

Abstract
Human fatty acid synthase (FASN) is the sole cytosolic enzyme responsible for de novo lipid synthesis. FASN is essential for cancer cell survival and contributes to drug and radiation resistance by up-regulating DNA damage repair but not required for most non-lipogenic tissues. Thus, FASN is an attractive target for drug discovery. However, despite decades of effort in targeting FASN, no FASN inhibitors have been approved due to poor pharmacokinetics or toxicities. Here, we show that the FDA-approved proton pump inhibitors (PPIs) effectively inhibit FASN and suppress breast cancer cell survival. PPI inhibition of FASN leads to suppression of non-homologous end joining repair of DNA damages by reducing FASN-mediated PARP1 expression, resulting in apoptosis from oxidative DNA damages and sensitization of cellular resistance to doxorubicin and ionizing radiation. Mining electronic medical records of 6,754 breast cancer patients showed that PPI usage significantly increased overall survival and reduced disease recurrence of these patients. Hence, PPIs may be repurposed as anticancer drugs for breast cancer treatments by targeting FASN and to overcome drug and radiation resistance.
Keywords: fatty acid synthase, proton pump inhibitor, enantiomer, DNA damage repair, PARP1
1. Introduction
Fatty acid synthase (FASN) is the sole mammalian cytosolic enzyme with multiple domains responsible for de-novo lipid synthesis [1, 2]. Most normal non-lipogenic tissues express little FASN due to high fat content in modern Western diet while FASN expression is required and universally increased in many cancers including breast and pancreatic cancers [2, 3]. The increased FASN expression not only increases oncogenic potential [4, 5], but also associates with poor prognosis and disease recurrence of many cancers [2]. Breast cancers with high level of FASN were four times more likely to recur and metastasize than the ones without detectable FASN [6, 7]. FASN expression has also been associated with poor relapse-free survival and distant metastasis-free survival of luminal A, basal, and Her2+, but not luminal B subtypes of breast cancer [8]. Furthermore, FASN expression has been shown to contribute to drug and radiation resistance [9–11] possibly by increasing DNA damage repair [11] via increased de novo synthesis of fatty acids [12]. Clearly, inhibiting FASN offers a unique opportunity to discover novel cancer therapeutics [2, 3, 13].
Indeed, many inhibitors targeting FASN have been discovered including the first generation inhibitors such as cerulenin [14, 15] and newer ones such as GSK2194069 [16] and Fasnall [17]. However, few inhibitors have entered clinical study and none has been approved despite years of efforts in targeting FASN. While some inhibitors have poor pharmacokinetic properties and systematic availability, others are toxic and cause weight loss [2, 3, 18, 19]. It, thus, was thought that repurposing FDA-approved drugs in targeting FASN would be an effective alternative strategy to avoid these issues in prior studies. To this end, it was found that proton pump inhibitors (PPIs), approved for treating digestive disorders, inhibited FASN by binding toits thioesterase (TE) domain and inhibited proliferation of pancreatic cancer cells [20].
However, the detailed mechanism of PPI inhibition of FASN leading to suppression of cancer cell proliferation is unclear.
Here, we tested the hypotheses that PPIs inhibit FASN and synergize with DNA - damaging treatments by inhibiting DNA damage repair via FASN and that PPI usage improves patient outcome using cell-based and biochemical assays and by mining electronic medical records (EMR) of a cohort of 6,754 breast cancer patients. We show that PPIs are effective in inhibiting FASN activity, leading to reduced expression of PARP1 and inhibition of DNA - damage repair in breast cancer cells. The reduction in repair activity leads to induction of apoptosis and sensitization of cancer cells to dox orubicin and IR. We also show that PPI usage significantly increases overall survival of breast cancer patients and reduces disease recurrence. Furthermore, the R-enantiomers are better than the S-enantiomers in inhibiting FASN and in improving patient outcome. We, thus, conclude that PPIs, especially the R-enantiomers, could be repurposed as effective anticancer drugs by targeting FASN and inhibiting DNA damage repair.
2. Methods
2.1. Cell lines.
Human breast cancer cell lines MCF7, MDA-MB-231, MDA-MB-468, and T47D were from ATCC and authenticated using STR on August 3, 2016. These cells were cultured at 37°C with 5% CO2 in DMEM supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. The stable FASN-over expressing MCF7 (M/FASN) and MCF7/A dV p3000 (M3K) cells with FASN-knockdown (M3K/Sh) and their control vector-transfected MCF7 cells (MCF7/V ec) and scrambled shRNA-transfected M3K cells (M3K/Scr) were previously generated [9–11] and maintained in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 mg/ml streptomycin, and 400 µg/ml G418.
2.2. FASN and TE activity assays.
FASN holoenzyme activity assay was performed as previously described [9, 10]. Briefly, MCF7 cells were harvested and washed with and lysed in ice-cold PBS containing 1 mM DTT, 1 mM EDTA, and 0.05% Brij35 with intermittent sonication. The lysate was cleared of debris by centrifugation at 12,000g for 20 min. Lysate proteins (250 µg) were incubated with PPIs in 200 mM K2HPO4 pH6.8, 1 mM DTT, 1 mM MgCl2, 240 µM NADPH, 30 µM acetyl-CoA for 10 min at 37°C followed by addition of 50 µM malonyl-CoA to initiate reaction and measurement of OD340nmevery minute for 20 min.
TE activity assay was performed also as previously described [20]. Briefly, recombinant TE (500 nM) was pre-incubated with PPIs at different concentrations in buffer A (100 mM Tris-HCl, 50 mM NaCl, 0.05% Brij35, pH 7.5) in opaque black, flat-bottom 96-well plates at 37°C for 30 min followed by addition of 300 μM 4-methylumbelliferyl heptanoate, incubation at 37°C for 1 h, and measurement of fluorescence at 355/460 nm.
2.3. Colony formation assay.
Colony formation assay was performed as previously described [20, 21]. Briefly, breast cancer cells were seeded in six-well plates at 200 cells/well, cultured overnight, and treated without or with PPIs at different concentrations for 14 days. The colonies formed were then washed with PBS, fixed in 100% methanol and stained with 0.5% crystal violet in 25% methanol. The stained colonies were counted manually and concentration-response curves were generated to derive IC50 using Prism (GraphPad, La Jolla, USA).
Combination studies were performed according to the Chou and Talalay method [22]. Briefly, colony formation assays were performed in the presence of different concentrations of lansoprazole and doxorubicin with three different potency ratios (lansoprazole/doxorubicin: 50/50, 25/75 and 75/25). For each potency ratio, combination index of three effective concentrations were calculated for synergism using Prism.
For the combination with ionizing radiation (IR), 1×106 breast cancer cells were seeded in 100-mm dish and pre-treated without or with 10 µM lansoprazole once a day for 3 days. Cells were then harvested and re-plated in 6-well plates at 200 cells/well followed by IR and colony formation assay or Western blot analysis. Concentration-response curves and IC50 values were generated using Prism.
2.4. Cell lysate preparation and Western blot analysis.
Cells were harvested and then lysed in TNN buffer with 1 mM DTT, 1 mM PMSF and 0.1% SDS for 30 min at 4°C with occasional agitation. The cells were sonicated briefly and total proteins were harvested after centrifugation of the lysate at 16,000g for 15 minutes followed by determination of protein concentration using Bio-Rad protein assay kit. Total cell lysates were then separated by SDS-PAGE and subjected to Western blot analysis probed using antibodies against FASN (610963, BD biosciences), PARP-1 (46D11, CST), γ-H2AX (05–636, Millipore), and β-Actin (A2228, Sigma). The signals were developed with Amersham ECL Reagent (RPN2106, GE) and captured on X-ray films.
2.5. Host cell reactivation (HCR) assay.
HCR assay of NHEJ and homologous recombination (HR) activities was carried out as previously described [11]. For NHEJ activity assay, 1×104 cells were seeded in 24-well plates, cultured overnight and treated with 10 µM lansoprazole or vehicle twice a day for three days before transfection with either Hind III-linearized or intact pGL3-luc firefly luciferase reporter plasmid (400 ng) using Lipofectamine 2000 (Invitrogen) together with pRL-TK (Promega) encoding renilla luciferase (400 ng) as a transfection efficiency control. At 8 hours after transfection, cells were harvested and assayed for luciferase activity with Dual Luciferase Assay kit (Promega). Overall NHEJ activity was calculated from firefly luciferase activity of the linearized plasmid relative to the intact plasmid with normalization to the renilla luciferase in each group.
For HR activity assay, two fragments were generated from pGL3-luc firefly luciferase reporter plasmid by restriction digestion with Not I-Xcm I and with Bsr GI-Bam HI with an overlapping region in the firefly luciferase open reading frame and purified. The Not I-Xcm I fragment contains the SV40 promoter and the 5’ portion of the firefly luciferase gene while the Bsr GI-Bam HI fragment contains the 3’ portion of the firefly luciferase gene with a 245-bp overlap between the two fragments. A third fragment consisting of the SV40 promoter and complete firefly luciferase gene was generate by Not I-Bam HI digestion and used as a control of total firefly luciferase activity. Briefly, 1×104 cells were seeded in 24-well plates, cultured overnight and treated with 10 µM lansoprazole or vehicle twice a day for three days before transfection with a mixture of Not I-Xcm I and Bsr GI-Bam HI fragments in equal ratio or with the Not I-Bam HI control fragment using Lipofectamine 2000 (Invitrogen). Not I-Xcm I or Bsr GI-Bam HI fragment alone were also tested as negative control. 40 ng pRL-TK (Promega) plasmid encoding renilla luciferase was co-transfected as a control for transfection efficiency. At 24 hours after transfection, cells were harvested and processed for luciferase activity assay as described above. Firefly luciferase signal were normalized to renilla luciferase signal in each group. Overall HR activity was calculated by firefly luciferase activity from cells transfected with both Not I-Xcm I and Bsr GI-Bam HI fragments relative to cells transfected with the control Not I-Bam HI fragment.
