

Abstract
This article reports the complete chloroplast genome of Achnatherum inebrians, a poisonous herb that is widely distributed in the rangelands of Northern China. The genome is 137 714 bp in total and consists of a large single‐copy (81 758 bp) region and small single‐copy (12 682 bp) region separated by a pair of inverted repeats (21 637 bp). The genome contains 130 genes, including 84 protein‐coding genes, 38 tRNA genes and 8 ribosomal RNA genes, and the guanine + cytosine content is 36.17%. We subsequently performed comparative analysis of complete genomes from A. inebrians and other Poaceae‐related species from GenBank. Thirty‐eight simple sequence repeats were identified, further demonstrating rapid evolution in Poaceae. Finally, the phylogenetic trees of 37 species of Poaceae and 2 species of Amaranthaceae were constructed by using maximum likelihood and Bayesian inference methods, based on the genes of the complete chloroplast genome. We identified hotspots that can be used as molecular markers and barcodes for phylogenetic analysis, as well as for species identification. Phylogenetic analysis indicated that A. inebrians is a member of the genus Stipa rather than Achnatherum.
Keywords: Achnatherum inebrians, chloroplast genomes, comparative analysis, phylogenetic analysis, Poaceae
The complete genomes from Achnatherum inebrians and other Poaceae‐related species from GenBank were subjected to comparative analysis. We not only identified hotspots that can be used as molecular markers and barcodes for phylogenetic analysis and species identification but also identified phylogenetic evidence that A. inebrians is a member of the genus Stipa sp. rather than Achnatherum sp.

Abbreviations
- BI
Bayesian inference
- GC
guanine + cytosine
- IR
inverted repeat
- IRa
inverted repeat region a
- IRb
inverted repeats region b
- LSC
large single copy
- ML
maximum likelihood
- NCBI
National Center for Biotechnology Information
- Pi
nucleotide variation
- RSCU
relative synonymous codon usage
- SSC
small single copy
- SSR
simple sequence repeat
Achnatherum inebrians is a common and widespread perennial toxic grass in the semiarid grassland regions of northern China [1]. In earlier classification, A. inebrians was named as Stipa inebrians, but Geng [2, 3] revised its classification from Stipa to Achnatherum (Gramineae, Pooideae, Stipeae), which is still used today. Chu and Yang [4] identified A. inebrians as the section [sect. Achnatheropsis (Tzvel.) Q.G.Chu.comb.nov.] according to the external morphology of the genus Achnatherum in 1990. This grass is majorly involved in reverse degradation and loss of biodiversity of overgrazed grasslands, while it serves as a diversity refuge for the soil fungal community [5, 6]. In Northwestern China, almost all the plants of A. inebrians are infected by a symptomless fungal endophyte, Epichloë (Epichloë gansuensis or Epichloë inebrian) [7, 8, 9]. Achnatherum inebrians is commonly referred to as drunken horse grass because of the presence of two alkaloids produced in Epichloë endophyte‐infected A. inebrians plants, ergonovine and ergine, which cause toxicity or death to horses and other livestock [10, 11, 12]. The presence of Epichloë endophytes in aboveground tissues can regulate the metabolic processes of host grasses, including promoting plant growth and enhancing the tolerance of host plants to various biotic and abiotic stresses, such as heavy metals, low temperature, drought and salinity [13, 14, 15, 16, 17, 18, 19].
Chloroplasts are small photosynthetic machinery and carbon fixation organelles that are present in algae and plant cells. Most chloroplast‐encoded proteins are responsible for photosynthesis and the synthesis of fatty acids and amino acids [20, 21]. Chloroplasts have their own genetic system, consisting of a closed circular structure ranging from 115 to 165 kb in length, a small single‐copy (SSC) region, a large single‐copy (LSC) region and a pair of inverted repeats (IRs) [22, 23, 24, 25]. Compared with nuclear genomes, chloroplast genomes have fewer nucleotide substitutions and rearrangements of genome structures, moderate genome size, and desirable collinear properties among different species, providing an ideal model to decipher genomic evolution and phylogenetic relationships in angiosperms [26, 27]. High‐throughput sequencing technology has stimulated the rapid development of chloroplast genome sequencing [28] and enabled the study of evolutionary dynamics at a more taxonomically complex level (species or lower level) [29].
Achnatherum species are poorly studied from a genomic perspective. To date, chloroplast genomes are available for only one representative, Achnatherum splendens [30]. This study for the first time reports the complete chloroplast genome sequence of A. inebrians, including a description of its general features, IR contraction and expansion, codon usage and analysis of simple sequence repeats (SSRs). In addition, we compared the gene contents, organization, and phylogenetic relationships with other chloroplast genomes in Poaceae, which will help improve the understanding of chloroplast genome characteristics, structural diversity and evolution within Poaceae.
Materials and methods
Sample collection and DNA extraction
Fresh A. inebrians leaves were collected from alpine grassland in Tianzhu county (37°11′N, 102°47′E), Gansu province, China. For chloroplast genome DNA extraction, the collected fresh pieces were immediately placed in liquid nitrogen and stored at −80°C until chloroplast genome DNA was extracted. The voucher specimen was stored at the Official Herbage and Turfgrass Seed Testing Centre, Ministry of Agriculture, Lanzhou, China. Total genomic DNA was extracted using the hexadecyltrimethyl ammonium bromide method, and the quality of chloroplast genome was measured by NanoDrop 2000 (Thermo Scientific, Wilmington, NC, USA) and agarose gel electrophoresis. The quantified DNA (260/280 value is 1.6–1.8, and the concentration is >20 ng·μL−1; the band is about 5K) was used for library construction.
Library preparation and sequencing and genome assembly
The qualified library was sequenced with Illumina NovaSeq (Wuhan Benagen Tech Solutions Company Limited, Wuhan, China). The raw sequencing data were filtered with low‐quality data to obtain effective data. soapnuke (Version: 2.1.0; Wuhan Benagen Tech Solutions Company Limited, Wuhan, Hubei, China) was used as the filtering software for the project, and the filtering standards were as follows: (a) remove reads with N base content exceeding 5%, (b) remove reads with low mass (Q score ≤ 5) and the number of bases reaches 50%, and (c) remove the adapter sequence contained in reads. The Illumina NovaSeq sequester was used for paired‐end sequencing, and the reads length was 150 bp, which in pieces was done by nucleic acid shear (Covaris M220; USA) apparatus [centrifuge at 3000 g (relative centrifugal force) for 1 min].
Chloroplast genome assembly was performed using novoplasty software (version 3.2; parameter: k‐mer = 39; https://github.com/ndierckx/novoplasty), and the published gene sequence of the target species was selected as the seed sequence (JF698225.1) to splice chloroplast genomes. The joining together with the relative chloroplast genome (NC_029390.1) was blastn (version: blast 2.9.0+; parameter: −e value, 1e−5; ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) alignment, which adjusts the order of target sequences based on alignment with related species. If the connected sequence contains gap (including N sequence), then gapcloser (version 1.12; https://github.com/aquaskyline/SOAPdenovo2) was used to further fill the hole to obtain the final stitching result.
