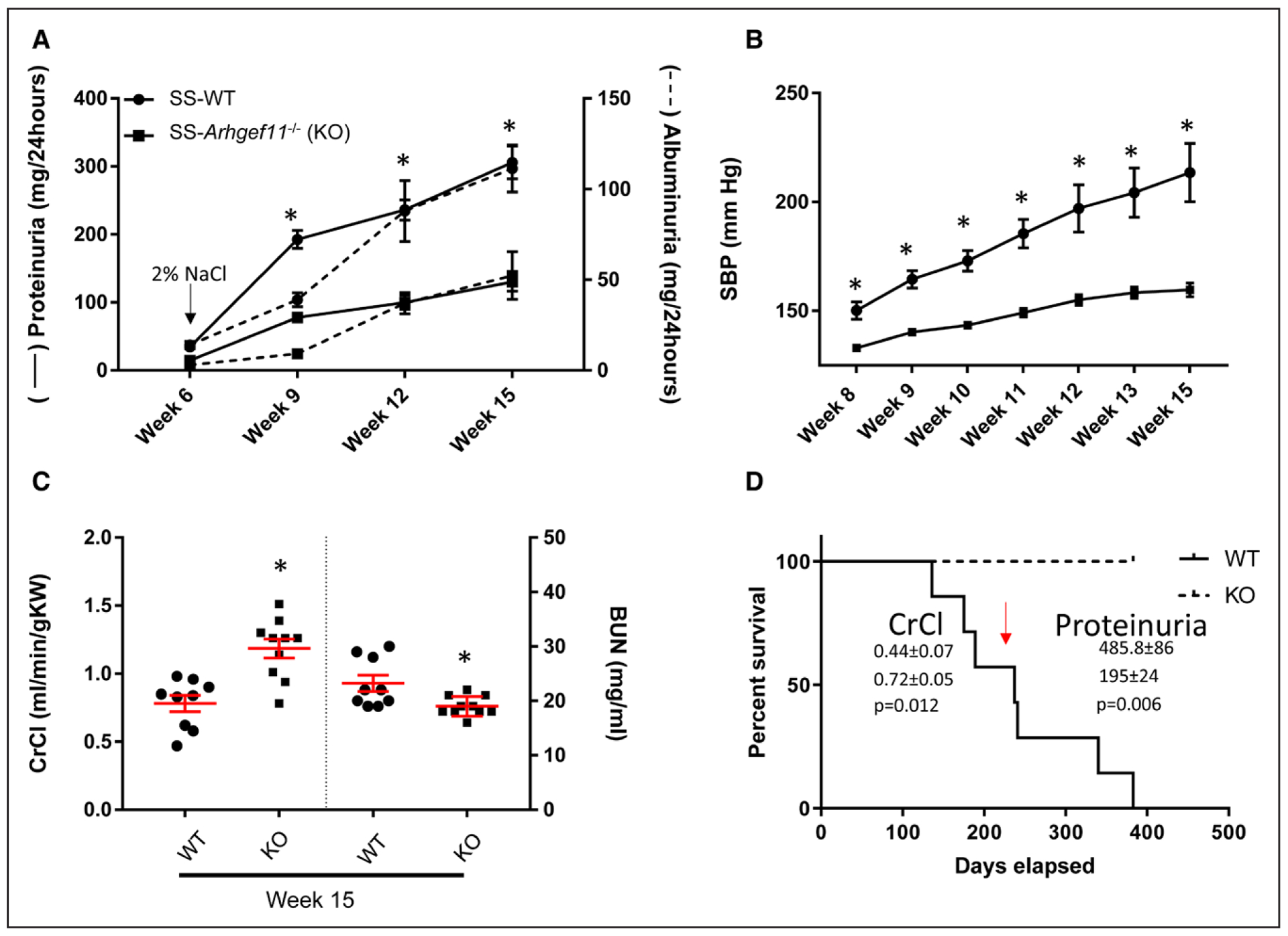
Reduced telemetry measured blood pressure (BP) and renal injury in Dahl salt-sensitive-(SS)-Arhgef11−/− (KO) rats compared with wild-type SS (SS-WT on elevated-salt diet (ES, 2% NaCl) from week 6 to week 15. A, Proteinuria and albuminuria assessed in the SS-WT (n=16) and KO (n=17) rats from 6 to 15 wk on ES diet. B, Telemetry measured systolic blood pressure from week 8 to week 15 (n=8 per group). C, Creatinine clearance (CrCl) normalized to gram kidney weight and blood urea nitrogen (BUN). D, Kaplan-Meier survival curve. Two way ANOVA using Sidak multiple comparison for proteinuria, albuminuria, or blood pressure (strain, time, and strain time all P<0.0001). For CrCl and BUN, unpaired t test; *P<0.05 KO vs WT. SBP indicates systolic blood pressure.
Renal function was measured at weeks 6, 9, 12, and 15 by determination of CrCl (mL/[min·100g BW]; Figure S3). KO animals demonstrated essentially no change in CrCl from week 9 to 15, whereas there were decline in SS-WT animals during the same time (Figure S3). At week 15, CrCl, normalized to kidney weight, and blood urea nitrogen were significantly improved in KO animals compared with SS-WT rats (Figure 3C). Similar to the findings of low salt and 2% NaCl comparison, no significant changes in clinical chemistries were observed for electrolytes (Na+, K+,) or liver enzymes, but KO animals demonstrated increased serum protein, albumin, and reduced total cholesterol and HDL compared with the SS-WT (Table S2).
SS-WT animals demonstrated increased mortality compared with KO animals, with no KO animals dying during the course of the study (Figure 3D). At approximately week 30 (or 210 days), the KO animals demonstrated 1.6-fold increase in renal function (SS-WT=0.4±0.07 and KO=0.7±0.05 mL/[min·100 gm BW]; P=0.012) and 2.5-fold decrease in proteinuria (SS-WT=486±86 and KO=195±24 mg/24 hours; P=0.006) compared with SS-WT animals.
Renal Hypertrophy, Pathology, and Immune Cell Flow Cytometry
At week 15, KO animals demonstrated a significant decrease in kidney weight compared with SS-WT, which was consistent with week 12 (Table S2). As expected, there was more substantial glomerular injury compared to week 12 for both groups, including greater amount of mesangial matrix expansion and more severe glomerulosclerosis in the SS-WT (SS-WT=1.6±0.09 and KO=1.0±0.07; Figure 4A). Glomerular hypertrophy was significantly greater in SS-WT (8384±295 um2) than in KO (7524±238 um2; Figure 4B). In addition, SS-WT glomeruli exhibited a greater number of glomeruli with more severe injury compared to those in KO animals (Figure 4C). Tubular injury was 2.3-fold higher in SS-WT (2.3±0.14) versus KO (1.0±0.07), with SS-WT demonstrating a 2-fold increase from week 12 and the KO essentially remaining at the same level. Interstitial fibrosis was significantly greater in SS-WT compared with KO in both the kidney cortex and medullary regions, with SS-WT animals exhibiting a greater amount of fibrosis in the medullary region (SS-WT=19.5%±2.4% versus KO=9.9%±1.5%; Figure 4E). Immune cell infiltration in the kidney was evaluated by flow cytometry. Macrophages (CD11b/c) were significantly elevated in the SS-WT kidney (15.7%±4.0) compared with KO (5.3%±1.1) as well as CD4+ T cells (5.8%±0.62 and 2.1±0.27, respectively; Figure S4). No significant differences were observed in CD8+ T cells.
Figure 4.