2.6. Immunofluorescence imaging.
Immunofluorescence staining was also performed as previously described [11]. About 50,000 cells/well were seeded on coverslip in 6-well plates, treated with 10 µM lansoprazole or DMSO twice a day for 3 days, and fixed with Acetone/Methanol (1:1) at room temperature for 10 minutes. Cells were then permeabilized using 0.5% Triton X-100 in PBS, blocked with 1% BSA, stained with γ-H2AX antibody (Millipore), followed by secondary anti-mouse-IgG-FITC antibody (Sigma). The cells were counter stained with DAPI and stored in the dark at 4°C before viewing on a confocal microscope (Olympus).
2.7. Flow cytometry assay.
Cells were seeded in 6-well plates at 2×105 cells/well, cultured overnight, and treated with 10 µM lansoprazole or vehicle twice a day for 3 days. Cells were washed with PBS and fixed in ice-cold 70% ethanol overnight. After centrifugation, pellets were resuspended in PBS, stained with propidium iodide solution for 30 min and analyzed using flow cytometry.
2.8. Electronic medical records (EMR).
The EMR database contains three components of 6,754 de-identified breast cancer patients from Indiana Network for Patient Care (INPC). The first component contains patient characteristics as shown in Supplemental Table S1. The second one contains lab test results and office visit information. The third one contains medication information. The use of this database was approved by IRB at Indiana University.
2.9. Data preprocessing.
To remove duplicated entrances and resolve contradicting records, we followed different rules for different conflicts. For conflicts on the first visit or diagnosis time, we used the earlier record time as the first diagnosis and considered the later one as system update. For all other information including diagnosis information such as cancer subtype, we used the latest record as we believe it is more accurate due to information or system update.
Next, plain text information from the database including ER, PR, and HER2 test results, treatment plans, and therapy types were digitized by assigning numerical values. Records that have ambiguity are assigned as unknown. For example, HER2 status with a 2+ score is a borderline score from IHC staining, which was usually confirmed with FISH test. For patient with a 2+ score without a FISH test result, a value of unknown is assigned in this study.
Thirdly, data were validated or corrected across components. For example, patient survival days from the first component were validated using prescription history of the same patient in the third component. When a conflict occurred, the last day of office visit or prescription refill, whichever is later, was recorded as the last day of visit or death.
2.10. Data collection.
All known generic and trade names of PPIs were identified within the database and used for this study, including omeprazole (Prilosec, Zegerid, Losec), pantoprazole (Protonix, Somac, Zurcal), esomeprazole (Nexium, Esotrex), lansoprazole (Prevacid, Zoton, Levant), rabeprazole (Zechin, Rabecid, AcipHex), and dexlansoprazole (Kapidex, Dexilant). For overall survival analysis, patients who took PPIs but stopped using them prior to their diagnosis of breast cancer were considered non-PPI users. For disease recurrence analysis, only patients who took PPIs after diagnosis of breast cancer but before recurrence were considered PPI users.
2.11. Statistical analysis.
The overall survival and recurrence events up to 4000 days were analyzed using the Kaplan-Meier method and the log-rank test. Cox proportional hazard models were employed with Hazard Ratios (HR) and 95% confidence intervals (CI) to quantify the magnitude and direction of the association analysis. The potential confounding factors including age, race, molecular marker (triple negative vs non-triple negative), tumor stage, and metastasis upon diagnosis were tested as single variable using Cox proportional hazard models and the factors that have significance were included in the multivariable regression analyses. The proportional hazard assumption was tested by examining scaled Schoenfeld residuals with p-values adjusted using Bonferroni’s correction. The corresponding covariate was stratified in the multivariate model in the case of violation of proportionality assumption. For secondary analysis of TNBC patients, different treatment and race subgroups, multivariate models were built to correct compounding factors in the same way as primary analysis except tumor stage are categorized as low (stage 0-II) or high (III-IV) or unknown to reach convergence. All p-values are reported corresponding to two-sided comparisons with p-value≤0.05 considered statistically significant. All statistical analyses were done in R version 3.1.2 on a Linux platform.
3. Results
3.1. PPIs inhibit FASN and suppress breast cancer cell survival.
To determine if PPIs inhibit FASN in breast cancer cells, we performed FASN activity assay using lysate isolated from MCF7 cells in the presence of four PPIs (lansoprazole, omeprazole, pantoprazole, and rabeprazole). As shown in Figure 1A and Supplemental Figure S1, all four PPIs dose-dependently inhibited FASN with IC 50’s ranging from 6.7±0.9 to 18.4±1.7 μM.
Figure 1. PPI inhibition of FASN activity and breast cancer cell survival.
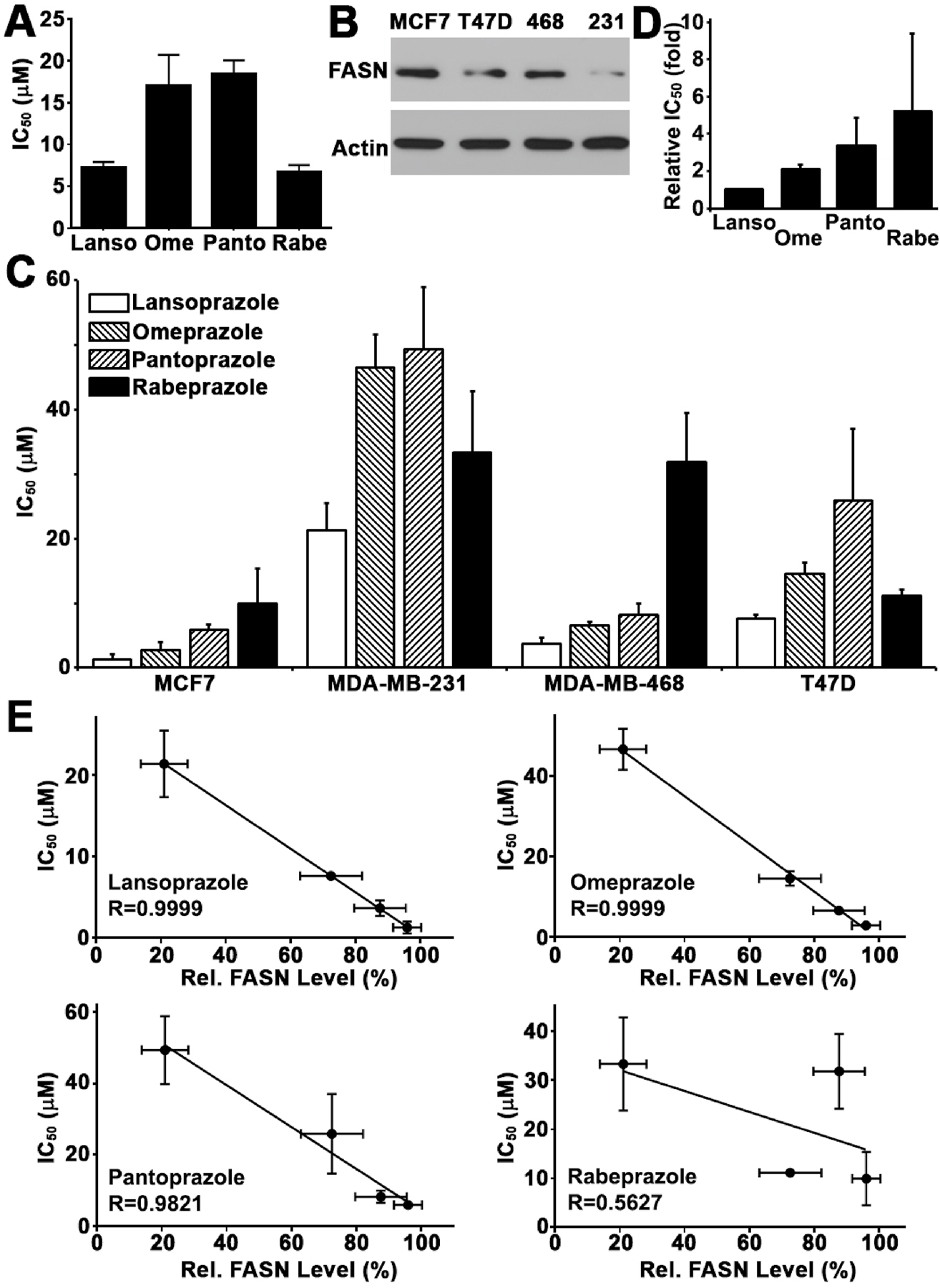
A, IC50 of PPIs in inhibiting FASN activity derived from dose-response curves (see Figure S1). Lanso, lansoprazole; Ome, omeprazole; Panto, pantoprazole; Rabe, rabeprazole. B, Western blot analysis of FASN expression in different human breast cancer cell lines. 468, MDA-MB-468; 231, MDA-MB-231. C, IC50 of PPIs in inhibiting breast cancer cell survival derived from dose response survival curves (see Figure S2). D, Relative average IC50 of PPIs in all four breast cancer cell lines tested. E, Correlation between PPI IC50 and the level of endogenous FASN in all 4 breast cancer cells shown in panel B.
Next, we tested if these PPIs inhibit survival of breast cancer cells with different levels of FASN expression (Figure 1B) using colony formation assay. As shown in Figure 1C and supplemental Figure S2, all four PPIs dose-dependently inhibited survival of four breast cancer cell lines with IC 50’s ranging from 1.3±0.7 to 49.4±9.5 μM and lansoprazole is the most effective one for all 4 cell lines (Figure 1D).