Genome annotation and comparative genome analyses
Chloroplast genome functional annotation includes encoding gene prediction and noncoding RNA annotation (rRNA and tRNA annotations). Gene annotation was performed using CPGAVAS2 [31], and the map of the circular A. inebrians chloroplast genome was drawn through the online tool Chloroplot [32].
The distribution of codon usage was detected by using codonw (version 1.4.4; https://sourceforge.net/projects/codonw/) with the relative synonymous codon usage (RSCU) ratio [33]. The codon of A. inebrians chloroplast was visually compared among species of 17 Poaceae with r language and tbtools [34].
The A. inebrians chloroplast genome was compared with the other five chloroplast genomes using the Shuffle–Lagan model of the mvista program [35]; Alopecurus japonicus served as the reference. irscope was used to visualize the boundaries between the IR and SC regions of A. inebrians, and the results were compared and analyzed with three other Poaceae species [36]. The four chloroplast genomes of Poaceae were initially compared using mafft [37] and then manually adjusted using bioedit [38]. Variable sites and nucleotide variations (Pi) in the entire chloroplast genome and LSC, IR and SSC regions of four species were calculated using dnasp [39].
Repeat sequence analyses
The SSRs of A. inebrians and three other chloroplast genomes were identified using the online web tool misa (version 2.1) [40]. The parameter sets of the minimum number of repetitions of SSRs for mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides and hexanucleotides were 10, 5, 4, 3, 3 and 3, respectively.
Phylogenetic analyses
Phylogenetic relationships were reconstructed by using the complete A. inebrians chloroplast genome and 36 other Poaceae chloroplast genomes submitted in the National Center for Biotechnology Information (NCBI); Cyperus rotundus and Eleocharis dulcis were used as outgroups. All species and accession numbers of the chloroplast genomes in NCBI are listed in Table S1. Phylogenetic analysis was conducted on the phylosuite version 1.2.2 platform [41]. The nucleotide sequence of the whole chloroplast genome was aligned in mafft based on default parameters [37]. Ambiguously aligned fragments were removed using gblocks [42], with the following parameter settings: minimum number of sequences for a conserved/flank position (20/20), maximum number of contiguous nonconserved positions (6), minimum length of a block (11) and allowed gap positions (0). ModelFinder [43] was used to select the best‐fit model using Akaike information criterion. Maximum‐likelihood (ML) phylogenies were inferred using iq‐tree [44] under the GTR+R4+F model for 5000 ultrafast [45] bootstraps, approximate Bayes test [46] and the Shimodaira–Hasegawa‐like approximate likelihood‐ratio test [47]. Bayesian inference (BI) phylogenies were inferred using mrbayes 3.2.0 [48] under the GTR+I+G+F model (two parallel runs and 1 000 000 generations), in which the initial 25% of sampled data were discarded as burn‐in. The generated trees were visualized using the online web tool iTOL [49].
Results
Chloroplast genome assembly and genome features
The genome size of the complete chloroplast genome of A. inebrians was 137 714 bp in length, with chloroplast circular molecules having quadripartite structures composed of IRa (21 637 bp) and IRb (21 637 bp) regions, separated by the LSC (81 758 bp) and SSC (12 682 bp) regions (Table 1; Fig. 1). The guanine + cytosine (GC) content of the complete chloroplast genomes was 38.8%, while LSC, SSC and IR regions showed 36.8%, 33.1% and 44.1% GC contents, respectively.
Table 1.
Summary of complete chloroplast genomes for Achnatherum inebrians, Achnatherum splendens, Stipa hymenoides, and Stipa purpurea.
| Item | Achnatherum inebrians | Achnatherum splendens | Stipa hymenoides | Stipa purpurea |
|---|---|---|---|---|
| Total size (bp) | 137 714 | 136 876 | 137 742 | 137 370 |
| LSC size (bp) | 81 758 | 80 958 | 81 709 | 81 202 |
| SSC size (bp) | 12 682 | 12 640 | 12 803 | 12 842 |
| IR size (bp) | 21 637 | 21 639 | 21 615 | 21 663 |
| Total GC content (%) | 38.8 | 38.9 | 38.8 | 38.8 |
| LSC GC content (%) | 36.8 | 36.7 | 36.9 | 36.9 |
| SSC GC content (%) | 33.1 | 33.3 | 33.6 | 32.9 |
| IR GC content (%) | 44.1 | 44.2 | 44.1 | 44.1 |
| Number of genes | 130 | 130 | 130 | 130 |
| Number of protein‐coding genes | 84 | 84 | 84 | 84 |
| Number of tRNA genes | 38 | 38 | 38 | 38 |
| Number of rRNA genes | 8 | 8 | 8 | 8 |
Fig. 1.
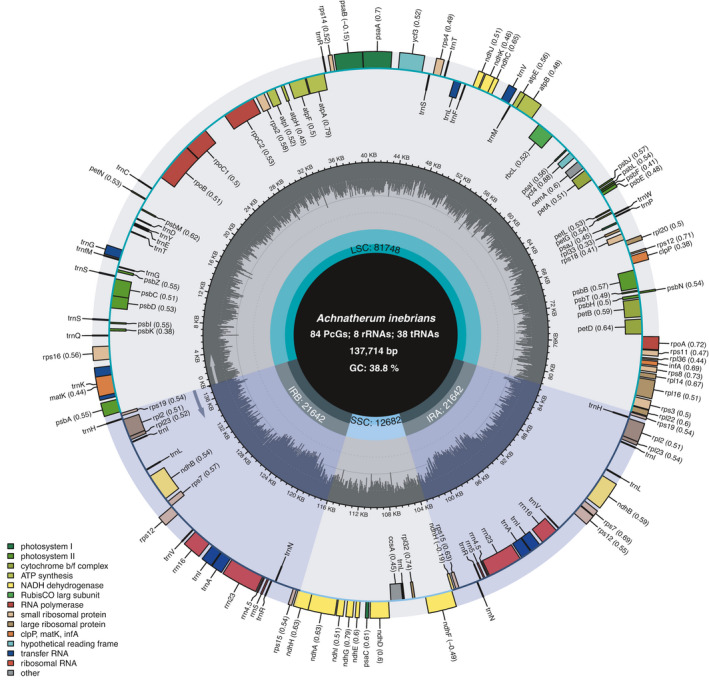
Chloroplast genome map of Achnatherum inebrians. The center of the figure provides the specific information (length, GC content and number of genes) of the A. inebrians chloroplast genome. In the first inner circle, the proportion of the shaded parts represents the GC content of each part. The lengths of the corresponding small single‐copy (SSC), IR (IRa and IRb) and LSC regions are also listed. The gene names and their optional codon usage bias are labeled on the outermost layer. The transcription directions for the inner and outer genes are listed clockwise and anticlockwise, respectively.