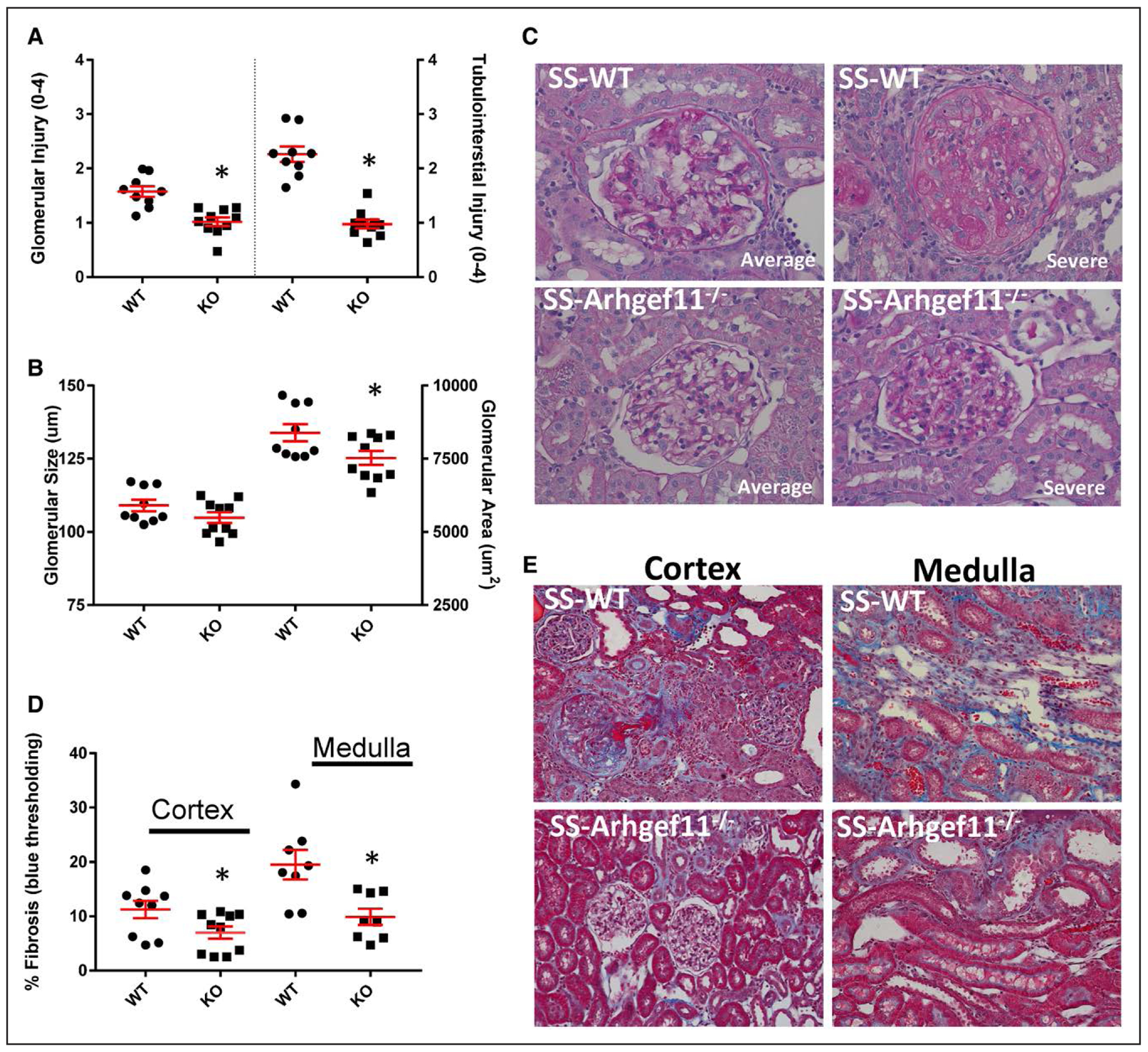
Reduced renal injury in Dahl salt-sensitive-(SS)-Arhgef11−/− (KO) rats compared with wild-type SS (SS-WT) on elevated-salt diet (ES, 2% NaCl) at week 15. A, Semi-quantitative scoring of glomerular and tubular injury. Each glomerulus was scored on a scale from 0 (normal) to 4 (global sclerosis). A minimum of 20 glomeruli were scored for each kidney section (n=9 per group/diet). Tubular injury was analyzed for degree of tubular atrophy, vacuolization, dilation, and proteinaceous casts on a scale from 0 (normal) to 4 (severe with >75% tubules demonstrating injury). B, Morphometric analysis of glomerular size (um) and glomerular area (um2) performed on 20 randomly selected images (Periodic acid Schiff [PAS] at 40×) per group/section. C, Representative images of glomeruli, one image represents the average injury observed based on semi-quantitative scoring and one image illustrates the severity of injury observed. D, Tubulointerstitial fibrosis (% blue staining) was evaluated by slides stained with Masson trichrome compared with background in 20 randomly selected images from renal cortex and medulla. E, Representative images of renal cortex and medulla. Unpaired t test, *P<0.05 KO vs SS-WT. Mean values±SE.
Transcriptome Analysis and Validation by Quantitative Real-Time PCR on Low Salt
Genes known to be associated with Arhgef11 and the associated RhoA signaling pathway (n=30) were evaluated by quantitative real-time PCR. There were few genes observed to be differentially expressed between WT and KO at either week 4 (Actr2, Cdc42, and Ppp1r12a) or week 12 (Pfn2, Plxnb, and Wasl). Of these, the extent of the fold changes were small at <20% (Figure S5). Subsequently, RNAseq was performed on SS-WT and KO kidneys at week 4 (preceding significant differences in renal injury/proteinuria between groups) and at week 12 to provide more mechanistic insight into the role of Arhgef11 in the kidney. At week 4, n=416 genes were observed to be differentially expressed between SS-WT and KO (>1.5-fold and Q<0.05), whereas n=1017 genes were observed at week 12 (Figure S6A through S6D). At week 4, 158 genes were upregulated in the KO and 258 were down regulated compared with SS-WT (Table S3). At week 12, 370 genes were upregulated and 647 were downregulated in the KO compared with SS-WT (Table S4). There was only a small overlap in the number of genes expressed at both time points (94 genes), with the majority in both groups unique to either week 4 or 12 (Figure S6).
At week 4, a gene set enrichment analysis was performed using 2 categories of enrichment, gene ontologies and pathway analysis (Reactome Pathway). At week 4, genes downregulated in the KO (vs SS-WT) were enriched for components of the plasma membrane, cytoskeleton, and intermediate filaments, whereas upregulated genes were enriched for brush border membrane, plasma membrane, and mitochondrial components. The upregulated genes were significantly associated with Reactome pathways linked to metabolism and solute carrier-mediated transport (Figure 5A and Table S5). Most of the differentially expressed genes observed at both week 4 and week 12 were either downregulated (n=39) or upregulated (n=22). However, a subset of genes were found to be downregulated early at week 4 and upregulated later at week 12 (n=9) or upregulated early and downregulated later (n=24; Figure 5B). The downregulated gene set was enriched for granule lumen and plasma membrane genes, with pathways associated with extracellular matrix organization. In contrast, the upregulated genes were enriched for mitochondria and plasma membrane, with metabolism as the major pathway. Similar to week 4, week 12 genes downregulated in KO (vs SS-WT) were enriched for the plasma membrane and cytoskeletal components, but different pathways were observed compared with week 4, including extracellular matrix organization, immune system, and integrin cell surface interactions (Figure 5C and Table S6). The upregulated genes were significantly associated with pathways linked to metabolism, biological oxidation, and solute carrier–mediated transport.
Figure 5.