3.2. FASN mediates PPI effects on breast cancer cell survival.
To determine if FASN mediates PPI inhibition of breast cancer cell survival, we first performed correlation analysis between the FASN protein level of the four breast cancer cell lines and IC50 of four PPIs. As shown in Figure 1E, the IC50’s of all 4 PPIs negatively associate with FASN level with correlation coefficient R=~0.6–1.0. These findings suggest that the expression level of endogenous FASN may contribute to the sensitivity of breast cancer cells to PPIs and that PPIs may suppress breast cancer cell proliferation via inhibiting FASN.
Next, we took advantage of the previously established stable MCF7 cells with ectopic FASN over-expression (MCF7/FASN) and MCF7/AdVp3000 cells with FASN knockdown (M3K/Sh) together with their respective (MCF7/Vec and M3K/Scr) control cells (Figure 2A) and treated them with lansoprazole followed by colony formation assay. As shown in Figure 2B, ectopic FASN over-expression and knockdown increased and reduced lansoprazole resistance, respectively. It is noteworthy that overexpressing or knocking down FASN did not affect the proliferation and survival of MCF7 and M3K cells, respectively (supplemental Figure S3). Thus, FASN expression affects lansoprazole sensitivity and, thus, is likely the target of lansoprazole.
Figure 2. Role of FASN in mediating PPI effect.
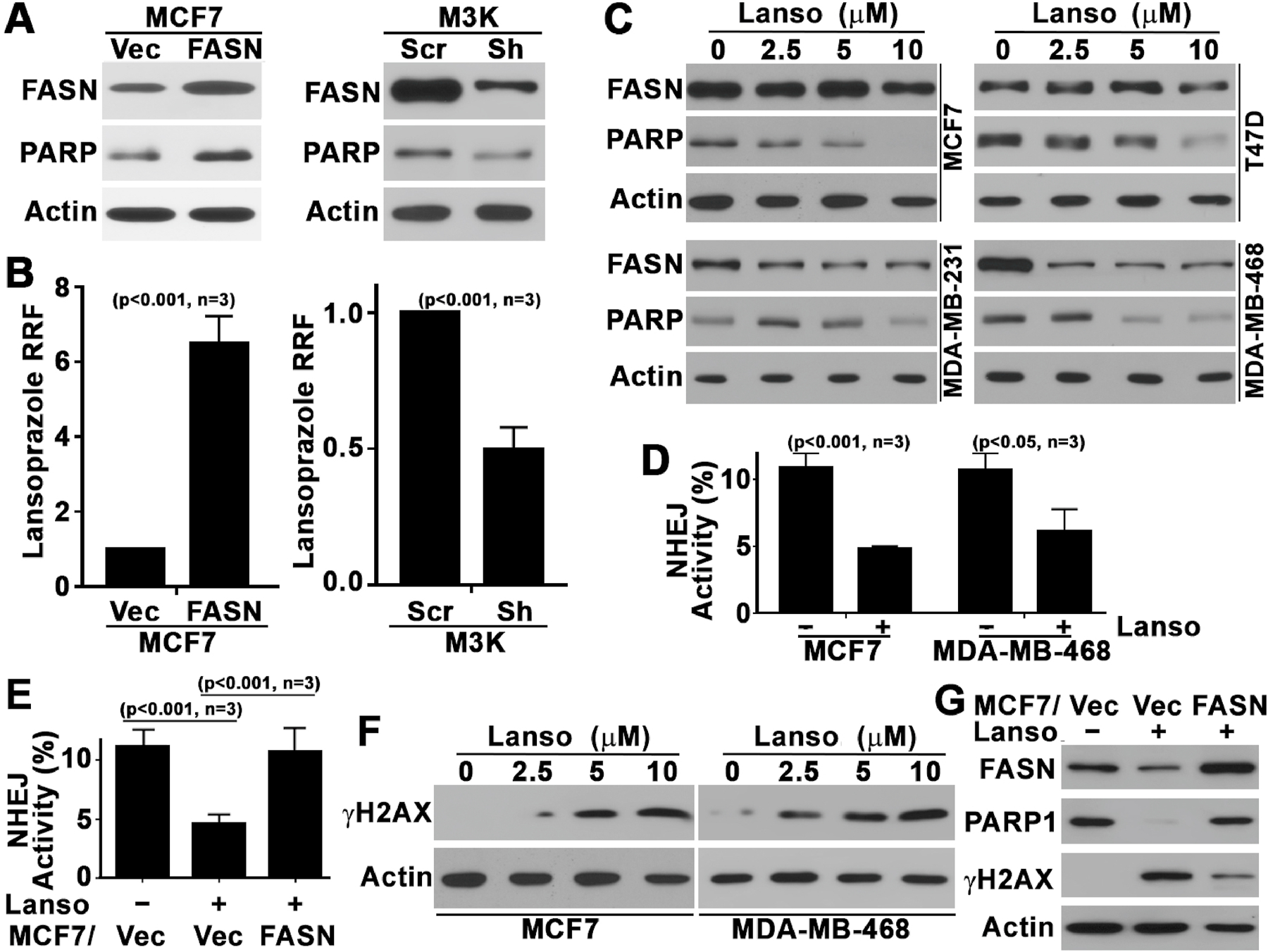
A, Western blot analysis of FASN and PARP1 expression in MCF7 cells with ectopic FASN over-expression and in drug resistant MCF7/AdVp3000 (M3K) cells with FASN knockdown. B, Relative resistance factor (RRF) to lansoprazole in MCF7 cells with FASN over-expression or M3K cells with FASN knockdown. RRF=Lansoprazole IC50(FASN or Sh)/IC50(Vec or Scr). C, Western blot analysis of endogenous FASN and PARP1 expression in MCF7, T47D, MDA-MB-231, and MDA-MB-468 cells following treatments with lansoprazole at different concentrations. D, Effect of lansoprazole on NHEJ activity in MCF7 and MDA-MB-468 cells. E, Rescue of lansoprazole inhibition of NHEJ activity by ectopic FASN over-expression. F, Western blot analysis of γH2AX level in MCF7 and MDA-MB-468 cells treated with lansoprazole at different concentrations. G, Ablation of lansoprazole-induced γH2AX increase by ectopic FASN over-expression as determined using Western blot analysis. Actin was used as a loading control for all Western blot analyses.
Next, we treated all 4 breast cancer cell lines with lansoprazole and determined the expression of PARP1, a known FASN downstream target gene (Figure 2A, also see [11]), using Western blot analysis. As shown in Figure 2C, lansoprazole dose-dependently reduced PARP1 expression in all 4 cell lines. Previously, it has been shown that FASN regulates NHEJ repair activity by regulating PARP1 expression [11]. Thus, we also tested if lansoprazole inhibits NHEJ activity in breast cancer cells using HCR assay. As shown in Figure 2D, lansoprazole significantly reduced NHEJ activity in both MCF7 and MDA-MB-468 cells. Furthermore, over-expressing ectopic FASN overcame lansoprazole inhibition of and rescued NHEJ activity in MCF7 cells (Figure 2E). However, lansoprazole did not change the homologous recombination activity (supplemental Figure S4), another major pathway in repairing double strand DNA breaks.
To determine if lansoprazole treatment causes damages to chromatin DNA by inhibiting NHEJ repair, we determined the level of γ-H2AX, an indicator of DNA damages, in MCF7 and MDA-MB-468 cells following lansoprazole treatment. As shown in Figure 2F, lansoprazole treatment increased γ-H2AX level in a dose-dependent manner in both cell lines. Lansoprazole-induced increase in γ-H2AX was confirmed by increased punctate staining of γ-H2AX foci in nucleus (supplemental Figure S5). Furthermore, the lansoprazole-induced γ-H2AX increase was ablated by over-expressing ectopic FASN (Figure 2G). Together, these findings suggest that lansoprazole likely inhibits breast cancer cell survival by increasing DNA damages via inhibiting FASN and NHEJ repair of DNA damages.
Previously, it has been shown that FASN regulates cell cycle progression [23, 24] and cell survival [25–28]. To further confirm that FASN mediates lansoprazole effect on breast cancer cells, we analyzed cell cycle distribution and apoptosis following lansoprazole treatment. As shown in Supplemental Figure S6A, lansoprazole dose-dependently induced apoptosis in both MCF7 and MDA-MB-468 cells. Lansoprazole also dose-dependently caused cell cycle arrest at G1 in MCF7 and S phase in MDA-MB-468 cells (supplemental Figure S6B), which is consistent with previous observations of FASN regulation of different cell cycle stages in different cells [2]. The difference in cell cycle arrest between MCF7 (wild type p53) and MDA-MB-468 (mutant p53) cells may be due to their differences in p53 status [29] and response to endogenous DNA damages when FASN and DNA repair are inhibited by lansoprazole (see below).
3.3. Lansoprazole and DNA-damaging treatments synergistically inhibit breast cancer cell survival.
Based on previous findings that FASN up-regulates NHEJ repair [11] and lansoprazole inhibits NHEJ via inhibiting FASN (Figure 2), we hypothesized that PPI might have synergistic effect with DNA-damaging treatments on breast cancer cells. To test this hypothesis, we performed colony formation survival assay using different potency combinations of lansoprazole and doxorubicin, that causes double strand DNA break. As shown in Figure 3A and Supplemental Figure S7, lansoprazole and doxorubicin with combinations at different potency ratios all synergistically inhibited both MCF7 and MDA-MB-468 cells with combination index <1.0. Similarly, lansoprazole also sensitized IR treatment of MCF7 and MDA-MB-468 cells (Figure 3B).
Figure 3. Synergistic effect of lansoprazole with doxorubicin and ionizing radiation (IR).
