

Abstract
Hepatocellular carcinoma (HCC) is a high mortality malignancy and the second leading cause of cancer-related deaths. Because the immune system plays a dual role by assisting the host barrier and tumor progression, there are complex interactions with considerable prognostic significance. Herein, we performed single-sample gene set enrichment (ssGSEA) to explore the tumor microenvironment (TME) and quantify the tumor-infiltrating immune cell (TIIC) subgroups of immune responses based on the HCC cohort of The Cancer Genome Atlas (TCGA) database. We evaluate molecular subpopulations, survival, function, and expression differential associations, as well as reveal potential targets, and biomarkers for immunotherapy. We combined the TME score and the 29 immune cell types in the low, medium, and high immunity groups. The stromal score, immune score, and ESTIMATE score were positively correlated with immune activity but negatively correlated with the tumor purity. There were 23 human leukocyte antigen (HLA)-related genes that were significantly different. However, KIAA1429 was not significant among the different immunity groups. Besides, programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expression increased with the increase of immune activity. This may provide valuable information for HCC immunotherapy. We also found that there was no significant difference in naïve B cells, macrophages M1, activated mast cells, resting natural killer (NK) cells, and T cells gamma delta among the different immunity groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that the differential proteins were mainly enriched in alpha-linolenic acid (ALA) metabolism, cytokine-cytokine receptor interaction, glycosaminoglycan biosynthesis-heparan sulfate/heparin, glycosphingolipid biosynthesis-ganglio series and proteasome. Our findings provide a deeper understanding of the immune scene, uncovering remarkable immune infiltration patterns of various subtypes of HCC using ssGSEA. This study advances the understanding of immune response and provides a basis for research to enhance immunotherapy.
Keywords: ssGSEA, HCC, data mining, gene expression
Introduction
Hepatocellular carcino ma (HCC) is a high mortality malignancy. It is the fifth most common cancer globally and the second leading cause of cancer-related deaths, with over 500,000 new cases annually.1 Viral hepatitis and nonalcoholic steatohepatitis are the most common causes of cirrhosis and approximately 80% of cases develop to HCC.2,3 Due to the recurrence of HCC, its prognosis remains dismal with an overall 5-year survival rate of 34% to 50%.4 Despite the rapid development of advanced medical technology, there are still no effective treatment strategies for HCC patients.5 Today, radical treatment can improve the survival rate of patients diagnosed with early-stage HCC and provide long-term potential treatment options.6 Although serum biomarkers, such as alpha-fetoprotein (AFP) and alkaline phosphatase (AKP or ALP), are frequently used in clinical practice, they lack sufficient sensitivity and specificity.7 Therefore, finding effective biomarkers is necessary for the diagnosis and treatment of HCC.
The tumor microenvironment (TME) contains cellular components (such as immune cells, fibroblasts, endothelial cells) and extracellular components (such as hormones, cytokines, extracellular matrix, growth factors) that cover the tumor cells with a network provided by the blood vessels.8 TME plays a critical role in tumorigenesis, progression and metastasis, and affects the therapeutic effect.9 Tumor-infiltrating immune cells (TIIC) are considered to be effective targets for drugs; they manage cancer progression by building an ecosystem in the TME and linking it with clinical outcomes and have shown potential prognostic value.10 Because the immune system plays a dual role by assisting the host barrier and tumor progression, there are complex interactions with considerable prognostic significance.11 TIIC upregulates suppressive immune checkpoints of tumor cells to prevent tumor progression in the host immune surveillance.12 Previous studies have estimated TIIC using flow cytometry and immunohistochemistry (IHC),13 which are limited by the number of fluorescent channels available, and rarely can assess the immune cell types immediately.14 In addition, previous studies have not revealed insights into the prognostic evaluation of the TIIC subgroups. CIBERSORT, a metagene instrument, uses deconvolution technology, and complex algorithms to quantify the cellular components and cell types of immune responses in heterogeneous samples, thereby greatly expanding the capabilities of the genome database.15,16 In this study, we performed a single-sample gene set enrichment (ssGSEA) to explore TME and quantify the TIIC subgroups of immune responses based on the HCC cohort of The Cancer Genome Atlas (TCGA) database. We evaluate molecular subpopulations, survival, function, and expression differential associations, as well as reveal potential targets and biomarkers for immunotherapy.