A total of 130 genes were found in the whole chloroplast genome of A. inebrians, including 84 protein‐coding genes, 38 tRNA genes, 8 rRNA genes, and 2 pseudogenes (ycf3 and ycf4; Table 1; Fig. 1). The protein‐coding genes include 11 genes for large ribosomal proteins [rpl32, rpl14, rpl22, rpl33, rpl20, rpl36, rpl23 (×2), rpl16, rpl2 (×2)], 16 for small ribosomal proteins [rps3, rps16, rps8, rps11, rps12 (×2), rps18, rps2, rps14, rps19 (×2), rps15 (×2), rps7 (×2), rps4], 5 for photosystem I (psaJ, psaA, psaB, psaC, psaI), 15 for photosystem II (psbB, psbK, psbH, psbL, psbA, psbI, psbM, psbJ, psbT, psbC, psbZ, psbF, psbD, psbE, psbN) and 6 for ATP synthase (Table 2).
Table 2.
List of annotated genes in the chloroplast of Achnatherum inebrians.
| Group | Gene group | Gene name |
|---|---|---|
| Self‐replication | Ribosomal proteins (LSU) | rpl32, rpl14, rpl22, rpl33, rpl20, rpl36, rpl23 a (×2), rpl16, b rpl2 a , b (×2) |
| Ribosomal proteins (SSU) | rps3, rps16, b rps8, rps11, rps12 a , b (×2), rps18, rps2, rps14, rps19 a (×2), rps15 a (×2), rps7 a (×2), rps4 | |
| RNA polymerase | rpoC2, rpoC1, rpoB, rpoA | |
| rRNA gene | rrn23 a (×2), rrn5 a (×2), rrn16 a (×2), rrn4.5 a (×2) | |
| tRNA genetrnC‐GCA | trnI‐CAU a (×2), trnS‐GGA, trnT‐GGU, trnC‐GCA, trnF‐GAA, trnN‐GUU a (×2), trnA‐UGC a , b (×2), trnP‐UGG,trnL‐CAA a (×2), trnI‐GAU a , b (×2), trnS‐GCU, trnG‐UCC,trnL‐UAG, trnR‐UCU, trnV‐GAC a (×2), trnT‐UGU, trnQ‐UUG, trnY‐GUA, trnR‐ACG a (×2), trnE‐UUC, trnW‐CCA, trnS‐UGA, trnH‐GUG a (×2), trnM‐CAU, trnK‐UUU, b trnD‐GUC, trnV‐UAC, b trnG‐GCC, trnfM‐CAU, trnL‐UAA b | |
| Gene for photosynthesis | Subunits of photosystem I | psaA, psaB, psaJ, psaI, psaC |
| Subunits of photosystem II | psbB, psbK, psbH, psbL, psbA, psbI, psbM, psbJ, psbT, psbC, psbZ, psbF, psbD, psbE, psbN | |
| Subunits of NADH dehydrogenase | ndhG, ndhB a , b (×2), ndhK, ndhD, ndhA, b ndhH a (×2), ndhF, ndhC, ndhI, ndhJ, ndhE | |
| Subunits of cytochrome b/f complex | petA, petG, petB, b petN, petD, b petL | |
| Subunits for ATP synthase | atpE, atpH, atpI, atpA, atpB, atpF b | |
| Large subunit RuBisCO | rbcL | |
| Other genes | Translational initiation factor | infA |
| Maturase | matK | |
| Protease | clpP | |
| Envelope membrane protein | cemA | |
| C‐type cytochrome synthesis gene | ccsA | |
| Hypothetical chloroplast reading frames (ycf) | ycf3, c ycf4 |
Genes located in the IRs.
Gene with one intron.
Gene with two introns.
In the chloroplast genome of A. inebrians, eight protein‐coding (rps19, rpl2, rpl23, ndhB, nadH, rps7, rps12 and rps15), four rRNA (rrn16, rrn23, rrn4.5 and rrn5) and eight tRNA genes (trnA‐UGC, trnH‐GUG, trnI‐GAU, trnI‐CAU, trnL‐CAA, trnN‐GUU, trnR‐ACG and trnV‐GAC) were duplicated in the IR regions (Fig. 1).
Introns play an important role in gene expression regulation. Many introns have the ability to enhance the high expression of exogenous genes at specific times and locations of plants, thus producing the desired agronomic traits. The chloroplast genome of A. inebrians includes 15 intron‐containing genes (Table S2). The pseudogene ycf3 has two introns, while all other genes contain a single intron. The intron of the trnK‐UUU gene is largest (2488 bp), and matK is located within its intron. The nadH gene is a transspliced gene with a 5′ exon located in an SSC region and two 3′ exons located in IR regions, as previously reported in other chloroplast genomes [50, 51].
Nucleotide sequences of protein‐coding genes usually start with ATG. However, there are some exceptions in the A. inebrians chloroplast genome in which the first nucleotide is changed from A to G or C, the second nucleotide is changed from T to C, and the third nucleotide is changed from G to C, such as rps19, which starts with GTG, rps12, starts with ACT, and rpl2, starts with ATA (Table S3). This is similar to the common features of many homologous genes reported in the chloroplast genomes of other plants [52, 53, 54, 55, 56, 57, 58].
Codon usage
The codon usage frequency and RSCU were analyzed based on the sequences of 84 protein‐coding genes in the A. inebrians chloroplast genome (Fig. 2). The highest frequency codon is ATT (leucine), which is the most abundant universal amino acid. The code usage pattern is similar to the reported patterns in other chloroplast genomes, with high A/T content. The codon used in the chloroplast genomes of 18 plants, including A. inebrians, was compared among all species to better understand the codon preference in Poaceae plants. As shown in Fig. 3, the distributions and the visualization of codon usage in the form of a heatmap of 18 species of Poaceae suggested that approximately one‐third of the codons was not frequently used. These codons are shown in blue, which indicates an RSCU value of less than 1 and weak codon bias. The results showed the codon usage preferences of the most chloroplast genome, among which TTA, AGA, GCT, TCT and ACT are used most frequently (Fig. 3). Approximately two‐thirds of all codons of A. inebrians that had high RSCU values showed a high A/T preference in the third codon. This phenomenon is common in the chloroplast genomes of higher plants [59, 60].
Fig. 2.
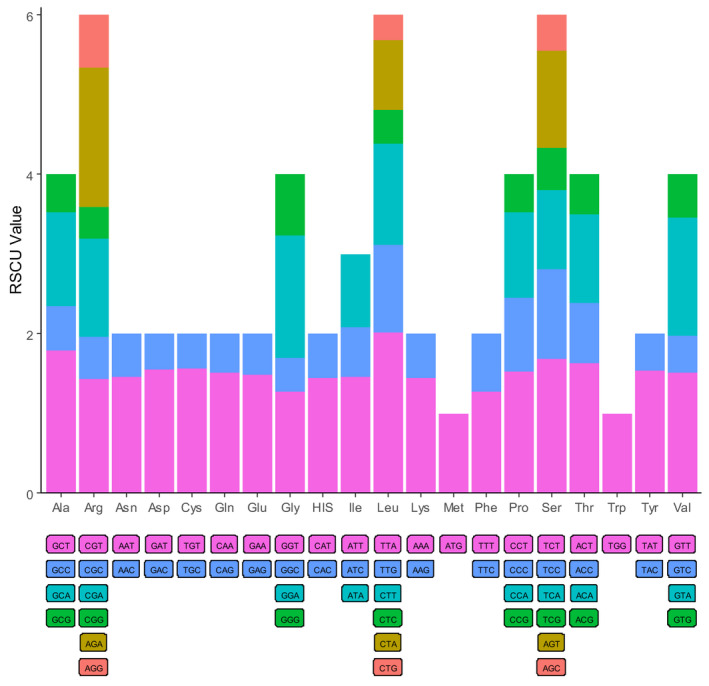
Codon content of 20 amino acids in all protein‐coding genes of the Achnatherum inebrians chloroplast genome.