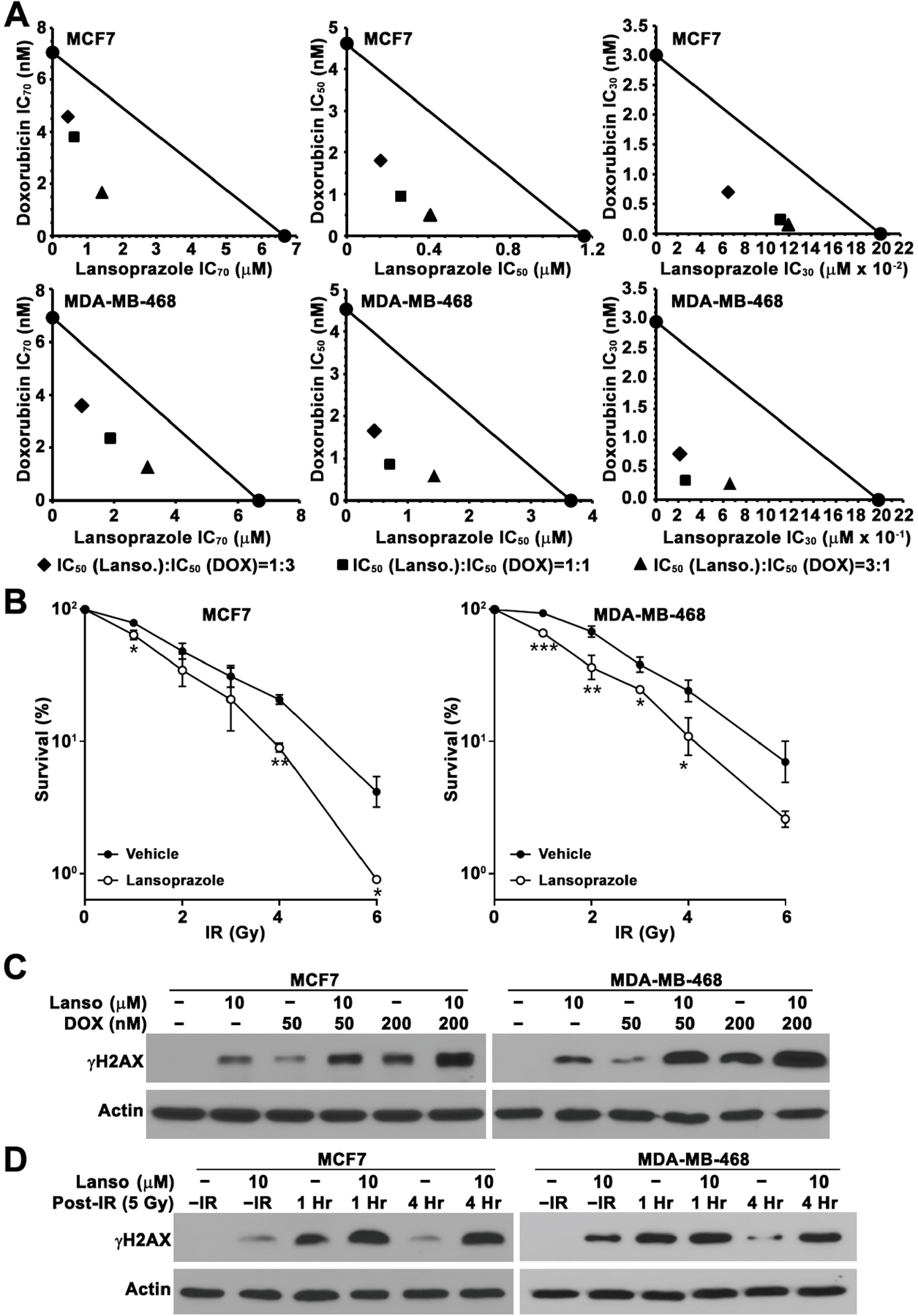
A, Isobologram of lansoprazole combination with doxorubicin with three different combination ratios. The solid circle connected by a solid line represents the IC70, IC50, or IC30 of lansoprazole as doxorubicin single agents in survival assay. Other solid symbols represents IC70, IC50, or IC30 of lansoprazole or doxorubicin in different combination treatments, which are below the line, indicating synergism. B, Lansoprazole sensitization of MCF7 and MDA-MB-468 cells to IR. (n=3, *p<0.05, **p<0.01). C and D, Western blot analysis of γH2AX in MCF7 and MDA- MB-468 cells following treatments with lansoprazole, doxorubicin, IR, or lansoprazole with doxorubicin or IR. Actin was used as a loading control.
To ensure that the above synergism is a result of lansoprazole inhibition of repair of DNA damages induced by doxorubicin or IR, we determined the effect of lansoprazole on DNA damages induced by doxorubicin. As shown in Figure 3C, lansoprazole synergistically increased doxorubicin-induced γ-H2AX production in both MCF7 and MDA-MB-468 cells.
We also took advantage of IR treatment, which allows monitoring DNA repair at different times following IR. We first pre-treated MCF7 and MDA-MB-468 cells with lansoprazole followed by 5 Gy IR treatment. Cells were collected at 1 or 4 hrs following IR for determination of γ-H2AX as an indicator of remaining DNA damages. As shown in Figure 3D, without IR, lansoprazole pre-treatment increased γ-H2AX compared with untreated control cells. At 1 hr following IR, dramatic increases in γ-H2AX level were observed in all cells. Interestingly, little γ-H2AX remained at 4 hrs post-IR in cells without lansoprazole pre-treatment whereas no reduction in γ-H2AX level was observed in the cells pre-treated by lansoprazole. These findings indicate that the majority of DNA damages induced by IR have been repaired at 4 hrs after IR in the control cells while the repair may have been inhibited in cells pre-treated by lansoprazole.
3.4. PPI usage improves overall survival of breast cancer patients.
To provide further evidence that PPIs may be repurposed as effective anticancer drugs, we next tested the benefit of PPI usage in breast cancer patient population for an association study. To achieve this goal, we performed retrospective studies of a cohort of 6,754 breast cancer patients diagnosed and treated between January 1st, 1995 and February 27th, 2014. The detailed patient characteristics and PPI usage of this cohort are shown in Supplemental Table S1.
These PPI users had significantly better overall survival than the non-PPI-users with an HR of 0.44 (log-rank p<2×10−16) from an uncorrected univariate analysis (Figure 4A and supplemental Table S2). In the multi-variate CoxPH model that corrects confounding factors (see Methods), the overall survival of PPI users improved further compared to the non-PPI-users with a HR of 0.40 (p<2×10−16). Note that being older or African American (AA), having an advanced or metastatic disease, or with disease of TNBC subtype all significantly increased the HR (Supplemental Table S2).
Figure 4.
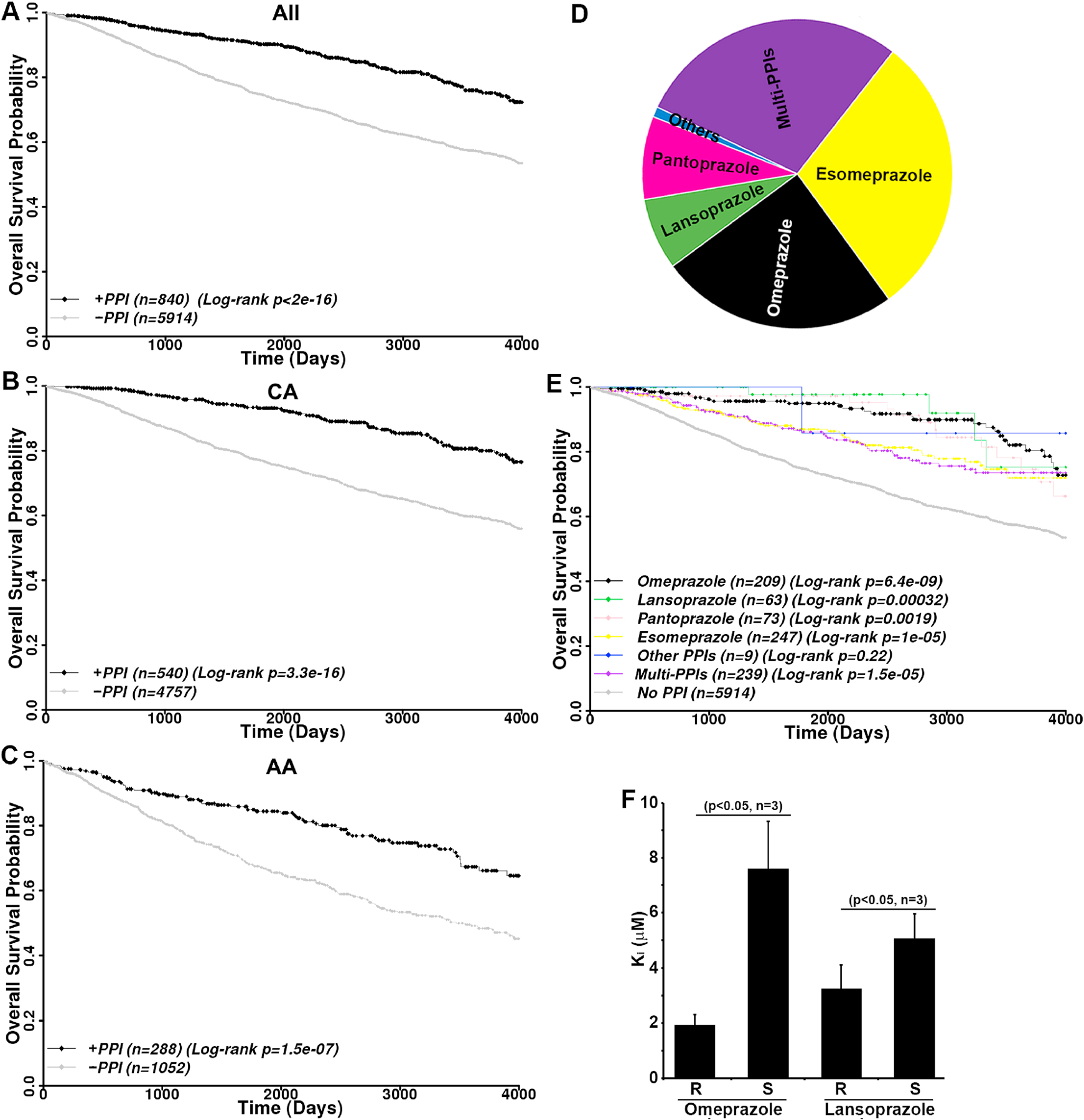
Benefits of PPI usage and differential effects of R- and S-enantiomers. A–C, Kaplan-Meier analysis of PPI usage on overall survival of all (A) or Caucasian American (B) and African American (C) breast cancer patients. D, User distribution of different PPIs. The other PPI user group includes dexlansoprazole and rabeprazole single users. E, Kaplan-Meier analysis of overall survival analysis of breast cancer patients using single and multiple PPIs. F, Inhibition of TE activity of FASN by R and S enantiomers of omeprazole and lansoprazole. Censored data are shown by diamonds.