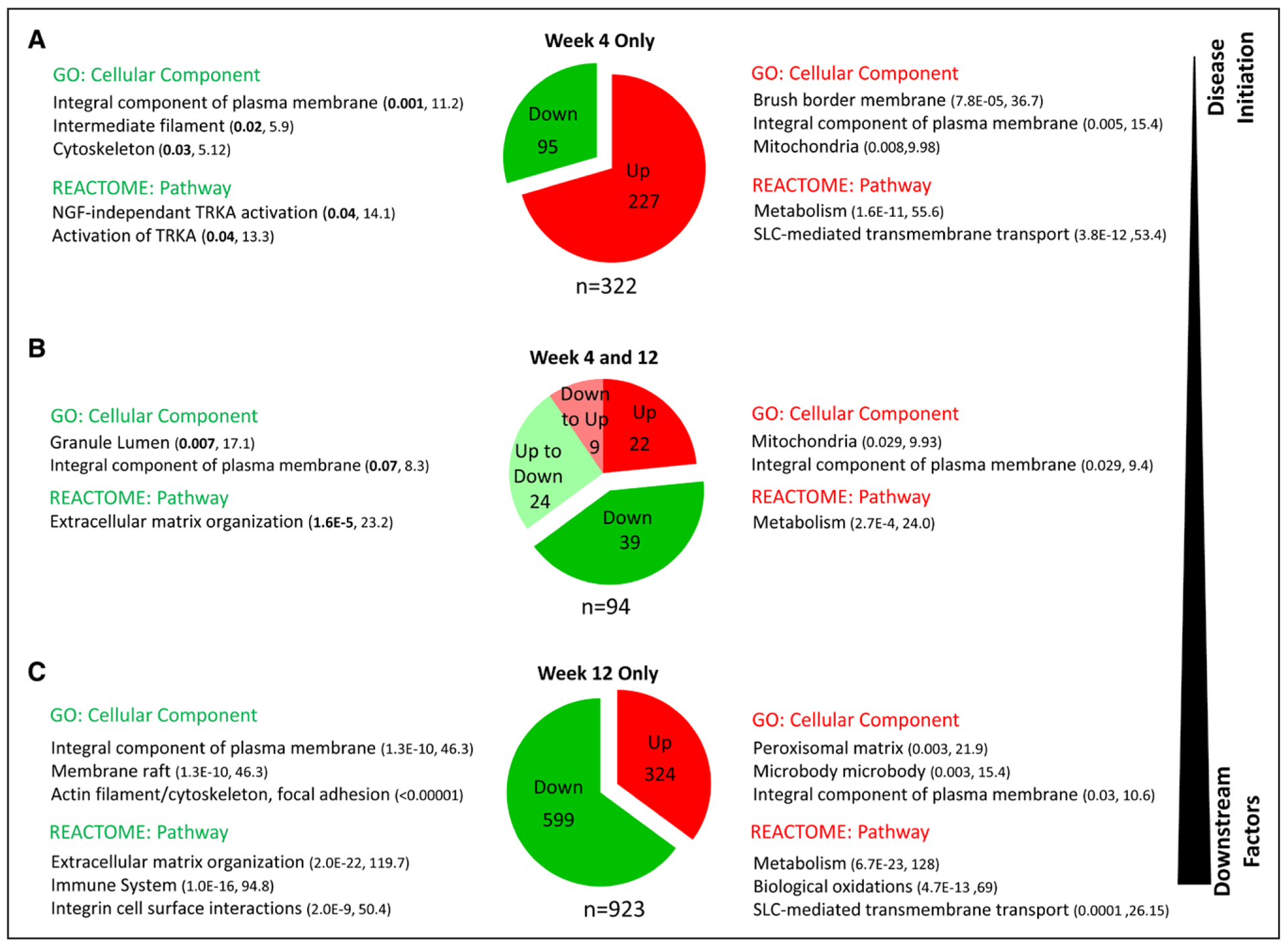
Pathway analysis of genes differentially expressed in kidney from wild-type Dahl salt-sensitive (SS-WT) and SS-Arhgef11−/− (KO) rats on low salt (0.3% NaCl) at week 4 and week 12. A, Top gene ontology (GO) terms and Reactome pathways of genes enriched at week 4 striated by upregulation/downregulation in KO vs WT. B, Top GO terms and Reactome pathways of genes enriched at both week 4 and 12. C, Top GO terms and Reactome pathways of genes enriched at week 12. For GO terms and Reactome pathways, the numbers within the parenthesis note the Q value (adjusted P value using the Benjamini-Hochberg method for correction for multiple hypotheses testing) and combined score (combination of the P value and z-score), both measures of significance for gene set enrichment. SLC indicates solute-carriers; and TRKA, Tropomyosin receptor kinase A.
A subset of genes from each comparison (week 4, week 12, or both) were validated using quantitative real-time PCR. There was a significant correlation between the RNAseq data and real-time PCR validation for both week 4 (n=18, r=0.91, P<0.0001) and week 12 (n=31, r=0.93, P<0.0001). Consistent with the differentially expressed genes identified by RNAseq, the majority of genes evaluated by real-time PCR were also found to be significantly different between KO and WT (Figure S7A through S7C). Several genes that demonstrated the largest difference between KO and WT at either week 4 or week 12, including Pmfbp, Rarres, Snap91, and Tnc were also successfully validated (Figure S7D). Genes used as markers of renal injury were queried from the RNAseq data and validated as well, including Actn1, Havcr1, Lcn2, S100a4, and Vim. As expected, all demonstrated reduced expression in the kidneys from KO compared with SS-WT, consistent with proteinuria measurements and histological analyses (Figure S7E). Finally, there were 2 gene families, MMP (matrix metalloproteinases) and RGS (regulator of G protein signaling), that demonstrated numerous genes dysregulated between KO and WT in the RNAseq data set. A subset of MMP (Mmp2, 7, 12, and 20), frequently dysregulated in fibrosis/extracellular matrix remodeling and RGS (Rgs16, 18, 20), was evaluated and confirmed via real-time PCR.
Proteomics Analysis on Low Salt
Discovery proteomics was performed on kidney homogenates from weeks 4 and 12 using the same tissue collected for the transcriptome experiments. At week 4, >6500 distinct peptide fragments from 1428 proteins were identified, with 121 proteins different between KO and SS-WT (Figure S8 and Table S7). Of these, 73 proteins were decreased and 48 increased in the KO compared with WT. At week 12, >5000 distinct peptide fragments were identified encompassing 1519 different proteins. Of these, 355 proteins were significantly different between groups, with 28 proteins decreased and 327 increased in the KO compared with WT (Figure S8 and Table S8). Approximately half (48%) of the proteins identified at weeks 4 and 12 were the same and the percent of uniquely observed at either time point was also similar (week 4=29% and week 12=23%). Of the 121 proteins different between KO and SS-WT at week 4, 32% or 36% were also different at week 12.
At week 4, proteins decreased in the KO (vs SS-WT) were enriched for components of the mitochondria and late endosome lumen, with pathways involving metabolism being significantly enriched (Figure S8 and S9). Proteins decreased in the KO were enriched for nuclear chromatin and secretory granule lumen. While not on the top of the GO terms (still significant), proteins associated with actin cytoskeleton and intermediate filaments, including CAP1, DES, TUBB, FSCN1, KRT, EEF1A1, were also enriched. The most significant enriched pathway involved cellular responses to stress (Table S9). At week 12, proteins decreased in the KO were enriched for components of the cytoskeleton/actin cytoskeleton (TUBB, MYH9, TAX1BP3, VIM, CNN3, and CTTNBP2NL). While no specific pathways were enriched using a stringent adjusted P value, several pathways, including Semaphorin interactions, TGFB signaling, and cytoskeleton regulation by Rho GTPases, were enriched using unadjusted P value (Table S10). Proteins increased in the KO at week 12 were enriched for large number of mitochondria components (n=89) and lysosome/vacuolar lumen. The most significant enriched pathways associated with proteins increased in KO were metabolism, including the citric acid cycle, respiratory electron transport, and metabolism of amino acids, carbohydrates, and lipids (Table S10).