Fig. 3.
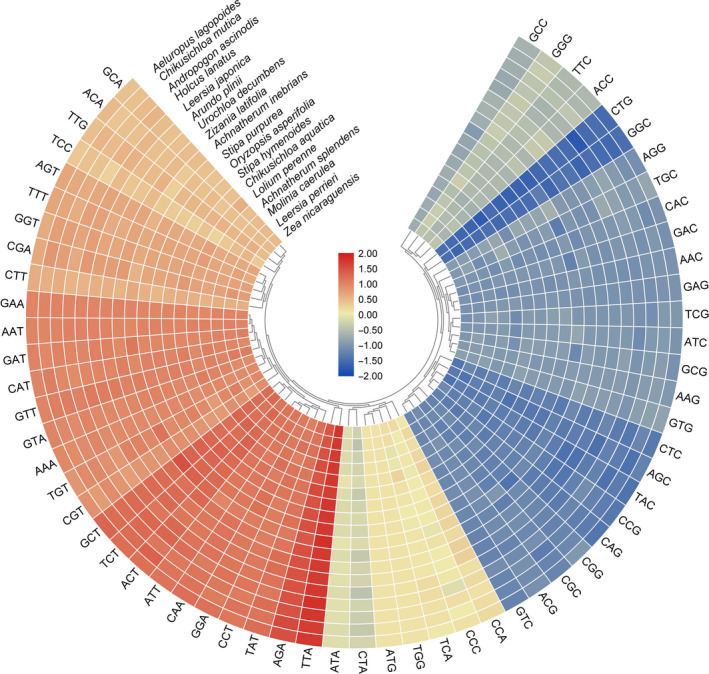
Heatmap analysis for codon distribution of all protein‐coding genes of 18 Poaceae species. Color key: higher red values indicate higher RSCU values, and lower blue values indicate lower RSCU values.
Repeat sequences and SSR analyses
SSRs, also known as microsatellites, a section of DNA in a genome consisting of the basic units of one to six and repeated many times, are widely distributed in chloroplast genomes. SSRs are often used as molecular markers for studying chloroplast genome evolution and population genetics [61, 62]. We investigated the distribution of SSRs in the A. inebrians chloroplast genome and found a total 38 SSRs, of which 31 were in the LSC region (82%), 3 were in the SSC region (8%) and 4 were in IR regions (10%; Fig. 4A). In total, four categories of SSRs, that is, mononucleotide, dinucleotide, trinucleotide and tetranucleotide, were detected. Mononucleotide repetition is most prevalent in each chloroplast genome, followed by dinucleotide, trinucleotide and tetranucleotide repetition. The most dominant SSRs are A/T mononucleotides (18%) from the frequency of the classified repeat types (Table S4). The SSR motifs in the A. inebrians and three other chloroplast genomes (A. splendens, Stipa hymenoides, Stipa purpurea) that are closely related to A. inebrians were analyzed (Fig. 4B). The study results showed little differences in the distribution pattern and number of SSRs among the four chloroplast genomes except the tetranucleotide repetition AAAG, which was detected in only A. inebrians (Fig. 4C).
Fig. 4.
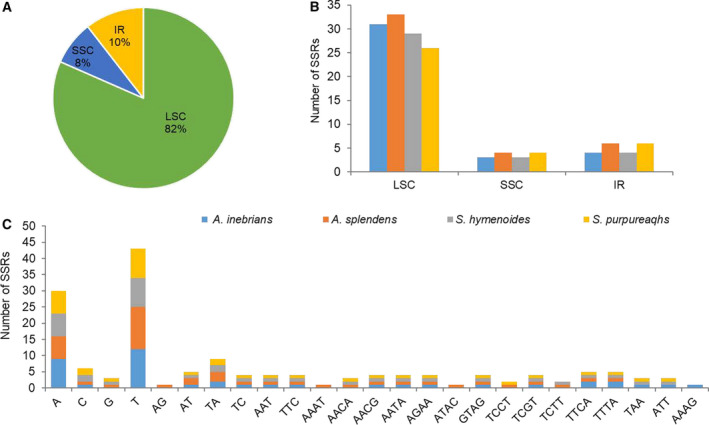
SSR analysis of the four Poaceae chloroplast genomes. (A) Presence of SSRs in the LSC, SSC and IR regions (A. inebrians). (B) The frequency of SSRs in LSC, IR and SSC regions. (C) The frequency of SSRs of different types.
Comparative genome analyses
In this study, the chloroplast genomes of eight Poaceae were analyzed using the mvista program, with S. hymenoides serving as a reference (Fig. 5). These species have considerable similarities in genome composition and size. The coding regions of the eight Poaceae species were almost identical, whereas the noncoding regions were more variable. The highly divergent regions were found among the intergenic spacers, including matk‐rps16, rps16‐trnQ‐UGG, trnG‐UGG‐trnT‐GGU, psbM‐petN, rbcl‐psal, ndhF‐rpl32, rps2‐rpl23 and psbE‐petL in LSC, and ndhF‐rpl32 and psaC‐ndhE in SSC, which might be regarded as potential molecular markers for Poaceae plants. In the whole chloroplast variable region, the A. inebrians share high sequence identity with those of S. purpurea more than A. splendens and relatively lower identity with those of Cynosurus cristatus and A. japonicus.
Fig. 5.
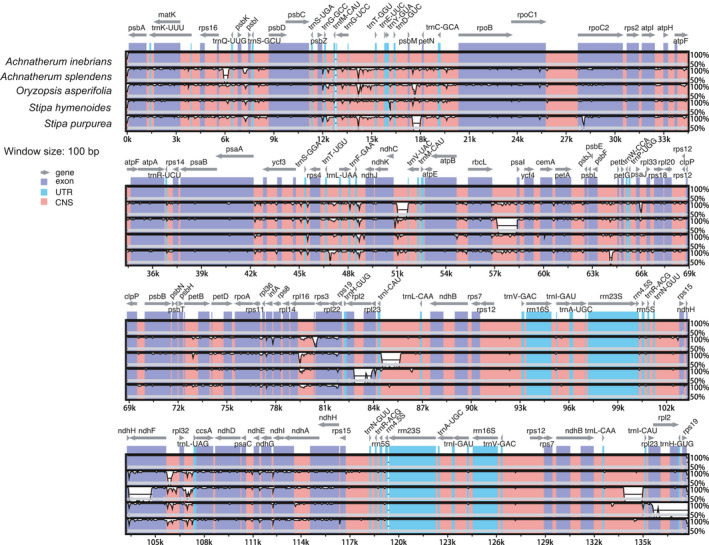
Sequence alignment of five Poaceae genomes in mvista. The x axis represents the coordinates in the chloroplast genome. The vertical scale indicates the identity percentage, ranging from 50% to 100%.