3.5. PPI usage increases the overall survival of both Caucasian and African American patients.
Next, we examined if the benefits of PPI usage vary among different racial groups of breast cancer patients. In this cohort, the vast majority of patients are Caucasians (CA, n=5297, 78.43%) followed by African Americans (AA, n=1340, 19.84%). Other races including Hispanics, Pacific Islanders, Native Americans, South or East Asians occupy a small portion (n=117, 1.73%) with only 12 PPI users. The sample size of these patients is too small for a meaningful analysis and, thus, they were excluded from further study. As shown in Figure 4B–C and Supplemental Table S3, PPI usage significantly improved the overall survival of both CA and AA populations with a univariate HR of 0.38 (log-rank p=3.3×10−16) and 0.50 (log-rank p=1.5×10−7), respectively. The multivariate HR for CA and AA are 0.34 (p<2×10−16) and 0.49 (p=1.5×10−7), respectively. Therefore, PPI usage likely benefits both AA and CA patients.
3.6. Effects of different PPIs on overall survival.
There are six FDA-approved PPIs and it is of interest to determine if they have different levels of benefits. Figure 4D shows that all 6 FDA-approved PPIs were used by this cohort with majority of patients using omeprazole and esomeprazole. Patients using rabeprazole (n=7) and dexlansoprazole (n=2) were grouped as other PPI users due to the small sample size. Patients using >1 PPI were grouped as multi-PPI users (n=239). As shown in Figure 4E and Supplemental Table S4, users of omeprazole, lansoprazole, pantoprazole, and esomeprazole all had significantly better overall survival than non-PPI users with a univariate HR of 0.33 (log-rank p=6.4×10−9), 0.20 (log-rank p=0.00032), 0.43 (log-rank p=0.0019), and 0.52 (log-rank p=1.0×10−5), respectively. The multivariate HR for these PPIs are 0.27 (p=3.91×10−11), 0.25 (p=0.006), 0.41 (p=0.0014), and 0.48 (p=9.75×10−7), respectively (Supplemental Table S4). However, the users of other PPIs did not show significant benefits with a multivariate HR of 0.49 (p=0.48), likely due to the small sample size. It is noteworthy that multi-PPI users also had significantly better overall survival compared with non-PPI users with a univariate HR of 0.53 (log-rank p=1.5×10−5) and multivariate HR of 0.48 (p=1.49×10−6).
To determine if there is any potential difference between different PPIs, we performed pairwise comparison of overall survival between single PPI users of omeprazole, lansoprazole, pantoprazole and esomeprazole. Of all 6 paired comparisons, only omeprazole-esomeprazole pair showed a significant difference. Esomeprazole users had worse overall survival than omeprazole users with a univariate HR of 1.62 (log-rank p<0.048) and a multivariate HR of 1.64 (p=0.065). Because omeprazole is racemic consisting of both R and S enantiomers and esomeprazole contains only the S enantiomer, this finding suggests that S-enantiomers may be less effective than R-enantiomers.
3.7. Differential effects of R- and S-enantiomers of PPIs on FASN TE activity.
To determine if different PPI enantiomers possibly have different activity in inhibiting FASN and, thus, resulting in different clinical outcomes, we performed TE activity assay in the presence of R and S enantiomers of omeprazole as we previously described [20]. As shown in Figure 4F, the R-omeprazole is significantly more effective than the S-omeprazole in inhibiting TE with lower Ki. Similarly, the R-lansoprazole is also significantly more effective than the S-lansoprazole. Thus, the S-enantiomers of PPIs are less effective than the R-enantiomers in inhibiting FASN, which may contribute to the worse clinical outcome for esomeprazole compared with the racemic omeprazole users.
3.8. PPI usage increases overall survival of patients with different disease subtypes.
We next analyzed the benefits of PPI usage in patients with different disease subtypes. As shown in Figure 5A and Supplemental Table S3, the TNBC patients had significant benefits of PPI usage with a univariate HR of 0.50 (log-rank p=0.015) and a multivariate HR of 0.49 (p=0.015). Similar to the TNBC patients, patients with luminal A subtype breast cancer also benefited from PPI usage with a univariate HR of 0.52 (log-rank p=0.00060) and multivariate HR of 0.46 (p=8.62×10−4) (Figure 5B and Supplemental Table S3). However, the PPI benefits for patients with luminal B and HER2 positive diseases are statistically insignificant, possibly due to small sample size, with univariant HR of 0.34 (log-rank p=0.072) and 0.49 (log-rank p=0.078) and multivariant HR of 0.29 p=(0.070) and 0.48 (p=0.076), respectively, although a clear trend of benefits of PPI usage in these sub-cohorts is observed (Figure 5C–D and Supplemental Table S3).
Figure 5.
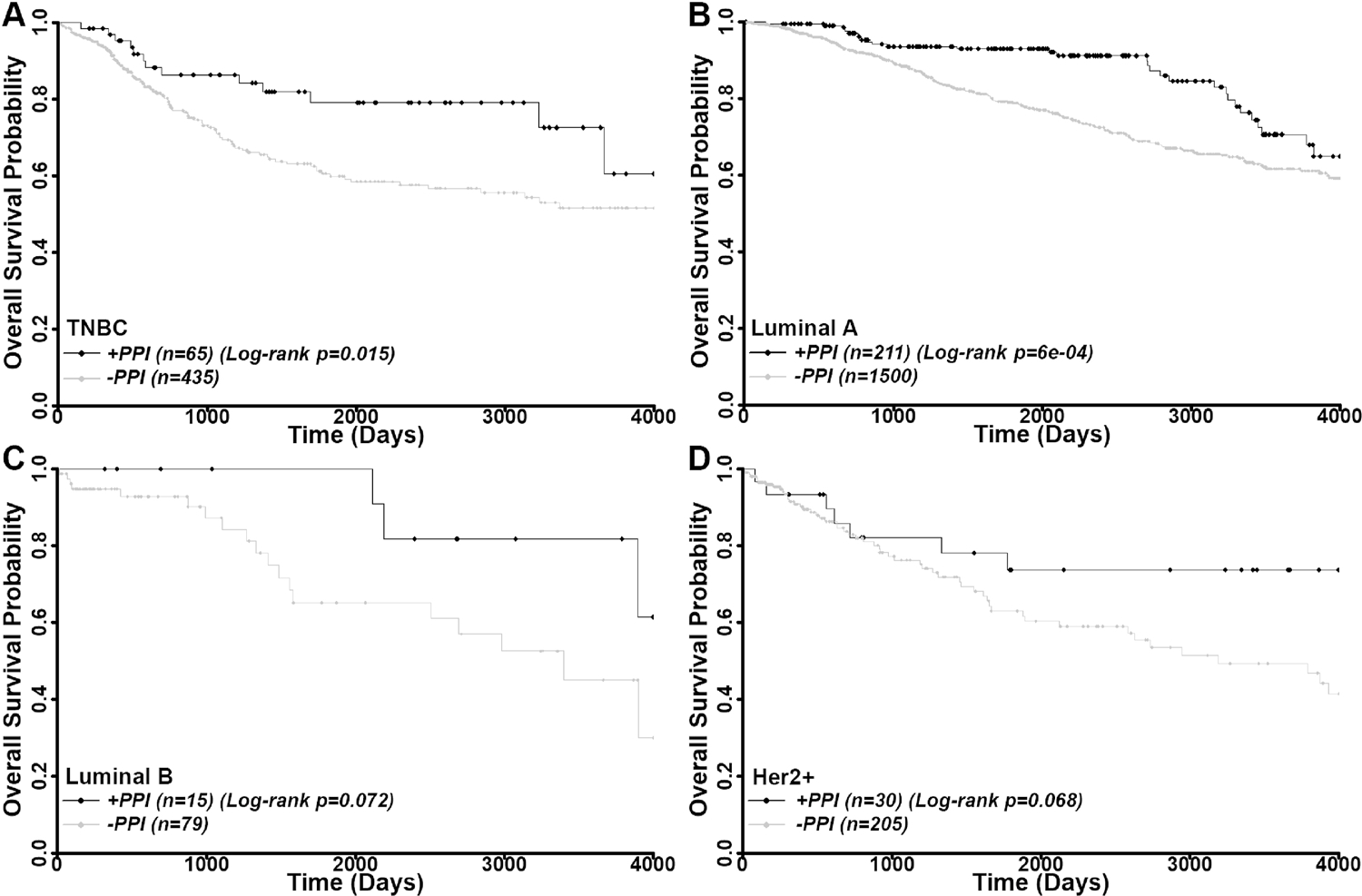
Benefits of PPI usage in different subtypes of breast cancer patients. Kaplan-Meier analyses of PPI usage on overall survival of TNBC (A), luminal A (B), luminal B (C), and Her2+ (D) subtype patients. Censored data are shown by diamonds.