Discussion
The major goal of this study was to investigate the physiological and molecular role of Arhgef11 in development of hypertension and renal injury in the Dahl SS rat. Previous genetic studies using the SS rat and the SHR identified a locus on chromosome 2 linked to proteinuria and histological renal injury, which contained Arhgef11 and a number of other genes.13 Subsequently, a small congenic strain was developed (SS-Arhgef11SHR) to test the impact of Arhgef11 (and surrounding genes/haplotype) donated by the SHR. The SS-Arhgef11SHR demonstrated decreased proteinuria, glomerular injury, and most notably tubulointerstitial injury/fibrosis. Additionally, the strain demonstrated improved renal hemodynamics (increased GFR, increased RBF, and decreased RVR) and greater survival compared with SS-WT (Figure S10).13 Most striking was the renal protection exhibited by the SS-Arhgef11SHR occurred independent of changes in blood pressure. Here, we report the physiological and molecular characterization of SS-Arhgef11−/− (KO) compared with the SS-WT which allows for the sole assessment of Arhgef11 gene.
Consistent with the phenotype of the SS-Arhgef11SHR congenic, the KO demonstrated reduced proteinuria, attenuated glomerular and tubular injury, improved renal function, and decreased mortality compared with the SS-WT (Figure S10). At 12 weeks of age, BP of the KO on low salt was only slightly lower to the SS-WT animals, even though these animals demonstrated a robust difference in proteinuria and improved renal injury measures, including reduced tubulointerstitial injury. This was similar to what was observed in the SS-Arhgef11SHR. As expected, SS-WT animals on 2% NaCl (for 6 weeks) exhibited a large increase in BP compared with low salt–fed animals, while the KO animals surprisingly exhibited no change in BP compared with the low-salt group. A subsequent study conducted on rats fed 2% NaCl for 9 weeks (started at 6 weeks of age) using telemetry measured BP confirmed that BP in KO animals changed only slightly, while BP in the SS-WT continued to increase out to 15 weeks of age. This suggests that while the KO demonstrates renal protection compared with SS-WT on low salt and a small lowering in BP, the loss of salt sensitivity (at least to a modest increase in salt) and reduced BP in the KO partly explains the renal protection observed on 2% NaCl. In total, the physiological characterization of the KO clearly demonstrates that loss of Arhgef11 has a major impact on the development of kidney injury in the Dahl SS, primarily through blunting tubulointerstial injury.
Molecularly, Arhgef11 is one of numerous (>70) GEF (guanine exchange factors) for Rho GTPases.23 ARHGEF11 contains several important domains, including a DH domain that facilitates GDP release and the PH domain that directs the GEF’s intracellular location.24 In addition, it contains a PDZ domain (a proline-rich domain), which is involved in protein-protein interactions, including anchoring cell surface receptors to the actin cytoskeleton via mediators like SLC9A3R1and Ezrin25 and an RGS (regulators of G protein signaling) domain26 important for interaction with an α subunits of heterotrimeric Gα12 and Gα13 proteins of the G12 family.27,28 Thus, the loss of function of any of these domain would likely impact diverse signaling pathways.
Activation of GPCR (G protein-coupled receptors) catalyzes the exchange of GDP for GTP on Gα12/13 subunit (or others such as Gαq and Gαi/o), resulting in the dissociation of the Gα12/13-GTP subunit from the Gβγ subunits.29 In particular, Gα12/13-GTP and ARHGEF11 interact through the RGS domain, which subsequently leads to activation of Rho proteins through exchange of GDP for GTP. ARHGEF11 demonstrates specificity for activating RhoA, but not other Rho family members, Rac1 and Cdc42.30 The activation of RhoA stimulates ROCK (Rho-associated kinase), which is known to have several different substrates, including actin-binding/regulating proteins (LIMK [LIM kinase], MLC [myosin light chain], and ezrin/radixin/moesin [ERM]), adducin, and calponin), microtubule-binding/regulating proteins (CRMP2/DPY12 and MAP2/Tau), and intermediate filaments (GFAP and Vimentin).31
In terms of actin dynamics, activation of RhoA/ROCK can lead to stress fiber formation (ie, changes in cell shape), gene expression changes, cell transformation (eg, epithelial-mesenchymal transition phenotype), and vesicle trafficking.32 Changes in actin dynamics (ratio G/F-actin pool) can impact SRF (serum response factor), a transcription factor that binds the serum response element in a number of genes associated with cytoskeletal proteins (eg, α-SMA), myofibroblast activation, and synthesis of extracellular matrix proteins.33,34 ROCK activation by RhoA also leads to phosphorylation of MLC and MLC phosphatase (inactivation) and can subsequently promote the development of stress fibers, impact cell contractility, and cell-cell contacts.35
As ARHGEF11 is known to act through RhoA, it was expected that many genes involved in the canonical (RhoA-ROCK) pathway would be dysregulated between WT and KO. However, based on the real-time PCR results, few genes were observed to be differentially expressed either at week 4 or 12. Therefore, an unbiased multi-omics strategy was used to identify other pathways that could contribute to the renal protection observed in the KO rats. Whole transcriptome and proteomic analyses demonstrated that loss of Arhgef11 did have an impact on genes involved in cytoskeleton regulation (actin, intermediate filaments, and microtubule components), but these were different than those in the canonical pathway. In contrast, previous work involving the SS-Arhgef11SHR did demonstrate altered RhoA/ROCK signaling at the protein level as assessed in whole kidney (Western blot).13 Primary proximal tubule cells cultured from the SS-WT exhibited increased expression of Arhgef11, increased RhoA activity, and upregulation of Rho-ROCK signaling compared with SS-Arhgef11SHR 14. The proximal tubule cells from the SS-WT were also more prone to TGFβ1-induced epithelial-mesenchymal transition, a hallmark feature of the development of renal interstitial fibrosis. The observed difference between the SS-Arhgef11SHR and KO could be the result of compensatory mechanisms (eg, increased expression/activation of other GEFs, RGS, etc) from the complete loss of Arhgef11 versus only reduced expression and functional differences conferred through the SHR allele.
Despite the lack of detectable differences in canonical RhoA/ROCK signaling pathway, there were transcriptome and proteomic changes in other genes/protein involved in actin filament regulation (Coro2a, Mylk, Actn1, Rac2, Marcksl1), intermediate filaments (eg, Des, Vim, and various keratins), and microtubules (Rarres1, Tubb6, Map1, 6, 7, and 9) as well as genes/proteins involved in actomyosin microtubule cross-talk (Myh9). In particular, there were differentially expressed genes/proteins for several interesting microtubule-associated factors. For example, Rarres1 was observed to be significantly dysregulated (>5-fold difference) at both week 4 (top gene) and at week 12. Rarres1 demonstrates high gene expression/protein levels in the kidney, particularly in the tubules (http://www.proteinatlas.org).36 Cell culture and animals studies have associated RARRES1 as a driver of fibrosis37 as well as the regulation of tubulin (component of the actin cytoskeleton) and impact mitochondrial bioenergetics.38 The association between loss of Arhgef11 and dysregulation of Rarres1 could potentially explain the fact that genes/proteins involved in metabolism and mitochondrial function were significantly enriched at both weeks 4 and 12.