Pis of four Poaceae were calculated to further demonstrate the differences in the chloroplast genomes of Gramineae at the sequence level. As shown in Fig. 6, the divergence values among S. purpurea, S. hymenoides, A. splendens and A. inebrians ranged from 0 to 0.06, with a mean of 0.00837, and the IR regions were more conserved than the LSC and SSC regions. The most divergent region, rps3‐rpl22, showed a divergence value of 0.06 in the LSC region, while the ccsA gene showed a high Pi (0.031) value in the SSC region. The intergenic regions among trnT‐GGU‐trnT‐GGU and rbcL‐psaI also showed a relatively high divergence value (>0.025). These regions may undergo rapid nucleotide replacement at the species level. These hotspots can be used as molecular markers and barcodes for phylogenetic analysis and species identification of Poaceae.
Fig. 6.
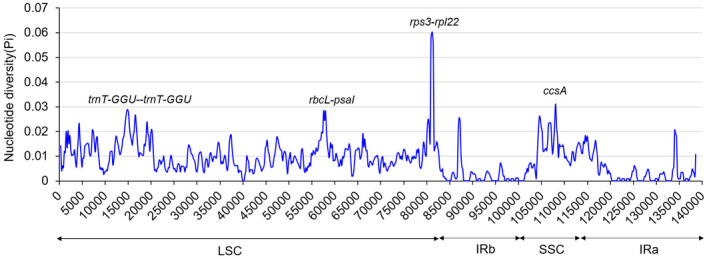
Sliding window analysis of nucleotide variability among the chloroplast genomes of four species (window length: 600 bp; step size: 200 bp).
Expansion and contraction at the borders of the IR regions are common evolutionary events that often result in genome size variations in chloroplast genomes. We investigated the position of genes at the junction regions of four chloroplast genomes: S. purpurea, S. hymenoides, A. splendens and A. inebrians. In the A. inebrians plastome, the boundary of IR–LSC extended into the rps19 gene; the boundary of IR–SSC extended into the ndhF gene, and 48 bp of ndhF extended into the IR region a (IRa); and the boundaries of IRs region b (IRb)–LSC and IRa–LSC extend into the rpl22 and psbA genes, respectively. Only 37 bp of rps22 was duplicated in the LSC region, while 48 bp of rps19 was duplicated in IRb. Similarly, the ndhH gene was located at the junction of SSC–IRa, and ndhH is 17, 28, 28 and 31 bp from the SSC and IRb borders in S. purpurea, S. hymenoides, A. splendens and A. inebrians, respectively. The connections between IR and SSC regions often vary in chloroplast genomes of higher plants and have been commonly reported in previous studies [63, 64]. In this study, a detailed comparison of the borders among the IR, LSC and SSC regions of the four Poaceae chloroplast genomes was explored and is presented in Fig. 7. Our results suggest that the IR–LSC boundary might be conserved among the chloroplast genomes of closely related family species.
Fig. 7.
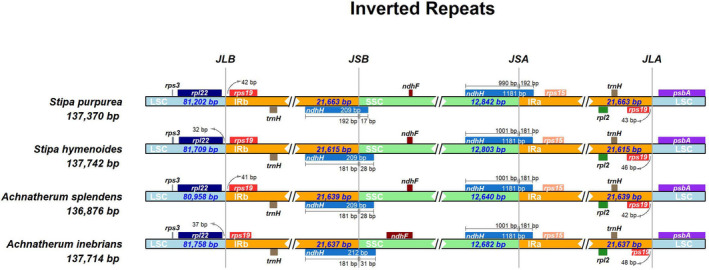
Comparison of the junction positions between the LSC, SSC and IR regions among the chloroplast genomes of four species.
Phylogenetic analysis
The phylogenetic tree was constructed based on 37 whole‐chloroplast genomes from the Poaceae family using C. rotundus and E. dulcis as outgroups (Fig. 8). The phylogenetic trees generated by BI (Fig. S1) and ML methods and their topology were nearly identical. The tree topology from ML analysis is shown in Fig. 8. The relevant data of phylogenetic trees are shown in the supplementary materials (Tables S5 and S6). According to the trees’ topology, the 37 species of Poaceae were divided into five subfamilies: Pooideae, Oryzoideae, Chloridoideae, Arundinoideae and Panicoideae. The ML (bootstraps value = 100) and BI (posterior probability values = 1) topology both supported that A. inebrians has a sister relationship to the genus S. hymenoides. The position of A. inebrians and all other nodes in the topology are supported with posterior probability values of 1.0, except three nodes. Our study provides valuable genetic information for genome‐scale phylogenetic studies in Poaceae plants.
Fig. 8.
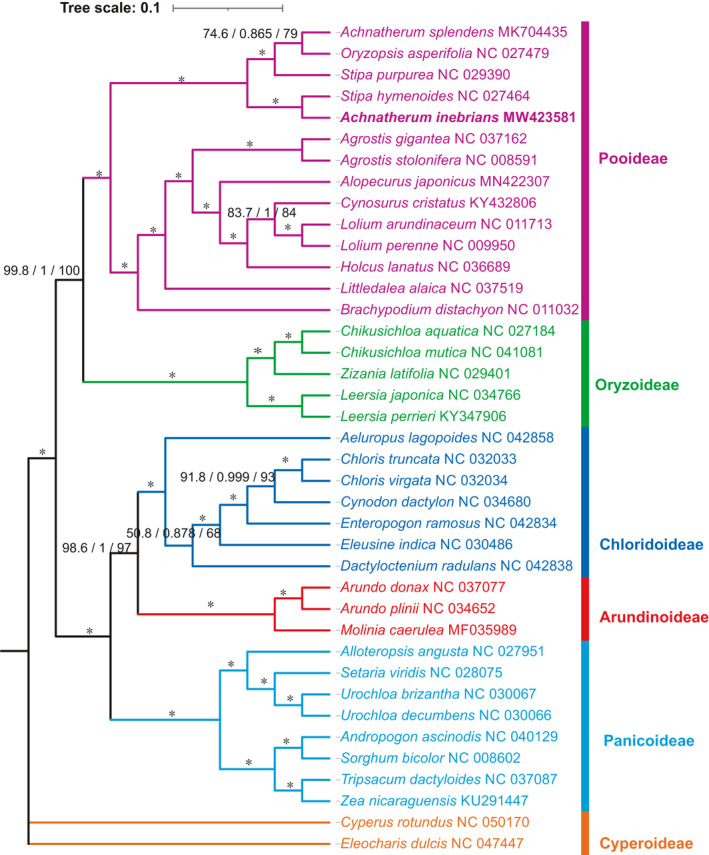
Phylogenetic tree reconstructed from the complete chloroplast genome sequences from 39 species. Statistical support values above the branches correspond to Shimodaira–Hasegawa‐like approximate likelihood‐ratio test (SH‐aLRT) values/approximate Bayes probabilities/ML bootstrap values. Asterisks (*) indicate branches with maximum values of the indices, except where noted.