3.9. Effects of PPI dosage on overall survival.
To determine if PPI dosage has any differential effects on the outcome, we analyzed data of patients who used only one PPI with available dosage information. Pantoprazole and rabeprazole were used at single dose with 40 mg and 20 mg, respectively. Lansoprazole, on the other hand, was used at two different doses (30 mg and 15 mg). However, of the 65 lansoprazole users, only 2 patients took 15 mg lansoprazole, which prohibits any meaningful analysis. Thus, pantoprazole, rabeprazole, and lansoprazole were excluded from dosage analysis.
Multiple dosing of 10 mg, 20 mg and 40 mg were used for omeprazole and 20 mg and 40 mg for esomeprazole. However, there are only 3 patients who used 10 mg omeprazole. Thus, we grouped 10 mg and 20 mg users into the 10–20 mg group for further study. As shown in Figure 6A and Supplemental Table S4, 10–20 mg and 40 mg omeprazole users have univariant HRs of 0.33 (log-rank p=2.1×10−7) and 0.21 (log-rank p=0.09), respectively. The multivariant HRs for these dosage groups are 0.28 (p=1.49×10−8) and 0.12 (p=0.038). Thus, both dosage groups significantly improved the overall survival. Despite only 15 patients were considered, the 40-mg omeprazole group may have benefited more than the 10–20 mg group with a lower hazard ratio. This difference is strikingly clear with esomeprazole where the 40 mg users significantly increased the overall survival with a univariant HR of 0.3 (log-rank p=4.2×10−7) and a multivariant HR of 0.33 (p=1.8×10−6), while the 20 mg esomeprazole users failed to do so with a univariant HR of 1.0 (log-rank p=0.98) and a multivariant HR of 0.73 (p=0.18) (Figure 6B and Table S4). This striking difference is likely derived from the fact that esomeprazole is less effective overall than the racemic omeprazole as discussed above, which makes the differential dose effect more clear-cut. Thus, we conclude that the dosage of PPIs may affect patient outcome.
Figure 6.
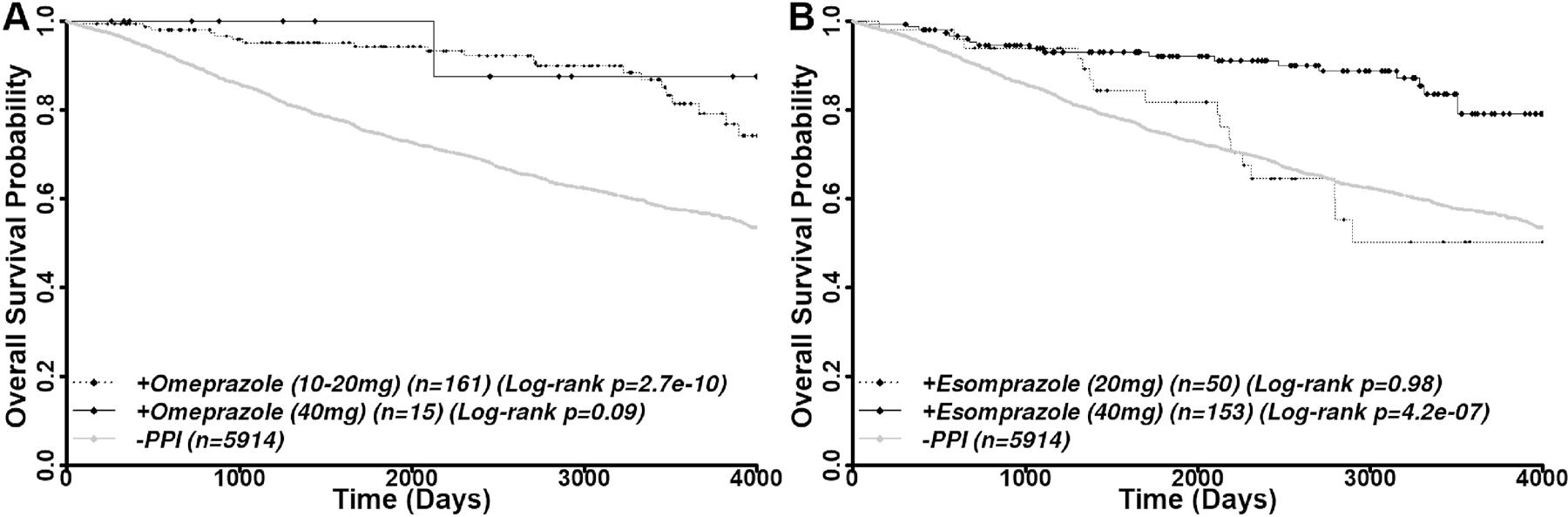
Different doses of PPIs and benefits. Kaplan-Meier analysis of different doses of omeprazole (A) and esomeprazole (B) on overall survival of breast cancer patients.
3.10. Potential synergistic effects of PPIs in combination with hormone, chemo, and radiation.
Although PPIs were not intended as a drug for treating breast cancer in this cohort, our finding that combination of PPIs with doxorubicin or IR synergistically inhibits breast cancer cell lines (see above) prompted us to examine if PPI usage benefits the subgroups of patients who were treated by hormone, chemo, or radiation alone differently. As shown in Figure 7A–C and Supplemental Table S3, PPI usage significantly increased the overall survival of patients who received hormone (univariate HR=0.60, log-rank p=0.01), chemo (univariate HR=0.53, log-rank p=0.0044), or radiation (univariate HR=0.33, log-rank p=4.3×10−5) treatment. In the multivariate analyses, PPI further increased overall survival with an HR of 0.57 (p=1.1×10−3), 0.46 (p=1.6×10−5), and 0.30 (p=2.5×10−5) for hormone-, chemo-, and radiation-treated patients, respectively. Although PPI significantly benefited all three treatment groups, the data suggest that PPI in combination with IR benefit patients the best, consistent with our finding that PPI inhibits FASN-mediated DNA repair.
Figure 7.
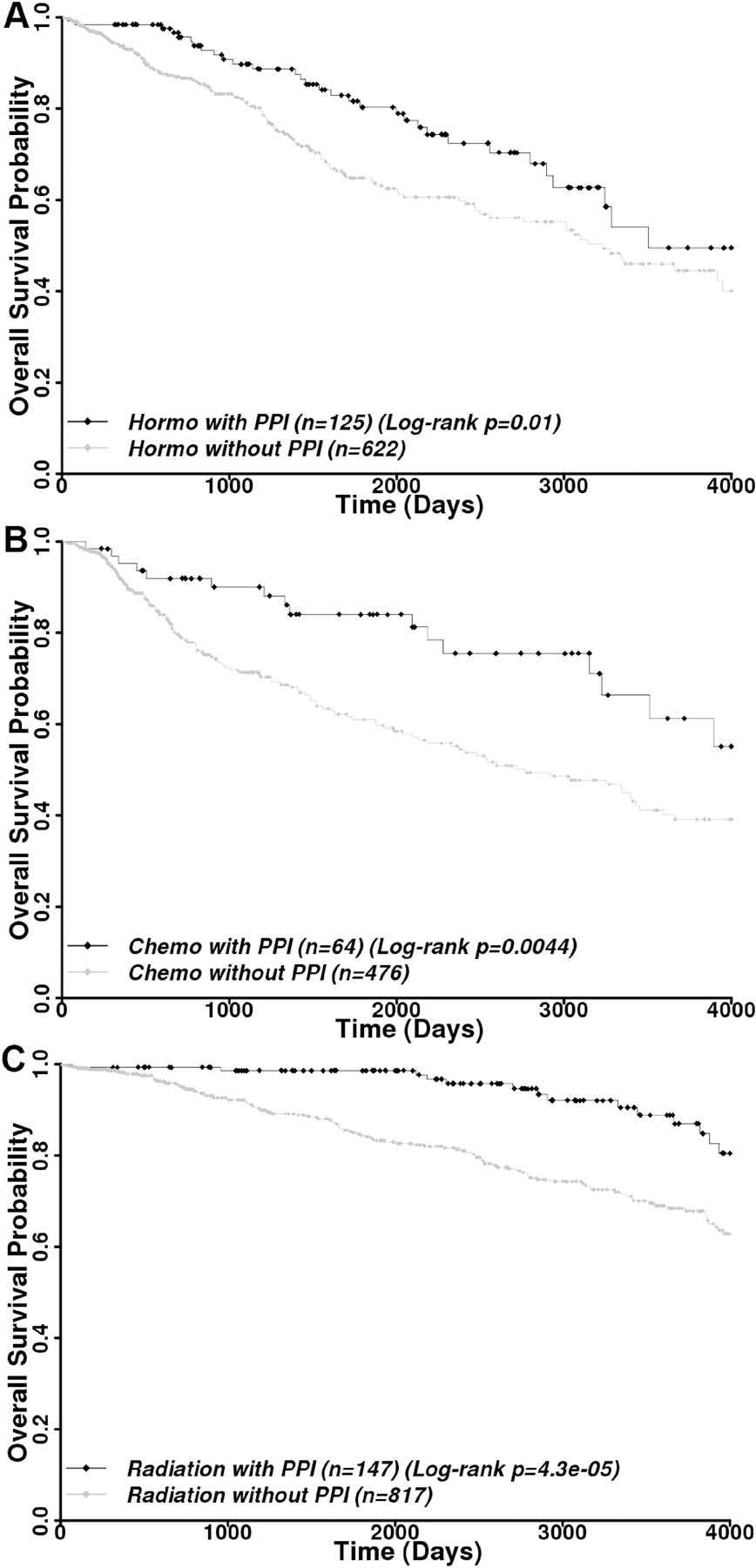
Benefits of PPI usage in combination with hormone, chemo, and radiation therapy. Kaplan-Meier analysis of overall survival among all breast cancer patients receiving hormone (A), chemo (B), or radiation (C) therapy treatment alone but with or without PPI usage. Censored data are shown by diamonds.