There also has been at least one study suggesting that RARRES1 is a potential novel regulator of lipid metabolism.39 The kidney is second only to the heart in mitochondrial count and oxygen consumption. Thus, the mitochondria play an important role in proper function of the kidney. In particular, fatty acid oxidation is an important energy source for the proximal tubule as it generates more ATP than does glucose.40 Alterations in the balance of fatty acid synthesis, uptake, and consumption can lead to the accumulation of excess oxidized lipids, which have been linked to increased proinflammatory pathways that contribute to renal injury.41 Overall, there were a large number of genes/proteins involved in lipid metabolism that were dysregulated between WT and KO at week 4 and week12. In summary, altered expression of Rarres1 could both be involved in mechanisms that lead to fibrosis in the SS-WT as well as altered mitochondrial bioenergetics.
There are a number of receptor mechanisms that activate the RhoA-signaling pathway (directly or indirectly), including stimulation of GPCR through LPA (lysophosphatidic acid),42 ET-1 (endothelin 1), Ang II (angiotensin II),43,44 thrombin, and S1P2 and S1P3 (sphingosine-1-phosphate,45 integrins (CTGF, Fibronectin)46,47 chemokines and growth factor receptors (TGFβ−1),48 many of which have been associated with kidney injury. In particular, LPA binds the LPAR (LPA receptor), of which there are at least 3 (LPAR1–3) that has been shown to interact with G12/13.49 LPA is a bioactive lysophospholipid that have been linked in the pathogenesis of kidney disease, especially renal fibrosis.50 The hydrolysis of lysophosphatidylcholine and other lysophospholipids to LPA results mainly from ATX (autotaxin, also known as EPP2). In particular, dysregulation of LPAR2–3, as well as EPP2, was observed at week 4. The activation of LPA1 and RhoA/ROCK-dependent signaling cascades facilitates binding of SRF to induce production of profibrotic cytokines, CTGF (connective tissue growth factor), and TGFβ (transforming growth factor) in the proximal tubules, which can promote epithelial-mesenchymal transition and development of fibrosis.49 In support of this role, LPA1−/− mice demonstrate an attenuation of fibrosis in the unilateral ureteral obstruction model of renal fibrosis.51
Similar to LPA, S1P functions through a GPCR mechanism, namely through various S1PR1–5 (S1P receptors), which also couples to G12/13.52 Initially, S1P is generated via SPHK1 (sphingosine kinase 1), exported out of the cell by a number of transporters, including the ATP-binding cassette transporters and SPNS1–2 transporters.53 While no expression differences were identified in Sphk1 between SS-WT and KO, there were a number of genes in this pathway dysregulated at week 12, including Abc1, Abcg2, and Spns2. The S1P-S1PRs axis has also been linked to renal fibrosis as S1P acting through S1PR2 induced differentiation of tubular epithelial cells into a myofibroblast/epithelial-mesenchymal transition, with changes in distribution of E-cadherin and increased α-SMA expression.45,54 Expression of SphK1 and S1PRs were increased in renal tissues of WT-unilateral ureteral obstruction mice, whereas the increase in renal SphK1 mRNA was blocked in KO-unilateral ureteral obstruction mice.55 Thus, the dysregulation of LPA-ATX and S1P-S1PR signaling pathway through ARHGEF11 may be linked to the attenuation of tubulointerstitial fibrosis observed in the KO as well as in the previous studies involving the SS-Arhgef11SHR.
Finally, pathways involved in solute carrier transport (especially Na+ cotransporters) were significantly enriched in-omics data sets, even before significant phenotype changes were observed. As sodium handling in the kidney plays a significant role in regulation of blood pressure, dysregulation of Na+ cotransporters between the WT and KO could provide some insight into loss of the salt sensitive phenotype observed in the SS-Arhgef11−/−. Many of the genes identified in the current analysis have been implicated at some level in the pathophysiology of kidney disease, including Slc13a1/2,56 Slc34a1/2,57 and Slc5a11.58 In particular, Slc8a1 (a Na+Ca2+ exchanger) which was dysregulated has been linked to hypertension and sodium sensitivity via human genetic studies.59 Genetic studies in mice demonstrated overexpressing Slc8a1 increased BP or alternatively reduced BP in Slc8a1+/− under DOCA-salt hypertension and salt-loading conditions.60 Interestingly, the linkage between G12 and G13 and RhoA with changes in the actin cytoskeleton was first suggested by stimulation of Slc9a1 (Na+–H+ exchanger).61 And while no expression of Slc9a1 or protein level changes were observed between SS-WT and SS-KO, this work supports a direct connection between ARHGEF11-RhoA and an important Na+ solute carrier. Another known association between RhoA and Na+-H+ exchangers occurs via Ezrin and SLC9A3R1,62 both of which were dysregulated between WT and KO at week 12. SLC9A3R1 participates as scaffold protein that connects plasma membrane proteins with ERM family members linking them to the actin cytoskeleton regulating SLC9A3 (Na+–H+ exchanger 3) and its subcellular location.63 Alternations in any number of Na+ cotransporters and SLC9A3, which plays an important role in regulation of water and salt homeostasis, could be involved in the observed loss of salt sensitive BP response in the KO.
In total, the physiological and molecular data generated suggests that there are a number of diverse molecular processes that account for the attenuated renal injury and blood pressure observed in the KO rats (Figure 6). Dysregulation of the LPA-ATX and S1P-S1PR signaling pathways, as well as important Na+ cotransporters functioning through ARHGEF11, could partially account for the development of hypertension and renal injury in the Dahl SS (or likewise protection in KO). The Dahl SS is highly permissive for the development of hypertension and renal injury, with genetic studies demonstrating at least 9 different loci contributing (compared with the SHR).12 With this in mind, it is important to note that even though the KO demonstrates significant attenuation of hypertension and renal injury, many other genes/factors that promote disease are still present. Thus, the mechanistic changes that result from the loss of Arhgef11 need to be considered in the context of these other susceptibility factors (eg, existing hypertension, vascular injury, activation of immune factors, etc), which independently or in concert with pathways associated with GPCR-ARHGEF11 axis leads to hypertension and kidney disease.
Figure 6.