Methods
Data acquisition
A freely accessible dataset with gene expression profiles and corresponding visualization data was identified and downloaded. Information on gene expression and corresponding clinical information were identified and downloaded from Level 4 gene expression data (standardized FPKM) of the TCGA HCC cohort. For the TCGA dataset, the RNA sequencing data (FPKM values) were changed to transcripts per million (TPM) values, which slightly resemble microarrays transcripts, and are easier to perform between samples. The clinicopathological data collected included gender, age, stage, grade, T-phase, M-phase, N-phase, survival status, and the number of days of survival. Data were analyzed using R (version 3.5.3) and R Bioconductor software packages. The Perl language was used to filter the immune cell-matrix based on a P-value of less than 0.05.
Assessment of immune infiltration
Infiltrating cells in TME and the proportion and cell type of immune cells in HCC heterogeneous samples were determined. A metagene tool, CIBERSORT, uses the deconvolution of large amounts of gene expression data and a complex algorithm. The selection was confirmed by fluorescence-activated cell sorting (FACS). TIICs included macrophages (M0/M1/M2 macrophages), resting memory CD4+ T cells of 7 T cell types (T follicular helper cells [Tfh], activated memory CD4+ T cells, γδ T cells, Tregs, CD8+ T cells and naive CD4+ T cells), resting natural killer (NK) cells, resting/activated mast cells, activated NK cells, resting dendritic cells (DCs), memory B cells, activated DCs, monocytes, naive B cells, plasma cells, eosinophils, and neutrophils. ssGSEA classified gene sets with common biological functions, chromosomal localization, and physiological regulation.17 The gene sets included 782 genes used to predict the abundance of the 29 TIICs in a single tissue sample are shown in Supplemental Table 1.
Statistical analysis
Statistical analysis was performed using R version 3.5.3 and Bioconductor. The unpaired Student’s t-test and the Wilcoxon test were used to evaluate the normal distribution variables and the non-normal distribution variables, respectively. The difference in the gene expression of the immune checkpoint molecules and the clinical information between tumors and normal tissues were analyzed by the Wilcoxon test. To identify the differential genes in differentially expressed genes (DEG) assessments, we applied the Benjamini-Hochberg method to convert the P-value to a false discovery rate (FDR). Using “maxstat” (R package), TIIC penetration was classified as low, medium or high abundance. ssGSEA-normalized HCC DEGs data were compared with a genome using “GSVA” (R-package). For each statistical analysis, a P-value of <0.05 was considered statistically significant.
Results
Immunity group and TME analysis
After standardization of the microarray results, we first classified immune infiltration into low, medium or high abundance clusters using the ssGSEA score. The immune score of each sample was normalized to between 0 and 1 for later analysis. The cluster map of the 29 immune cell types in each cluster enrichment is shown in Figure 1. The results showed that the expression of immune cells increased with the increase of immune activity. Therefore, cluster 1/2/3 was defined as low/medium/high immunity group, respectively. Next, the TME was scored (stromal score, immune score, ESTIMATE score, and tumor purity) in each HCC sample using “estimate” (R package). The detailed information of each sample is shown in Supplemental Table 2. Meanwhile, the TME score was combined with the 29 immune cell types in the low, medium and high immunity groups. The results showed that the stromal score, immune score and ESTIMATE score were positively correlated with immune activity but negatively correlated with tumor purity (Figure 2). In addition, the connection between the TME set and the immunity group was significantly different (P < 0.01, Figure 3). At the same time, the heatmap of CIBERSORT, ESTIMATE, and ssGSEA algorithms were compared to evaluate cellular components or cellular immune responses between normal and tumor tissue samples (Figure 4).