3.11. PPI usage reduces recurrence rate and improves 5-year survival of patients with recurrent disease.
Previously, it was reported that breast cancer patients with high FASN level are 4 times more likely to have recurrent disease [6, 7]. We next analyzed the potential effect of PPI usage on breast cancer recurrence. Of this cohort, 266 patients had disease recurrence within 4000 days of follow up. As shown in Figure 8A and Supplemental Table S3, PPI users are much less likely to have disease recurrence compared with non-PPI-users with a univariate HR of 0.087 (log-rank p=7.7×10−14) and a multivariate HR of 0.088 (p=4.1×10−9).
Figure 8. Association between PPI usage and breast cancer recurrence.
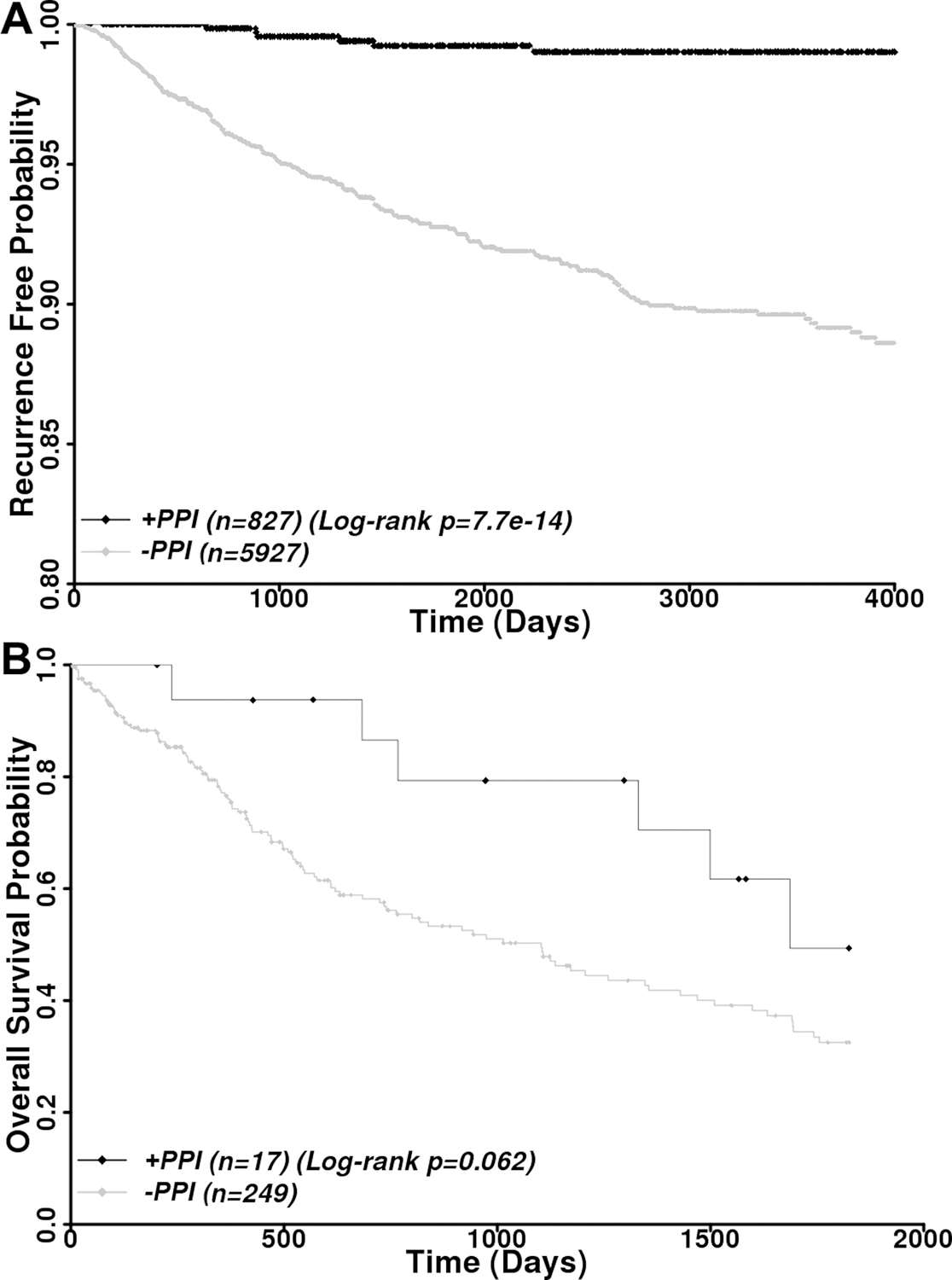
A, Kaplan-Meier analysis of recurrence free disease among all patients with and without PPI usage. B, Kaplan-Meier analysis of PPI effects on 5-year survival of patients with recurrent disease. Censored data are shown by diamonds.
To determine if PPI usage benefited patients who already had cancer recurrence, we analyzed 5-year overall survival of PPI users vs non-PPI users of 266 patients with recurrent disease. For this analysis, only patients who took PPIs after disease recurrence were considered PPI users. As shown in Figure 8B and Supplemental Table S3, the 5-year overall survival of PPI users is much better than non-PPI users with a univariate HR of 0.47 (log-rank p=0.062) and a multivariate HR of 0.41 (p=0.038). Thus, PPI usage likely helps prevent disease recurrence and improve the 5-year survival of patients with recurrent breast cancer. However, it is noteworthy that we were unable to stratify the patients with recurrent disease into different subtypes for analyses due to small sample size in each subtype. For example, there is only 1 HER2+ and 1 TNBC patient with current disease who used PPIs, which will certainly prevent any meaningful analysis.
4. Discussion
The identification of PPIs as effective inhibitors of FASN, the mechanism of their actions, and the findings of their significant benefits to breast cancer patients suggest that PPIs may be repurposed as FASN inhibitors for combinational treatment of breast cancers, especially TNBC that lacks effective and targeted treatments. Indeed, the findings from this study have led to an investigator-initiated multicenter clinical trial testing PPIs in combination treatments of TNBC patients (NCT02595372).
We showed that lansoprazole treatment reduced PARP1 expression and NHEJ activity and resulted in increased DNA damage in breast cancer cells, which can be rescued by over-expressing FASN. These findings suggest that PPI suppression of cancer cell proliferation and induction of apoptosis is likely due to inhibition of FASN and NHEJ repair activity. The finding that lansoprazole treatment alone induced increased expression of γH2AX suggests that these cancer cells cannot maintain genomic integrity and that the endogenous oxidative DNA damages may cause cell death. Based on these findings, it is tempting to speculate that combining PPIs with PARPi’s may also generate synergistic effect on BRCA mutant breast cancers and perhaps help expand the use of PARPi’s to BRCA wild-type breast cancers due to PPI inhibition of PARP1 expression via FASN.
The finding that artificially increasing FASN level decreases the potency of lansoprazole while artificially knocking down FASN increases the potency appears to contradict with the finding that the potency of lansoprazole negatively associates with the level of endogenous FASN in breast cancer cells. It is possible that cancer cells with high endogenous FASN level may require and be addicted to the high level of FASN and, thus, are more sensitive to PPI than cells with less FASN. However, the overexpressed ectopic FASN are not required for MCF7 cell proliferation but may sink lansoprazole and, thus, reduce lansoprazole potency. Similarly, M3K cells express excess endogenous FASN, which are required to resist drugs such as doxorubicin and cisplatin but not required for M3K cell proliferation. FASN knockdown in M3K cells reduced the excess level of FASN that serves as sink of lansoprazole by binding to lansoprazole, reducing lansoprazole potency. Hence, we believe that both findings suggest that PPI likely inhibit breast cancer cells by acting on FASN.
The finding that the S-enantiomer of PPIs is less effective in inhibiting the thioesterase activity of FASN and in benefiting breast cancer patient outcome support the conclusion that the benefit of PPI usage in patients is due to PPI inhibition of FASN. These findings also suggest that the FDA-approved dexlansoprazole (R-enantiomer of lansoprazole) may be chosen as a leading candidate for future studies in further developing and repurposing PPIs as anticancer drugs.
It is noteworthy, however, that the conclusion from the data analyses of the EMR should be taken with caution. Firstly, the retrospective data mining provides only an association between PPI usage and overall survival. It does not provide evidence on if PPI works via inhibiting FASN although the cell-based studies suggest that FASN may be the target. Nevertheless, a recent multi-center clinical trial showed that addition of high dose omeprazole to neoadjuvant TNBC treatment yielded a ~74% pCR rate without adding toxicity and FASN activities in tumors were inhibited [30]. This finding suggests that PPIs likely inhibit FASN in human breast cancer with added benefits. Furthermore, the findings that PPIs lowers disease recurrence and that the S-enantiomers are less effective in inhibiting FASN and in overall survival both support FASN as the PPI target in patient outcome. Secondly, patients who took PPIs were likely due to gastric acid hyperactivity. Whether the gastric acid hyperactivity not the PPI use per se that improved survival outcome remains to be determined. However, our observation that patients who used lower doses of PPIs performed worse than the high dose users argues against this possibility. Finally, although we have considered many compounding factors, other potential bias with complex and missing data or undetected errors in the EMR could affect the outcome of the data analyses. One example is body mass index, which varies from time to time for some patients while the majority of patients do not have these data for a meaningful analysis. Unfortunately, we regret that we have exhausted the information in this dataset and are unable to delineate other potential compounding factors.