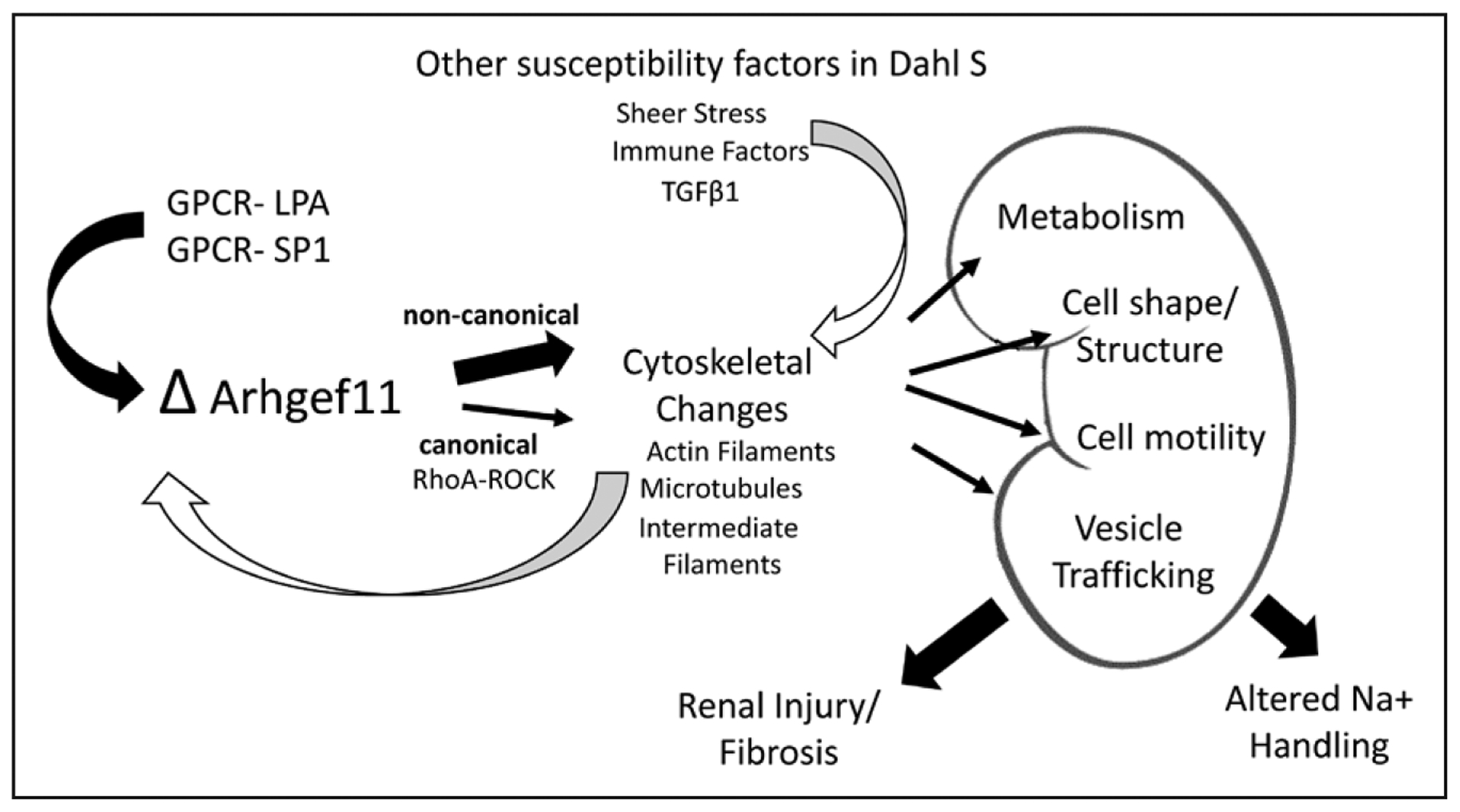
Overview of proposed physio-molecular mechanism of the role of Arhgef11 in onset and progression of kidney injury. GPCR indicates G protein-coupled receptor; LPA, lysophosphatidic acid; SP1, sphingosine-1-phosphate; and TGFβ, transforming growth factor-β.
There are some limitations to our study. For example, the use of a systemic knockout of Arhgef11 confounds our ability to exclusively determine the sole impact of Arhgef11 in the kidney. It is likely that the loss of Arhgef11 in other tissue/cell types, such as the systemic vasculature, contributes to the regulation of blood pressure and plays a role in the reduced BP observed in the KO. Unfortunately, rat gene-editing/genetic models are not as developed as mouse models, and future studies will be needed to better dissect tissue and cell types–specific influences of loss of Arhgef11. Specifically, the impact of Arhgef11 in different cell types of the kidney can be investigated using Arhgef11 flox/flox and various Cre recombinase lines (SGLT2-Cre, etc) in mice. Of course, the limitation of these models are a lack of genetic hypertensive mouse model that exhibit susceptibility to kidney injury and the need to use experimental models of hypertension (Ang II, DOCA, etc) which may or may not provide the same context as studying the role in the Dahl SS model that spontaneously develops hypertension and renal disease. Additionally, while the whole transcriptome analysis can provide strong insight into pathway changes, it also does not provide context of which kidney cell types (endothelial, podocytes, mesangial, proximal tubule [S1–S3], distal tubule, etc) may be most impacted by loss of Arhgef11. Future studies applying single-cell RNA sequencing technology will provide a more detailed view of molecular changes at the level of major cell types in the kidney.
Perspectives
There have been several human studies that have identified an association between ARHGEF11 and altered metabolism/impaired glucose tolerance and diabetes mellitus,64,65 susceptibility to intracranial aneurysm,66 bipolar disorder,67 and schizophrenia.68 In total, the precise molecular mechanism that links ARHGEF11 to these disease processes is lacking, but it is expected to work through the RhoA/ROCK pathway. The current study provides strong evidence that Arhgef11 also plays an important role in cardiovascular disease processes, including hypertension and renal injury. We hypothesize that Arhgef11 has a central role in the regulation of GPCR/Gα12 and Gα13, particularly signaling through the LPA-ATX and S1P-S1PR pathways, which have a strong association with the development of fibrosis. In addition, there appears to be a diverse number of cellular functions, including mitochondrial metabolism, solute carrier transporters, and vesicle trafficking, that work through Arhgef11 to promote kidney injury, hypertension, and decline in kidney function in the Dahl SS model. In summary, the current findings provide several molecular factors that contribute to hypertensive CKD and upon further investigation may provide utility as novel therapeutics targets.
Supplementary Material
Novelty and Significance.
What Is New?
The current study is the first to assess the role of Arhgef11 in the development of hypertension-related kidney injury and associated molecular pathways.
A multi-omics approach identified that regulation of the LPA (lysophosphatidic acid)-ATX (autotaxin) and S1P-S1PR (sphingosine-1-phosphate) signaling pathways as well as important Na+ cotransporters function via ARHGEF11 and partially account for the development of hypertension and renal injury in the Dahl salt-sensitive rats.
What Is Relevant?
Hypertension is the second leading cause of renal failure in humans. Identification of novel genes (such as Arhgef11) and pathways causative to kidney injury in the context of hypertension could serve as novel therapeutic targets.
Summary
Previous genetic studies using the Dahl salt-sensitive rats identified broad genomic intervals linked with cardiovascular and kidney disease; subsequent congenic strain analysis implicated a small subset of genes. This work provides a significant advance by identifying a single novel gene, Arhgef11, and pathways that play a significant role in development of onset and progress of kidney disease, which is highly relevant for human hypertensive kidney disease.
Acknowledgments
We thank Joshua Jefferson for processing, sectioning, and staining of the histology samples.
Sources of Funding
This work was supported by R01HL137673 (M.R. Garrett), R01HL134711 (J.M. Sasser), and HL075360, HL129823, and HL137319 and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Number 5I01BX000505 (M.L. Lindsey). The work performed through the University of Mississippi Medical Center Molecular and Genomics Facility is supported, in part, by funds from the NIGMS, including Mississippi INBRE (P20GM103476), Obesity, Cardiorenal and Metabolic Diseases-COBRE (P20GM104357), and Mississippi Center of Excellence in Perinatal Research (MS-CEPR)-COBRE (P20GM121334).
Footnotes
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/HYPERTENSIONAHA.119.14338.
Disclosures
None.
References
- 1.Centers for Disease Control and Prevention. Chronic Kidney Disease Surveillance System—United States. 2019. http://www.cdc.gov/ckd.