Discussion
In this study, next‐generation sequencing technology was used to sequence the chloroplast genome of A. inebrians, and its genetic information was reported for the first time. The comparative analysis of gene composition and structure revealed that A. inebrians has a conserved chloroplast genome like other grassland plants [65, 66].
A total of 130 genes were found in the A. inebrians chloroplast genome, including 84 protein‐coding genes, 38 tRNA genes and 8 rRNA genes. The ycf1, ycf2 and accD were lost, which is a common trend in many Poaceae plants [67], indicating that genetic degeneration occurred during the process of gene evolution.
A total of 38 SSRs were identified in the A. inebrians chloroplast genome. The most dominant SSRs were A/T mononucleotides (18%) from the frequency of classified repeat types. SSRs can be regarded as good markers in plant populations for addressing genetic diversity among closely related taxa. Therefore, improved ability to study interspecies differences can be used in conjunction with SSR markers developed by nuclear genomes to address phylogenetic relationships among closely related species [68].
During the genome evolution process, the sequence marginal region of the IR region was changed [69]. With the expansion and contraction of the IR boundary, some genes entered the IR region and some entered the single‐copy region, resulting in changes in the number of genes among different species. The chloroplast genome size is mainly dependent on the expansion and contraction of IR and SSC boundary regions [70].
The comparative analysis of A. inebrians and other species showed that, except for the high conservation of complete chloroplast, there are some significant differences among them. For example, the mvista program and Pi analysis both determined that rbcl‐psal and psbE‐petL can be used for the development of phylogenetic markers. A. inebrians share high sequence identity with those of S. purpurea more than A. splendens and the same as phylogenetic tree. It is a major finding and will be helpful for researchers in getting more information about genetic resources.
Phylogenetic studies of plants mainly use the chloroplast and nuclear genome to analyze the genome structure and modifications [66, 70]. The Poaceae family not only has an economic importance but also it is one of the major families on which international cooperative molecular phylogenetic studies were conducted [71, 72]. Our results support Poaceae being composed by two big clades: BOP (Bambusoideae, Oryzoideae, and Pooideae) and PACCAD (Panicoideae, Aristidoideae, Chloridoideae, Micrairoideae, Arundinoideae, and Danthonioidea), which is similar to the findings reported in previous research [72, 73]. In this study, for the first time, we reconstructed phylogenetic trees based on the chloroplast genome of 37 Poaceae plants, including A. inebrians. In terms of evolutionary relationships, our study results strongly support that A. inebrians belongs to the genus Stipa.
As for the division and classification of Achnatherum, there is an unavoidable relationship between it and Stipa. In the past, many scholars did not recognize or use the genus Achnatherum and still used Stipa in their studies [74, 75, 76, 77]. But at the same time, other scholars used Achnatherum in their studies [3, 78, 79, 80]. According to the comparison of the morphological characteristics (Table S7), A. inebrians is inclined to the Achnatherum, but there are some (awn, fruit, basal disc) morphologically similar to Stipa. Our study provides support only for relevant classification at the molecular level and does not fully represent the real classification status. Specific follow‐up studies can make use of mitochondrial genes, nuclear genes and other genetic markers for further classification.
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Author contributions
XW, XL and CL designed experiments. XW, ZC and YJ carried out the experiments. XW and ZC analyzed experimental results. XW, TC and KM wrote the manuscript.
Supporting information
Fig. S1. Phylogenetic tree generated by BI.
Table S1. All information of species and the accession numbers of their chloroplast genomes in NCBI.
Table S2. List of intron‐containing genes in the CP genomes of Achnatherum inebrians.
Table S3. Nucleotide sequences of protein‐coding genes of Achnatherum inebrians chloroplast genome.
Table S4. Frequency of classified repeat types (considering sequence complementary).
Table S5. The relevant data of phylogenetic tree generated by BI.
Table S6. The relevant data of phylogenetic tree generated by maximum likelihood.
Table S7. The morphological characteristics of Achnatherum inebrians, genus Achnatherum, genus Stipa.
Acknowledgements
This study was funded by the National Basic Research Program of China (2014CB138702); the Natural Science Foundation of China (31971756); Program for Changjiang Scholars and Innovative Research Team in University, China (IRT17R50); the Strategic Priority Research Program of Chinese Academy of Sciences (XDA20100102); the Second Tibetan Plateau Scientific Expedition and Research (STEP) Program (2019QZKK0302); Fundamental Research Funds for the Central Universities (LZUJBKY‐2021‐kb10); 111 Project (B12002); and the Fundamental Research Funds for the Central Universities (LZUJBKY‐2021‐kb36).
Data accessibility
The annotated complete chloroplast genome sequences were submitted to the NCBI database with accession number MW423581.