The fact that PPIs inhibit FASN indicate that PPIs may also be repurposed for other diseases where FASN over-expression is a culprit in addition to breast cancer. For example, it was recently found that increased FASN expression in adipose tissues are l inked to obesity, type 2 diabetes, and insulin resistance [31] and that chronic use of a FASN inhibitor, platensimycin, led to improved insulin sensitivity in db/+ mice fed with high-fructose diet and reduced ambient glucose levels in db/db mice [32]. Increased FASN expression may also contribute to nonalcoholic fatty liver disease or steatosis [33, 34]. Thus, PPIs may be used to benefit these in addition to cancer patients.
Supplementary Material
Highlights.
FASN contributes to poor prognosis, disease recurrence, and drug/IR resistance
PPIs inhibit FASN activity, PARP1 expression, and NHEJ repair activity
PPI synergizes with doxorubicin and IR
S-enantiomers of PPIs are less effective than R-enantiomers
PPI usage increases overall survival and reduces disease recurrence
5. Acknowledgement.
The authors wish to thank the Regenstrief Institute at Indiana University School of Medicine for the use of patient database. This work was supported in part by grants from DoD (W81XWH-16-1-0030) to JTZ, from NIH (R 01 GM127656) to JYL, and from Breast Cancer Research Foundation to KM.
Abbreviations used:
- FASN
fatty acid synthase
- H2AX
H2A histone family member X
- IR
ionizing radiation
- NHEJ
non-homologous end joining
- PARP
poly (ADP-ribose) polymerase
- PPI
proton pump inhibitor
- TE
thioesterase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. Conflict of Interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Buckley D, Duke G, Heuer TS, O’Farrell M, Wagman AS, McCulloch W, Kemble G, Fatty acid synthase - Modern tumor cell biology insights into a classical oncology target, Pharmacol. Ther, 177 (2017) 23–31. [DOI] [PubMed] [Google Scholar]
- [2].Liu H, Liu JY, Wu X, Zhang JT, Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker, Int. J. Biochem. Mol. Biol, 1 (2010) 69–89. [PMC free article] [PubMed] [Google Scholar]
- [3].Menendez JA, Lupu R, Fatty acid synthase (FASN) as a therapeutic target in breast cancer, Expert Opin. Ther. Targets, 21 (2017) 1001–1016. [DOI] [PubMed] [Google Scholar]
- [4].Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA, Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells, Cell Prolif, 41 (2008) 59–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M, Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer, J. Natl. Cancer Inst, 101 (2009) 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kuhajda FP, Piantadosi S, Pasternack GR, Haptoglobin-related protein (Hpr) epitopes in breast cancer as a predictor of recurrence of the disease, N. Engl. J. Med, 321 (1989) 636–641. [DOI] [PubMed] [Google Scholar]
- [7].Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR, Fatty acid synthesis: a potential selective target for antineoplastic therapy, Proc. Natl. Acad. Sci. U. S. A, 91 (1994) 6379–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Corominas-Faja B, Vellon L, Cuyas E, Buxo M, Martin-Castillo B, Serra D, Garcia J, Lupu R, Menendez JA, Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in HER2+ breast cancer, Histol. Histopathol, 32 (2017) 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu H, Liu Y, Zhang JT, A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction, Mol. Cancer Ther, 7 (2008) 263–270. [DOI] [PubMed] [Google Scholar]
- [10].Liu H, Wu X, Dong Z, Luo Z, Zhao Z, Xu Y, Zhang JT, Fatty acid synthase causes drug resistance by inhibiting TNF-alpha and ceramide production, J. Lipid Res, 54 (2013) 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu X, Dong Z, Wang CJ, Barlow LJ, Fako V, Serrano MA, Zou Y, Liu JY, Zhang JT, FASN regulates cellular response to genotoxic treatments by increasing PARP-1 expression and DNA repair activity via NF-kappaB and SP1, Proc. Natl. Acad. Sci. U. S. A, 113 (2016) E6965–E6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mashima T, Seimiya H, Tsuruo T, De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy, Br. J. Cancer, 100 (2009) 1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Flavin R, Peluso S, Nguyen PL, Loda M, Fatty acid synthase as a potential therapeutic target in cancer, Future Oncol, 6 (2010) 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vance D, Goldberg I, Mitsuhashi O, Bloch K, Inhibition of fatty acid synthetases by the antibiotic cerulenin, Biochem Biophys Res Commun, 48 (1972) 649–656. [DOI] [PubMed] [Google Scholar]
- [15].D’Agnolo G, Rosenfeld IS, Awaya J, Omura S, Vagelos PR, Inhibition of fatty acid synthesis by the antibiotic cerulenin. Specific inactivation of beta-ketoacyl-acyl carrier protein synthetase, Biochim Biophys Acta, 326 (1973) 155–156. [DOI] [PubMed] [Google Scholar]
- [16].Hardwicke MA, Rendina AR, Williams SP, Moore ML, Wang L, Krueger JA, Plant RN, Totoritis RD, Zhang G, Briand J, Burkhart WA, Brown KK, Parrish CA, A human fatty acid synthase inhibitor binds beta-ketoacyl reductase in the keto-substrate site, Nat. Chem. Biol, 10 (2014) 774–779. [DOI] [PubMed] [Google Scholar]
- [17].Alwarawrah Y, Hughes P, Loiselle D, Carlson DA, Darr DB, Jordan JL, Xiong J, Hunter LM, Dubois LG, Thompson JW, Kulkarni MM, Ratcliff AN, Kwiek JJ, Haystead TA, Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer, Cell Chem Biol, 23 (2016) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Angeles TS, Hudkins RL, Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors, Expert Opin Drug Dis, 11 (2016) 1187–1199. [DOI] [PubMed] [Google Scholar]
- [19].Mullen GE, Yet L, Progress in the development of fatty acid synthase inhibitors as anticancer targets, Bioorg. Med. Chem. Lett, 25 (2015) 4363–4369. [DOI] [PubMed] [Google Scholar]
- [20].Fako VE, Wu X, Pflug B, Liu JY, Zhang JT, Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human Fatty Acid synthase, J. Med. Chem, 58 (2015) 778–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Z, Dong Z, Myer D, Yip-Schneider M, Liu J, Cui P, Schmidt CM, Zhang JT, Role of 14-3-3sigma in poor prognosis and in radiation and drug resistance of human pancreatic cancers, BMC Cancer, 10 (2010) 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chou TC, Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul, 22 (1984) 27–55. [DOI] [PubMed] [Google Scholar]
- [23].Pizer ES, Chrest FJ, DiGiuseppe JA, Han WF, Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines, Cancer Res, 58 (1998) 4611–4615. [PubMed] [Google Scholar]
- [24].Knowles LM, Axelrod F, Browne CD, Smith JW, A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2, J Biol Chem, 279 (2004) 30540–30545. [DOI] [PubMed] [Google Scholar]
- [25].De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV, RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells, Cancer Res, 63 (2003) 3799–3804. [PubMed] [Google Scholar]
- [26].Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP, Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells, Cancer Res, 63 (2003) 7330–7337. [PubMed] [Google Scholar]
- [27].Menendez JA, Colomer R, Lupu R, Inhibition of tumor-associated fatty acid synthase activity enhances vinorelbine (Navelbine)-induced cytotoxicity and apoptotic cell death in human breast cancer cells, Oncol Rep, 12 (2004) 411–422. [PubMed] [Google Scholar]
- [28].Menendez JA, Mehmi I, Atlas E, Colomer R, Lupu R, Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-kappaB, Int J Oncol, 24 (2004) 591–608. [PubMed] [Google Scholar]
- [29].Schwartz D, Rotter V, p53-dependent cell cycle control: response to genotoxic stress, Semin. Cancer Biol, 8 (1998) 325–336. [DOI] [PubMed] [Google Scholar]
- [30].Sardesai SD, Thomas A, Gallagher C, Lynce F, Ottaviano YL, Ballinger TJ, Schneider BP, Storniolo AM, Bauchle A, Althouse SK, Perkins S, Zhang J-T, Miller K, Inhibiting fatty acid synthase in operable triple negative breast cancer, J. Clin. Oncol, 38 (2020) 584–584.31821065 [Google Scholar]
- [31].Menendez JA, Vazquez-Martin A, Ortega FJ, Fernandez-Real JM, Fatty acid synthase: association with insulin resistance, type 2 diabetes, and cancer, Clin. Chem, 55 (2009) 425–438. [DOI] [PubMed] [Google Scholar]
- [32].Wu M, Singh SB, Wang J, Chung CC, Salituro G, Karanam BV, Lee SH, Powles M, Ellsworth KP, Lassman ME, Miller C, Myers RW, Tota MR, Zhang BB, Li C, Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes, Proc. Natl. Acad. Sci. U. S. A, 108 (2011) 5378–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dorn C, Riener MO, Kirovski G, Saugspier M, Steib K, Weiss TS, Gabele E, Kristiansen G, Hartmann A, Hellerbrand C, Expression of fatty acid synthase in nonalcoholic fatty liver disease, Int. J. Clin. Exp. Pathol, 3 (2010) 505–514. [PMC free article] [PubMed] [Google Scholar]
- [34].Postic C, Girard J, Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice, J. Clin. Invest, 118 (2008) 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.