- 2.United States Renal Data System. 2018 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2018. https://www.usrds.org/. [Google Scholar]
- 3.Bash LD, Coresh J, Köttgen A, Parekh RS, Fulop T, Wang Y, Astor BC. Defining incident chronic kidney disease in the research setting: the ARIC Study. Am J Epidemiol. 2009;170:414–424. doi: 10.1093/aje/kwp151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazancioğlu R Risk factors for chronic kidney disease: an update. Kidney Int Suppl (2011). 2013;3:368–371. doi: 10.1038/kisup.2013.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, Conlon E, Nakayama M, van der Ven AT, Ityel H, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019;95:914–928. doi: 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cañadas-Garre M, Anderson K, Cappa R, Skelly R, Smyth LJ, McKnight AJ, Maxwell AP. Genetic susceptibility to chronic kidney disease – some more pieces for the heritability puzzle. Front Genet. 2019;10:453. doi: 10.3389/fgene.2019.00453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wuttke M, Köttgen A. Insights into kidney diseases from genome-wide association studies. Nat Rev Nephrol. 2016;12:549–562. doi: 10.1038/nrneph.2016.107 [DOI] [PubMed] [Google Scholar]
- 8.Limou S, Vince N, Parsa A. Lessons from CKD-Related Genetic Association studies–moving forward. Clin J Am Soc Nephrol. 2018;13:140. doi: 10.2215/CJN.09030817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teumer A, Li Y, Ghasemi S, Prins BP, Wuttke M, Hermle T, Giri A, Sieber KB, Qiu C, Kirsten H, et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat Commun. 2019;10:4130. doi: 10.1038/s41467-019-11576-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1985;7(3 pt 1):340–349. [PubMed] [Google Scholar]
- 11.Rapp JP, Garrett MR. Will the real dahl S rat please stand up? Am J Physiol Renal Physiol. 2019;317:F1231–F1240. doi: 10.1152/ajprenal.00359.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrett MR, Dene H, Rapp JP. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J Am Soc Nephrol. 2003;14:1175–1187. doi: 10.1097/01.asn.0000060572.13794.58 [DOI] [PubMed] [Google Scholar]
- 13.Williams JM, Johnson AC, Stelloh C, Dreisbach AW, Franceschini N, Regner KR, Townsend RR, Roman RJ, Garrett MR. Genetic variants in arhgef11 are associated with kidney injury in the Dahl salt-sensitive rat. Hypertension. 2012;60:1157–1168. doi: 10.1161/HYPERTENSIONAHA.112.199240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia Z, Johnson AC, Wang X, Guo Z, Dreisbach AW, Lewin JR, Kyle PB, Garrett MR. Allelic variants in arhgef11 via the rho-rock pathway are linked to rpithelial-mesenchymal transition and contributes to kidney injury in the Dahl salt-sensitive rat. PLoS One. 2015;10:e0132553. doi: 10.1371/journal.pone.0132553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Johnson AC, Williams JM, White T, Chade AR, Zhang J, Liu R, Roman RJ, Lee JW, Kyle PB, et al. Nephron deficiency and predisposition to renal injury in a novel one-kidney genetic model. J Am Soc Nephrol. 2015;26:1634. doi: 10.1681/ASN.2014040328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Johnson AC, Sasser JM, Williams JM, Solberg Woods LC, Garrett MR. Spontaneous one-kidney rats are more susceptible to develop hypertension by DOCA-NaCl and subsequent kidney injury compared with uninephrectomized rats. Am J Physiol Renal Physiol. 2016;310:F1054–F1064. doi: 10.1152/ajprenal.00555.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regner KR, Harmon AC, Williams JM, Stelloh C, Johnson AC, Kyle PB, Lerch-Gaggl A, White SM, Garrett MR. Increased susceptibility to kidney injury by transfer of genomic segment from SHR onto Dahl S genetic background. Physiol Genomics. 2012;44:629–637. doi: 10.1152/physiolgenomics.00015.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ 2nd, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, Lindsey ML. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. 2019;114:6. doi: 10.1007/s00395-019-0715-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turbeville HR, Taylor EB, Garrett MR, Didion SP, Ryan MJ, Sasser JM. Superimposed preeclampsia exacerbates postpartum renal injury despite lack of long-term blood pressure difference in the Dahl salt-sensitive rat. Hypertension. 2019;73:650–658. doi: 10.1161/HYPERTENSIONAHA.118.12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindsey ML, Gray GA, Wood SK, Curran-Everett D. Statistical considerations in reporting cardiovascular research. Am J Physiol Heart Circ Physiol. 2018;315:H303–H313. doi: 10.1152/ajpheart.00309.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goicoechea SM, Awadia S, Garcia-Mata R. I’m coming to GEF you: regulation of rhoGEFs during cell migration. Cell Adh Migr. 2014;8:535–549. doi: 10.4161/cam.28721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis SA, Shen X, Young JB, Kaul P, Lerner DJ. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood. 2006;107:1627–1635. doi: 10.1182/blood-2005-03-1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868 [DOI] [PubMed] [Google Scholar]
- 26.Kimple AJ, Bosch DE, Giguère PM, Siderovski DP. Regulators of G-protein signaling and their Gα substrates: promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63:728–749. doi: 10.1124/pr.110.003038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riobo NA, Manning DR. Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol Sci. 2005;26:146–154. doi: 10.1016/j.tips.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 28.Suzuki N, Hajicek N, Kozasa T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals. 2009;17:55–70. doi: 10.1159/000186690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park F Accessory proteins for heterotrimeric G-proteins in the kidney. Front Physiol. 2015;6:219. doi: 10.3389/fphys.2015.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol. 2018;217:447–457. doi: 10.1083/jcb.201612069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken). 2010;67:545–554. doi: 10.1002/cm.20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sah VP, Seasholtz TM, Sagi SA, Brown JH. The role of rho in G protein-coupled receptor signal transduction. Annu Rev Pharmacol Toxicol. 2000;40:459–489. doi: 10.1146/annurev.pharmtox.40.1.459 [DOI] [PubMed] [Google Scholar]
- 33.Tsou PS, Haak AJ, Khanna D, Neubig RR. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on rho GTPase-regulated gene transcription. Am J Physiol Cell Physiol. 2014;307:C2–C13. doi: 10.1152/ajpcell.00060.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chai J, Norng M, Tarnawski AS, Chow J. A critical role of serum response factor in myofibroblast differentiation during experimental oesophageal ulcer healing in rats. Gut. 2007;56:621–630. doi: 10.1136/gut.2006.106674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itoh M, Tsukita S, Yamazaki Y, Sugimoto H. Rho GTP exchange factor ARHGEF11 regulates the integrity of epithelial junctions by connecting ZO-1 and rhoA-myosin II signaling. Proc Natl Acad Sci U S A. 2012;109:9905–9910. doi: 10.1073/pnas.1115063109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjöstedt E, Asplund A. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 37.Teufel A, Becker D, Weber SN, Dooley S, Breitkopf-Heinlein K, Maass T, Hochrath K, Krupp M, Marquardt JU, Kolb M, et al. Identification of RARRES1 as a core regulator in liver fibrosis. J Mol Med (Berl). 2012;90:1439–1447. doi: 10.1007/s00109-012-0919-7 [DOI] [PubMed] [Google Scholar]