Figure 1.

Heatmap of the 29 immune cell types in each cluster enrichment.
Figure 2.

Combination of the TME score and the 29 immune cell types in the low, medium and high immunity groups.
Figure 3.

Correlation between the TME set and the immunity groups.
Figure 4.

The heatmap of CIBERSORT, ESTIMATE, and ssGSEA algorithms were compared to assess cellular components or cell immune responses between normal and tumor tissue samples.
Immunity group and gene expression
Human leukocyte antigen (HLA) is the most complex human genetic system by far and plays a vital role in medical research. We explored the relationship between HLA-related gene expression and the different immunity groups. The results showed that 23 HLA-related genes were significantly different. However, there was no significant difference in KIAA1429 among the different immunity groups (Figure 5). HLA-related gene expression increased with the increase of immune activity. Thus, we hypothesize that HLA expression is related to the survival of HCC patients, however, this requires further investigation. Meanwhile, we explored the differential expression of programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) among the different immunity groups. We found that PD-L1 and CTLA-4 expression increased with the increase of immune activity, and this may provide information for HCC immunotherapy (Figure 6). Next, Kaplan–Meier curve was used to analyze the survival rate of patients in each immunity group. The results demonstrated that the immunity group had a significant effect on the patients’ overall survival (P = 0.03, Figure 7(a)). Next, the association between the clinicopathological factor and the immune subgroups was explored. The results indicated that the immune subgroups were related to the tumor stage (P = 0.006, 95% CI: 0.07–0.41, stage I vs stage III), suggesting that patients with high immune activity are inclined to progress to an advanced stage.
Figure 5.
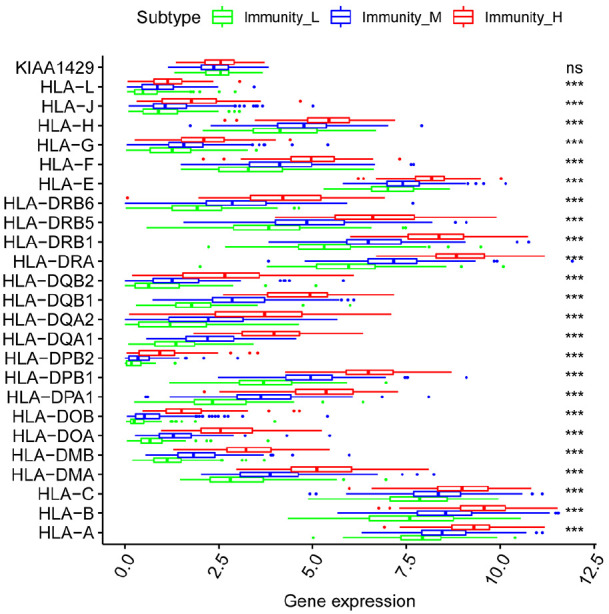
Relationship between HLA-related gene expression and the immunity groups.
Figure 6.

Differential expression of programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in different immunity groups.
Figure 7.

CIBERSORT analysis of tumor-infiltrating immune cells in TME and the proportion of immune cells and cell types in HCC heterogeneous samples. (a) Survival analysis in immunity group. (b) Tumor-infiltrating immune cells analysis.
Tumor-infiltrating immune cells analysis
Next, we used CIBERSORT to analyze tumor-infiltrating immune cells in the TME and measured the proportion of immune cells and cell types in HCC heterogeneous samples. As shown in Figure 7(b), there was no significant differences in naïve B cells, macrophages M1, activated mast cells, resting NK cells and T cells gamma delta among the different immunity groups.