References
- 1. Shi ZC (1997) Important Poisonous Plants of China Grassland, pp. 166–176.China Agriculture Press, Beijing. [Google Scholar]
- 2. Hance HF (1876) On a Mongolian grass producing intoxicating in cattle. J Botany 14, 210–212. [Google Scholar]
- 3. Geng YL (1959) Illustration of China Main Plants—Poaceae, pp. 579–618.Science Press, Beijing. [Google Scholar]
- 4. Chu QG and Yang YC (1990) Study on classification and morphological evolution of the genus Achnatherum in China. J Liyang Agric Coll 7, 282–290. [Google Scholar]
- 5. Yao X, Christensen MJ, Bao GS, Zhang CP, Li XZ, Li CJ and Nan ZB (2015) A toxic endophyte‐infected grass helps reverse degradation and loss of biodiversity of over‐grazed grasslands in northwest China. Sci Rep 5, 18527–18534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yao X, Chen ZJ, Wei XK, Chen SH, White J, Huang X, Li CJ and Nan ZB (2020) A toxic grass Achnatherum inebrians serves as a diversity refuge for the soil fungal community in rangelands of northern China. Plant Soil 448, 425–438. [Google Scholar]
- 7. Nan ZB and Li CJ (27‐29 Sep. 2000) Neotyphodium in native grasses in China and observations on endophyte/host interactions. In Proceedings of Proceedings of the 4th International Neotyphodium/Grass Interactions Symposium (Paul VH and Dapprich PD eds). Soest. [Google Scholar]
- 8. Li CJ, Nan ZB, Paul VH, Dapprich PD and Liu Y (2004) A new Neotyphodium species symbiotic with drunken horse grass (Achnatherum inebrians) in China. Mycotaxon 90, 141–147. [Google Scholar]
- 9. Chen L, Li X, Li C, Swoboda GA, Young CA, Sugawara K, Leuchtmann A and Schardl CL (2015) Two distinct Epichloe species symbiotic with Achnatherum inebrians, drunken horse grass. Mycologia 107, 863–873. [DOI] [PubMed] [Google Scholar]
- 10. Li CJ, Nan ZB, Zhang CJ, Zhang CY and Zhang YH (2009) Effects of drunken horse grass infected with endophyte on Chinese rabbit. J Agric Tech 11, 84–90.(in Chinese with English Abstract). [Google Scholar]
- 11. Liang Y, Wang H, Li C, Nan Z and Li F (2017) Effects of feeding drunken horse grass infected with Epichloe gansuensis endophyte on animal performance, clinical symptoms and physiological parameters in sheep. BMC Vet Res 13, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang XX, Li CJ, Nan ZB and Matthew C (2012) Neotyphodium endophyte increases Achnatherum inebrians (drunken horse grass) resistance to herbivores and seed predators. Weed Res 52, 70–78. [Google Scholar]
- 13. Johnson LJ, Bonth ACM, Briggs LR, Caradus JR, Finch SC, Fleetwood DJ, Fletcher LR, Hume DE, Johnson RD and Popay AJ (2013) The exploitation of Epichloë endophytes for agricultural benefit. Fungal Divers 60, 171–188. [Google Scholar]
- 14. Kauppinen M, Saikkonen K, Helander M, Pirttila AM and Wali PR (2016) Epichloe grass endophytes in sustainable agriculture. Nat Plants 2, 15224–15230. [DOI] [PubMed] [Google Scholar]
- 15. Xia C, Li N, Zhang Y, Li C, Zhang X and Nan Z (2018) Role of Epichloe endophytes in defense responses of cool‐season grasses to pathogens: a review. Plant Dis 102, 2061–2073. [DOI] [PubMed] [Google Scholar]
- 16. Zhang X, Li C and Nan Z (2010) Effects of cadmium stress on growth and anti‐oxidative systems in Achnatherum inebrians symbiotic with Neotyphodium gansuense . J Hazard Mater 175, 703–709. [DOI] [PubMed] [Google Scholar]
- 17. Zhou L, Li C, Zhang X, Johnson R, Bao G, Yao X and Chai Q (2015) Effects of cold shocked Epichloe infected Festuca sinensis on ergot alkaloid accumulation. Fungal Ecol 14, 99–104. [Google Scholar]
- 18. Xia C, Christensen MJ, Zhang X and Nan Z (2018) Effect of Epichloe gansuensis endophyte and transgenerational effects on the water use efficiency, nutrient and biomass accumulation of Achnatherum inebrians under soil water deficit. Plant Soil 424, 555–571. [Google Scholar]
- 19. Wang J, Nan Z, Christensen MJ, Zhang X, Tian P, Zhang Z, Niu X, Gao P, Chen T and Ma L (2018) Effect of Epichloe gansuensis endophyte on the nitrogen metabolism, nitrogen use efficiency, and stoichiometry of Achnatherum inebrians under nitrogen limitation. J Agric Food Chem 66, 4022–4031. [DOI] [PubMed] [Google Scholar]
- 20. Xia E‐H, Tong W, Wu Q, Wei S, Zhao J, Zhang Z‐Z, Wei C‐L and Wan X‐C (2020) Tea plant genomics: achievements, challenges and perspectives. Hortic Res 7, 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoham RW and Remias D (2020) Snow and glacial algae: a review. J Phycol 56, 264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Y, Zhou T, Duan D, Yang J, Feng L and Zhao G (2016) Comparative analysis of the complete chloroplast genomes of five quercus species. Front Plant Sci 7, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang JB, Tang M, Li HT, Zhang ZR and Li DZ (2013) Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol Biol 13, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Green BR (2011) Chloroplast genomes of photosynthetic eukaryotes. Plant J 66, 34–44. [DOI] [PubMed] [Google Scholar]
- 25. Daniell H, Lin CS, Yu M and Chang WJ (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olmstead RG and Palmer JD (1994) Chloroplast DNA systematics ‐ a review of methods and data‐analysis. Am J Bot 81, 1205–1224. [Google Scholar]
- 27. Kelchner SA (2000) The evolution of non‐coding chloroplast DNA and its application in plant systematics. Ann Mo Bot Gard 87, 482–498. [Google Scholar]
- 28. Wicke S, Mueller KF, de Pamphilis CW, Quandt D, Wickett NJ, Zhang Y, Renner SS and Schneeweiss GM (2013) Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the Broomrape family. Plant Cell 25, 3711–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barrett CF, Wicke S and Sass C (2018) Dense infraspecific sampling reveals rapid and independent trajectories of plastome degradation in a heterotrophic orchid complex. New Phytol 218, 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Z, Jia G and Ni X (2019) The complete chloroplast genome sequence of Achnatherum splendens (Pooideae), a high‐quality forage grass in Northern China. Mitochondrial DNA Part B‐Resources 4, 1841–1843. [Google Scholar]
- 31. Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L and Liu C (2019) CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res 47, W65–W73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng S, Poczai P, Hyvonen J, Tang J and Amiryousefi A (2020) Chloroplot: an online program for the versatile plotting of organelle genomes. Front Genet 11, 576124–576131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharp PM, Tuohy TMF and Mosurski KR (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14, 5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y and Xia R (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- 35. Frazer KA, Pachter L, Poliakov A, Rubin EM and Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32, W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amiryousefi A, Hyvonen J and Poczai P (2018) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. [DOI] [PubMed] [Google Scholar]
- 37. Katoh K and Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hall TA (1999) Bioedit: a user‐friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucleic Acids Symp Ser 41, 95–98. [Google Scholar]
- 39. Rozas J, Ferrer‐Mata A, Carlos Sanchez‐DelBarrio J, Guirao‐Rico S, Librado P, Ramos‐Onsins SE and Sanchez‐Gracia A (2017) DnaSP 6: dna sequence polymorphism analysis of large data sets. Mol Biol Evol 34, 3299–3302. [DOI] [PubMed] [Google Scholar]
- 40. Beier S, Thiel T, Muench T, Scholz U and Mascher M (2017) MISA‐web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang D, Gao F, Jakovlic I, Zou H, Zhang J, Li WX and Wang GT (2020) PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 20, 348–355. [DOI] [PubMed] [Google Scholar]
- 42. Talavera G and Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56, 564–577. [DOI] [PubMed] [Google Scholar]