- 38.Maimouni S, Lee MH, Sung YM, Hall M, Roy A, Ouaari C, Hwang YS, Spivak J, Glasgow E, Swift M, et al. Tumor suppressor RARRES1 links tubulin deglutamylation to mitochondrial metabolism and cell survival. Oncotarget. 2019;10:1606–1624. doi: 10.18632/oncotarget.26600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maimouni S, Issa N, Cheng S, Ouaari C, Cheema A, Kumar D, Byers S. Tumor suppressor RARRES1- A novel regulator of fatty acid metabolism in epithelial cells. PLoS One. 2018;13:e0208756. doi: 10.1371/journal.pone.0208756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13:629–646. doi: 10.1038/nrneph.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakai N, Chun J, Duffield JS, Wada T, Luster AD, Tager AM. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013;27:1830–1846. doi: 10.1096/fj.12-219378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ushio-Fukai M, Alexander RW, Akers M, Lyons PR, Lassègue B, Griendling KK. Angiotensin II receptor coupling to phospholipase D is mediated by the betagamma subunits of heterotrimeric G proteins in vascular smooth muscle cells. Mol Pharmacol. 1999;55:142–149. doi: 10.1124/mol.55.1.142 [DOI] [PubMed] [Google Scholar]
- 44.Domazet I, Holleran BJ, Richard A, Vandenberghe C, Lavigne P, Escher E, Leduc R, Guillemette G. Characterization of angiotensin II molecular determinants involved in AT1 receptor functional selectivity. Mol Pharmacol. 2015;87:982–995. doi: 10.1124/mol.114.097337 [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Ritter JK, Li N. Sphingosine-1-phosphate pathway in renal fibrosis. Am J Physiol Renal Physiol. 2018;315:F752–F756. doi: 10.1152/ajprenal.00596.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu J, Nguyen D, Ouyang H, Zhang XH, Chen XM, Zhang K. Inhibition of rhoA/rho-kinase pathway suppresses the expression of extracellular matrix induced by CTGF or TGF-β in ARPE-19. Int J Ophthalmol. 2013;6:8–14. doi: 10.3980/j.issn.2222-3959.2013.01.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renshaw MW, Toksoz D, Schwartz MA. Involvement of the small GTPase rho in integrin-mediated activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:21691–21694. doi: 10.1074/jbc.271.36.21691 [DOI] [PubMed] [Google Scholar]
- 48.Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am J Physiol Lung Cell Mol Physiol. 2011;301:L656–L666. doi: 10.1152/ajplung.00166.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park F, Miller DD. Role of lysophosphatidic acid and its receptors in the kidney. Physiol Genomics. 2017;49:659–666. doi: 10.1152/physiolgenomics.00070.2017 [DOI] [PubMed] [Google Scholar]
- 50.Sakai N, Bain G, Furuichi K, Iwata Y, Nakamura M, Hara A, Kitajima S, Sagara A, Miyake T, Toyama T, et al. The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci Rep. 2019;9:7414. doi: 10.1038/s41598-019-43576-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pradère JP, Klein J, Grès S, Guignéc C, Neau E, Valet P, Calise D, Chun J, Bascands JL, Saulnier-Blache JS. LPA1 receptor activation promotes renal interstitial fibrosis. Journal Am Soc Nephrol. 2007;18:3110–3118. doi: 10.1681/ASN.2007020196 [DOI] [PubMed] [Google Scholar]
- 52.Zhang X, Cai Y, Zhang W, Chen X. Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P signaling. Biochem Cell Biol. 2018;96:742–751. doi: 10.1139/bcb-2017-0302 [DOI] [PubMed] [Google Scholar]
- 53.Zhu X, Ren K, Zeng YZ, Zheng Z, Yi GH. Biological function of SPNS2: from zebrafish to human. Molecular Immunology. 2018;103:55–62. [DOI] [PubMed] [Google Scholar]
- 54.Ishizawa S, Takahashi-Fujigasaki J, Kanazawa Y, Matoba K, Kawanami D, Yokota T, Iwamoto T, Tajima N, Manome Y, Utsunomiya K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under rho kinase activation via the S1P2 receptor. Clin Exp Nephrol. 2014;18:844–852. doi: 10.1007/s10157-014-0933-x [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Wang W, Ji XY, Ritter JK, Li N. Knockout of sphingosine kinase 1 attenuates renal fibrosis in unilateral obstruction model. Am J Nephrol. 2019;50:196–203. doi: 10.1159/000502448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marques FZ, Campain AE, Tomaszewski M, Zukowska-Szczechowska E, Yang YHJ, Charchar FJ, Morris BJ. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension. 2011;58:1093–1098. doi: 10.1161/HYPERTENSIONAHA.111.180729 [DOI] [PubMed] [Google Scholar]
- 57.Lederer E, Wagner CA. Clinical aspects of the phosphate transporters naPi-IIa and naPi-IIb: mutations and disease associations. Pflugers Arch. 2019;471:137–148. doi: 10.1007/s00424-018-2246-5 [DOI] [PubMed] [Google Scholar]
- 58.Gil RB, Ortiz A, Sanchez-Niño MD, Markoska K, Schepers E, Vanholder R, Glorieux G, Schmitt-Kopplin P, Heinzmann SS. Increased urinary osmolyte excretion indicates chronic kidney disease severity and progression rate. Nephrol Dial Transplant. 2018;33:2156–2164. doi: 10.1093/ndt/gfy020 [DOI] [PubMed] [Google Scholar]
- 59.Citterio L, Simonini M, Zagato L, Salvi E, Delli Carpini S, Lanzani C, Messaggio E, Casamassima N, Frau F, D’Avila F, et al. Genes involved in vasoconstriction and vasodilation system affect salt-sensitive hypertension. PLoS One. 2011;6:e19620. doi: 10.1371/journal.pone.0019620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med. 2004;10:1193–1199. doi: 10.1038/nm1118 [DOI] [PubMed] [Google Scholar]
- 61.Hooley R, Yu CY, Symons M, Barber DL. G alpha 13 stimulates Na+–H+ exchange through distinct Cdc42-dependent and RhoA-dependent pathways. J Biol Chem. 1996;271:6152–6158. doi: 10.1074/jbc.271.11.6152 [DOI] [PubMed] [Google Scholar]
- 62.Morales FC, Takahashi Y, Kreimann EL, Georgescu MM. Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc Natl Acad Sci U S A. 2004;101:17705–17710. doi: 10.1073/pnas.0407974101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Babich V, Di Sole F. The Na+/H+ Exchanger-3 (NHE3) activity requires ezrin binding to phosphoinositide and its phosphorylation. PLoS One. 2015;10:e0129306. doi: 10.1371/journal.pone.0129306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu M, Sabra MM, Damcott C, Pollin TI, Ma L, Ott S, Shelton JC, Shi X, Reinhart L, O’Connell J, et al. Evidence that rho guanine nucleotide exchange factor 11 (ARHGEF11) on 1q21 is a type 2 diabetes susceptibility gene in the old order amish. Diabetes. 2007;56:1363–1368. doi: 10.2337/db06-1421 [DOI] [PubMed] [Google Scholar]
- 65.Jin QS, Kim SH, Piao SJ, Lim HA, Lee SY, Hong SB, Kim YS, Lee HJ, Nam M. R1467H Variants of Rho Guanine Nucleotide Exchange Factor 11 (ARHGEF11) are associated with type 2 diabetes mellitus in Koreans. Korean Diabetes J. 2010;34:368–373. doi: 10.4093/kdj.2010.34.6.368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akiyama K, Narita A, Nakaoka H, Cui T, Takahashi T, Yasuno K, Tajima A, Krischek B, Yamamoto K, Kasuya H, et al. Genome-wide association study to identify genetic variants present in Japanese patients harboring intracranial aneurysms. J Hum Genet. 2010;55:656–661. doi: 10.1038/jhg.2010.82 [DOI] [PubMed] [Google Scholar]
- 67.Kermath BA, Vanderplow AM, Cahill ME. Dysregulated prefrontal cortical rhoA signal transduction in bipolar disorder with psychosis: new implications for disease pathophysiology. Cerebral Cortex. 2019;01:1–13. [DOI] [PubMed] [Google Scholar]
- 68.Mizuki Y, Takaki M, Okahisa Y, Sakamoto S, Kodama M, Ujike H, Uchitomi Y. Human Rho guanine nucleotide exchange factor 11 gene is associated with schizophrenia in a Japanese population. Hum Psychopharmacol. 2014;29:552–558. doi: 10.1002/hup.2435 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.