GO and KEGG analysis
Finally, we analyzed the function of the differentially expressed genes identified in different immunity groups. Biological analyses were performed using gene ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The GO analysis results showed that differentially expressed genes were enriched in complement activation, classical pathway; immunoglobulin complex, circulating; immunoglobulin receptor binding; and major histocompatibility complex (MHC) class II protein complex. The KEGG pathway analysis revealed that the differential proteins were mainly enriched in alpha-linolenic (ALA) acid metabolism, cytokine-cytokine receptor interaction, glycosaminoglycan biosynthesis-heparan sulfate/heparin, glycosphingolipid biosynthesis-ganglio series and proteasome (Figure 8 and Supplemental Tables 3 and 4).
Figure 8.

GO enrichment and KEGG pathway analyses of differentially expressed genes in different immunity groups.
Discussion
ssGSEA is an implementation method proposed for GSEA, which can be performed on a single sample. It calculates the rank value of each gene based on the expression profile and then performs subsequent statistical analysis. In our study, this method scored 29 immune-related gene sets in each sample (the immune gene set included immune cell types, functions, and pathways). Through cluster analysis, the samples were divided into low/medium/high immunity groups. The correlations between the immune groups and the TME, gene expression, TIIC and biological/pathway analyses were analyzed to verify the grouping accuracy.
The significance of TME and TIIC for cancer prognosis has been demonstrated. Malignant solid tumor tissues consist of tumor cells, tumor-associated normal epithelial, immune, stromal, and vascular cells. TME is of great significance not only for understanding the occurrence, development and metastasis of tumors but also for the diagnosis, prevention and prognosis of tumors. Lipid metabolism disorders and related signaling pathways may suggest new strategies for treating HCC by reprogramming cellular lipid metabolism or regulating TME.18 Currently, cancer-associated fibroblasts (CAF) are the main stromal cell type in the HCC microenvironment, which have promoted the development of HCC and gradually become a hot research topic in HCC targeted therapy.19 Mesenchymal stromal cells (MSCs) may migrate to the tumor sites and play a role in HCC through paracrine interactions.20 MSC transplantation can serve as an effective immunomodulatory strategy to induce tolerance to a variety of immune-related diseases.21 Yoshihara et al.22 determined immune and stromal scores via gene expression signatures and used these scores to infer immune and stromal cell fractions in tumor samples. In our study, we first classified immune infiltration into low, medium or high abundance immunity groups and scored the TME in each HCC sample. We found that stromal score and immune score were positively correlated with immune activity, thus providing strong evidence for TME study using ssGSEA. We also analyzed the tumor-infiltrating immune cells in the TME and measured the proportion of immune cells and cell types in different immunity groups. However, more related explorations should be conducted in future studies.
We also explored the relationship between gene expression and the immunity group. Engineered T-cell immunotherapy against HLA-A*02:01-restricted AFP peptide-specific T cell receptor (TCR) has shown encouraging results in in vitro experiment to induce anti-HCC response.23 Previous study has also found that HLA class I highly expressed in cancer cells compared with the surrounding normal tissue cells in early-stage solid cancers (including HCC), which may lead to a more sensitive anti-tumor effect of cytotoxic T lymphocyte (CTL)-based cancer immunotherapy.24 HCC cells strongly upregulated HLA class I but remained negative for HLA class II and PD-L1 was expressed in cells with monocyte-macrophage morphology that primarily located at the tumor margins, but not in the tumor cells.25 Similarly, studies have also reported that CD8+ CTL significantly upregulated PD-L1 expression in tumor cell lines through HLA class I specificity.26 The higher the population mutation frequency in TCGA tumor patients, the weaker the ability of HLA antigens to present the mutant peptides, thereby enhancing immune escape of tumor clones carrying mutations.27 In view of aforementioned studies, we can infer that HLA is a potential biomarker in tumor immunotherapy in addition to tumor mutational burden (TMB). Therefore, we explored the relationship between HLA-related gene expression and the different immunity groups. We found that 23 HLA-related genes were significantly different in different immunity groups. This may provide crucial information for further in-depth investigations.