- 43. Kalyaanamoorthy S, Bui Quang M, Wong TKF, von Haeseler A and Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lam‐Tung N, Schmidt HA, von Haeseler A and Bui Quang M (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bui Quang M, Minh Anh Thi N and von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30, 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anisimova M, Gil M, Dufayard J‐F, Dessimoz C and Gascuel O (2011) Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood‐based approximation schemes. Syst Biol 60, 685–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guindon S, Dufayard J‐F, Lefort V, Anisimova M, Hordijk W and Gascuel O (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321. [DOI] [PubMed] [Google Scholar]
- 48. Zhao MJ, Ren Q, Wang YL, Deng RK, Ren MM, Wang G and Liu XG (2017) A Three‐Level Parallel Algorithm For MrBayes 3.2. 2017 IEEE International Symposium on Parallel and Distributed Processing with Applications and 2017 IEEE International Conference on Ubiquitous Computing and Communications (ISPA/IUCC), pp. 12–15. [Google Scholar]
- 49. Letunic I and Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang D, Li K, Gao J, Liu Y and Gao L‐Z (2016) The complete plastid genome sequence of the wild rice Zizania latifolia and comparative chloroplast genomics of the rice tribe Oryzeae, Poaceae. Front Ecol Evol 4, 4–17. [Google Scholar]
- 51. Zhang Y‐J, Ma PF and Li DZ (2011) High‐throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS One 6, e20596–e20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yi D‐K and Kim K‐J (2016) Two complete chloroplast genome sequences of genus Paulownia (Paulowniaceae): Paulownia coreana and P. tomentosa . Mitochondrial DNA Part B‐Resources 1, 627–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park I, Kim WJ, Yeo SM, Choi G, Kang YM, Piao R and Moon BC (2017) The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 22, 982–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shen X, Wu M, Liao B, Liu Z, Bai R, Xiao S, Li X, Zhang B, Xu J and Chen S (2017) Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua . Molecules 22, 1330–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu M, Li Q, Hu Z, Li X and Chen S (2017) The complete Amomum kravanh chloroplast genome sequence and phylogenetic analysis of the commelinids. Molecules 22, 1875–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zeng S, Zhou T, Han K, Yang Y, Zhao J and Liu Z‐L (2017) The complete chloroplast genome sequences of six Rehmannia species. Genes 8, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chi X, Wang J, Gao Q, Zhang F and Chen S (2018) The complete chloroplast genomes of two Lancea species with comparative analysis. Molecules 23, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao B, Yuan L, Tang T, Hou J, Pan K and Wei N (2019) The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family. PLoS One 14, e0218817–e0218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim KJ and Lee HL (2004) Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res 11, 247–261. [DOI] [PubMed] [Google Scholar]
- 60. Zhou J, Cui Y, Chen X, Li Y, Xu Z, Duan B, Li Y, Song J and Yao H (2018) Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: molecular structures, comparative analysis, and phylogenetic analysis. Molecules 23, 437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pauwels M, Vekemans X, Gode C, Frerot H, Castric V and Saumitou‐Laprade P (2012) Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol 193, 916–928. [DOI] [PubMed] [Google Scholar]
- 62. Powell W, Morgante M, McDevitt R, Vendramin GG and Rafalski JA (1995) Polymorphic simple sequence repeat regions in chloroplast genomes ‐ applications to the population‐genetics of Pines. Proc Natl Acad Sci USA 92, 7759–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang S, Shi C and Gao LZ (2013) Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into evolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS One 8, e73946–e73957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Palmer JD, Nugent JM and Herbon LA (1987) Unusual structure of geranium chloroplast DNA: a triple‐sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc Natl Acad Sci USA 84, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. MasonGamer RJ and Kellogg EA (1996) Testing for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae). Syst Biol 45, 524–545. [Google Scholar]
- 66. Maier RM, Neckermann K, Igloi GL and Kossel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251, 614–628. [DOI] [PubMed] [Google Scholar]
- 67. Downie SR, Katzdownie DS, Wolfe KH, Calie PJ and Palmer JD (1994) Structure and evolution of the largest chloroplast gene (ORF2280): internal plasticity and multiple gene loss during angiosperm evolution. Curr Genet 25, 367–378. [DOI] [PubMed] [Google Scholar]
- 68. Song Y, Chen Y, Lv J, Xu J, Zhu S and Li M (2019) Comparative chloroplast genomes of sorghum species: sequence divergence and phylogenetic relationships. Biomed Res Int 2019, 5046958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Oldenburg DJ and Bendich AJ (2004) Most chloroplast DNA of maize seedlings in linear molecules with defined ends and branched forms. J Mol Biol 335, 953–970. [DOI] [PubMed] [Google Scholar]
- 70. Wakasugi T, Tsudzuki T and Sugiura M (2001) The genomics of land plant chloroplasts: gene content and alteration of genomic information by RNA editing. Photosynth Res 70, 107–118. [DOI] [PubMed] [Google Scholar]
- 71. Barker NP, Clark LG, Davis JI, Duvall MR, Guala GF, Hsiao C, Kellogg EA, Linder HP, Mason‐Gamer RJ, Mathews SY et al. (2001) Phylogeny and subfamilial classification of the grasses (Poaceae). Ann Mo Bot Gard 88, 373–457. [Google Scholar]
- 72. Aliscioni S, Bell HL, Besnard G, Christin P‐A, Columbus JT, Duvall MR, Edwards EJ, Giussani L, Hasenstab‐Lehman K, Hilu KW et al. (2012) New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol 193, 304–312. [DOI] [PubMed] [Google Scholar]
- 73. Cotton JL, Wysocki WP, Clark LG, Kelchner SA, Pires JC, Edger PP, Mayfield‐Jones D and Duvall MR (2015) Resolving deep relationships of PACMAD grasses: a phylogenomic approach. BMC Plant Biol 15, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hitchcock AS (1997) Manual of the Grasses of the United States, 2nd edn. Miscellaneous publications, Washington. [Google Scholar]
- 75. Winter BD (1965) The South African Stipeae and Aristideae (Gramineae). Bothalia 8, 201–404. [Google Scholar]
- 76. Bor NL (1960) The Grasses of Burma, Ceylon, India and Pakistan, pp. 637–647.Pergamon Press Ltd, London: Oxford. [Google Scholar]
- 77. Freitag H (1985) The Genus Stipa (Gramineae) in Southwest and South Asia. Notes RBG Edinb 42, 355–489. [Google Scholar]
- 78. Pilrer R (1954) Das system der Gramineae. Bot Jahrb Syst 76, 281–384. [Google Scholar]
- 79. Tzvelev NN (1989) The system of grasses (Poaceae) and their evolution. Bot Rev 55, 141–204. [Google Scholar]
- 80. Guo BZ (1987) Flora of China, Vol. 9, p. 326. Science Press, Beijing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phylogenetic tree generated by BI.
Table S1. All information of species and the accession numbers of their chloroplast genomes in NCBI.
Table S2. List of intron‐containing genes in the CP genomes of Achnatherum inebrians.
Table S3. Nucleotide sequences of protein‐coding genes of Achnatherum inebrians chloroplast genome.
Table S4. Frequency of classified repeat types (considering sequence complementary).
Table S5. The relevant data of phylogenetic tree generated by BI.
Table S6. The relevant data of phylogenetic tree generated by maximum likelihood.
Table S7. The morphological characteristics of Achnatherum inebrians, genus Achnatherum, genus Stipa.
Data Availability Statement
The annotated complete chloroplast genome sequences were submitted to the NCBI database with accession number MW423581.