The programmed cell death protein 1 (PD-1) pathway has received huge attention due to its role in eliciting the immune checkpoint response of the T cells. HCC-specific Tfh failure may be caused by elevated PD-1 and PD-L1 signals.28 In addition, in HCC patients with hepatitis B virus (HBV) infection, the upregulation of circulating PD-L1/PD-1 was associated with poor prognosis after cryoablation.29 CTLA-4 has important therapeutic effects on transplant rejection and various autoimmune diseases. Studies have found that blocking CTLA-4 can reveal tumor-associated antigen-specific immune responses by altering cytokines.30 Previous studies have also provided evidence of the association of enhanced regulatory T cells (Tregs) activity with poor immune responses to tumor antigens.31 Moreover, the elevated levels of DcR3, Foxp3 and CTLA-4 in tissue were positively correlated with tumor growth.32 Anti-CTLA-4 mAb can improve anti-tumor immunity by abrogating tumor-infiltrating Treg-mediated suppression in HCC.33 Meanwhile, we explored the differential expression of PD-L1 and CTLA-4 in different immunity groups and found that PD-L1 and CTLA-4 expression increased with the increase of immune activity.
We finally analyze the function of the identified differential proteins among different immunity groups. KEGG pathway analysis revealed that differential proteins were mainly enriched in ALA acid metabolism, cytokine-cytokine receptor interaction, glycosaminoglycan biosynthesis-heparan sulfate/heparin, glycosphingolipid biosynthesis-ganglio series and proteasome. ALA is an essential polyunsaturated fatty acid that can mediate mitochondrial apoptosis, reduce the microenvironment of hypoxia and inhibit the de novo synthesis of fatty acids, thereby conferring anticancer effects.34 Moreover, ALA can reduce the growth of prostate tumors, reduce histopathological processes and increase survival rates.35 A previous study also showed that ALA can effectively inhibit HER2+ overexpression in breast cancer.36 Cytokines can interact with each other by regulating the expression of the cell surface receptors. Studies have found that the interaction of cytokines with cytokines is involved in the assembly of the dodecyl complex, thereby linking cytokine binding to receptor activation.37 Glycosaminoglycans are part of a dynamic extracellular matrix (ECM) network that control important biochemical and biomechanical signals required for tissue morphogenesis, differentiation, homeostasis, and cancer development.38 Therefore, we suppose that the differentially expressed genes identified in our study may play a critical role in the metabolism signaling pathway. However, more researches should be conducted to ratify this proposition. Nonetheless, this study has some limitations. Firstly, our results have not been validated in clinical samples. Secondly, our results do not provide accurate clinical data due to the relatively small number of patients. Hence, more comprehensive studies with large sample sizes should be conducted to validate these results and promote the development of novel strategies for precision cancer medicine.
Conclusion
Our findings provide a deeper understanding of the immune scene, uncovering remarkable immune infiltration patterns of various subtypes of HCC using ssGSEA. This study advances the understanding of immune response and provides a basis for research to enhance immunotherapy.
Supplemental Material
Supplemental material, sj-xlsx-1-iji-10.1177_20587384211018389 for Single sample scoring of hepatocellular carcinoma: A study based on data mining by Dan Zhu, Zeng-Hong Wu, Ling Xu and Dong-Liang Yang in International Journal of Immunopathology and Pharmacology
Footnotes
Author contributions: ZHW and DZ designed and analyzed the research study; DZ, ZHW and LX wrote and revised the manuscript; ZHW, DZ and DLY collected the data and all authors contributed to and approved the final version of manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Availability: The datasets analyzed in the current study are available in the TCGA repository (http://cancergenome.nih.gov/).
ORCID iDs: Zeng-Hong Wu  https://orcid.org/0000-0003-4977-6478
https://orcid.org/0000-0003-4977-6478
Dong-Liang Yang
https://orcid.org/0000-0002-5923-4560
Supplemental material: Supplemental material for this article is available online.
References
- 1. Torre LA, Bray F, Siegel RL, et al. (2015) Global cancer statistics, 2012. CA: A Cancer Journal for Clinicians 65(2): 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Tang Y, Wang H, Ma L, et al. (2016) Diffusion-weighted imaging of hepatocellular carcinomas: A retrospective analysis of correlation between apparent diffusion coefficients and histological grade. Abdominal Radiology (New York) 41(8): 1539–1545. [DOI] [PubMed] [Google Scholar]
- 3. Coskun M. (2017) Hepatocellular carcinoma in the cirrhotic liver: Evaluation using computed tomography and magnetic resonance imaging. Experimental and Clinical Transplantation: Official Journal of the Middle East Society for Organ Transplantation 15(Suppl. 2): 36–44. [DOI] [PubMed] [Google Scholar]
- 4. Lang H, Sotiropoulos GC, Brokalaki EI, et al. (2007) Survival and recurrence rates after resection for hepatocellular carcinoma in noncirrhotic livers. Journal of the American College of Surgeons 205(1): 27–36. [DOI] [PubMed] [Google Scholar]
- 5. Jiao Y, Fu Z, Li Y, et al. (2018) High EIF2B5 mRNA expression and its prognostic significance in liver cancer: A study based on the TCGA and GEO database. Cancer Management and Research 10: 6003–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Lope CR, Tremosini S, Forner A, et al. (2012) Management of HCC. Journal of Hepatology 56(Suppl. 1): S75–S87. [DOI] [PubMed] [Google Scholar]
- 7. Shen Y, Bu L, Li R, et al. (2017) Screening effective differential expression genes for hepatic carcinoma with metastasis in the peripheral blood mononuclear cells by RNA-seq. Oncotarget 8(17): 27976–27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu T, Dai Y. (2017) Tumor microenvironment and therapeutic response. Cancer Letters 387: 61–68. [DOI] [PubMed] [Google Scholar]
- 9. Pitt JM, Marabelle A, Eggermont A, et al. (2016) Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Annals of Oncology 27(8): 1482–1492. [DOI] [PubMed] [Google Scholar]
- 10. Xiong Y, Wang K, Zhou H, et al. (2018) Profiles of immune infiltration in colorectal cancer and their clinical significant: A gene expression-based study. Cancer Medicine 7(9): 4496–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varun SN, Toor SM, Taha RZ, et al. (2018) DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clinical Epigenetics 10(1): 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vougiouklakis T, Bao R, Nakamura Y, et al. (2017) Protein methyltransferases and demethylases dictate CD8+ T-cell exclusion in squamous cell carcinoma of the head and neck. Oncotarget 8(68): 112797–112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chevrier S, Levine JH, Zanotelli VRT, et al. (2017) An immune atlas of clear cell renal cell carcinoma. Cell 169(4): 736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang S, Zhang E, Long J, et al. (2019) Immune infiltration in renal cell carcinoma. Cancer Science 110(5): 1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman AM, Liu CL, Green MR, et al. (2015) Robust enumeration of cell subsets from tissue expression profiles. Nature Methods 12(5): 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Charoentong P, Finotello F, Angelova M, et al. (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Reports 18(1): 248–262. [DOI] [PubMed] [Google Scholar]
- 17. Subramanian A, Tamayo P, Mootha VK, et al. (2005) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102(43): 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu B, Lin JZ, Yang XB, et al. (2020) Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Proliferation 53(3): e12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yin Z, Dong C, Jiang K, et al. (2019) Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. Journal of Hematology & Oncology 12(1): 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ai J, Ketabchi N, Verdi J, et al. (2019) Mesenchymal stromal cells induce inhibitory effects on hepatocellular carcinoma through various signaling pathways. Cancer Cell International 19: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu C, Li L. (2019) The immunoregulation of mesenchymal stem cells plays a critical role in improving the prognosis of liver transplantation. Journal of Translational Medicine 17(1): 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshihara K, Shahmoradgoli M, Martínez E, et al. (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications 4(1): 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen H, Shao HW, Chen XH, et al. (2013) Identification of a novel HLA-A2-restricted mutated Survivin epitope and induction of specific anti-HCC CTLs that could effectively cross-recognize wild-type Survivin antigen. Cancer Immunology Immunotherapy 62(2): 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akazawa Y, Nobuoka D, Takahashi M, et al. (2019) Higher human lymphocyte antigen class I expression in early-stage cancer cells leads to high sensitivity for cytotoxic T lymphocytes. Cancer Science 110(6): 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramia E, Chiaravalli AM, Bou Nasser Eddine F, et al. (2018) CIITA-related block of HLA class II expression, upregulation of HLA class I, and heterogeneous expression of immune checkpoints in hepatocarcinomas: Implications for new therapeutic approaches. Oncoimmunology 8(3): 1548243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang CY, Wang Y, Luo GY, et al. (2017) Relationship between PD-L1 expression and CD8+ T-cell immune responses in hepatocellular carcinoma. Journal of Immunotherapy 40(9): 323–333. [DOI] [PubMed] [Google Scholar]
- 27. Marty R, Kaabinejadian S, Rossell D, et al. (2017) MHC-I genotype restricts the oncogenic mutational landscape. Cell 171(6): 1272–1283.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou ZQ, Tong DN, Guan J, et al. (2016) Follicular helper T cell exhaustion induced by PD-L1 expression in hepatocellular carcinoma results in impaired cytokine expression and B cell help, and is associated with advanced tumor stages. American Journal of Translational Research 8(7): 2926–2936. [PMC free article] [PubMed] [Google Scholar]
- 29. Zhen Z, Feng S, Lin Z, et al. (2011) Upregulation of circulating PD-L1/PD-1 is associated with poor post-cryoablation prognosis in patients with HBV-related hepatocellular carcinoma. PLoS One 6(9): e23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizukoshi E, Nakamoto Y, Arai K, et al. (2011) Comparative analysis of various tumor-associated antigen-specific T-cell responses in patients with hepatocellular carcinoma. Hepatology 53(4): 1206–1216. [DOI] [PubMed] [Google Scholar]
- 31. Thakur S, Singla A, Chawla Y, et al. (2011) Expansion of peripheral and intratumoral regulatory T-cells in hepatocellular carcinoma: A case-control study. Indian Journal of Pathology and Microbiology 54(3): 448–453. [DOI] [PubMed] [Google Scholar]
- 32. Chen C, Zhang C, Zhuang G, et al. (2008) Decoy receptor 3 overexpression and immunologic tolerance in hepatocellular carcinoma (HCC) development. Cancer Investigation 26(10): 965–974. [DOI] [PubMed] [Google Scholar]
- 33. Pedroza-Gonzalez A, Zhou G, Singh SP, et al. (2015) GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology 4(12): e1051297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy S, Rawat AK, Singh M. (2017) Alpha-linolenic acid stabilizes HIF-1α and downregulates FASN to promote mitochondrial apoptosis for mammary gland chemoprevention. Oncotarget 8(41): 70049–70071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander W. (2013) Prostate cancer risk and omega-3 fatty acid intake from fish oil. P & T: A Peer-Reviewed Journal for Formulary Management 38: 561–564. [PMC free article] [PubMed] [Google Scholar]
- 36. MacLennan M, Ma DW. (2010) Role of dietary fatty acids in mammary gland development and breast cancer. Breast Cancer Research: BCR 12: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dey R, Ji K, Liu Z, et al. (2009) A cytokine–cytokine interaction in the assembly of higher-order structure and activation of the interleukine-3: Receptor complex. PLoS One 4: e5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Afratis NA, Karamanou K, Piperigkou Z, et al. (2017) The role of heparins and nano-heparins as therapeutic tool in breast cancer. Glycoconjugate Journal 34(3): 299–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-xlsx-1-iji-10.1177_20587384211018389 for Single sample scoring of hepatocellular carcinoma: A study based on data mining by Dan Zhu, Zeng-Hong Wu, Ling Xu and Dong-Liang Yang in International Journal of Immunopathology and Pharmacology